DOI:
10.1039/D3AN00336A
(Paper)
Analyst, 2023,
148, 3584-3593
Infrared nanospectroscopy depth-dependent study of modern materials: morpho-chemical analysis of polyurethane/fibroin binary meshes†
Received
3rd March 2023
, Accepted 26th June 2023
First published on 29th June 2023
Abstract
Infrared scattering-type scanning near-field optical microscopy (IR s-SNOM) and imaging is here exploited together with attenuated total reflection (ATR) IR imaging and scanning electron microscopy (SEM) to depict the chemical composition of fibers in hybrid electrospun meshes. The focus is on a recently developed bio-hybrid material for vascular tissue engineering applications, named Silkothane®, obtained in the form of nanofibrous matrices from the processing of a silk fibroin-polyurethane (SFPU) blend via electrospinning. Morphology and chemistry of single fibers, at both surface and subsurface level, have been successfully characterized with nanoscale resolution, taking advantage of the IR s-SNOM capability to portray the nanoscale depth profile of this modern material working at diverse harmonics of the signal. The applied methodology allowed to describe the superficial characteristics of the mesh up to a depth of about 100 nm, showing that SF and PU do not tend to co-aggregate to form hybrid fibers, at least at the length scale of hundreds of nanometers, and that subdomains other than the fibrillar ones can be present. More generally, in the present contribution, the depth profiling capabilities of IR s-SNOM, so far theoretically predicted and experimentally proven only on model systems, have been corroborated on a real material in its natural conditions with respect to production, opening the room for the exploitation of IR s-SNOM as valuable technique to support the production and the engineering of nanostructured materials by the precise understanding of their chemistry at the interface with the environment.
1. Introduction
Tissue engineering (TE) consists in conjugating principles and methods of engineering and life sciences in order to explore the relationships between the morphology and the function of new designed materials in physiological and pathological tissues and to develop constructs that aim at restoring, maintaining and improving biological function by mimicking native structures.1In situ TE is gaining attention over the in vitro approach due to its undeniable advantages, mostly the possibility to fabricate “off-the-shelf” devices which do not require cellular seeding, then reducing manufacturing times and costs.2 Besides being highly biocompatible, implanted scaffolds are generally made of totally or partially degradable materials able to guide and control cellular recruitment, adhesion and proliferation to obtain a functional endogenous living tissue while progressively reducing their structural and mechanical properties.3 Composite materials, also known as hybrid materials, which are composed of two or more constituents, are good candidates for TE applications since they combine the strengths of each one maximizing devices performances from the implantation phase until its absorption. Indeed, a clever design and the choice of the most suitable materials allow to conjugate the benefits of both natural and synthetic polymers, thus obtaining scaffolds with biological functionality and adequate mechanical properties.4–6
In other words, the engineering process requires the controlled tuning of morphology and physico-chemical properties of the hybrid material, for enhancing its functionality and biocompatibility for TE applications. The extensive development over the last decades of nanotechnological approaches for both production and characterization of new materials offered the opportunity to develop high performance materials with functionalities tunable down to the molecular level.
As a consequence, characterization techniques resolved at the nanoscale are highly demanded and constantly evolving. While nano-resolved microscopies, as electron and atomic force microscopies, have been widely demonstrated to be extremely useful for morphological characterization, the nano-resolved chemical analysis represents a more complex goal to be achieved. Indeed, routine methodologies used for nanocomposites chemical characterization, i.e. conventional spectroscopies, do not typically reach the spatial resolution needed for single nano-fiber detection and could only look at composites as quasi-homogeneous materials.7,8 In the present study, we propose the use of nano-resolved IR microscopy, namely infrared scattering-type scanning near field optical microscopy (IR s-SNOM), supported by micro Attenuated Total Reflection (ATR) Fourier Transform Infrared (FTIR) imaging, to address the study of a binary fibrous material named Silkothane®.9 In Silkothane® the combination of natural silk fibroin (SF) and synthetic medical grade polyurethane (PU) proved to be highly suitable for TE applications.5,6 Of note, SF is a biodegradable and biocompatible protein with remarkable mechanical properties (i.e. stiffness), while PU is characterized by good deformability in response to external solicitations, proving to be suitable for multiple applications, e.g. vascular prostheses7,10 and cardiac patches.8,11 Silkothane® is obtained in the form of nanofibrous matrices from the processing of a SFPU blend via electrospinning.12,13 The latter is an innovative technology based on the application of high voltages to draw a charged solution from a nozzle to a grounded target, thus obtaining fibrous meshes that resemble the morphology of biological extracellular matrices. The thorough functional characterization performed on Silkothane® demonstrated that this composite material balances the advantages of SF and PU simultaneously displaying an excellent biocompatibility, ascribable to the natural component, and intermediate mechanical properties given by the presence of PU, that enhances the deformability of the construct.12,13 van Uden et al.12,13 previously highlighted that Silkothane® is composed of a nano-fibrous structure with a randomized arrangement of fibers. Nevertheless, since routine techniques did not allow to conduct any spatially resolved chemical characterization at fiber level, the chemical nature of the individual fibers remains elusive and the deep comprehension of how the different compounds arrange within meshes is still debated. Here we present the analyses of a Silkothane® nanofibrous matrix obtained from the blend of SF and PU. The chemistry of the material and the composition of single nano-fibers is investigated with the specific aim to demonstrate whether and to which extent the two compounds in the mixture separate during electrospinning. To this purpose, the analyses were compared to the data collected on a control matrix, where fibers are unequivocally made of pure SF or pure PU by production, since two different nozzles were used to simultaneously and separately spin the two compounds. The chemical characterization by FTIR ATR imaging and IR s-SNOM allowed to depict physio-chemical details from micro down to the nanoscale. Indeed, micro-ATR imaging provides details about the spatial and chemical distribution of the component materials over a wide area of the sample down to the microscale with a penetration depth in the range of few hundreds of nanometers up to a few micrometers.14,15 Nevertheless, micro-ATR presents some drawbacks on the analysis of these fibrous materials: first the penetration depth of the evanescent field can be larger than the average wire diameter and then, the pressure applied onto the sample by the tip of the internal refractive element can induce the deformation of the wires. With IR nano-spectroscopic and imaging analyses, characterised by a lateral resolution around 20 nm, it was possible to reveal nanoscopic details of the fibers and the chemistry beneath their surface. As well-known, IR s-SNOM enables high-resolution spectroscopy and imaging that couples morphological details with chemical-vibrational ones in a non-destructive way.16–19 For the purpose of this work, the profiling capabilities of IR s-SNOM have been exploited, comparing nano-IR maps at the second (O2) and third (O3) harmonic of the optical signal, providing diverse probing depths, up to around 100 nm below the sample surface20,21 with O3 being shallower, more related to the surface of the sample, while O2 containing chemical information from depths up to hundred nanometers. Nevertheless this process is not free of limitations since the signal to noise decreases as well with the probing depth. This peculiarity, which will be discussed extensively in the manuscript, is especially relevant to access volumetric information of the superficial corona on untreated samples, avoiding complex preparation protocols necessary for cross-sectional spectroscopy and imaging21 and providing, to the best of our knowledge, the very first correlated morpho-chemical characterization of this class of materials.
2. Materials and methods
2.1. Mesh preparation
2.1.1. Materials.
Silk Bombyx mori cocoons were supplied by the Council of Research and Experiments in Agriculture, Apiculture and Sericulture Unit (CREA-API; Padua, Italy) and processed as previously described to obtain regenerated SF films.12 Commercial poly-carbonate-urethane pellets (Carbothane Aromatic, AC-4075A) were provided by Lubrizol (Wickliffe, OH, United States), and formic acid (FA) and dichloromethane (DCM) by Carlo Erba Reagents (Cornaredo, Italy).
2.1.2. Electrospinning.
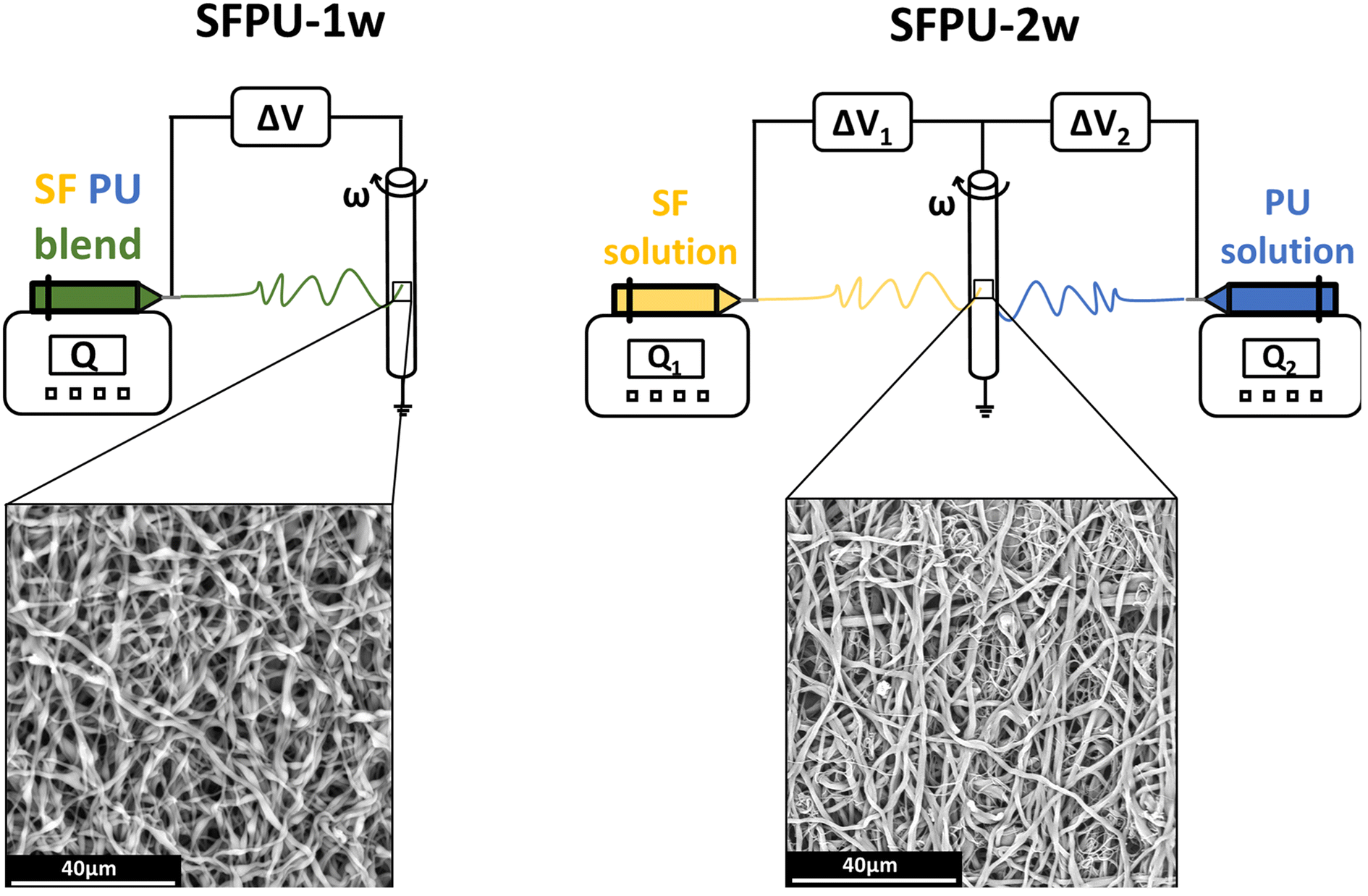
Two meshes of Silkothane® were manufactured through two different electrospinning methods according to the parameters reported in Table 1. The first method involved the one-wire electrospinning of a SF/PU blend (SFPU-1w) (Fig. 1, left). Initially, SF was dissolved in (5% w/v) and PU in a mixture of FA and DCM (3% w/v) with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 volume ratio, stirred separately for 1 hour. A resulting blend (4% w/v) of SF and PU (1:1) in FA and DCM (3:2) was obtained by merging SF and PU solutions and stirring them for 15 additional minutes. The Silkothane® blend was processed via electrospinning on a tubular (6 mm in diameter) rotating target, setting the process parameters as described in Table 1.12 The second manufacturing method, used to produce the SFPU two-wire mesh (SFPU-2w), dealt with the electrospinning of SF and PU as two independent wires (Fig. 1, right). SF was dissolved in FA (7% w/v) and PU in a mixture of FA and DCM (3% w/v) with 1:2 volume ratio, stirred for 1 hour each. These two solutions were simultaneously electrospun on the same tubular (6 mm in diameter) rotating collector by means of two mirrored setups, setting process parameters as described in Table 1. Once removed from the collectors, both tubular matrices underwent a crystallisation process into five descending series of ethanol and demineralized water according to the standard protocol.12 Both samples were then vacuum-dried and sterilised with ethylene oxide.
:2 volume ratio, stirred separately for 1 hour. A resulting blend (4% w/v) of SF and PU (1:1) in FA and DCM (3:2) was obtained by merging SF and PU solutions and stirring them for 15 additional minutes. The Silkothane® blend was processed via electrospinning on a tubular (6 mm in diameter) rotating target, setting the process parameters as described in Table 1.12 The second manufacturing method, used to produce the SFPU two-wire mesh (SFPU-2w), dealt with the electrospinning of SF and PU as two independent wires (Fig. 1, right). SF was dissolved in FA (7% w/v) and PU in a mixture of FA and DCM (3% w/v) with 1:2 volume ratio, stirred for 1 hour each. These two solutions were simultaneously electrospun on the same tubular (6 mm in diameter) rotating collector by means of two mirrored setups, setting process parameters as described in Table 1. Once removed from the collectors, both tubular matrices underwent a crystallisation process into five descending series of ethanol and demineralized water according to the standard protocol.12 Both samples were then vacuum-dried and sterilised with ethylene oxide.
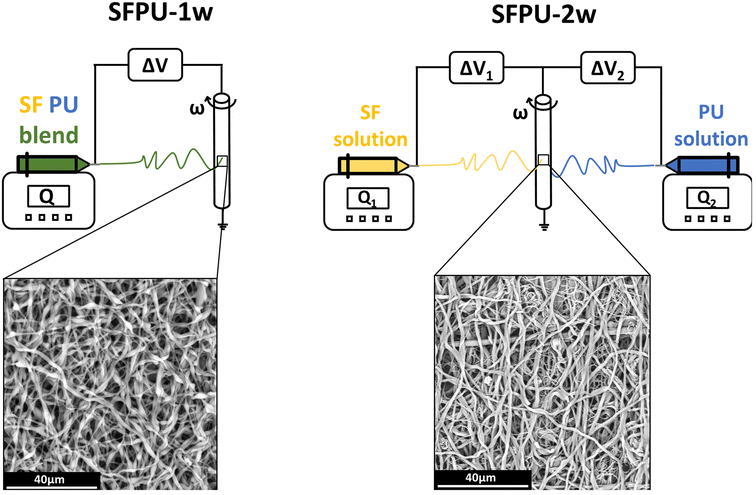 |
| | Fig. 1 Schematic representation of the manufacturing methods adopted to prepare SFPU-1w and SFPU-2w meshes, and corresponding morphologies observed at SEM. On the left, SFPU-1w is obtained by electrospinning a SF/PU blend (1:1 w:w); on the right, SFPU-2w is obtained by simultaneously electrospinning pure SF and pure PU solutions. | |
Table 1 Electrospinning process parameters set to manufacture SFPU-1w and SFPU-2w meshes. ΔV: applied voltage, Q: solution flow-rate, ω: mandrel rotational speed
| |
Material |
ΔV (kV) |
Q (ml h−1) |
ω (rpm) |
| SFPU-1w |
SF/PU blend |
17 |
2.5 |
1333 |
| SFPU-2w |
SF |
24 |
1.5 |
500 |
| PU |
19 |
3.5 |
500 |
2.2. SEM imaging
SEM imaging was performed on 5 × 5 mm2 samples extracted from each tubular matrix, respectively. Squared samples were subject to metallization procedure via vapour sputtering to prevent fiber movement due to electric repulsion, and to obtain images with high contrast. The vapour sputtering was performed for 105 s by means of the SC7620 Mini Sputter Coater (Quorum, UK), using a leak current of 18 mA, Argon as the inert conductive mean and Gold as the coating element. Subsequently, SEM analyses were carried out using the PhenomProX Desktop SEM from Thermo Fisher Scientific (Waltham, Massachusetts, USA) setting an acceleration voltage of 10–15 kV in backscattered electron detector (BSD) modality.
2.3. Micro-ATR imaging
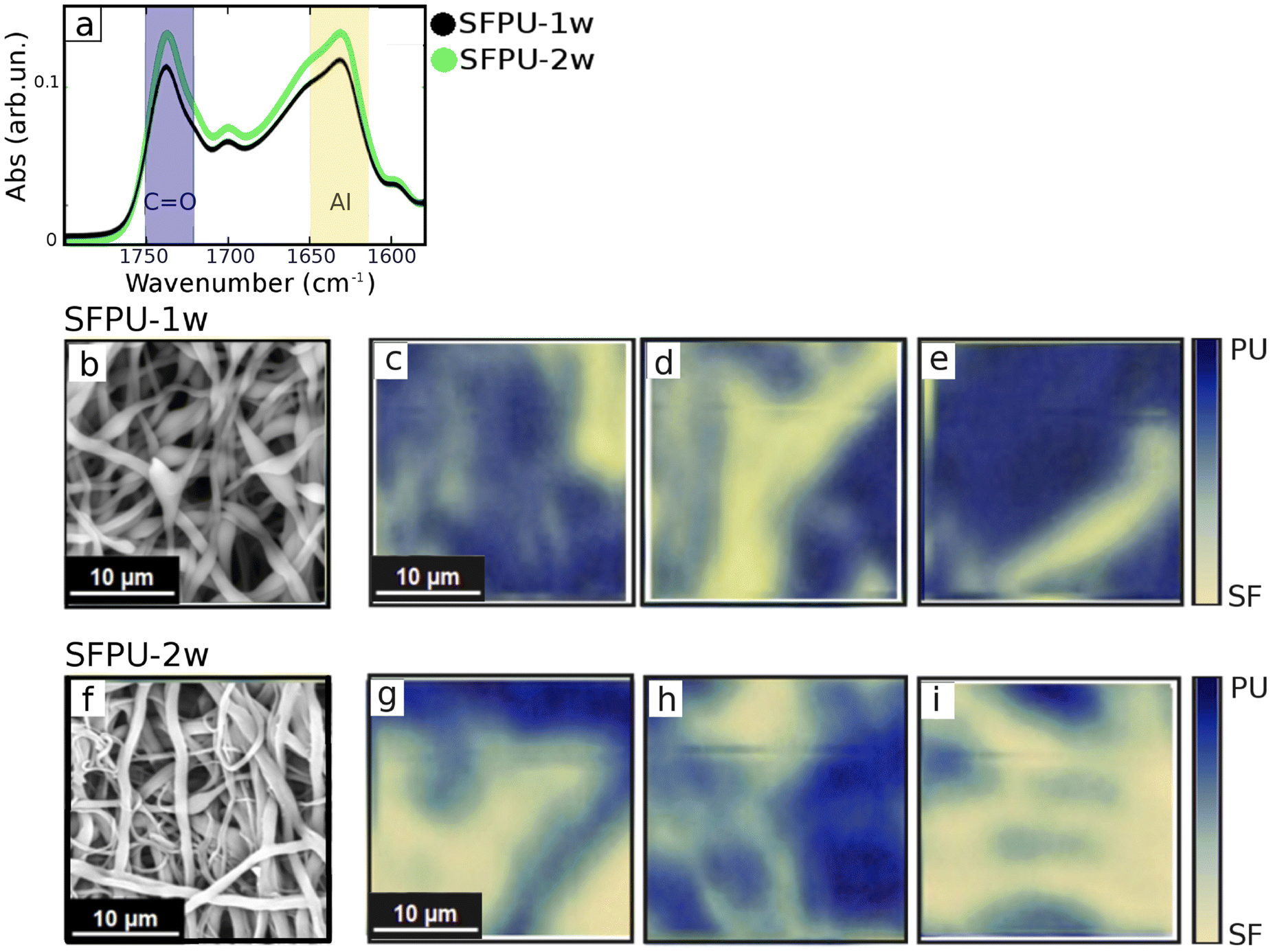
For micro-ATR analysis small fragments of samples, 5 × 5 mm2, were glued on a clean silicon wafer and measured without any further treatment. Micro-ATR imaging measurements were performed at the Chemical and Life Sciences branch of SISSI beamline (SISSI-Bio) at Elettra Sincrotrone Trieste22 with a Bruker Vertex 70v interferometer, coupled to the Hyperion3000 microscope and the Focal Plane Array (FPA) detector (64 × 64 pixels). A micro-ATR objective equipped with a germanium crystal 100 μm in diameter was used. Measurements were performed in the region between 4000 and 1000 cm−1 and more than 50 maps were acquired for each matrix at 2.5 kHz. 256 scans were accumulated and averaged. Data analysis was performed with Opus 8.5 (Bruker). Absorbance was calculated by using air as a background. Spectra were baseline corrected and normalized to the full absorption range. We considered distinctive signals for the two compounds, Amide I of β-sheet structures (peaked at 1630 cm−1 (ref. 23 and 24)) for SF and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode (peaked at 1730 cm−1) for PU. Color maps were built by superimposing the integrated SF (1645–1615 cm−1) and PU (1745–1715 cm−1) absorption as follows: the edge colors of the maps representing 0% and 100% of SF, or PU, were evaluated by considering the maximum and minimum integrated SF (1615–1645 cm−1) and PU (1745–1715 cm−1) absorption found on a set of 16 different maps. The 50% values for both maps, defining the level at which SF and PU are present with same extent, were set to the values found for average spectra, plotted in Fig. 2a, obtained averaging all the micro-ATR images for each sample.
O stretching mode (peaked at 1730 cm−1) for PU. Color maps were built by superimposing the integrated SF (1645–1615 cm−1) and PU (1745–1715 cm−1) absorption as follows: the edge colors of the maps representing 0% and 100% of SF, or PU, were evaluated by considering the maximum and minimum integrated SF (1615–1645 cm−1) and PU (1745–1715 cm−1) absorption found on a set of 16 different maps. The 50% values for both maps, defining the level at which SF and PU are present with same extent, were set to the values found for average spectra, plotted in Fig. 2a, obtained averaging all the micro-ATR images for each sample.
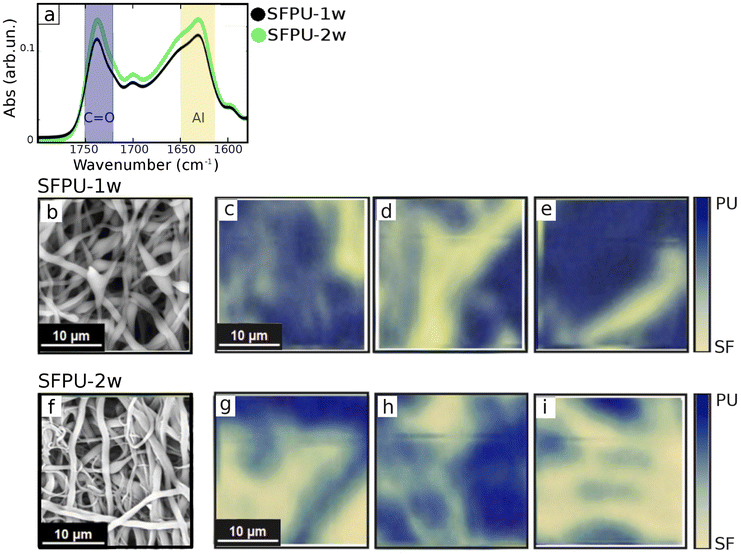 |
| | Fig. 2 (a) Average IR absorbance of SFPU-1w and SFPU-2w in the spectral region of Amide I absorption of SF (yellow shadowed) and CO bonds of PU (blue shadowed). SEM image (b) and micro-ATR two-color maps (c–e) of SFPU-1w. SEM image (f) and micro-ATR two-color maps (g–i) of SFPU-2w. micro-ATR imaging maps were reconstructed by plotting in false colors the integrated absorption of Amide I band (representative for SF) and carbon double bond CO (representative for PU) as described in micro-ATR imaging section. | |
2.4. IR s-SNOM spectroscopy and imaging
The same samples prepared for micro-ATR imaging were measured with the IR s-SNOM system. Spectroscopy and imaging at the nanoscale were performed at the SISSI-Bio beamline of Elettra Sincrotrone Trieste22 with the IR-neaSCOPE microscope (AttoCube GmbH), equipped with a spectroscopy and an imaging module. The spectroscopy module exploites a Difference Frequency Generation (DFG) laser (FemtoFiber dichro midIR, Toptica Photonics AG) and a Mercury Cadmium Telluride (MCT) detector (Infrared Associates, Inc.). The DFG laser covers the spectral range between 1780 cm−1 and 1280 cm−1 allowing to probe simultaneously Amide I band of SF and CO stretching of PU. For each spectrum acquired, 10 scans were averaged at a spectral resolution of 8 cm−1. The spectral resolution was selected finding a compromise among the spectral resolution needed for discriminating the PU and SF bands of interest and the measurement time. AFM conductive tips, 20 nm wide, were used in tapping mode, at a driving frequency of 268 kHz, with free amplitude of 110 nm and with an approach set point at 80% of the free tapping amplitude. Spectroscopy data was processed with Neaplotter software (Neaspec) by correcting the phase of the signal. IR s-SNOM absorbance was calculated using clean silicon as a reference. Imaging measurements were done with the Neaspec imaging module (equipped with the PseudoHeterodyne, PsHet, detection system), coupled to a quantum cascade laser (QCL, from MIRcat, Daylight Solutions). Two lines of the QCL laser were alternatively used in order to perform measurements at 1628 and 1740 cm−1, with a power of 1 mW. Two colors chemical images were built, similarly to micro-ATR imaging analysis, by considering as edge colors of the maps representing 0% and 100% of SF, or PU, the maximum and minimum integrated SF (at 1628 cm−1) and PU (1740 cm−1) signal found on the maps acquired during a single experimental session (since the absolute value of the nano-IR signal strictly depends on the specific experimental conditions as for instance tip, optical alignment and laser phase, just to mention a few). Unwanted far-field scattering evaluation was done through the following procedure. First, phase discontinuities were corrected when necessary by adding a constant phase to O2 and O3 values. The pixels more affected by scattering were thus identified through comparison of O2 and O3 maps with their linear combination.25 For the images more affected by unwanted scattering effects (Fig. 5c and d), the identified pixels were filtered by applying an erosion filter (performed with 5 points and 50% threshold, Gwyddion 2.53). The mentioned procedure allowed us to identify and reduce unwanted scattering effects and better highlights chemical differences.
3. Results and discussion
Hereafter we present and discuss the study of Silkothane® matrices made by SF and PU, according to the manufacturing methods sketched in Fig. 1, and performed at both micro and nanoscale, aiming to highlight the importance of nano-resolved microscopies to support the design and development of hybrid materials for tissue engineering.
3.1. Morphological and chemical heterogeneity of SFPU-1w and SFPU-2w at the microscale
Fig. 2 shows SEM maps and ATR-FTIR results, obtained on SFPU-1w (a–e) and SFPU-2w (a, f–i) matrices, being the latter a control material, in which fibers are made either of pure SF or pure PU (see Mesh preparation). Average micro-ATR spectra of SFPU-1w and SFPU-2w, shown in Fig. 2a, reveal similar chemical profiles of the two matrices. Therefore, in order to investigate whether fibers are made of SF and PU alone, or a SF/PU mixture, and to give a picture of how the two compounds are arranged along each fiber, we first performed micro-ATR imaging and compared it to what observed with SEM microscopy. SEM microscopy (see Fig. 1 and 2b) shows that the SFPU-1w sample is composed of fibers extending in length beyond the visual field, with lateral size (parallel to the material surface) ranging from 0.2 to 1.2 μm. As it can be perceived by SEM images, and as it will be better highlighted by AFM morphological analysis, the fibers volume can be schematized with cylinders with elliptical section, appearing thus more like ribbons than wires with lateral size larger than transverse one (perpendicular to the material surface). In this sample, the fibers form a porous mesh that displays the same recurrent profile characterized by the presence of swellings. Micro-ATR imaging allowed to probe areas of an extent comparable to SEM imaging but with lower lateral resolution (around 400 nm for the specific materials).14 In micro-ATR maps at Fig. 2c–e and g–i both PU (blue, representing CO absorption peak area) and SF (yellow, representing Amide I absorption peak area) signals are visible. The matrix surface appears quite heterogeneous and the fibrillar structure is spotted by the presence of wire-like features. The same analysis was performed on a control material, i.e. SFPU-2w matrix, in which individual fibers were certainly made of pure SF or pure PU (see Mesh preparation). The SEM image of the SFPU-2w matrix, reported at Fig. 2f, reveals that SFPU-2w has a fibrous porous architecture similar to SFPU-1w and the elliptical section of the fibers is preserved. However, the swellings observed for one-wire matrix are not detectable in the two-wire matrix and, overall, SFPU-2w looks denser than SFPU-1w and composed of two distinct families of fibers differing in diameter. Such phenomenon was expected as intrinsically related to the manufacturing process where SF and PU were electrospun separately (see Mesh preparation for details). Micro-ATR maps of SFPU-2w appear, conversely, very similar to the ones acquired for SFPU-1w. However, by comparing SEM images to micro-ATR maps, it is immediately evident that the latter show, for both matrices, the presence of fibers bigger than the ones observed with SEM. This mismatch between the two techniques is ascribable to the axial pressure applied by the ATR crystal (about 5000 Pa) to the sample surface, that induces the deformation of the fibers that result eventually enlarged and flattened. Despite micro-ATR being, among conventional far-field IR microscopy, the technique that is more used in the study of thin films and surfaces, the compression effect is unavoidable and makes this approach barely informative on the chemistry of the individual fibers.
3.2. Chemical composition of SFPU-1w and SFPU-2w fibers at the nanoscale
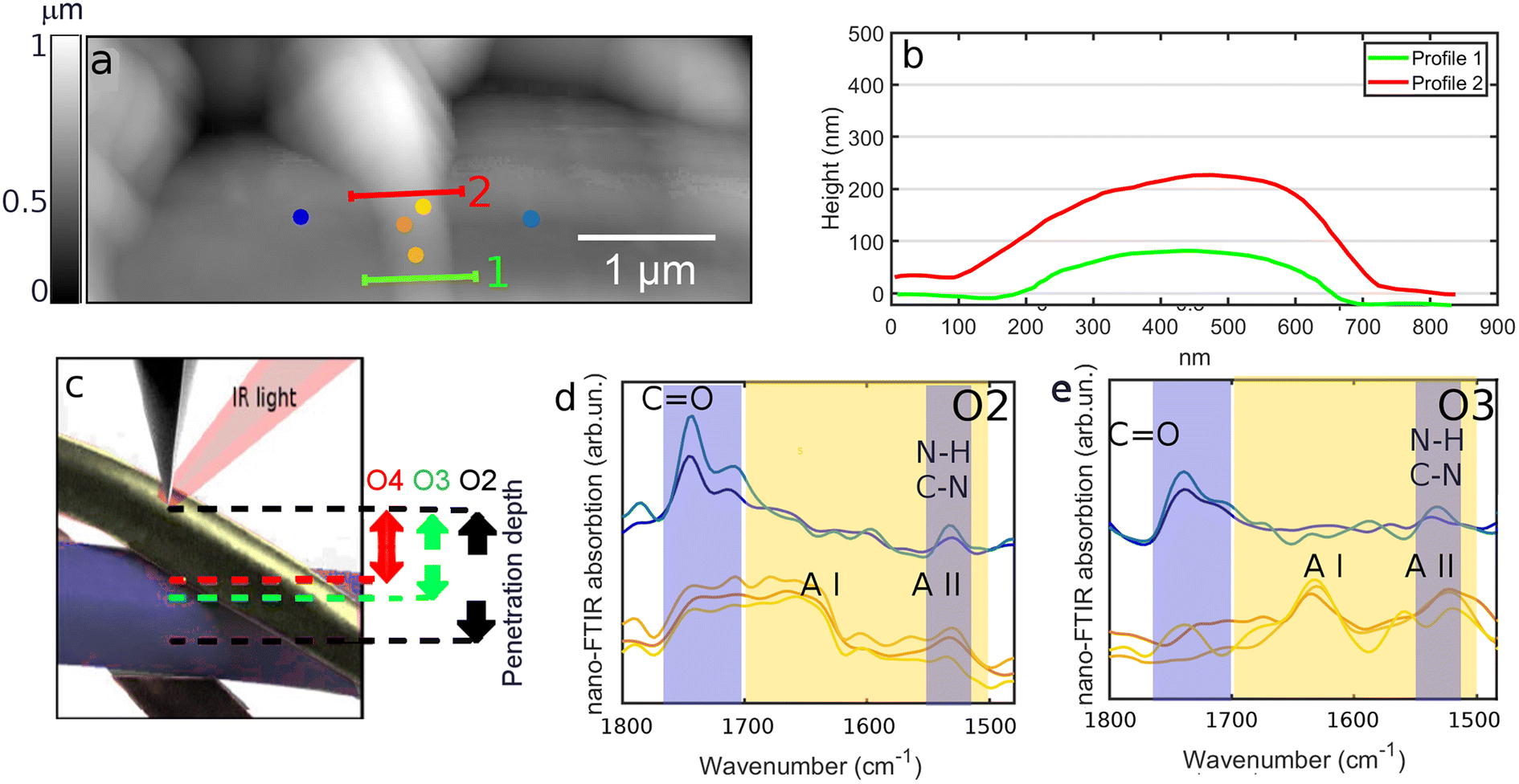
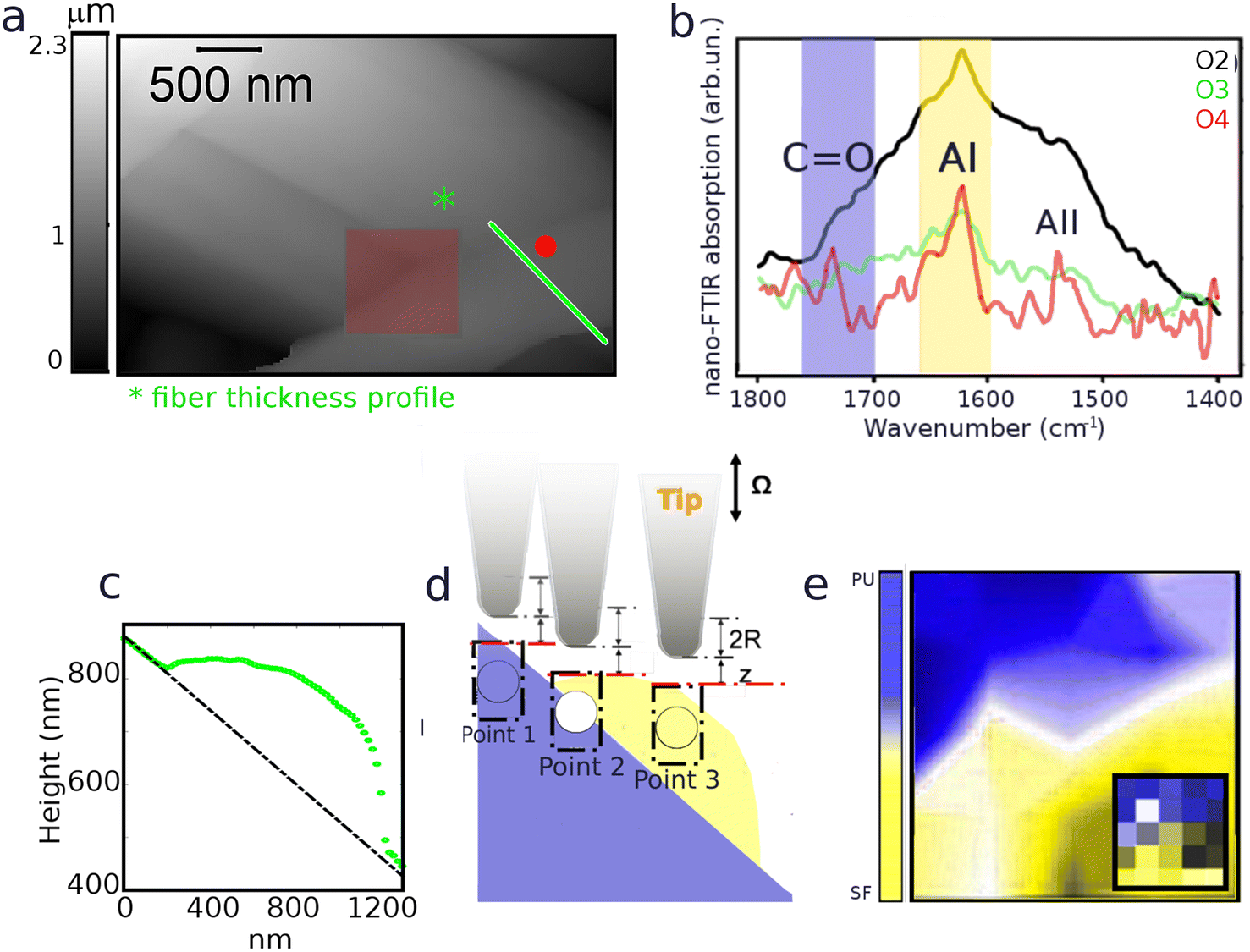
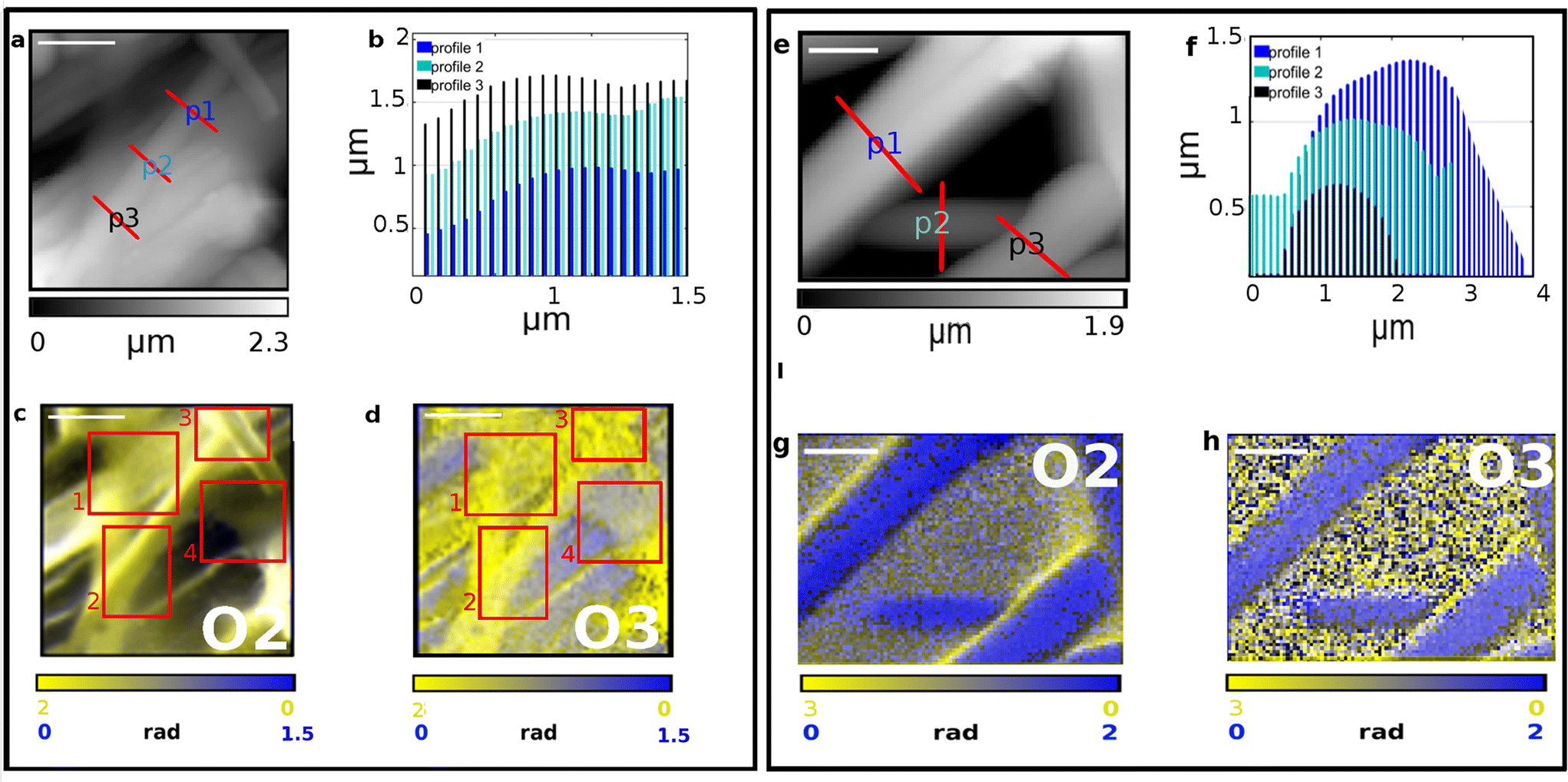
Single fibers chemical composition was investigated by means of IR s-SNOM spectroscopy and PsHet imaging taking advantage of the IR tunable lasers operating in mid infrared range (see section 2 for details). Scattering based IR near-field techniques have indeed limited penetration capabilities and enhanced surface sensitivity allowing the characterization of Silkothane® at the superficial corona, down to a depth of around 100 nm.21,25,26 Moreover, since the signal extraction is performed taking advantage of harmonics demodulation and it is well established from theory that the higher the harmonic order the lower the sample depth reached by the IR s-SNOM signal,25–27 through the observation of different harmonics is possible to acquire information up to different penetration extent in a single measurement. Taking also into account that the deformation induced by the scanning tip is negligible, to the specific purpose of the presented study this is reflected in the possibility; (i) to isolate the chemical signature of thinner fibers, having transverse size in the range of the penetration depth, from the one belonging to the material below them; (ii) to characterize the chemical homogeneity of the surface corona of thicker fibers, with transverse size larger than the maximum reachable penetration, spotting the possible chemical mixing of the two compounds. Indeed, subsurface chemical sensitivity of IR s-SNOM is widely proven for model multi-layer systems26,28 even if yet scarcely observed and exploited on real systems, where the complexity of morphology and chemistry can hamper data acquisition and analysis. In order to tune imaging frequencies and in depth profiling settings we performed a preliminary spectroscopic analysis. To calculate nano-IR absorption, second (O2) and third harmonics (O3) were used here. First harmonic is not typically taken into consideration since it is massively affected by far-field contributions that hide the near-fields signals and hinder a proper data analysis. The results are reported in Fig. 3 and 4. Fig. 3a shows the topography map of a specific region of SFPU-2w, where a fiber with lateral size of about 500 nm and, as shown in transverse profiles at Fig. 3b, with transverse size between 50–150 nm, lays on a bundle of larger fibers. According to the near-field theory, predicting a probing depth for IR s-SNOM 2nd harmonic around 100 nm,21,26,27 comparable to the free tapping amplitude of AFM, the IR s-SNOM measurement on the selected Silkothane® fiber can be schematized as in Fig. 3c: here, while O2 of IR s-SNOM can probe the fiber entirely, in its whole transverse section, and possibly collect signal also from the fiber below, higher order harmonics, can offer the opportunity to suppress the latter contributions, at least for fibers with transverse size around 100 nm or thinner. Therefore we performed a spectroscopic punctual study on the top fiber and on the fibers below it, by collecting the spectra reported in Fig. 3d and e at the exact positions marked with spots of same color in Fig. 3a. As expected, O2 spectra on the top fiber, plotted in yellow in Fig. 3d, are characterized by a broad absorption band extended from 1760 to 1580 cm−1, with shoulders at about 1740 and 1630 cm−1 ascribed to CO of PU and Amide I of SF, and a peak in the region 1580–1520 cm−1, possibly due to Amide II of SF and C–N and N–H groups of PU. Considering O3 signal on the top fiber, distinct peaks appear at Amide I and Amide II band positions (1710–1580 cm−1 and 1580–1480 cm−1). Since the two-wires fabrication strategy produces one-compound fibers, and given the transverse size of the top filament, it is conceivable that the top fiber is made of pure SF, while the PU features observed in O2 derive from the fiber below. Indeed, when spectra are acquired on the fibers in background (spots and spectra reported in blue), PU features are found in O2 and O3 spectra with no contribution of SF Amide bands, suggesting that a PU thicker fiber is localized at the measured positions. Fig. 4 shows the application to SFPU-1w of the developed spectroscopic methodology. The AFM topography map at Fig. 4a highlights one fiber with lateral size of about 1 μm on top of other fibers. The spectra acquired at different harmonics (O2 to O4) at the position marked with a red spot in AFM map, similarly to what observed for SFPU-2w, show that the signals of SF Amide bands are progressively more clearly detectable while going from O2 to O4, while the spectral features associated to PU groups became less prominent until they disappear at the higher harmonic O4. Since the transverse profile of the top fiber, as shown in the profile reported at Fig. 4c measured along the line drawn with same light green color at Fig. 4a is around 100 nm, we associate the spectral differences between O2 and O3 to subsurface absorption, similarly to what observed for SFPU-2w. In order to verify the spatial distribution of the two components of the mesh we performed IR s-SNOM hyper-spectral mapping on the squared region highlighted as red shadowed box in Fig. 4a. The map region has a lateral size of 1 μm and it was sampled as a 4 × 4 matrix of spectra. The chemical map was built through the superposition of Amide I (1740–1580 cm−1) and CO (1750–1710 cm−1) peak areas, highlighted as yellow and blue shadows respectively in Fig. 4b. Taking into consideration the schematic representation of the measurement at Fig. 4d and the chemical map at Fig. 4e, the blue regions in the map correspond to sampled points as point 1 and the yellow regions to measured points as point 3. At those positions pure compounds are detected, respectively PU and SF. The white region corresponds to sampled points as point 2, where the penetration of the measurements allows to sample both top and bottom fibers. Indeed, the transverse profile shows that the fiber has an elliptical section and it is indeed thinner at the lateral edges. Therefore, the white color should not be confused with a mixture of the two components, but instead to subsurface contributions participating to the signal. The map suggests thus that the thin fiber in foreground is made of pure SF and it lays onto PU fibers standing on the background. Based on the results obtained with IR s-SNOM spectroscopy and hyper-spectral mapping we further investigated the chemical nature of fibers through IR s-SNOM PsHet imaging. Imaging was performed by acquiring single frequency IR maps at the spectral position of Amide I and CO peaks, respectively of SF and PU, previously obtained through IR s-SNOM analysis. Fig. 5 and 6 show AFM maps (Fig. 5a, e and 6a, e), with fibers transverse profiles (Fig. 5b, f and 6b, f) and IR s-SNOM phase maps (O2 at Fig. 5c, g and 6c, g and O3 at Fig. 5d, h and 6d, h) respectively, of SFPU-2w and SFPU-1w respectively. Left panels (Fig. 5a–d and 6a–d) show thinner fibers, with transverse size comparable to the penetration depth, while right panels (Fig. 5e–h and 6e–h) show thicker ones. For a sake of simplicity, the right panel of Fig. 5 is first commented, where a region of SFPU-2w characterized by larger fibers is presented. As can be appreciated by the AFM sample morphology in Fig. 5e, and associated p1 to p3 line profiles in Fig. 5f, the fiber's thickness is in the range of several hundreds of nanometers and exceeds the penetration depth of IR s-SNOM. O2 and O3 maps, in Fig. 5g and h respectively, clearly highlight the presence of fibers made by PU up to a depth comparable to the maximum penetration depth of the technique, about 100 nm. Since by production individual SFPU-2w fibers are made by a single component, it is possible to conclude that the imaged fibers are pure PU ones. The maps show also a faint yellowish region in between the PU fibers; the increased contrast in O3 image, proves that this is a thin SF layer, possibly due to a veil of non-fibrillar amorphous SF onto the entire scanned area. It is interesting to notice, at this stage, that the PU wires show a marked yellow contrast at the edges, particularly evident in O2. This result must not be interpreted as a thicker SF layer at the PU fiber edge, while it is associated to a background scattering effect, due to indirect illumination, particularly evident for sample with a complex and uneven topography, as the case of this real matrices.25 To support this interpretation, the correction procedure proposed by Mester et
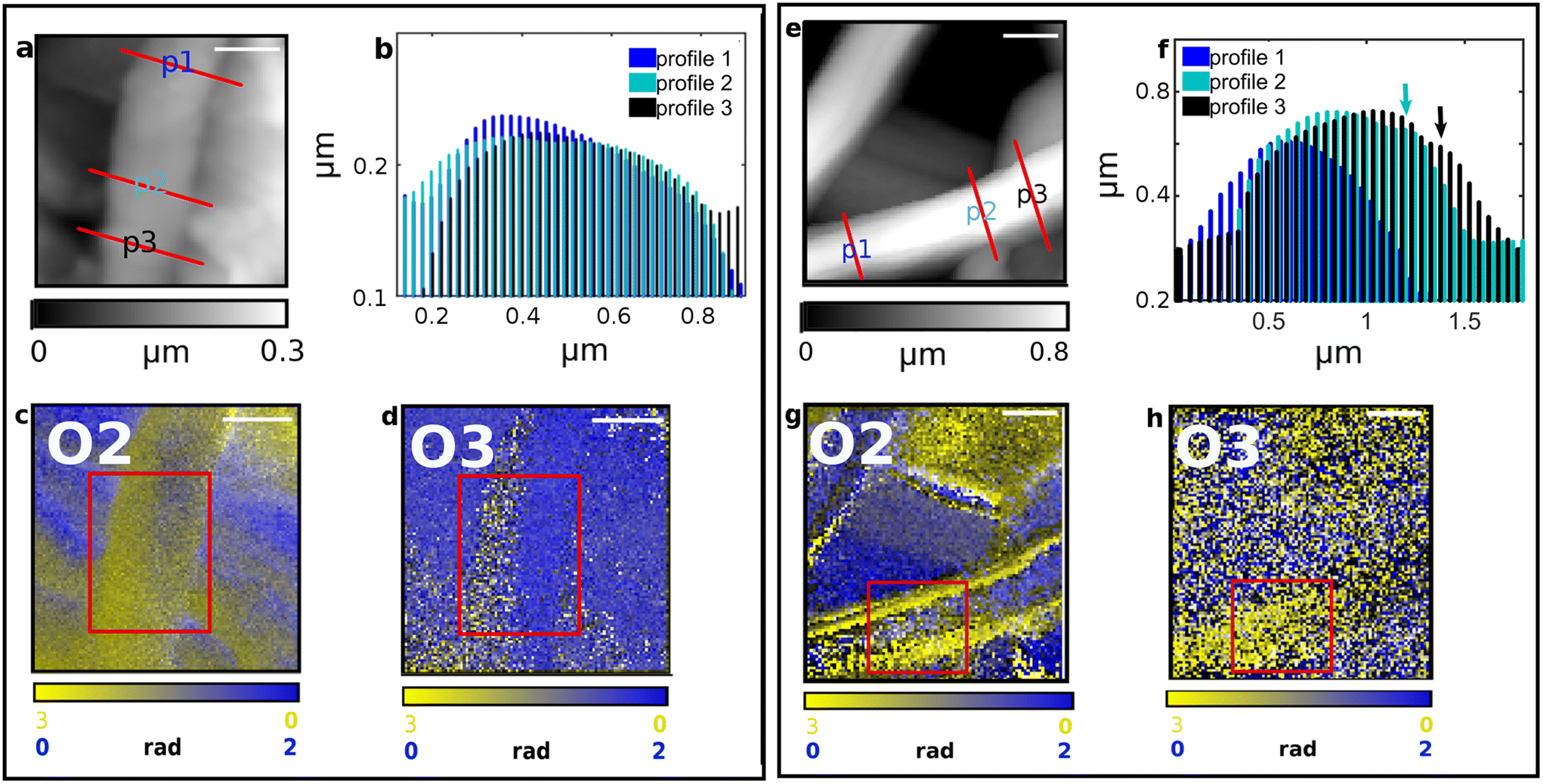
al.25 was applied for obtaining the O3–O2 maps, as reported in Fig. S1,† where the edge effect is much suppressed (more details on the correction algorithm and on its applicability are reported in the Fig. S1† caption). Overall, very similar considerations can be drawn for the SFPU-2w matrix regions characterized by thinner fibers. Fig. 5a shows a region of SFPU-2w, where thin fibers lay on top of other bundled structures. This architecture can be clearly appreciated looking at the central top thin fibers. The transverse p1 to p3 profiles of the latter, marked in Fig. 5a and plotted in Fig. 5b, highlight that the central fiber is around 120 nm thick. The O2 map of the region 1 (i.e.: underlying large bundle) in Fig. 5c shows a clear yellowish region with some gray contributions. In the O3 map of Fig. 5d, the same region appears more homogenous and yellower. Since the sampling depth of O3 is shallower than O2, it is possible to conclude that the large selected bundle is made by individual PU and SF fibers, with the latter layering on top of the PU ones. Conversely, at area 4 in red square, the faint yellowish region may be interpreted as a veil of non-fibrillar amorphous SF, as already commented for the larger fibers. Considering the central top-fiber in the region marked as 2, O2 maps shows a yellowish fiber having a brighter yellow left edge. This result must not be interpreted as a mixed SFPU fiber, that is impossible by construction, while the bright edge is again due to a background scattering effect, also visible in the O3 map of the same region. Due to the thickness of the fiber and the production method, we can conclude that the selected top fiber is a pure SF one. The same conclusion drawn for region 2 can be extended to region 3, where similar bright edges can be observed on the thin selected fiber. For what concerns the SFPU-1w sample, in left panel of Fig. 6a a thin fiber of SFPU-1w is shown with transverse size of about 100 nm, as deduced from transverse profiles at Fig. 5b. O2 map at Fig. 6c shows that, while the left side of the top fiber is mainly composed of SF, in the right side a detectable extent of PU is also found. Since reducing the penetration depth, i.e. by observing O3 map, the image contrast of the left side does not change while the right side became bluer, we can assume that the top fiber is likely composed of two wires assembled along their longitudinal axis, with PU at the right side and SF at the left side. Most of the images acquired on thin fibers of SFPU-1w showed single compound domains with SF and PU fibers entangled together along their longitudinal axis, similar to the fibers just described, supporting the hypothesis by Zhang et al.5 that the two compounds do not aggregate homogeneously upon electrospinning as a blend. For thicker fibers of SFPU-1w, shown at Fig. 6e with transverse profiles at Fig. 6f, we observed at O2 fibers with high content of SF at the lateral edges, all along the longitudinal axis, and a PU rich region in the inner part. For these structures, the map acquired at lower penetration depth, i.e. the O3 map, revealed that SF is not present only at the edges by also on top of the fiber. This situation is consistent with a core–shell fiber. However, such structures were present to a minor extent in the sample. All in all, the analysis of SFPU-1w highlights that the matrix is mainly composed of one-compound SF and PU thin fibers boundled together along the longitudinal axis and of core–shell structures with a corona made of PU covered with SF not mixing together.
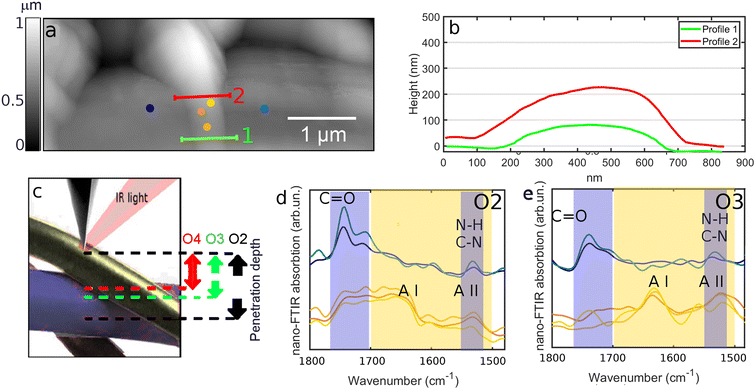 |
| | Fig. 3 (a) AFM topography map acquired on SFPU-2w. (b) Height profiles of the top fiber extracted along the lines marked with same color at (a). Height profiles reveal that the top fiber has a transverse size in the range of 100–150 nm. (c) Schematic representation of probing depth dependence of IR s-SNOM versus harmonic order demodulation of IR s-SNOM phase. (d and e) 2nd (O2) and 3rd (O3) harmonic maps of IR s-SNOM phase acquired on SFPU-2w at the position marked as red spot at (a). | |
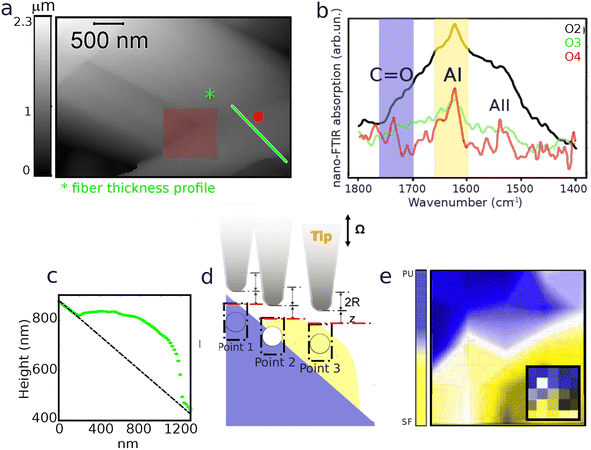 |
| | Fig. 4 (a) AFM topography map acquired on SFPU-1w showing a top fiber (right-bottom side) layng on a fibers bundle. The red spot marks the position at which the spectra, showing 2nd, 3rd and 4th harmonics of the phase (respectively O2, O3 and O4) reported at panel (b) were acquired. (c) Transverse profile of the top fiber extracted along the line marked with same color at (a). The transverse profile reveals that the top fiber has a size in the range of 100–150 nm. (d) Schematic representation of probing depth of IR s-SNOM at three representative positions on a SF fiber, with exact same profile shown at panel (c), on PU substrate: at point 1, when only PU is crossed we expect to observe the spectrum of pure PU; at point 2, when both compounds are crossed we expect to see a mix of PU and SF vibrational modes and a relative variation of the two contributions dependent on the harmonic of the signal; at point 3 when only SF is crossed we expect to acquire the spectrum of pure SF. The dotted rectangles are a schematization of the range of the probed volume in comparison to the tip radius. The colored circles, placed below the tip, gives a representation of how the measured point would appear in a chemical imaging map obtained similarly to the ones reported at Fig. 2. (e) Two colors smoothed hyper-spectral map (with raw map shown in the inset) reporting on the spatial distribution of SF (yellow: peak between 1600 and 1660 cm−1) and PU (blue: area between 1700 and 1760 cm−1) in the region marked as red square in panel (a). | |
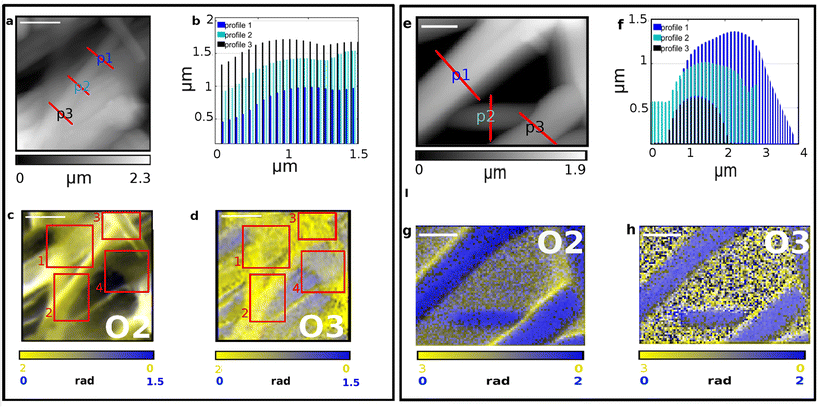 |
| | Fig. 5 IR s-SNOM Imaging analysis performed on two distinct regions (a–d and e–h) of SFPU-2w. Scale bar: a–d 2 μm, e–h 1 μm. AFM morphology maps are reported in panels (a) and (e) with red lines highlighting the transverse profiles shown at b and f. IR s-SNOM two-colors imaging maps are reported in panels c, g (O2, i.e. 2nd-harmonics of phase) and d, h (O3, i.e. 3rd-harmonics of phase)., with SF shown in yellow (acquired at 1628 cm−1) and PU in blue at (acquired at 1740 cm−1). The areas highlighted in red square at panels (c) and (d) show at: 1. a SF boundle, 2. and 3. SF fibers and 4. a SF thin veil. | |
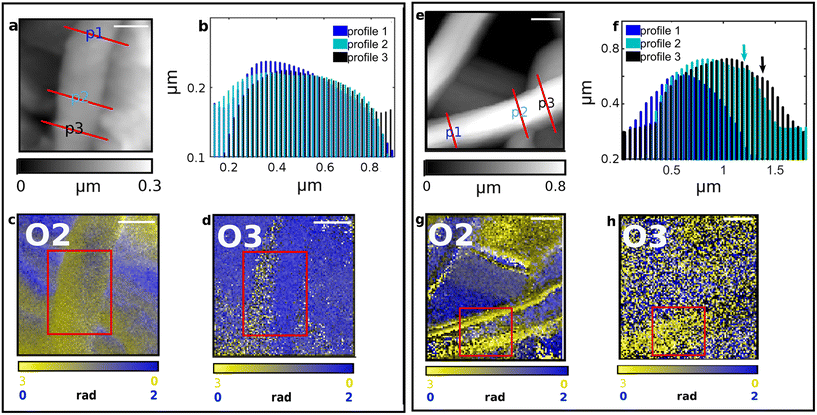 |
| | Fig. 6 IR s-SNOM Imaging analysis performed on two distinct regions (a–d and e–h) of SFPU-1w. Scale bar: a–d 500 nm, e–h 1 μm. AFM morphology maps are reported in panels (a) and (e) with red lines highlighting the transverse profiles shown at b and f. IR s-SNOM two-colors imaging maps are reported in panels c, g (O2, i.e. 2nd-harmonic of phase) and d, h (O3, i.e. 3rd-harmonic of phase), with SF shown in yellow (acquired at 1628 cm−1) and PU in blue at (acquired at 1740 cm−1). | |
4. Conclusions
The present study shows how the synergic use of micro- and nano-resolved infrared techniques can be successfully applied to the study of hybrid nanostructured materials. Mixed silk SF and PU meshes, engineered in the framework of semi-degradable materials for tissue scaffolding, were studied by means of a combination of scanning electron microscopy, micro-ATR imaging to IR s-SNOM. In particular, we demonstrated here how IR s-SNOM can be applied for the nanoscale morpho-chemical profiling of the corona the fibers constituting the hybrid meshes, up to a depth of around 100 nm. The obtained results demonstrate that the surface of the fibers exposed to the environment is not a mixture of the two fiber constituents, even when SF and PU are spun as a blend. The two meshes, obtained by spinning of both separate compounds and as a blend, are characterized by the co-existence of main fibrillar domains made of individual fibers, either completely made by a single component or with a corona of about 100 nm made by a pure component. For the meshes obtained by electrospinning of the blend, only occasional we revealed the presence of core–shell surface structures, where the layers tend to be well separated with not clear evidences of chemical mixing. In the case of the meshes obtained by electrospinning the two matrix constituents, non-fibrillary nanosized domains made of a very thin SF veil have also been detected. The present study is a practical innovative example on how to answer the ever-increasing demand for robust and precise morpho-chemical characterization of nanocomposites in tissue engineering, a requirement that is nowadays essential to support the production of newly conceived materials. Clearly, the limitation of the penetration depth guaranteed by the approach limits the considerations to the surface of the material up to about 100 nm from its exposed surface, that however is extremely important in order to understand the behavior of the material at the interface with the environment.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was part of the SILKELASTOGRAFT project, partially funded by Fondazione Cariplo and Regione Lombardia within the framework of the grant-Avviso congiunto per la concessione di contributi a sostegno del trasferimento della conoscenza nel settore dei Materiali avanzati- (project number 2018-1777). This work has been partly performed in the framework of the Nanoscience Foundry and Fine Analysis (NFFA-MIUR Italy) facility. The authors acknowledge the CERIC-ERIC Consortium for the access to experimental facilities and financial support (proposal # 20207142).
References
- E. Salernitano and C. Migliaresi, J. Appl. Biomater. Biomech., 2003, 1, 3–18 CAS.
- F. Zhang, T. Bambharoliya, Y. Xie, L. Liu, H. Celik, L. Wang, O. Akkus and M. W. King, Mater. Sci. Eng., C, 2021, 118, 111418 CrossRef CAS PubMed.
- A. M. J. Coenen, K. V. Bernaerts, J. A. W. Harings, S. Jockenhoevel and S. Ghazanfari, Acta Biomater., 2018, 79, 60–82 CrossRef CAS PubMed.
- A. Pryadko, M. A. Surmeneva and R. A. Surmenev, Polymer, 2021, 13, 1738 CAS.
- E. Yu, J. Zhang, J. A. Thomson and L. S. Turng, Int. Polym. Process., 2016, 31, 638–646 CrossRef CAS PubMed.
- K. Shimada, A. Higuchi, R. Kubo, T. Murakami, Y. Nakazawa and R. Tanaka, Organogenesis, 2017, 13, 115–124 CrossRef CAS PubMed.
-
P. Kumar, C. S. Ram, J. P. Srivastava, A. Kumar and A. K. Behura, in Natural and Synthetic Fiber Reinforced Composites, Wiley, 2019, pp. 137–153 Search PubMed.
-
J. Zhu, S. Jasper and X. Zhang, in Electrospun Nanofibers, Elsevier, 2017, pp. 181–206 Search PubMed.
- A. Caldiroli, E. Pederzani, M. Pezzotta, N. Azzollini, S. Fiori, M. Tironi, P. Rizzo, F. Sangalli, M. Figliuzzi, G. B. Fiore, A. Remuzzi, S. A. Riboldi, M. Soncini and A. Redaelli, Biomed. Mater., 2022, 17, 35944550 Search PubMed.
- N. Dehghan-Manshadi, S. Fattahi, M. Hadizadeh, H. Nikukar, S. M. Moshtaghioun and B. Aflatoonian, Eur. Polym. J., 2019, 121, 109294 CrossRef CAS.
- S. Das, V. K. Singh, A. K. Chaudhari, A. K. Dwivedy and N. K. Dubey, Int. J. Biol. Macromol., 2021, 188, 751–763 CrossRef CAS PubMed.
- S. van Uden, V. Catto, G. Perotto, A. Athanassiou, A. C. L. Redaelli, F. G. Greco and S. A. Riboldi, J. Biomed. Mater. Res., Part B, 2019, 107, 807–817 CrossRef CAS PubMed.
- S. van Uden, N. Vanerio, V. Catto, B. Bonandrini, M. Tironi, M. Figliuzzi, A. Remuzzi, L. Kock, A. C. L. Redaelli, F. C. Greco and S. A. Riboldi, Biomed. Mater., 2019, 30, 025007 CrossRef PubMed.
- M. Bertasa, E. Possenti, A. Botteon, C. Conti, A. Sansonetti, R. Fontana, J. Striova and D. Sali, Analyst, 2017, 142, 4801–4811 RSC.
- M. N. Blum and H. John, Drug Test. Anal., 2012, 4, 298–302 CrossRef CAS PubMed.
- P. Hermann, B. Kästner, A. Hoehl, V. Kashcheyevs, P. Patoka, G. Ulrich, J. Feikes, M. Ries, T. Tydecks, B. Beckhoff, E. Rühl and G. Ulm, Opt. Express, 2017, 25, 16574 CrossRef CAS PubMed.
- H. A. Bechtel, S. C. Johnson, O. Khatib, E. A. Muller and M. B. Raschke, Surf. Sci. Rep., 2020, 75, 100493 CrossRef CAS.
- E. Yoxall, M. Schnell, S. Mastel and R. Hillenbrand, Opt. Express, 2015, 23, 13358 CrossRef CAS PubMed.
- F. Piccirilli, F. Tardani, A. D'arco, G. Birarda, L. Vaccari, S. Sennato, S. Casciardi and S. Lupi, Nanomaterials, 2021, 11, 1103 CrossRef CAS PubMed.
- F. Huth, A. Govyadinov, S. Amarie, W. Nuansing, F. Keilmann and R. Hillenbrand, Nano Lett., 2012, 12, 3973–3978 CrossRef CAS PubMed.
- L. Mester, A. A. Govyadinov, S. Chen, M. Goikoetxea and R. Hillenbrand, Nat. Commun., 2020, 11, 3359 CrossRef CAS PubMed.
-
G. Birarda, D. Bedolla, F. Piccirilli, C. Stani, H. Vondracek and L. Vaccari, in Biomedical Vibrational Spectroscopy 2022: Advances in Research and Industry, ed. Z. Huang, SPIE, 2022, p. 31 Search PubMed.
- D. F. Galparsoro, X. Zhou, A. Jaaloul, F. Piccirilli, V. Vetri and V. Foderà, ACS Appl. Bio Mater., 2021, 4, 1876–1887 CrossRef PubMed.
- F. Piccirilli, N. Plotegher, M. G. Ortore, I. Tessari, M. Brucale, F. Spinozzi, M. Beltramini, P. Mariani, V. Militello, S. Lupi, A. Perucchi and L. Bubacco, Biophys. J., 2017, 113, 1685–1696 CrossRef CAS PubMed.
- L. Mester, A. A. Govyadinov and R. Hillenbrand, Nano Lett., 2022, 11, 377–390 CAS.
- R. Krutokhvostov, A. A. Govyadinov, J. M. Stiegler, F. Huth, A. Chuvilin, P. S. Carney and R. Hillenbrand, Opt. Express, 2012, 20(1), 593–600 CrossRef PubMed.
- S. Mastel, A. A. Govyadinov, T. V. A. G. de Oliveira, I. Amenabar and R. Hillenbrand, Appl. Phys. Lett., 2015, 106, 023113 CrossRef.
- T. Taubner, F. Keilmann and R. Hillenbrand, Nano Lett., 2005, 13, 8893 CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Alberto
Redaelli
c,
Alberto
Redaelli