
Establishing stereochemical comparability in phosphorothioate oligonucleotides with nuclease P1 digestion coupled with LCMS analysis†
Zifan
Li
 *,
Fei
Tong
,
Li
Xiao
,
Nicholas R.
Larson
,
Xuan
Zhou
,
Yueheng
Zhang
,
Jonas P.
Immel-Brown
and
George M.
Bou-Assaf
*
*,
Fei
Tong
,
Li
Xiao
,
Nicholas R.
Larson
,
Xuan
Zhou
,
Yueheng
Zhang
,
Jonas P.
Immel-Brown
and
George M.
Bou-Assaf
*
Pharmaceutical Operations & Technology, Biogen, 225 Binney Street, Cambridge, MA 02142, USA. E-mail: zifan.li@biogen.com; george.bou-assaf@biogen.com
First published on 20th September 2023
Abstract
Stereochemical comparability is critical for ensuring manufacturing consistency in therapeutic phosphorothioate oligonucleotides. Currently, analytical methods for this assessment are limited. We hereby report on a novel protocol capable of detecting a stereochemistry change in a single phosphorothioate linkage by employing nuclease P1 digestion of the oligonucleotide with subsequent LCMS analysis of the resulting fragments. The method proves valuable for establishing stereochemical comparability and for ensuring manufacturing consistency of oligonucleotide therapeutics.
Therapeutic oligonucleotides gained attention in recent years for their potential in treating a wide range of diseases.1–4 Common oligonucleotide modalities include antisense oligonucleotides (ASO),5 short interference RNA (siRNA),6 microRNA (miRNA),7 aptamers,8etc. To improve their pharmacokinetic properties, therapeutic oligonucleotides have been designed in a way that some (or all) phosphodiester (PO) linkages in an oligonucleotide sequence are substituted for phosphorothioate (PS) linkages.9–12 With the introduction of PS linkages, phosphorus becomes a new chiral center as a result of the substitution of non-bridging oxygen for sulfur, thus generating two distinct configurations, Rp and Sp.13 Aside from a few reports describing the stereo-controlled incorporation of PS linkages,14–17 the majority of PS forming reactions are not stereospecific.4 The chirality of the phosphorus center in an oligonucleotide sequence may affect its pharmacological activity, including nuclease resistance, binding affinity, and toxicities.14,18–20 Batch-to-batch consistency in diastereoisomeric composition is critical for ensuring drug safety and efficacy.15,20,21 Therefore, it is imperative to develop sensitive analytical methods that are fit for the purpose of establishing stereochemical comparability between batches for clinical and commercial purposes.
Demonstration of stereochemical comparability for PS oligonucleotides is challenging because the number of stereoisomers increases exponentially with the number of PS linkages. For example, an 18-mer ASO with 17 PS linkages will contain 217 (131072) distinct diastereoisomers. Given the current state-of-the-art analytical technologies, developing a method capable of distinguishing every diastereoisomer in full-length oligonucleotides is virtually impossible. Several strategies have been developed to establish stereochemical comparability between PS oligonucleotides without characterizing individual diastereoisomers. For example, methods such as 31P nuclear magnetic resonance (NMR) and circular dichroism (CD) could potentially monitor overall diastereoisomeric distribution change, but these methods are challenged when the characterization of individual stereocenters is required because they report on the global population of stereoisomers in the mixture.22,23 One method that can determine the diastereoisomeric composition at each PS linkage consists of the analysis of crude dimethoxytrityl-on (DMT-on) samples collected after each coupling cycle by high performance liquid chromatography (HPLC).19 This approach is labor intensive, not appropriate for use in a good manufacturing practice (GMP) environment and does not evaluate the diastereoisomeric content of the full-length oligonucleotide after (but rather during) its synthesis. The establishment of stereochemical comparability at each PS linkage for full-length therapeutic oligonucleotides remains unexplored until the current study.
Here, we describe a method based on the nuclease digestion of oligonucleotides in combination with liquid chromatography mass spectrometry (LCMS) to separate diastereoisomers of the digested fragments. Enzymatic digestion truncates full-length oligonucleotides into shorter fragments containing a couple of PS linkages. The subsequent chromatographic step enables separation and quantitation of the shortmers whereas mass spectrometry allows their identification. Among enzymes used for oligonucleotide digestion, nuclease P1 (NP1) stands out as the enzyme of choice because of its outstanding performance and its superior digestion efficiency for oligonucleotides with PO and PS linkages.24,25 NP1 also preferentially digests Sp diastereoisomers of oligonucleotides.26 Therefore, PS oligonucleotides with comparable stereochemistry are digested at a similar rate and produce the same unique fragments with similar diastereoisomeric composition after a defined incubation period. The strategy adopted here is similar to the “bottom-up” approach commonly used in the proteomics field and referred to as “peptide mapping”.27 The chromatogram of the separated peptide fragments which were digested by exposure of the protein to an enzyme (a protease) constitutes a unique fingerprint that distinguishes one protein from another. Oligonucleotides with identical sequences but different diastereoisomeric composition at one or more PS linkages will exhibit a change in their “fingerprint” as measured by ion-pairing reversed-phase-high performance liquid chromatography (IP-RP-HPLC).28 Therefore, batch-to-batch comparability can be evaluated by comparing the chromatograms of the digested fragments.
In this study, three 18-mer PS/PO mixed backbone ASOs (Fig. 1) were synthesized from the 3′- to the 5′-end using solid phase oligonucleotide synthesis. Structurally, these three ASOs share the same gapmer sequence, with 2′-methoxyethyl (MOE) nucleotides on both the 3′- and 5′-ends flanking deoxy nucleotides in the middle. It was reported by Ravikumar and Cole that employing different activators during the coupling step can influence the diastereoisomeric distribution, since the stereoelectronic effects and steric hindrance between the activator and the incoming phosphoramidite dictate the stereochemistry of the newly formed PS linkage.29–31 To generate ASOs with different diastereoisomeric compositions (Rp/Sp ratios), different combinations of activators were used as follows:
• For ASO1, 4,5-dicyanoimidazole (DCI) was used for coupling at every PS linkage.
• For ASO2, DCI was used as activator for the first 16 PS linkages and 5-(benzylthio)-1H-tetrazole (BTT) was used for the 17th PS linkage (the 5′-most PS linkage).
• For ASO3, BTT was used for every PS linkage.
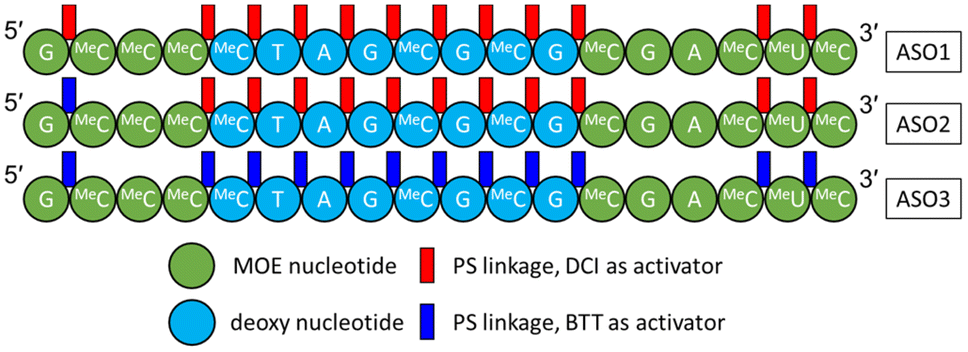 | ||
| Fig. 1 Three ASO constructs used in this study. A = adenine; G = guanine; MeC = 5-methylcytosine; T = thymine; MeU = 5-methyluridine. PO linkages are omitted. | ||
After the synthesis, the crude ASOs (DMT-on) were analyzed using IP-RP-HPLC without any digestion and the relative ratios of the main peaks represented the diastereoisomeric ratios at the 5′-most PS linkage (ESI, Fig. S1†). The earlier eluting peak is tentatively assigned to the Sp stereoisomer, whereas the later eluting peak is assigned to the Rp stereoisomer.19,32 The UV chromatogram of ASO1 showed almost identical amounts of Sp and Rp (51% and 49%), whereas those of ASO2 and ASO3 showed approximately 39% Sp and 61% Rp at the 5′-most PS linkage (ESI, Fig. S1†). The comparable Rp/Sp ratios obtained for ASO2 and ASO3 support the experimental design hypothesis that the different activators caused the reproducible stereochemistry changes on the 5′-most PS linkage.23
The crude ASOs were then purified by hydrophobic interaction chromatography (HIC), followed by detritylation and desalting. The optimized NP1 digestion conditions consisted of using 1 U of NP1 (New England Biolabs) for every 2.5 nmol of full-length ASO. The mixture was incubated at 37 °C for 1 day and heated to 100 °C for 10 minutes to inactivate NP1 and avoid further digestion before or during sample analysis by LCMS. The digested oligonucleotide fragments are separated on a C18 column, by a slow gradient and 10 mM triethylamine acetate (TEAA) in water and methanol as mobile phases, known for its excellent separation efficiency for PS oligonucleotide diastereoisomers.33,34 The resulting UV and MS chromatograms showed the undigested ASO and the truncated fragments of different lengths. As expected, the UV chromatograms of ASO1 and ASO2 showed similar “fingerprints” because they were synthesized with the same activator (DCI) except for the last coupling cycle for ASO2, whereas ASO3 displayed a notably different “fingerprint” given that it was synthesized with a completely different activator (BTT) (ESI, Fig. S2†).
While UV chromatograms enable a qualitative assessment of diastereoisomeric composition similarity by comparison of the “fingerprints”, MS provides detailed sequence information and identification of each fragment. The extracted ion-chromatograms (EIC) of three representative fragments selected based on their observed monoisotopic m/z are shown in Fig. 2A, C, and E. ASO1 and ASO2 only differ in the activator used during the last coupling cycle (DCI versus BTT), therefore, the 5′-end-containing fragments could be compared to assess the sensitivity of the NP1 digestion method to distinguish between two shortmers with identical sequence but with a different diastereoisomeric distribution at a single PS linkage. Particularly, in Fig. 2A, the peak intensities before 24.3 min in the EIC were similar between ASO1 and ASO2, thus corresponding to PS linkages between MOE MeC and deoxy MeC and/or deoxy MeC and deoxy T at the 3′-end of the fragment. The variance in peak intensities observed after 24.3 min is attributed to the difference in diastereoisomeric composition caused by the activator change at the 5′-most PS linkage between MOE G and MOE MeC. Peak intensities of ASO3 were easily distinguishable from those of ASO1 and ASO2, presumably due to the different activator used throughout the synthesis. The specificity of the NP1 digestion method was further demonstrated by examining the 3′ fragment EICs shown in Fig. 2E. EICs of ASO1 and ASO2 were almost identical, whereas that of ASO3 was significantly different. In addition, a fragment that originates from the middle of the sequence exhibited similar chromatographic fingerprints, thus suggesting that the diastereoisomeric content of these PS linkages is similar among all three ASOs (Fig. 2C). This observation aligns with a previous report which indicates that the diastereoisomeric distribution in PS linkage is little affected by the type of activator used to couple deoxy nucleotides.29,35
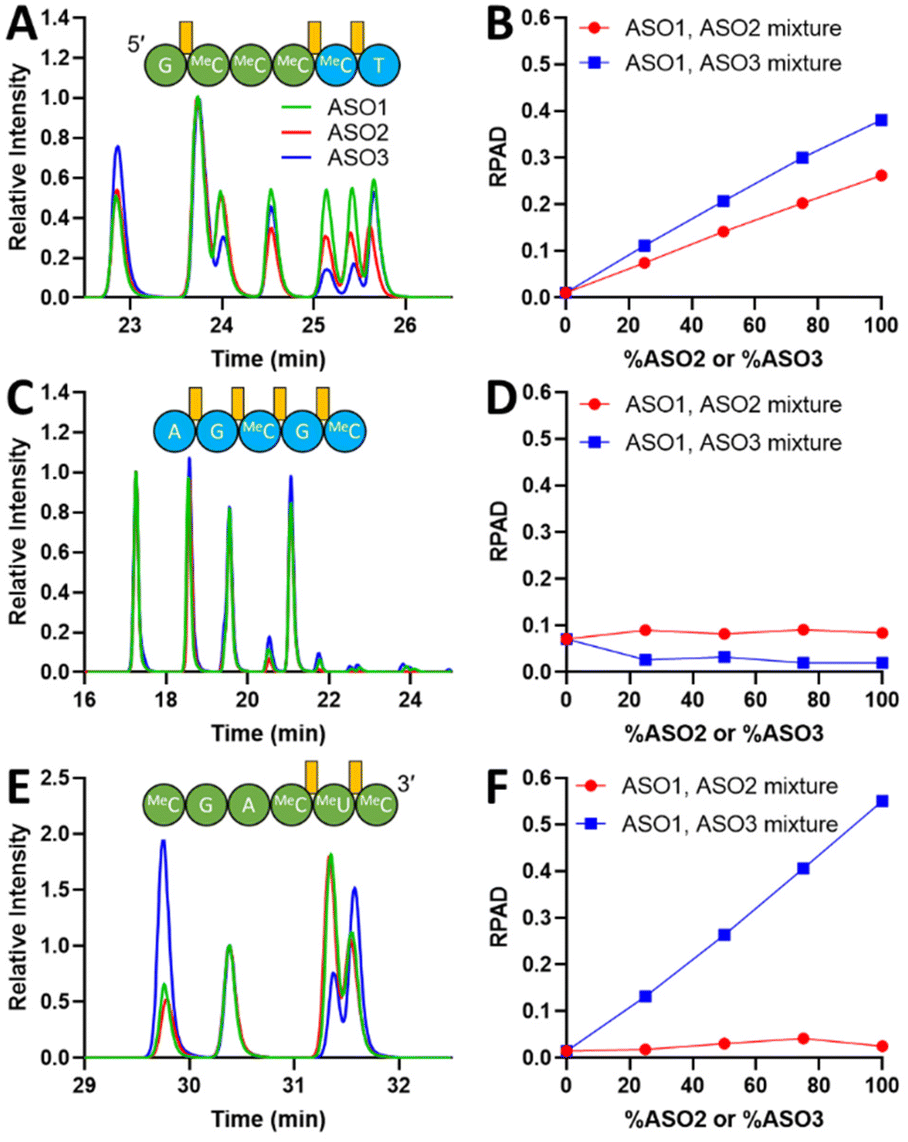 | ||
| Fig. 2 EIC overlays of representative fragments from ASO1, ASO2, and ASO3, and calculated RPAD. (A) EIC overlay of a 5′ fragment; (B) RPAD of the selected 5′ fragment; (C) EIC overlay of a middle fragment; (D) RPAD of the selected middle fragment; (E) EIC overlay of a 3′ fragment; (F) RPAD of the selected 3′ fragment. Green: ASO1; Red: ASO2; Blue: ASO3. PS bonds are labeled in orange. EICs were aligned to minimize retention time shift. Chromatograms were normalized to a selected peak. ASO1, ASO2, and ASO3 digestions were performed in triplicates. Error bars are included in the figures. | ||
Further digestion can be achieved with extended incubation time or increased NP1-to-oligonucleotide ratio. With the extended incubation time, fragments with Sp configurations are further shortened, thus reducing the number of diastereoisomers to be detected. For instance, the all-deoxy full PS fragment AGMeCGMeC standard (Integrated DNA Technologies) showed 13 distinguishable EIC peaks (ESI, Fig. S3†) when injected neat, but the same fragment fingerprint generated by NP1 digestion of ASO1–3 for 1 day showed only 4 major peaks (Fig. 2C). The results suggest that the current LC method is capable of separating multiple diastereoisomers as evidenced in Fig. S3 (ESI†), and that the fewer peaks detected in Fig. 2C are a result of the extended digestion period. Therefore, it is essential to maintain the same digestion conditions for the direct comparison between different ASO samples. Method optimization is required to ensure the entire ASO sequence is covered by fragments generated during digestion, with Fig. S4 (ESI†) being one example.
One advantage of the extended digestion incubation period is the generation of multiple overlapping fragments that enable better localization of the differences in diastereoisomeric content, potentially to the single PS linkage level. For instance, if a 4-mer does not show any differences in diastereoisomeric content between two ASO samples but its overlapping 5-mer with the same 5′ or 3′ end does, then the difference in diastereoisomeric content could be narrowed down to the overhang PS linkage.
Fragments containing 5′ or 3′ MOE nucleotides generally exhibited more peaks compared to the middle deoxy fragments. This observation suggests that PS linkages which couple MOE-modified nucleotides undergo slower digestion by NP1 than their deoxy counterparts, which is consistent with the previously reported observation that the rate of digesting 2′-methoxy modified RNAs is slower than that of unmodified nucleotides.36
To quantitatively assess the method capability, we considered employing peak ratios to determine changes in diastereoisomeric content. It is worth mentioning that peak ratios do not represent the true Rp/Sp ratios due to NP1's preferential digestion towards Sp diastereoisomers. Although peak ratios could potentially be used as indicators of diastereoisomeric content change, choosing appropriate peaks in a given fragment EIC for the calculation could be challenging. Here, we introduce the calculation of a relative peak area difference (RPAD) to compare the normalized peak areas in a sample to those in a reference:
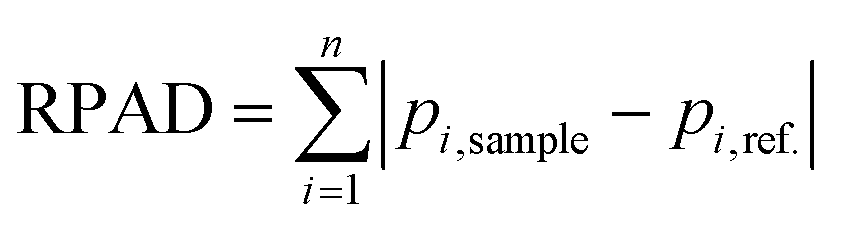
To determine the sensitivity of the method, its linearity, and its limit of quantitation (LOQ), i.e., the smallest difference in Rp/Sp ratio that can be unequivocally deemed as a true change in diastereoisomeric content, a series of ASO mixtures with a linear change in molar composition was created: 100% ASO1, 75% ASO1: 25% ASO2, 50% ASO1: 50% ASO2, 25% ASO1: 75% ASO2, and 100% ASO2. A similar mixture series of ASO1 and ASO3 was also prepared. All sample mixtures were digested by NP1 and analyzed with the same method conditions mentioned above. If two ASO fragments have different diastereoisomeric contents, we would expect that the calculated RPAD value would correlate with the change in the corresponding ASOs molar ratios. On the other hand, if the fragments diastereoisomeric content is the same, the RPAD value would not change with the increasing ASOs molar ratios. RPAD values for the three ASO fragments discussed above are plotted against % ASO2 or % ASO3 in Fig. 2B, D, and F.
A linear correlation between the RPAD value of the 5′ fragment and the percentage of ASO2 is demonstrated in Fig. 2B. The trend indicates that the differences in EIC peak pattern between the individual digestion of ASO1 and ASO2 after 24.3 min (Fig. 2A) is caused by a different diastereoisomeric distribution at the 5′-most PS linkage. Compared to the ASO1/ASO2 mixture, the linear correlation observed for the ASO1/ASO3 mixture has a larger slope, likely because of the more significant change in the diastereoisomeric distribution at two more PS linkages formed by BTT in ASO3. Similarly, in the 3′ fragment (Fig. 2E), the RPAD of ASO1/ASO3 was much higher than that of ASO1/ASO2, because a different activator was used throughout the syntheses of ASO1 and ASO3. However, because the same activator was used for the syntheses of ASO1 and ASO2 (except for the last PS linkage which is not part of the 3′ fragment), no correlation between the RPAD value and the ASO mixture molar ratios was observed (Fig. 2F). For the middle fragment, we do not expect to see any differences in diastereoisomeric content as described above. Indeed, no correlation was observed between the calculated RPAD values and the ASOs molar ratios in each of the mixtures (Fig. 2D). The residual RPAD values calculated are attributed to assay variations.
The method LOQ was calculated based on the residual standard deviation and slope of the calibration curve derived from the RPAD between ASO1 and ASO2 against the actual % Rp of the crude DMT-on ASOs. The lowest LOQ achieved corresponds to 1% change in Rp content at the 5′-most PS linkage after examining each observed 5′ fragment.
To ensure stereochemical comparability at each PS linkage can be achieved, it is crucial to select the right fragments when analyzing PS ASOs. Method development is necessary to ensure accurate and reproducible results for a new ASO, because the efficiency of NP1 digestion may differ. Inefficient digestion may produce long fragments that exceed the chromatographic separation capacity, thus lowering the method sensitivity and spatial resolution. Higher NP1/ASO ratio and longer digestion time may be necessary to produce shorter fragments for effective comparability purposes. Factors such as ASO purity and buffer composition may also impact the analytical results. Maintaining consistent sample quality and preparation is important to ensure reliable and reproducible results, even if the method seems robust during preliminary evaluations.
As outlined here, the quantitative approach for dealing with the NP1 digestion data is suitable not only for establishing batch-to-batch comparability in ASO products with PS linkages, but also for demonstration of sameness between generic and innovator products. During process and analytical development, setting suitable acceptance criteria for the RPAD indicator is critical for defining an appropriate stereochemical comparability assay. Acceptance criteria may be established based on batch history experience and the intrinsic assay variability. Each fragment fingerprint may require its individual RPAD limit.
In conclusion, we hereby report on the development of a method for the establishment of stereochemical comparability in PS containing oligonucleotides using NP1 digestion followed by LCMS analysis. We also propose the implementation of RPAD in the EICs of digested fragments as indicators to quantitatively assess the diastereoisomeric content change in two different ASO samples. The method is potentially capable of distinguishing a diastereoisomeric distribution change as low as 1% at a single PS linkage. This method provides a novel insight into batch-to-batch stereochemical comparability in oligonucleotide products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Kayla Mello and Jackson Struble for providing synthesis and purification support in this project. We thank Brad Robertson for the support in editing. We are grateful to all our Biogen colleagues of the ASO team for their support and input.References
- S. Thakur, et al. , Front. Pharmacol., 2022, 13, 1006304 CrossRef CAS PubMed.
- C. I. E. Smith, et al. , Annu. Rev. Pharmacol. Toxicol., 2019, 59, 605–630 CrossRef CAS PubMed.
- T. C. Roberts, et al. , Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed.
- A. Khvorova, et al. , Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS PubMed.
- C. F. Bennett, et al. , Annu. Rev. Pharmacol. Toxicol., 2010, 50, 259–293 CrossRef CAS PubMed.
- J. Neumeier, et al. , Front. Plant Sci., 2021, 11, 526455 CrossRef PubMed.
- R. Rupaimoole, et al. , Nat. Rev. Drug Discovery, 2017, 16, 203–222 CrossRef CAS PubMed.
- A. D. Keefe, et al. , Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS PubMed.
- H. Takakusa, et al. , Nucleic Acid Ther., 2023, 33, 83–94 CrossRef CAS PubMed.
- F. Eckstein, Nucleic Acid Ther., 2014, 24, 374–387 CrossRef CAS PubMed.
- H. L. Lightfoot, et al. , Nucleic Acids Res., 2012, 40, 10585–10595 CrossRef CAS PubMed.
- S. T. Crooke, et al. , Nucleic Acids Res., 2020, 48, 5235–5253 CrossRef PubMed.
- T. Chen, et al. , J. Chromatogr. A, 2022, 1678, 463349 CrossRef CAS PubMed.
- M. Byrne, et al. , Transl. Vis. Sci. Technol., 2021, 10, 23 CrossRef PubMed.
- N. Iwamoto, et al. , Nat. Biotechnol., 2017, 35, 845–851 CrossRef CAS PubMed.
- P. Kandasamy, et al. , Nucleic Acids Res., 2022, 50, 5401–5423 CrossRef CAS PubMed.
- M. Li, et al. , Chem. Commun., 2017, 53, 541–544 RSC.
- M. Koziolkiewicz, et al. , Nucleic Acids Res., 1995, 23, 5000–5005 CrossRef CAS PubMed.
- W. B. Wan, et al. , Nucleic Acids Res., 2014, 42, 13456–13468 CrossRef CAS PubMed.
- E. D. Funder, et al. , PLoS One, 2020, 15, e0232603 CrossRef CAS PubMed.
- H. Jahns, et al. , Nucleic Acids Res., 2022, 50, 1221–1240 CrossRef CAS PubMed.
- S. G. Roussis, et al. , Anal. Chem., 2021, 93, 16035–16042 CrossRef CAS PubMed.
- A. Murakami, et al. , Anal. Biochem., 1994, 223, 285–290 CrossRef CAS PubMed.
- J. D. Jones, et al. , Anal. Chem., 2023, 95, 4404–4411 CrossRef CAS PubMed.
- H. Jahns, et al. , Nat. Commun., 2015, 6, 6317 CrossRef CAS PubMed.
- B. V. L. Potter, et al. , Biochemistry, 1983, 22, 1369–1377 CrossRef CAS PubMed.
- I. Sokolowska, et al. , Anal. Chem., 2020, 92, 2369–2373 CrossRef CAS PubMed.
- Y. Tamura, et al. , Nucleosides Nucleotides, 1998, 17, 269–282 CrossRef CAS PubMed.
- V. T. Ravikumar, et al. , Nucleosides Nucleotides Nucleic Acids, 2003, 22, 1415–1419 CrossRef CAS PubMed.
- V. T. Ravikumar, et al. , Nucleosides Nucleotides Nucleic Acids, 2003, 22, 1639–1645 CrossRef CAS PubMed.
- V. T. Ravikumar, et al. , Org. Process Res. Dev., 2002, 6, 798–806 CrossRef CAS.
- A. Wilk, et al. , Nucleic Acids Res., 1995, 23, 530–534 CrossRef CAS PubMed.
- L. Li, et al. , J. Chromatogr. A, 2017, 1500, 84–88 CrossRef CAS PubMed.
- M. Enmark, et al. , Anal. Bioanal. Chem., 2019, 411, 3383–3394 CrossRef CAS PubMed.
- Z. S. Cheruvallath, et al. , Nucleosides Nucleotides Nucleic Acids, 2000, 19, 533–543 CrossRef CAS PubMed.
- H. Grosjean, et al. , Methods Enzymol., 2007, 425, 55–101 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an01392h |
| This journal is © The Royal Society of Chemistry 2023 |