
Different approaches for estimation of the expanded uncertainty of an analytical method developed for determining pharmaceutical active compounds in wastewater using solid-phase extraction and a liquid chromatography coupled with tandem mass spectrometry method†
João A.
Rodrigues
a,
Sofia
Silva
 a,
Vitor Vale
Cardoso
a,
Maria João
Benoliel
a and
Cristina M. M.
Almeida
*bc
a,
Vitor Vale
Cardoso
a,
Maria João
Benoliel
a and
Cristina M. M.
Almeida
*bc
aEmpresa Portuguesa das Águas Livres, S.A. – Direção de Laboratórios e de Controlo de Qualidade da Água, Av. de Berlim, 15, 1800-031 Lisboa, Portugal
biMed.UL (Institute for Medicines and Pharmaceutical Sciences, Portugal), Faculty of Pharmacy, University of Lisboa, Av. Prof. Gama Pinto, 2, 1649-003 Lisboa, Portugal. E-mail: calmeida@ff.ulisboa.pt; Fax: +351-217-946-470; Tel: +351-217-946-400
cLaboratory of Bromatology and Water Quality, Faculty of Pharmacy, University of Lisbon, Av. Prof. Gama Pinto, 2, 1649-003 Lisboa, Portugal
First published on 28th November 2022
Abstract
Although the evaluation of the uncertainty of an analytical method is a mandatory step in the method's validation, its applicability to the monitoring of trace compounds in complex samples is not simple, nor is it part of the routine of most laboratories, namely those dedicated to research. This manuscript focuses on the full validation of an analytical procedure for determining trace concentrations of twenty-four pharmaceutical active compounds (PhACs) in wastewaters using solid-phase extraction (SPE) and ultra-high performance liquid chromatography coupled with tandem mass spectrometry (UHPLC-MS/MS). The method optimization was performed on different wastewater matrices, namely influents and final effluents from two distinct wastewater treatment plants (WWTPs). Matrix effects and extraction efficiency (absolute recovery) of the developed method were determined. Validation was performed to obtain the method's linearity/working range, precision, trueness, method detection limits (MDLs) and method quantification limits (MQLs). The expanded uncertainty of the data obtained was estimated according to the requirements of international procedures dedicated to the expression of uncertainty. Different approaches for the estimation of uncertainty were applied. The validated method was used in the analysis of target PhACs in wastewater samples collected at two WWTPs. The obtained results facilitated the introduction of a validated method for routine measurement of PhACs in wastewater samples and allowed method accreditation by the competent national authority.
Introduction
The adequate treatment of wastewaters is a perpetually growing challenge. Due to demographic and social evolution, the diversity of effluents that are discharged into the sewer systems has increased drastically, with a variety of sources such as domestic, industrial or medical/clinical waste that can eliminate a large number of different contaminant compounds.1 Moreover, there is an ever-increasing number of compounds of emerging concern (CECs), mainly because of the continuous scientific advances and the manufacture of new products, which can hinder the quality requirements for treated effluents.Wastewater treatment plants (WWTPs) must evolve to keep up with these new challenges to allow the treated effluents to be disposed of without any or minimal danger to human health or damage to the environment. Currently, most WWTPs have at least secondary treatment to remove organic matter and solids through biological processes followed by secondary sedimentation. In addition, a separate preliminary treatment stage is also performed, aiming the removal of coarse suspended solids1,2 and a primary treatment can be in place to remove about 50% of settleable organic and inorganic solids by sedimentation. A tertiary treatment stage may also be necessary to comply with the required phosphorus and nitrogen levels for the discharge of the final treated effluents into water resources that are considered sensitive (susceptible to eutrophication). This step also ensures the removal of any remaining pathogens that were not efficiently eliminated in the prior treatment stages.1,3
After tertiary treatment, the water has undergone sufficient purification to be clean and suitable for numerous operations that require clean water, such as industrial and manufacturing processes, oil and gas extraction and refining, utility cooling and agricultural practices such as irrigation. The technologies in tertiary treatment are used to further improve the water quality and the types of technologies to be used will depend on the expected use.4,5
The prevailing European legislation for wastewater quality requirements for discharge dates to 1991 (ref. 6) and has suffered only minor updates since then, reflecting the public mistrust concerning water reuse and safety issues, with treated effluents being viewed just as waste that requires safe discharge into the environment rather than a potentially valuable water resource.7 The EU directive states a set of three parameters: biochemical oxygen demand (BOD), chemical oxygen demand (COD) and total suspended solids (TSS), to be monitored in normal discharge, and two additional parameters, total phosphorus and total nitrogen, for discharge areas considered sensitive. These limited monitoring parameters reflect the degree to which wastewater is reused in Europe. As of 2007, the overall European Union effluent reclamation percentage was 2.4%, in contrast with the 14% registered for the USA or the 87% registered for Israel.8
In 2020 a report published by the European Commission, Regulation (EU) 2020/741, established a general guideline for the minimum quality requirements that would allow for treated effluents to be used for agricultural irrigation.9 However, as the report states, there are still several gaps in knowledge, especially concerning CECs. This term refers to compounds that are not only new and now being discovered, but also to those that have been used for decades but only recently have raised cause for concern, mostly due to advances in analytical techniques that now allow for their detection and quantification. Such examples include PhACs, water treatment by-products, nanoparticles, and personal care products, among many others.9,10 Wastewaters can also lead to strong contaminating effects on natural aquatic systems, even after treatment, as several organic compounds are not eliminated by conventional wastewater treatments, and some of them may become ubiquitous in the environment and even increased in toxicity. In this way, to use reclaimed effluents safely, comprehensive characterization of wastewater contaminants is indispensable.11
The widespread use of PhACs, whether for human or veterinary applications, has increased research regarding their occurrence in aquatic matrices. PhACs reach WWTPs through human and animal' excretion (considering that they may not be completely metabolized) or by being improperly disposed of. Studies have reported that PhACs belonging to different therapeutic classes have been detected worldwide in WWTP streams, in concentrations ranging from nanograms to micrograms per liter, with the most commonly PhACs cited classes being hormones, analgesics, non-steroidal anti-inflammatories (NSAIDs) and antibiotics.12–15
Due to the low concentrations of these target analytes and the complexity of matrices, such as wastewater, the use of extraction/concentration and cleanup processes is necessary prior to instrumental quantification. For aqueous samples, due to the wide range of sorbents (packing materials) and method simplicity, solid-phase extraction is nowadays the most common pre-treatment procedure. The HLB (hydrophilic–lipophilic-balance) is the SPE material most often used in the extraction of PhACs belonging to different therapeutic classes.14,16–20 Relative to quantification, liquid chromatography tandem mass spectrometry is the most common method. Regardless of the sensitivity and capability of this hyphenated method, several restrictions remain. Some of them are associated with ion enrichment or suppression problems in UHPLC-MS/MS.21 Therefore, there are many problems associated with the reliable identification and quantification of these analytes in such complex matrices.22
Currently, the characterization of the water environment concerning any contaminant must be accompanied not only by the evaluation of the methods' performance but also by the validation of the results obtained. Without this assessment, it is impossible to have quality and reliable results, which are essential to a meaningful evaluation of environmental hazards. On the other hand, it is vital to establish standard protocols for its determination and compare the results between different laboratories to select the most appropriate one. The decision-making power in several areas of science and in other areas of society is based on the results of analytical studies, which should not be questioned. Consequently, the implementation and validation of the methods are of paramount importance.21,23 Considering all the above information, the main aims of this study were:
(i) To develop and optimize an analytical method for the simultaneous determination of 24 PhACs in wastewaters by solid-phase extraction (SPE) coupled with the UHPLC-MS/MS method through the determination of the following parameters: linearity, working range, instrumental analytical limits, and threshold limits of the global method (MDL and MQL). Simultaneously, the precision, trueness, matrix effects (ME), and absolute recovery (AR) were also studied, according to the requirements of national and international regulations or legislation;24–26
(ii) To present a methodology for estimating the expanded uncertainty of the results of the analytical procedure according to the international regulation, such as ISO 11352:2012,27 and to define the acceptance criteria of all statistical tests performed to be applied in routine analysis, according to the accreditation laboratory rules.
(iii) To apply the validated method to the determination of target PhACs in wastewater influents and effluents from two Portuguese WWTPs and to compare the results expressed as result ± expanded uncertainty determined by three approaches with those presented as a result ± standard deviation to highlight the difference between these different approaches.
Experimental
PhAC standards and reagents
The selection of the studied PhACs was based on Portuguese consumption data provided by Infarmed,28,29 environmental occurrence,30 toxicity and persistence in the environment,31–34 and compounds proposed for inclusion in the Water Framework Directive.35The PhACs are grouped by their therapeutic classes, namely: non-steroidal anti-inflammatory drugs, NSAIDs (diclofenac – DCF, ibuprofen – IBUP and naproxen – NPX), beta-blockers (atenolol – ATN, metoprolol – MTPL and propranolol – PPNL), anticonvulsants (carbamazepine – CBZ), analgesics (acetaminophen – APAP), antidepressants (fluoxetine – FLX), antibiotics (erythromycin – ERT, sulfadiazine – SDZ, sulfamethoxazole – SMX and sulfapyridine – SPD), lipid regulators (clofibric acid – CFA and bezafibrate – BZF), sexual hormones (17-α-ethinylestradiol – EE2, β-estradiol – E2, estrone – E1, estriol – E3, diethylstilbestrol – DES, gestodene – GTD and testosterone – TTE), psychostimulants (caffeine – CAF) and corticosteroids (cortisone – CTS).
All PhAC standards were of analytical grade (highest purity available, ≥95%), although provided by different brands: Sigma-Aldrich (acetaminophen, atenolol, bezafibrate, carbamazepine, clofibric acid, cortisone, diclofenac, erythromycin, fluoxetine, ibuprofen, naproxen, propranolol, sulfadiazine, and sulfapyridine), Fluka (caffeine, sulfamethoxazole, and testosterone), and LGC (gestodene and metoprolol); all three brands from Spain, and Dr. Ehrenstorfer, GmbH (diethylstilbestrol, estradiol, estriol, estrone, and 17α-ethinylestradiol) from Germany.
Individual stock solutions of PhACs were prepared in methanol. A standard solution mixture was prepared by diluting each individual standard solution in methanol to a concentration of around 1 mg L−1 for PhACs, except ibuprofen and hormones, with 2 mg L−1 and 6 mg L−1, respectively (Std 1). All solutions were prepared in a glass material and stored at −20 ± 3 °C in the absence of light.
Methanol (liquid chromatography grade, 99.9%), formic acid (liquid chromatography grade, ≥98%) and ammonium acetate (98%, p.a.) were provided by Merck (Germany).
Wastewater sampling
The wastewater samples were taken from two Portuguese WWTPs, one located in the east part of the Algarve region, in the south of Portugal (Faro Nw WWTP) and the second one located in Lisbon (Beirolas WWTP).The Faro Nw WWTP was designed for 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 inhabitants, corresponding to 13200 m3 day−1. The plant layout includes screening, degritting/degreasing, two extended aeration oxidation ditches, with surface aerators, designed for 9–10 days of solids retention time, two secondary settlers, UV disinfection and chemical scrubbers for odor control. Currently the WWTP receives about 4700 m3 day−1 and an organic loading of 1500 kg BOD5 per day, corresponding to 25100 inhabitants. The effluent usually has the following mean annual quality values: <10 mg L−1 BOD5, 35 mg L−1 COD, 4 mg L−1 TSS, 8 mg L−1 N, 1.3 mg L−1 P, 2.5 NTU turbidity, and <300 MPN/100 mL fecal coliforms.13
500 inhabitants, corresponding to 13200 m3 day−1. The plant layout includes screening, degritting/degreasing, two extended aeration oxidation ditches, with surface aerators, designed for 9–10 days of solids retention time, two secondary settlers, UV disinfection and chemical scrubbers for odor control. Currently the WWTP receives about 4700 m3 day−1 and an organic loading of 1500 kg BOD5 per day, corresponding to 25100 inhabitants. The effluent usually has the following mean annual quality values: <10 mg L−1 BOD5, 35 mg L−1 COD, 4 mg L−1 TSS, 8 mg L−1 N, 1.3 mg L−1 P, 2.5 NTU turbidity, and <300 MPN/100 mL fecal coliforms.13
The Beirolas WWTP has an installed capacity for 213510 population equivalent which corresponds to an average flow rate of 54500 m3 day−1. The wastewater treatment line consists of the following steps: (i) preliminary treatment, by sieving and degritting/degreasing, for removal of coarse materials; (ii) primary treatment, performed in two parallel settlers to remove floatable materials, reducing the load for subsequent treatment steps; (iii) secondary treatment in a sequence of anaerobic/anoxic/aerobic reactors; (iv) secondary settling; (v) filtration of the secondary effluent, and if necessary UV disinfection, to comply with legal discharge requirements.13
For each WWTP, Faro Nw and Beirolas, two sampling points were considered: wastewater influents (WWIs) and effluents (WWEs). All wastewater samples consisted of 24 hours composite samples, with a fifteen minute sampling frequency (200 mL h−1). A total volume of 4.8 L was collected in an amber glass bottle and, to obtain a representative sample, 500 mL of this volume was filtered consecutively through quantitative paper filter, a 1.0 μm glass microfilter and a 0.45 μm polytetrafluoroethylene (PTFE) membrane. In this way, the measurement of PhAC concentrations in each sample corresponded only to the concentration in the dissolved fraction. The filtered samples were stored at 5 ± 3 °C until extraction, which occurred within 7 days.
UHPLC-MS/MS analysis
The UHPLC-MS/MS system consisted of a Dionex Ultimate 3000 system equipped with a binary pump, an automatic injector and a thermostatted column compartment coupled to a mass spectrometer TSQ Endura triple quadrupole model, from Thermo Scientific (USA) equipped with an electrospray ionization (ESI) source. Xcalibur 4.0 software was used for data acquisition. Analytes were separated on a Kinetex EVO C18 column (2.1 × 50 mm, 2.6 μm pore size) from Phenomenex (USA). Due to the wide range of polarities of the target PhACs (0.61 < pKa < 18.52; −0.09 < logKOW < 5.19), two chromatographic methods were applied, one in an acidic medium and another in a basic medium.36 In the acidic method, mobile phase A was a mixture of H2O + 0.01 mM ammonium acetate (NH4Ac) + 0.5% formic acid (HCOOH) (V/V) and mobile phase B was 100% methanol (MeOH). The gradient program started with 95% mobile phase A, followed by a linear decrease to 50% until 3.0 min, 30% until 5.5 min, and 10% until 8.0 min (held for 2.0 min). To re-equilibrate the system, an increase of mobile phase A to 95% was performed in 1.0 min (held for 3.0 min). In the basic method, mobile phase A was an ammonium solution 0.05% (V/V) and mobile phase B was MeOH 100%. The program started with 70% of mobile phase A, which was reduced to 30% within 3.0 min and 10% within 5.0 min (held for 2.0 min). For system re-equilibration, an increase of mobile phase A to 70% was performed in 1.0 min (held for 3.0 min). The injection volume was 20 μL, and the flow rate was 0.3 mL min−1 and 0.5 mL min−1 for acidic and basic methods, respectively.
Both methods were applied for the quantification of all target analytes: 14 PhACs were quantified by the acidic method (ATN, APAP, SDZ, SPD, CAF, SMX, MTPL, PPNL, CTS, CBZ, CFA, NPX, GTD and TTE) and 10 PhACs were quantified by the basic method (BZF, IBUP, DCF, DES, E1, E2, E3, EE2, ERT and FLX).
Mass spectrometry analysis was performed using an electrospray ionization (ESI) source, in positive and negative modes. The operation conditions and the final MS/MS conditions are detailed in previous studies.13,14,37
SPE-UHPLC-MS/MS method
Extraction was performed by the solid-phase extraction (SPE) technique using an automated AutoTrace 280 SPE workstation, Thermo Scientific Dionex (USA), equipped with an adjustable nitrogen stream to dry the cartridge before elution. Oasis HLB (200 mg, 6 mL) cartridges, from Waters (USA) were used for extraction. The SPE method used for extraction of PhACs in wastewaters was based on previous studies in natural (surface and groundwater) and drinking waters.18,37,38 Briefly, the Oasis HLB (200 mg, 6 mL) cartridge was conditioned by passing through 6 mL of methanol and 6 mL of ultra-pure water. Fifty mL of wastewater sample (after filtration steps) was applied to the wet cartridge at a flow rate lower than 5 mL min−1. Then, the cartridge was rinsed with 5 mL water (5 mL min−1) and dried using a nitrogen stream for 15 min. Analytes were eluted with 8 mL of methanol (two elution steps of 4 mL) at a flow rate of 2 mL min−1. The organic extract was evaporated to dryness under a gentle stream of nitrogen (5 psi, 35 °C) in a TurboVap system, reconstituted with 1 mL of ultra-pure water and filtered through a PTFE syringe filter of 0.45 μm.For quantification purposes, the standard addition method was selected. For each sample, a calibration curve with a minimum of six calibration points was constructed by adding 100 μL of the sample to 100 μL of each of the standard solutions prepared in ultra-pure water or 100 μL of ultra-pure water, in the case of the non-spiked calibration point.
The matrix effects and recovery calculations were also evaluated for target matrices (wastewaters).
Validation studies
UHPLC-MS/MS validation
For the selected 24 PhACs, the chromatographic linear range was first studied by analysing 24 concentration levels, in the 0.3–800 μg L−1 range. Calibration curves were plotted for each compound and least-squares regression analysis for measured values of MRM1 transition was applied.Linearity by an external calibration method was evaluated using the coefficient of determination (r2 ≥ 0.9950) and coefficient of variation of the method (CVm ≤ 5%). Furthermore, several statistical tests, such as residual analysis (±15%), the Mandel test (PG ≤ F(1; N − 3; 95%)), the RIKILT test (±10%) and the normalized area test (±15%), were also applied to assess the linearity in the selected concentration range.24,25 These results were treated to comply with all the established requirements for each statistical test. The final choice of the working range was made after the evaluation of the mentioned tests.24,25 In addition, the full concentration range was analysed in three shorter ranges, reflecting a lower, an intermediate and a higher working range.
The instrumental analytical thresholds, namely the limit of detection (LOD) and the limit of quantification (LOQ), were determined based on (i) the calibration curve (LOD = 3 × relative standard deviation of calibration curve (SY/X)/slope; LOQ = 10 × SY/X/slope), (ii) repeatability and (iii) intermediate precision conditions (n = 10 standard samples of each lowest concentration, 1.5–9.3 μg L−1, of the linear range analysed in the same day for repeatability and during a week for intermediate precision). The values of the LOD and LOQ for repeatability and intermediate precision were calculated as 3 × SD (standard deviation) and 10 × SD, respectively.
The repeatability and intermediate precision (n = 10) of the chromatographic method were also evaluated by analysing three concentration levels of the working range: the lowest (1.5–9.3 μg L−1), intermediate (5.1–31 μg L−1) and highest standard concentration (9.2–57 μg L−1) of the linearity range. The repeatability and intermediate precision of the instrumental method were expressed as relative standard deviation, RSDr and RSDR, respectively.
Analysis of variance (ANOVA) was used to evaluate the analysis data (repeatability, reproducibility, and recovery) and significant differences among means were determined by one-way analysis of variance.
Matrix effects and recovery calculations
Matrix effects play a relevant role in the UHPLC-MS/MS analysis, hindering method sensitivity, linearity, precision, and trueness. Matrix effects, such as signal enhancement or suppression, may vary due to matrix complexity, potentially leading to the occurrence of different interferences. To evaluate these effects, wastewater samples were spiked, and the obtained areas (Asw) were compared with those of the injected standard at the same spiking level (As), varying between 102 and 630 μg L−1. The matrix effects were determined using eqn (1).39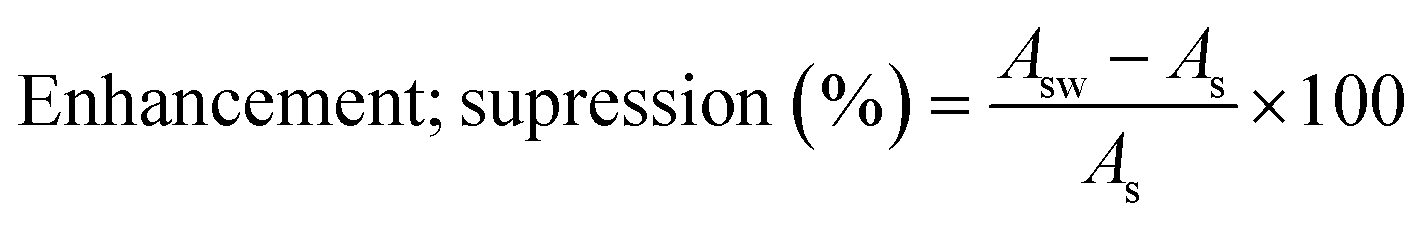 | (1) |
The recovery studies were performed by spiking target wastewaters with two levels of concentration, a low (2.6–16 μg L−1) and high concentration (9.2–57 μg L−1) value within the working range, each condition with ten replicates. During recovery studies, the wastewater before and after spiking was analysed.
The precision of the global SPE-UHPLC-MS/MS method was assessed under repeatability conditions, and it is expressed as a relative standard deviation, RSDr (n = 10).
Analysis of variance (ANOVA) was used to evaluate the analysis data (LODs, LOQs, MDLs, MQLs, repeatability and recovery), and significant differences among means were determined by one-way analysis of variance. Differences corresponding to p-values below 0.05 were statistically significant.
SPE-UHPLC-MS/MS validation
The quantification by the external standard method is not adequate due to matrix interferences. Some target PhACs showed suppression or enhancement effects. To minimize those effects in UHPLC-MS/MS, the standard addition method of calibration (SAC) was chosen for quantification. Even under these conditions, instrumental analysis has problems of response linearity over wide concentration ranges in wastewater matrices. Therefore, the SPE-UHPLC-MS/MS method was validated using wastewater samples at ten spiking levels. All statistical tests described for external calibration were performed again.24,25To complete the validation studies, the uncertainty of the global method was measured.
Uncertainty evaluation
The main uncertainty sources of the method were identified and quantified, followed by the determination of the combined standard uncertainty (uc) using a Gauss propagation model. The last step for uncertainty evaluation of an analytical result was the calculation of the expanded uncertainty (U), using a coverage factor k = 2 (95% of confidence level). Two of the approaches to calculate uncertainty are the ‘bottom-up’ and the ‘top-down’ methods. The ‘bottom-up’ method was proposed by ISO27 to quantify uncertainty in physical measurements and was subsequently adopted by Eurachem.40 On the other hand, the ‘top-down’ method uses validation data or data from proficiency test schemes to estimate the uncertainty of the method.41–43Using the ‘bottom-up’ approach we combined the components of uncertainty related to the calibration curve (ucurve), the standard solution preparation (ustandard), and the intermediate precision of the method (uprecision). The combined uncertainty (uc) was calculated using the following equation:
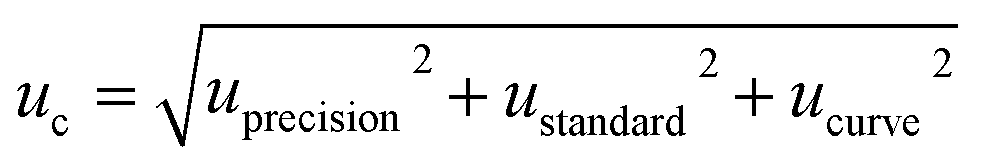 | (2) |
The component uncertainty of the intermediate precision of the analytical method was calculated with eqn (3).
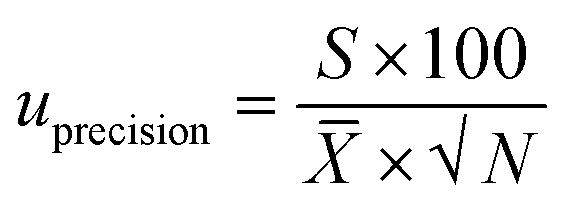 | (3) |
![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) is the mean value of the laboratory's test results.
is the mean value of the laboratory's test results.
The standard uncertainty of the standard solution preparation is a combination of other components of uncertainty such as the purity of the commercial standard and the volumetric and the weight measurements (eqn (4)).
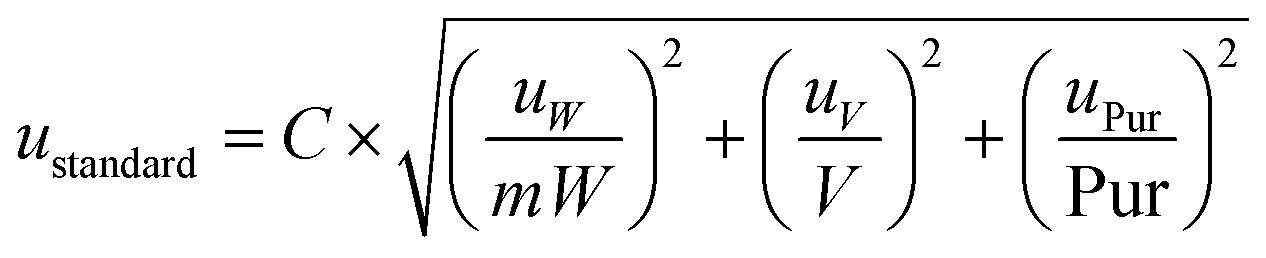 | (4) |
The standard uncertainty of the calibration curve was calculated with eqn (5):
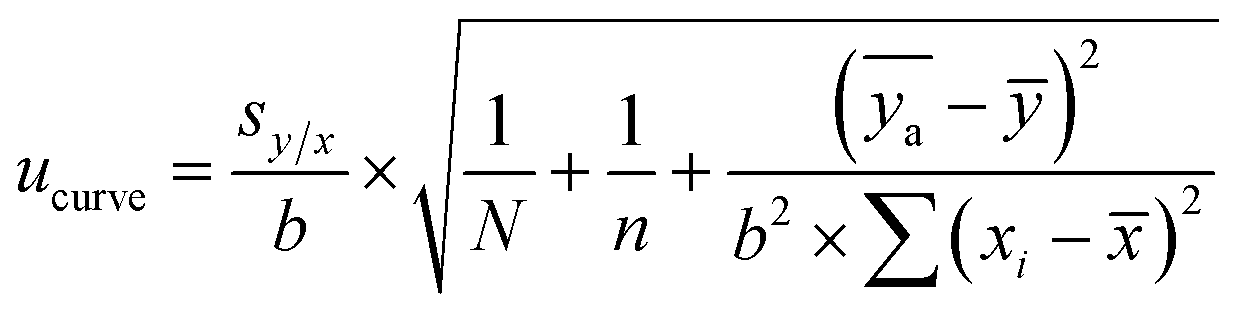 | (5) |
![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) is the mean of the xi values, ȳ is the mean of the y values for the calibration standards, and
is the mean of the xi values, ȳ is the mean of the y values for the calibration standards, and 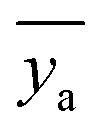 is the mean of N repeat measurements of y for the sample.
is the mean of N repeat measurements of y for the sample.
The ‘top-down’ methodology combined the components of uncertainty related to precision and trueness of the recovery studies (matrix effect). In this approach, the estimation of measurement uncertainty is based on analytical quality control results and validation data, which represent the within-laboratory reproducibility, and the method and laboratory bias. The combined uncertainty (uc) was calculated using eqn (6):
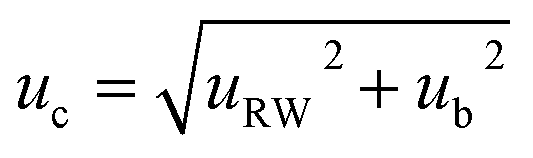 | (6) |
The uncertainty component for the within-laboratory reproducibility (uRW) was calculated as follows:
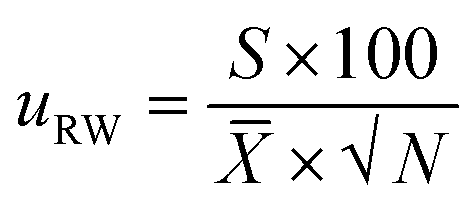 | (7) |
is the mean value of the laboratory's test results.
The uncertainty component from method and laboratory bias (ub) was calculated using eqn (8):
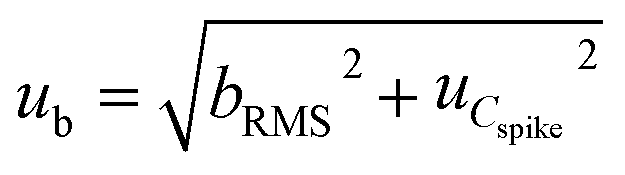 | (8) |
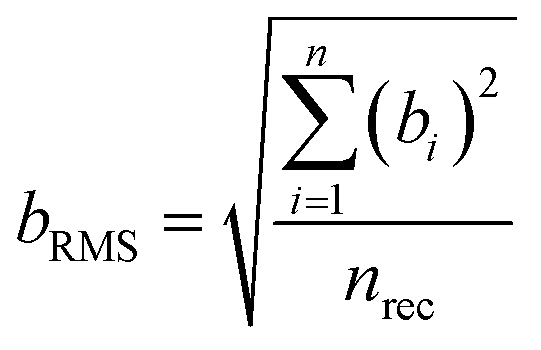 | (9) |
The uncertainty of the concentration of the analyte in the target matrix, (uCspike), consists of three components: the uncertainty of the sample volume (uV); the uncertainty of the added volume, (uV′); and the uncertainty of the concentration solution added (uspike). V and V′ are the sample volume (mL) and the spiked standard volume (mL), respectively.
The uncertainty of the added analyte concentration was determined using eqn (10).
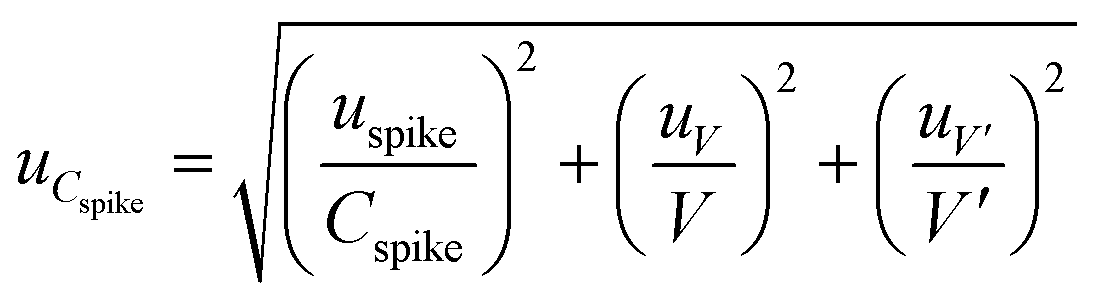 | (10) |
According to the Nordtest approach, the uncertainty of the bias based on recovery assays was determined as follows:44,45
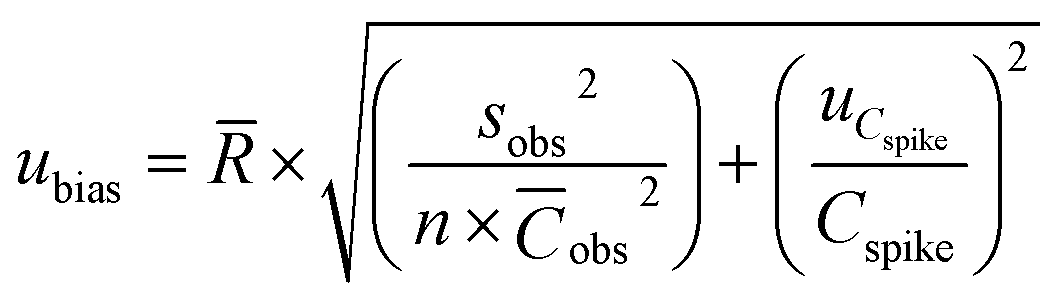 | (11) |
![[R with combining macron]](https://www.rsc.org/images/entities/i_char_0052_0304.gif) is the mean recovery of the analyte in the spiked samples for a target matrix, Sobs is the standard deviation of n recovery data for a certain concentration level, n is the number of recovery assays performed in a target matrix, is the mean concentration of the analyte in the spiked samples (μg L−1), uCspike is the uncertainty of the analyte concentration in the target matrix and Cspike is the analyte concentration in the spiked sample (μg L−1).
is the mean recovery of the analyte in the spiked samples for a target matrix, Sobs is the standard deviation of n recovery data for a certain concentration level, n is the number of recovery assays performed in a target matrix, is the mean concentration of the analyte in the spiked samples (μg L−1), uCspike is the uncertainty of the analyte concentration in the target matrix and Cspike is the analyte concentration in the spiked sample (μg L−1).
In both approaches, the expanded uncertainty (U) was obtained by multiplying uc by a coverage factor (k) using the following equation:27
| U = k × uc | (12) |
Usually, the value of the coverage factor k is chosen based on the desired level of confidence to be associated with the interval defined by uc. Typically, k is in the range of 2 to 3. When the normal distribution applies and uc is a reliable estimate of the standard deviation, U = 2 × uc (i.e., k = 2) defines an interval having a level of confidence of approximately 95%, and U = 3 × uc (i.e., k = 3) defines an interval having a level of confidence greater than 99%.27
Results and discussion
Chromatographic validation
For the low concentration range (1.5–10 μg L−1, Table S2†), all PhACs showed r2 ≥ 0.9980 (above the acceptable limit of 0.9950) and CVm ≤ 2.5% (limit of 5.0%). In addition, all the requirements of the statistical tests performed were fulfilled, confirming the methods' linearity in the low concentration range. Despite the excellent results, ERT and FLX showed an irregular behavior (peak shape) for two concentration levels, leading to peak area fluctuations that hindered the linearity. Therefore, these two concentration levels were removed.
Similar results were obtained for the medium and high concentration ranges (10–100 μg L−1 and 100–800 μg L−1, Tables S3 and S4† respectively). Most of the studied PhACs showed r2 ≥ 0.9980 and CVm ≤ 2.5. The only exception was ERT (r2 = 0.9977; CVm = 5.22), which had the issue of peak shape mentioned earlier. Observing the results obtained in the applied statistical tests, several compounds failed to meet the established requirements in the high concentration range, particularly the RIKILT test (APAP, SPD, MTPL, DCF, EE2 and FLX) and the normalized area test (APAP and FLX). These results suggest that this concentration range is not suitable, and it should be reduced in the future if one intends to use this concentration range. In the medium concentration range, the statistical requirements were fulfilled for most of the compounds, confirming the linearity of the methods in this concentration range. The only exceptions were APAP (failed the RIKILT and the normalized area tests), ERT (failed the normalized area test) and FLX (failed the RIKILT test). Therefore, in these working ranges, for these compounds, we need to perform a calibration using a calibration curve and avoid the use of a response factor. Overall, the results showed that the selected low concentration range is the best suited linear range for PhAC analysis, showing excellent values of r2 and CVm, and fulfilling all the requirements in terms of statistical tests.24,25 Therefore, this concentration range will be our platform for all the subsequent studies. Furthermore, the medium concentration range also proved its suitability for quantifying most of the target PhACs, if one needs to measure higher concentrations.
| PhACs | Lowest conc. of the working range (μg L−1) | Calibration curve | Repeatability | Intermediate precision | |||
|---|---|---|---|---|---|---|---|
| LOD (μg L−1) | LOQ (μg L−1) | LOD (μg L−1) | LOQ (μg L−1) | LOD (μg L−1) | LOQ (μg L−1) | ||
| ATN | 1.60 | 0.20 | 0.68 | 0.20 | 0.65 | 0.19 | 0.63 |
| APAP | 1.54 | 0.11 | 0.37 | 0.09 | 0.31 | 0.13 | 0.42 |
| SDZ | 1.56 | 0.25 | 0.84 | 0.11 | 0.36 | 0.12 | 0.40 |
| SPD | 1.53 | 0.22 | 0.72 | 0.11 | 0.37 | 0.17 | 0.47 |
| CAF | 1.57 | 0.23 | 0.78 | 0.09 | 0.32 | 0.17 | 0.58 |
| SMX | 1.59 | 0.33 | 1.09 | 0.07 | 0.24 | 0.15 | 0.51 |
| MTPL | 1.56 | 0.35 | 1.16 | 0.19 | 0.64 | 0.24 | 0.78 |
| PPNL | 1.55 | 0.25 | 0.82 | 0.25 | 0.82 | 0.25 | 0.82 |
| CTS | 1.54 | 0.21 | 0.69 | 0.21 | 0.69 | 0.21 | 0.71 |
| CBZ | 1.58 | 0.15 | 0.49 | 0.11 | 0.38 | 0.09 | 0.29 |
| CFA | 9.26 | 1.95 | 6.49 | 2.17 | 7.23 | 2.16 | 7.19 |
| NPX | 1.70 | 0.16 | 0.54 | 0.12 | 0.41 | 0.15 | 0.49 |
| GTD | 1.61 | 0.36 | 1.19 | 0.16 | 0.53 | 0.19 | 0.63 |
| TTE | 1.57 | 0.15 | 0.48 | 0.12 | 0.38 | 0.11 | 0.38 |
| BZF | 1.66 | 0.10 | 0.33 | 0.12 | 0.39 | 0.14 | 0.47 |
| IBUP | 3.23 | 0.20 | 0.67 | 0.29 | 0.97 | 0.58 | 1.93 |
| DCF | 1.72 | 0.13 | 0.43 | 0.13 | 0.44 | 0.27 | 0.91 |
| E3 | 8.57 | 0.76 | 2.53 | 0.86 | 2.87 | 1.04 | 3.46 |
| E1 | 8.66 | 1.04 | 3.48 | 0.65 | 2.18 | 0.76 | 2.54 |
| E2 | 9.43 | 0.67 | 2.22 | 1.08 | 3.59 | 0.70 | 2.34 |
| EE2 | 9.00 | 1.23 | 4.11 | 1.24 | 4.15 | 0.94 | 3.13 |
| DES | 8.49 | 1.42 | 4.75 | 1.02 | 3.41 | 1.23 | 4.10 |
| ERT | 1.61 | 0.31 | 1.04 | 0.40 | 1.34 | 0.47 | 1.58 |
| FLX | 1.64 | 0.15 | 0.51 | 0.29 | 0.97 | 0.32 | 1.08 |
All LOQs were lower than the first concentration level of the working range and no statistically significant differences were observed between the LOQs obtained by the different approaches (p < 0.05). Although the requirement of the determined LOQs was accomplished (LOQ lower than the first level of the working range), routinely the value of the LOQ is established at the first concentration level, because quantification can only be performed inside the working range to ensure higher accuracy in the results.
| PhACs | Low concentration | Intermediate concentration | High concentration | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Conc. (μg L−1) | r (RSDr, %) | R (RSDR, %) | Conc. (μg L−1) | r (RSDr, %) | R (RSDR, %) | Conc. (μg L−1) | r (RSDr, %) | R (RSDR, %) | |
| ATN | 1.6 | 3.98 | 3.91 | 5.35 | 3.32 | 3.29 | 9.63 | 1.39 | 2.36 |
| APAP | 1.5 | 2.07 | 2.75 | 5.12 | 1.43 | 1.61 | 9.22 | 0.39 | 0.94 |
| SDZ | 1.56 | 2.30 | 2.54 | 5.20 | 0.84 | 0.87 | 9.36 | 0.55 | 0.90 |
| SPD | 1.53 | 2.39 | 2.97 | 5.10 | 0.96 | 0.82 | 9.18 | 0.61 | 0.99 |
| CAF | 1.57 | 1.98 | 3.58 | 5.24 | 0.89 | 1.31 | 9.44 | 0.60 | 1.36 |
| SMX | 1.59 | 1.45 | 3.20 | 5.32 | 0.96 | 1.35 | 9.57 | 0.55 | 1.36 |
| MTPL | 1.56 | 4.49 | 5.22 | 5.21 | 2.62 | 2.11 | 9.39 | 1.32 | 1.17 |
| PPNL | 1.55 | 5.75 | 5.48 | 5.16 | 4.20 | 3.09 | 9.29 | 1.87 | 2.47 |
| CTS | 1.54 | 4.23 | 4.51 | 5.12 | 2.78 | 2.13 | 9.22 | 1.55 | 1.28 |
| CBZ | 1.58 | 2.50 | 1.90 | 5.28 | 1.17 | 1.50 | 9.50 | 0.91 | 2.75 |
| CFA | 9.26 | 8.63 | 7.88 | 30.9 | 2.81 | 4.57 | 55.5 | 2.43 | 2.53 |
| NPX | 1.70 | 2.31 | 2.79 | 5.67 | 2.57 | 3.17 | 10.2 | 0.82 | 0.89 |
| GTD | 1.61 | 3.13 | 3.72 | 5.38 | 1.56 | 1.38 | 9.69 | 1.11 | 2.43 |
| TTE | 1.57 | 2.39 | 2.34 | 5.25 | 1.60 | 1.74 | 9.45 | 0.68 | 1.06 |
| BZF | 1.66 | 2.47 | 2.90 | 5.54 | 1.39 | 2.12 | 9.97 | 0.66 | 1.83 |
| IBUP | 3.23 | 3.34 | 6.12 | 10.8 | 2.80 | 2.29 | 19.4 | 1.03 | 2.66 |
| DCF | 1.72 | 2.36 | 5.08 | 5.72 | 1.00 | 2.44 | 10.3 | 0.34 | 1.68 |
| E3 | 8.57 | 3.50 | 4.08 | 28.6 | 1.83 | 4.09 | 51.4 | 1.28 | 7.64 |
| E1 | 8.66 | 2.54 | 2.90 | 28.9 | 1.34 | 2.30 | 51.9 | 0.91 | 5.03 |
| E2 | 9.43 | 3.70 | 2.38 | 31.4 | 1.49 | 3.37 | 56.6 | 1.45 | 5.35 |
| EE2 | 9.00 | 4.52 | 3.45 | 30.0 | 2.58 | 3.48 | 54.0 | 1.86 | 4.00 |
| DES | 8.49 | 4.13 | 4.79 | 28.3 | 4.28 | 3.49 | 50.9 | 1.68 | 6.03 |
| ERT | 1.61 | 10.6 | 10.1 | 5.36 | 10.1 | 9.48 | 9.64 | 4.85 | 4.47 |
| FLX | 1.64 | 8.14 | 6.94 | 5.46 | 10.4 | 4.18 | 9.82 | 6.57 | 2.98 |
In conclusion, the optimized SPE-UHPLC-MS/MS methods proved suitable for the quantification of the target PhACs. Moreover, the selected working range (low concentration range: between 1.5 and 10 μg L−1 for PhCs; and 9–57 μg L−1 for hormones and CFA), showed excellent linearity with determination coefficients (r2) between 0.9984 and 0.9999 and coefficients of variation (CVm) lower than 3%. In addition, the methods are precise, with a relative standard deviation below 11% and 10% under repeatability and intermediate precision conditions, respectively. Therefore, the optimized UHPLC-MS/MS methods were validated and considered suitable for their application to wastewater analysis. Further studies were performed to evaluate the matrix effects.
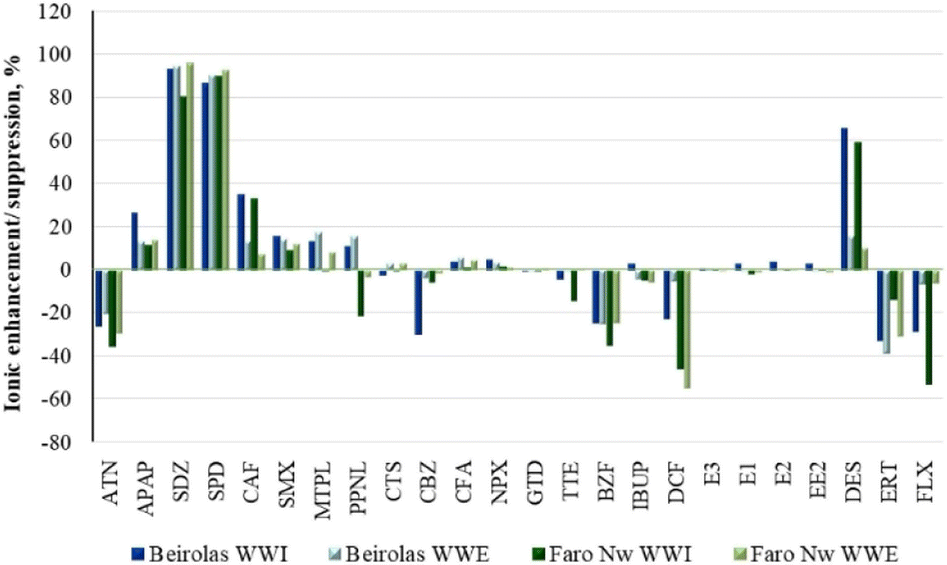 | ||
| Fig. 1 Ionic enhancement/suppression of PhACs in wastewaters by UHPLC-MS/MS. | ||
APAP, SDZ, SPD, CAF, SMX, PPNL, and DES were subjected to ionic enhancement. The highest values (>80%) were obtained with DZ and SPD. In addition, higher enhancement values were also observed for CAF and DES in the influents of both WWTPs (BEI-WWI and FNw-WWI).
Suppression effects were observed for ATN, BZF, DCF, ERT and FLX, with values below 55%. Except for DES, no apparent ionic effects were detected for hormones.
No statistically significant differences were observed between target wastewaters in ion enhancement/suppression (p > 0.05).
To minimize these matrix effects, the quantification of PhACs was performed with standard addition calibration curves.46 Although deuterated compounds are the most common approach, the number of target compounds limits their application in this study. Furthermore, the deuterated compound approach is not enough to overcome all interferences due to the complexity and variability of the influent matrices.
| PhACs | N | Conc. (μg L−1) | r 2 | CVm (%) | Norm (%) [85–115] | Residues (%) [85–115] | RIKILT (%) [90–110] | Mandel test (PG ≤ F(0.05; 1; N − 3)) |
|---|---|---|---|---|---|---|---|---|
| ATN | 7 | 1.6–9.6 | 0.9989 | 0.80 | [98–100] | [−1.1; 1.1] | [99–101] | 1.51 ≤ 7.71 |
| APAP | 5 | 1.5–9.2 | 0.9998 | 1.05 | [99–102] | [−1.2; 0.9] | [98–101] | 4.61 ≤ 18.51 |
| SDZ | 6 | 1.6–9.4 | 0.9998 | 0.96 | [97–100] | [−1.4; 1.6] | [99–102] | 0.05 ≤ 10.13 |
| SPD | 7 | 1.5–9.2 | 0.9984 | 1.37 | [99–103] | [−1.6; 2.3] | [98–102] | 1.02 ≤ 7.71 |
| CAF | 6 | 1.6–9.4 | 0.9998 | 0.59 | [99–100] | [−0.9; 0.7] | [99–101] | 0.87 ≤ 10.13 |
| SMX | 7 | 1.6–9.6 | 0.9992 | 0.67 | [100–102] | [−0.9; 0.9] | [99–101] | 5.02 ≤ 7.71 |
| MTPL | 7 | 1.6–9.4 | 0.9999 | 0.59 | [99–101] | [−1.4; 0.8] | [99–101] | 0.46 ≤ 7.71 |
| PPNL | 7 | 1.6–9.3 | 0.9987 | 2.19 | [92–109] | [−7.5; 3.5] | [93–104] | 3.24 ≤ 7.71 |
| CTS | 6 | 1.5–9.2 | 0.9984 | 1.69 | [98–108] | [−2.1; 1.9] | [94–104] | 3.13 ≤ 10.13 |
| CBZ | 6 | 1.6–9.5 | 0.9989 | 0.71 | [100–102] | [−0.7; 0.9] | [99–101] | 2.69 ≤ 10.13 |
| CFA | 6 | 9.3–56 | 0.9992 | 1.84 | [93–103] | [−2.3; 1.9] | [96–106] | 1.12 ≤ 10.13 |
| NPX | 6 | 1.7–10 | 0.9999 | 0.77 | [98–101] | [−1.3; 2.4] | [98–102] | 6.14 ≤ 10.13 |
| GTD | 5 | 1.6–9.7 | 0.9991 | 2.19 | [96–103] | [−2.2; 2.8] | [97–104] | 1.15 ≤ 18.51 |
| TTE | 6 | 1.6–9.5 | 0.9990 | 1.81 | [100–109] | [−3.0; 2.3] | [95–103] | 0.39 ≤ 10.13 |
| BZF | 7 | 1.7–10 | 0.9987 | 1.31 | [99–102] | [−2.3; 1.4] | [98–101] | 0.82 ≤ 7.71 |
| IBUP | 6 | 3.2–19 | 0.9989 | 2.23 | [92–100] | [−2.2; 6.3] | [97–106] | 0.72 ≤ 10.13 |
| DCF | 5 | 1.7–10 | 0.9997 | 1.31 | [97–104] | [−4.8; 2.3] | [96–103] | 1.07 ≤ 18.51 |
| E3 | 7 | 8.6–51 | 0.9994 | 1.77 | [97–105] | [−3.2; 4.8] | [96–104] | 0.04 ≤ 7.71 |
| E1 | 7 | 8.7–52 | 0.9994 | 1.88 | [88–100] | [−2.5; 10.2] | [96–109] | 0.00 ≤ 7.71 |
| E2 | 6 | 9.4–57 | 0.9989 | 2.13 | [98–105] | [−3.4; 3.8] | [97–104] | 0.65 ≤ 10.13 |
| EE2 | 5 | 9.0–54 | 0.9972 | 3.36 | [100–108] | [−3.6; 3.5] | [97–104] | 0.02 ≤ 18.51 |
| DES | 6 | 8.5–51 | 0.9984 | 1.53 | [95–100] | [−1.5; 3.6] | [99–103] | 2.06 ≤ 10.13 |
| ERT | 5 | 1.6–9.6 | 0.9975 | 3.34 | [91–106] | [−6.9; 8.1] | [93–108] | 0.09 ≤ 18.51 |
| FLX | 6 | 1.6–9.8 | 0.9976 | 3.10 | [97–110] | [−6.0; 5.9] | [93–105] | 0.27 ≤ 10.13 |
| PhACs | N | Conc. (μg L−1) | r 2 | CVm (%) | Norm (%) [85–115] | Residues (%) [85–115] | RIKILT (%) [90–110] | Mandel test (PG ≤ F(0,05; 1; N − 3)) |
|---|---|---|---|---|---|---|---|---|
| ATN | 6 | 1.6–9.6 | 0.9993 | 2.1 | [99–110] | [−8.7; 1.6] | [93–103] | 5.49 ≤ 10.13 |
| APAP | 5 | 1.5–9.2 | 0.9998 | 0.59 | [100–101] | [−1.0; 0.5] | [99–100] | 0.04 ≤ 18.51 |
| SDZ | 7 | 1.6–9.4 | 0.9988 | 1.7 | [95–100] | [−2.0; 3.0] | [98–103] | 0.01 ≤ 7.71 |
| SPD | 6 | 1.5–9.2 | 0.9997 | 0.38 | [100–101] | [−0.5; 0.3] | [99–100] | 0.00 ≤ 10.13 |
| CAF | 6 | 1.6–9.4 | 0.9998 | 0.56 | [99–101] | [−0.5; 0.6] | [100–101] | 2.12 ≤ 10.13 |
| SMX | 7 | 1.6–9.6 | 0.9991 | 0.74 | [98–100] | [−0.9; 1.5] | [99–102] | 4.51 ≤ 7.71 |
| MTPL | 7 | 1.6–9.4 | 0.9998 | 0.67 | [99–102] | [−1.2; 1.6] | [99–102] | 0.16 ≤ 7.71 |
| PPNL | 7 | 1.6–9.3 | 0.9993 | 1.7 | [95–102] | [−2.2; 5.1] | [97–105] | 0.00 ≤ 7.71 |
| CTS | 7 | 1.5–9.2 | 0.9975 | 2.2 | [91–100] | [−4.0; 5.1] | [96–105] | 1.10 ≤ 7.71 |
| CBZ | 7 | 1.6–9.5 | 0.9990 | 0.79 | [99–101] | [−0.8; 0.9] | [99–101] | 0.38 ≤ 7.71 |
| CFA | 7 | 9.3–56 | 0.9990 | 1.7 | [95–104] | [−4.0; 5.9] | [96–106] | 0.14 ≤ 7.71 |
| NPX | 6 | 1.7–10 | 0.9993 | 1.5 | [97–104] | [−3.2; 3.6] | [97–103] | 0.54 ≤ 10.13 |
| GTD | 6 | 1.6–9.7 | 0.9991 | 1.0 | [98–100] | [−0.8; 1.2] | [99–101] | 0.25 ≤ 10.13 |
| TTE | 7 | 1.6–9.5 | 0.9988 | 2.2 | [91–102] | [−2.6; 10.1] | [97–109] | 1.60 ≤ 7.71 |
| BZF | 7 | 1.7–10 | 0.9974 | 1.2 | [97–100] | [−1.0; 2.1] | [99–102] | 1.49 ≤ 7.71 |
| IBUP | 6 | 3.2–19 | 0.9994 | 1.3 | [97–103] | [−2.4; 3.5] | [97–103] | 1.88 ≤ 10.13 |
| DCF | 6 | 1.7–10 | 0.9997 | 1.5 | [100–103] | [−1.2; 1.6] | [99–102] | 1.51 ≤ 10.13 |
| E3 | 7 | 8.6–51 | 0.9990 | 2.2 | [98–104] | [−3.3; 2.6] | [97–103] | 0.26 ≤ 7.71 |
| E1 | 7 | 8.7–52 | 0.9991 | 1.9 | [97–101] | [−1.7; 2.7] | [98–103] | 0.22 ≤ 7.71 |
| E2 | 7 | 9.4–57 | 0.9994 | 1.6 | [94–104] | [−3.6; 6.4] | [96–106] | 0.21 ≤ 7.71 |
| EE2 | 6 | 9.0–54 | 0.9984 | 2.7 | [96–105] | [−4.4; 4.2] | [95–105] | 2.55 ≤ 10.13 |
| DES | 6 | 8.5–51 | 0.9993 | 1.7 | [97–105] | [−4.6; 3.6] | [95–101] | 0.10 ≤ 10.13 |
| ERT | 5 | 1.6–9.6 | 0.9972 | 2.4 | [100–106] | [−3.0; 2.4] | [97–102] | 0.13 ≤ 18.51 |
| FLX | 6 | 1.6–9.8 | 0.9992 | 3.10 | [97–110] | [−6.0; 5.9] | [93–105] | 0.27 ≤ 10.13 |
The concentration range of the standard addition calibration curves applied to the wastewater extracts fulfilled all the statistical requirements for a linear method. For all compounds, a well-defined working range was obtained, characterized by r2 ≥ 0.9972, CVm ≤ 4.0%, PG ≤ F(95%, 1, N − 3), residual analysis and normalized values < 15%, and RIKILT test values < 10%.24,25
Tables S9–S10† compare the analytical limits (LOD and LOQ) based on calibration curves and repeatability. Due to the high values of standard deviation of PhACs by SPE-UHPLC-MS/MS (15% ≤ RSD ≤ 30%), the LOD and LOQ values determined under repeatability conditions were much higher than those obtained with calibration curves. The LOQ based on calibration curves is close to or lower than the lowest concentration level of the working range. Therefore, these limits are the correct ones but, in routine analysis, the LOQ value was defined at the first point of the calibration curve. The instrumental LOQs of UHPLC-MS/MS for wastewater influents (WWIs) were corrected with a concentration factor of 50, due to the wastewater concentration by SPE.
These corrected values, corresponding to the method quantification limit (MQL) of the SPE-UHPLC-MS/MS method, were tested in routine analysis with a daily control standard. The method detection limit was obtained with the ratio MQL/3.3. Both limits are in Table 5.
| Compound | Lowest conc. of working range (μg L−1) | MDL (ng L−1) | MQL (ng L−1) | Compound | Lowest conc. of working range (μg L−1) | MDL (ng L−1) | MQL (ng L−1) |
|---|---|---|---|---|---|---|---|
| ATN | 1.60 | 9.7 | 32 | GTD | 1.61 | 9.8 | 32 |
| APAP | 1.54 | 9.3 | 31 | TTE | 1.57 | 9.5 | 31 |
| SDZ | 1.56 | 9.5 | 31 | BZF | 1.66 | 10 | 33 |
| SPD | 1.53 | 9.3 | 31 | IBUP | 3.23 | 20 | 65 |
| CAF | 1.57 | 9.5 | 31 | DCF | 1.72 | 10 | 34 |
| SMX | 1.59 | 9.6 | 32 | E3 | 8.57 | 52 | 171 |
| MTPL | 1.56 | 9.5 | 31 | E1 | 8.66 | 52 | 173 |
| PPNL | 1.55 | 9.4 | 31 | E2 | 9.43 | 57 | 189 |
| CTS | 1.54 | 9.3 | 31 | EE2 | 9.00 | 55 | 180 |
| CBZ | 1.58 | 9.6 | 32 | DES | 8.49 | 51 | 170 |
| CFA | 9.26 | 56 | 185 | ERT | 1.61 | 9.8 | 32 |
| NPX | 1.70 | 10 | 34 | FLX | 1.64 | 9.9 | 33 |
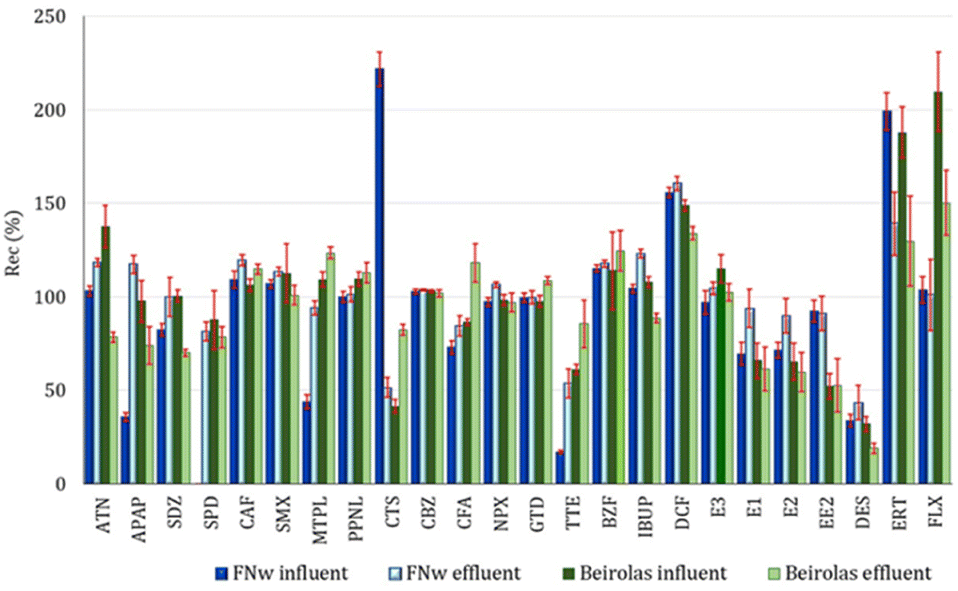 | ||
| Fig. 2 Absolute recovery of PhACs in wastewaters from Beirolas and FNw WWTPs by UHPLC-MS/MS at low spiking concentrations (n = 10). | ||
At Beirolas, ERT and FLX were the PhACs with the highest recoveries for both wastewater matrices (influent and effluent) and for both spiking concentration levels. The signal and peak forms were not constant, mainly for erythromycin. Due to this different behaviour, ERT and FLX were not statistically evaluated with the remaining PhCs and hormones. The average recoveries were 86%, 87%, 85% and 84% for the influent and effluent of a low spiking concentration and influent and effluent of a high spiking concentration, respectively. The hormones (TTE, E1, E2 and EE2) showed recovery values lower than those of the remaining target compounds, with recoveries lower than 60% for all studied matrices.
Regarding the Beirolas influent (low spiking concentrations), the average recovery values were between 28.3% and 158% for TTE and ATN, respectively. For a high spiking concentration, the recoveries varied between 34.5% and 147% for TTE and DCF, respectively.
In the Beirolas effluent, the lowest and highest recovery values belonged to the same PhCs, EE2 and DCF, with values varying between 30.6% and 139% and 22.2% and 129%, for low and high spiking concentrations, respectively (Table S11†).
Some compounds (APAP, SDZ, CTS, CFA, TTE, and BZF) showed higher recoveries in effluents than influents due to the lower matrix effects.
Although there were no significant statistical differences between the RSD values obtained in influent and effluent wastewater at both spiking concentrations, the influent with a low spiking concentration showed an average RSD slightly higher (12.6%) than the average RSD of the influent with a high spiking concentration (8.2%).
Homogeneity of variance analysis was applied to the best (effluents at both spiking concentrations) and the worst (influents at both spiking concentrations) recovery data from different matrices. However, the results showed no significant differences. All obtained test values (PG) were lower than the tabulated value of the Fisher/Snedecor distribution (F(95%; N − 1; N − 1)).
Regarding Faro Nw (Table S12†), the average recoveries were 93%, 86%, 90% and 89% for the influent and effluent of a low spiking concentration and influent and effluent of a high spiking concentration, respectively.
As observed in Beirolas wastewater samples, the hormones (TTE, E1, E2 and EE2) showed recovery values lower than the remaining target compounds, lower than 60%, for all studied matrices, except for effluent wastewater with high spiking concentration.
The recovery profile was quite similar at both spiking concentrations. At low spiking concentrations, the recoveries varied between 17.2% (TTE) and 221% (CTS). Most PhACs (ATN, SDZ, CAF, SMX, MTPL, PPNL, CBZ, CFA, NPX, GTD, BZF, IBUP, E1, E2, E3, EE2 and FLX) showed good recovery percentages, with values between 60% and 120%. The hormones (TTE and DES), APAP, SPD and MTPL showed recovery values lower than 60%, whereas DCF, ERT and CTS showed recoveries higher than 140%.
At high spiking concentrations, the recoveries ranged between 26.0% (TTE) and 255% (FLX). The PhACs, ATN, APAP, SDZ, CAF, SMX, PPNL, CTS, CBZ, CFA, NPX, GTD, BZF, IBUP, E2, E1, EE2 and ERT showed good recoveries with values between 60% and 125%. The hormones (TTE, E1, and DES), MTPL and SPD showed recovery values lower than 60%. DCF and FLX showed recoveries higher than 140%.
Homogeneity of variance analysis was applied to the best (effluents with both spiking concentrations) and the worst (influents with both spiking concentrations) recovery data from different matrices. However, the results showed no significant differences between the recoveries. All test values (PG) obtained were lower than the tabulated value of the Fisher/Snedecor distribution (F(95%; N − 1; N − 1)). Homogeneity of variance analysis was also applied to the wastewaters with the same spiking concentration (influent and effluent). The results showed no significant differences between the recoveries obtained with low spiking concentrations. Still, there were significant differences between the recoveries obtained for influent and effluent wastewater with high spiking concentrations (PG > tabulated value of Fisher/Snedecor distribution).
Although recoveries higher than 60% are desirable, lower recoveries, even those lower than 40%, can be acceptable if their repeatability is good enough to obtain as much information as possible about the presence of PhACs in wastewaters.
Most of the target compounds showed RSD values lower than 15% and only erythromycin presented an RSD higher than 25% (28.2%). Therefore, the optimized SPE-UHPLC-MS/MS method fulfills the above requirements.
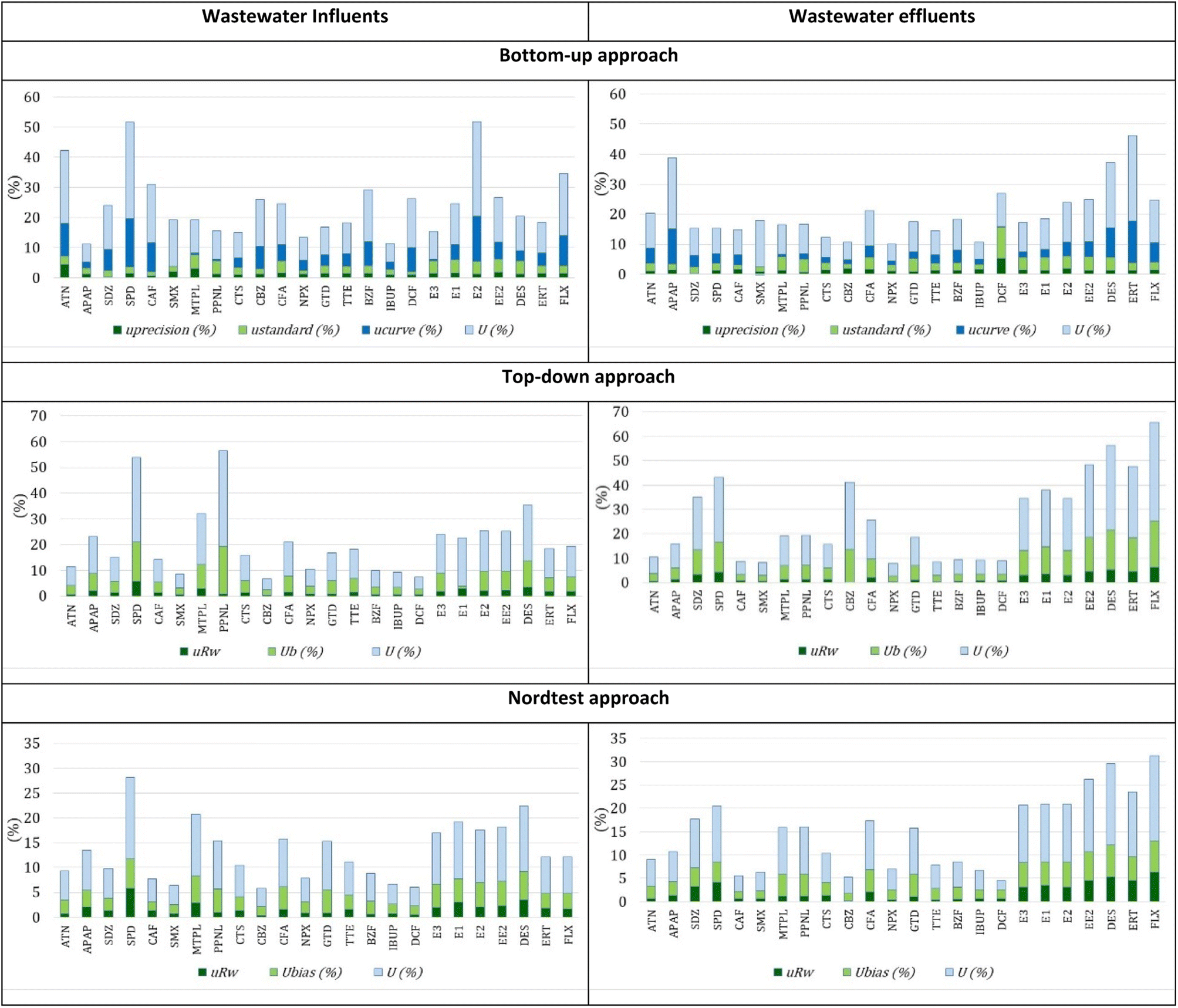 | ||
| Fig. 3 Individual uncertainty components and respective expanded uncertainty estimated in the analysis of the 24 PhACs in wastewater influents and effluents of the Faro WWTP by SPE-UHPLC-MS/MS using three approaches for uncertainty quantification. | ||
Regarding wastewater influents from Beirolas WWTPs, the estimation of uncertainty of SPE-UHPLC-MS/MS by bottom-up, top-down and Nordtest approaches was between 3.8–22.5%, 5.2–46.3% and 3.8–22.1%, respectively. For wastewater effluents, the estimation of uncertainty for these three approaches was between 4.7–24.1%, 5.1–41.9% and 3.3–21.0%, respectively.
Regarding wastewater influents from Faro WWTPs, the estimation of uncertainty of SPE-UHPLC-MS/MS by bottom-up, top-down and Nordtest approaches was between 6.1–32.2%, 4.2–37.1% and 3.6–16.5%, respectively. For wastewater effluents, the estimation of uncertainty for these three approaches was between 5.5–28.4%, 5.0–40.2% and 2.0–18.2%, respectively. Regardless of the approach used, the combined uncertainty is very similar for the two types of wastewaters, influents and effluents, from both WWTPs.
The uncertainty measurements also fulfill the 50% criteria established by the document “Analytical quality control and method validation procedures for pesticide residues analysis in food and feed”, SANTE 11312/2021. This document can also be used as a reference in water analysis of organic compounds, such as PhACs, at trace concentrations.47
These results are much lower than those reported for NSAIDs in natural waters based on on-line (U = 15–36%) and off-line SPE (U = 18–64%) using the LC-DAD-MS technique,48 and those reported for antibiotics in seawater using SPE disks and LC-MS/MS (U = 4.2–69.7%).22
However, they were quite similar to those reported by other authors.48 These authors obtained values from 8.5 to 29.0% for NSAIDs and antiepileptic drugs in rivers by solid-phase microextraction coupled to LC-DAD.
Our previously uncertain data for endocrine hormone disrupters (hormones and alkylphenols) with bottom-up and top-down approaches are obtained in quantifying these target compounds in natural waters (groundwater and surface water) and drinking water by SPE-LC-MS/MS. The expanded uncertainty of the method was also evaluated using top-bottom methodology for the same target PhACs and methodology (SPE-UHPLC-MS/MS) but with different wastewaters.15 The uncertainty values (U) ranged between 13 and 41%.
These differences are due to the different approaches used to estimate this metrological parameter, different target PhACs, water matrices or analytical methods, for sample preparation and/or for quantification.
Regardless of the approach used, the combined uncertainty is very similar for the two types of wastewaters, influent and effluent, from both WWTPs. The main components of uncertainty are identical for both types of water, with the main discrimination factor being the type of PhAC.
The differences between values were due to the main component of uncertainty in each approach. These differences are consistent with those obtained by other authors, since the main uncertainty differences are due to the sample matrices, method quantification or different uncertainty approaches which included different sources of uncertainty for calculating the combined value.48–50
In the bottom-up approach, the most significant uncertainty component is the calibration curve. Although the quantification protocol is carried out using the standard addition method, the matrix significantly influences the behavior of the curves throughout the various work series, namely on the slope.
In the top-down approach, the most significant component of uncertainty is bias. The effects of the matrix and consequently of the recovery results are the most considerable components of the uncertainty.
In the Nordtest approach, although the most significant uncertainty component is also associated with matrix effects (recovery), the values are lower due to the bias determination formula. The minor uncertainty component is always associated with precision for any approaches used.
| PhAC | Influents from Beirolas WWTP | Effluents from Beirolas WWTP | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μg L−1 | Uncertainty approach | SD | μg L−1 | Uncertainty approach | SD | |||||||
| Bottom-up | Top-down | Nordtest | Bottom-up | Top-down | Nordtest | |||||||
| a n.d. – not detected. | ||||||||||||
| ATN | 0.521 | ± | 0.056 | 0.091 | 0.049 | 0.004 | 0.0612 | ± | 0.0042 | 0.0052 | 0.0038 | 0.0006 |
| APAP | 28.6 | ± | 1.1 | 6.7 | 3.9 | 0.2 | n.d. | — | — | — | — | |
| SDZ | 0.0136 | ± | 0.0007 | 0.0011 | 0.0007 | 0.0001 | 0.0124 | ± | 0.0008 | 0.0009 | 0.0006 | 0.00002 |
| SPD | 0.259 | ± | 0.032 | 0.082 | 0.043 | 0.008 | 0.216 | ± | 0.014 | 0.043 | 0.020 | 0.001 |
| CAF | 22.7 | ± | 3.3 | 1.6 | 0.9 | 0.136 | 0.329 | ± | 0.070 | 0.017 | 0.011 | 0.003 |
| SMX | 0.45 | ± | 0.04 | 0.13 | 0.06 | 0.010 | 0.206 | ± | 0.010 | 0.022 | 0.012 | 0.002 |
| MTPL | 0.032 | ± | 0.002 | 0.004 | 0.003 | 0.0004 | 0.0278 | ± | 0.0028 | 0.0029 | 0.0026 | 0.0002 |
| PPNL | 0.034 | ± | 0.003 | 0.004 | 0.003 | 0.0003 | 0.0293 | ± | 0.0028 | 0.0078 | 0.0043 | 0.0003 |
| CTS | 0.035 | ± | 0.006 | 0.003 | 0.002 | 0.0004 | n.d. | — | — | — | — | |
| CBZ | 0.337 | ± | 0.016 | 0.091 | 0.012 | 0.005 | 0.368 | ± | 0.022 | 0.100 | 0.013 | 0.007 |
| CFA | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| NPX | 3.35 | ± | 0.25 | 0.20 | 0.16 | 0.037 | 0.078 | ± | 0.004 | 0.009 | 0.005 | 0.001 |
| GTD | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| TTE | 0.014 | ± | 0.001 | 0.002 | 0.001 | 0.0002 | n.d. | — | — | — | — | |
| BZF | 0.216 | ± | 0.029 | 0.014 | 0.012 | 0.011 | 0.0380 | ± | 0.0092 | 0.0067 | 0.0035 | 0.0003 |
| IBUP | 5.58 | ± | 0.35 | 0.37 | 0.24 | 0.056 | n.d. | — | — | — | — | |
| DCF | 1.007 | ± | 0.182 | 0.052 | 0.039 | 0.010 | 1.39 | ± | 0.27 | 0.08 | 0.06 | 0.01 |
| E3 | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| E1 | 0.084 | ± | 0.019 | 0.036 | 0.017 | 0.001 | n.d. | — | — | — | — | |
| E2 | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| EE2 | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| DES | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| ERT | 0.072 | ± | 0.010 | 0.013 | 0.007 | 0.001 | 0.159 | ± | 0.026 | 0.066 | 0.031 | 0.002 |
| FLX | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| PhAC | Influents from Faro Nw WWTP | Effluents from Faro Nw WWTP | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μg L−1 | Uncertainty approach | SD | μg L−1 | Uncertainty approach | SD | |||||||
| Bottom-up | Top-down | Nordtest | Bottom-up | Top-down | Nordtest | |||||||
| a n.d. – not detected. | ||||||||||||
| ATN | 0.452 | ± | 0.109 | 0.033 | 0.026 | 0.003 | 0.181 | ± | 0.021 | 0.012 | 0.010 | 0.002 |
| APAP | 28.6 | ± | 1.7 | 4.1 | 2.3 | 0.6 | 0.257 | ± | 0.061 | 0.025 | 0.017 | 0.004 |
| SDZ | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| SPD | 0.167 | ± | 0.054 | 0.055 | 0.028 | 0.010 | 0.067 | ± | 0.006 | 0.018 | 0.008 | 0.001 |
| CAF | 32.3 | ± | 6.3 | 2.8 | 1.5 | 0.4 | 0.430 | ± | 0.036 | 0.023 | 0.015 | 0.007 |
| SMX | 0.272 | ± | 0.042 | 0.015 | 0.011 | 0.002 | n.d. | — | — | — | — | |
| MTPL | 0.071 | ± | 0.008 | 0.014 | 0.009 | 0.002 | 0.061 | ± | 0.006 | 0.007 | 0.006 | 0.001 |
| PPNL | 0.0277 | ± | 0.0026 | 0.0103 | 0.0027 | 0.0002 | 0.0254 | ± | 0.0025 | 0.0031 | 0.0025 | 0.0002 |
| CTS | 0.061 | ± | 0.005 | 0.006 | 0.004 | 0.001 | n.d. | |||||
| CBZ | 0.293 | ± | 0.046 | 0.012 | 0.011 | 0.001 | 0.299 | ± | 0.017 | 0.082 | 0.010 | 0.005 |
| CFA | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| NPX | 3.17 | ± | 0.03 | 0.21 | 0.15 | 0.03 | 0.085 | ± | 0.005 | 0.004 | 0.004 | 0.001 |
| GTD | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| TTE | 0.0283 | ± | 0.0029 | 0.0032 | 0.0019 | 0.0005 | n.d. | — | — | — | — | |
| BZF | 0.152 | ± | 0.026 | 0.010 | 0.008 | 0.001 | 0.0284 | ± | 0.0029 | 0.0017 | 0.0015 | 0.0004 |
| IBUP | 5.63 | ± | 0.35 | 0.32 | 0.23 | 0.04 | 0.0573 | ± | 0.0033 | 0.0033 | 0.0023 | 0.0009 |
| DCF | 0.690 | ± | 0.112 | 0.032 | 0.026 | 0.003 | 0.574 | ± | 0.063 | 0.032 | 0.011 | 0.030 |
| E3 | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| E1 | 0.116 | ± | 0.016 | 0.022 | 0.013 | 0.003 | n.d. | — | — | — | — | |
| E2 | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| EE2 | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| DES | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
| ERT | 0.305 | ± | 0.031 | 0.034 | 0.022 | 0.005 | 0.210 | ± | 0.060 | 0.061 | 0.029 | 0.003 |
| FLX | n.d. | — | — | — | — | n.d. | — | — | — | — | ||
As expected, the PhAC concentration in wastewater influents was higher than that in wastewater effluents, with the higher concentrations in the influents belonging to APAP, CAF, IBUP, NPX and DCF; whereas in the effluents, the higher concentrations belong to DCF, thus confirming its recalcitrant characteristics.
The results were expressed as result ± expanded uncertainty using the different uncertainty approaches to highlight the differences between them. The results were also expressed as result ± standard deviation (obtained under intermediate precision conditions of the standard control). The value of SD is lower than that of U. These results are consistent with those obtained by other authors.22 The lower the PhAC concentrations, the greater the uncertainty of the results. This aspect is crucial in quantifying compounds in trace concentrations, such as PhACs, mainly in wastewater effluents. As they are usually present in such samples at concentrations close to the detection limit, the uncertainty of such results will always be relatively high. This demonstrates the importance of calculating the expanded uncertainty in analytical procedures; failure to do so can lead to underestimating the results.21
Conclusions
The quantification of PhACs in complex matrices such as wastewater samples is still challenging and demanding. Analysts are constantly dealing with a wide range of challenges associated with the need to perform reliable identification and quantification of these compounds in complex matrices.In this work, a SPE-UHPLC-MS/MS methodology is presented as a good approach for the measurement of PhACs in wastewaters, allowing overcoming problems related to the complexity of wastewaters, which causes significant matrix effects, and the presence of some target PhACs at trace concentrations. Additionally, quantification by the standard addition method is also selected, allowing further minimization of the matrix effects. Due to the physico-chemical properties of the target PhACs, two chromatographic methods, an acidic and a basic method, were optimised and validated allowing the identification and measurement of 14 (acidic) and 10 (basic) PhACs, respectively.
The SPE-UHPLC-MS/MS methodology was validated, considering the assessment and evaluation of linearity, precision, global analytical thresholds, matrix effects and recoveries. For each target PhAC, the expanded uncertainty of the method was also evaluated in the analysis of wastewater influents and effluents of two distinct WWTPs (Beirolas and Faro Nw). The estimation of the expanded uncertainty of the analytical method was based on different approaches, namely bottom-up, top-down and Nordtest, in line with the recommendations of the International Standard Organization (ISO) to the Expression of Uncertainty in Measurement. The measured PhAC concentrations in wastewaters were expressed as the result ± expanded uncertainty by the three different approaches. It was observed that precision is associated with the minor uncertainty component whereas the most significant component is related to matrix effects.
This study demonstrates that the expanded uncertainty evaluation is a powerful tool for the optimization and validation of analytical methods since it allows us to assess and evaluate the parameters where we should concentrate our efforts to improve the reliability of our results. The information presented here will facilitate the introduction of uncertainty estimation in chromatographic measurements on a much broader scale than is the case at present.
Author contributions
Conceptualization, João Rodrigues, Vitor Cardoso, and Cristina Almeida; formal analysis, João Rodrigues and Sofia Silva; methodology, Vitor Vale Cardoso and Cristina Almeida; resources, Vitor Vale Cardoso and Maria João Benoliel; supervision, Vitor Vale Cardoso and Maria João Benoliel; validation, João Rodrigues, Sofia Silva, Vitor Vale Cardoso and Cristina Almeida; writing – original draft, João Rodrigues and Cristina Almeida; writing – review & editing, João Rodrigues, Vitor Vale Cardoso and Cristina Almeida.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support from the project co-funded by the European Commission, LIFE Environment programme, “LIFE Impetus – Improving current barriers for controlling pharmaceutical compounds in urban wastewater treatment plants” (LIFE14 ENV/PT/000739). The contribution of other partners of the LIFE Impetus project is deeply acknowledged, namely colleagues from Faro NW WWTP and Beirolas WWTP responsible for wastewater sampling.Notes and references
- S. Naidoo and A. Olaniran, Int. J. Environ. Res. Public Health, 2014, 11, 249–270 CrossRef.
- J. Paul Guyer, An Introduction to Preliminary Wastewater Treatment, Guyer Partners, 2018 Search PubMed.
- Department for Environment, Food & Rural Affairs (DEFRA), Sewage Treatment in the UK, UK Implementation of the EC Urban Waste Water Treatment Directive, 2002.
- S. Kaviya, in Urban Water Crisis and Management, ed. A. L. Srivastav, S. Madhav, A. K. Bhardwaj and E. Valsami-Jones, Elsevier, 2022, vol. 6, pp. 361–379 Search PubMed.
- B. Bihadassen, M. Hassi, F. Hamadi, A. Aitalla, M. Bourouache, A. El Boulani and R. Mimouni, Appl. Water Sci., 2020, 10, 7 CrossRef.
- 91/271/EEC, J. Eur. Commun., 34, 40.
- E. A. Victor, D. O. Omole and D. E. Bassey, Heliyon, 2020, 6, e05246 CrossRef.
- EC SWD(2018) 249 final/2, Proposal for a Regulation of the European Parliament and of the Council on Minimum Requirements for Water Reuse, 2018.
- L. Alcalde-Sanz and B. M. Gawlik, Minimum quality requirements for water reuse in agricultural irrigation and aquifer recharge – towards a water reuse regulatory instrument at EU level, EUR28962 EN, Report PUBSY No. 109291, Luxembourg, 2017 Search PubMed.
- S. Sauve and M. Desrosiers, Chem. Cent. J., 2014, 8, 15 CrossRef PubMed.
- O. Koba, O. Golovko, R. Kodesova, A. Klement and R. Grabic, Environ. Pollut., 2016, 218, 574–585 CrossRef.
- D. Gašo-Sokač, M. Habuda-Stanić, V. Bušić and D. Zobundžija, Croatian Journal of Food Science and Technology, 2017, 9, 204–210 CrossRef.
- C. Silva, C. Almeida, J. Rodrigues, S. Silva, M. Coelho, A. Martins, R. Lourinho, E. Cardoso, V. Cardoso, M. Benoliel and M. Rosa, Urban Water J., 2021, 465–478 CrossRef.
- V. Gaffney, V. Cardoso, E. Cardoso, A. Teixeira, J. Martins, M. Benoliel and C. Almeida, Environ. Sci. Pollut. Res., 2017, 24, 14717–14734 CrossRef.
- S. Silva, V. V. Cardoso, L. Duarte, R. N. Carneiro and C. M. M. Almeida, Appl. Sci., 2021, 11, 1–19 Search PubMed.
- M. Gros, M. Petrovic and D. Barcelo, Anal. Chem., 2009, 81, 898–912 CrossRef PubMed.
- E. Garcia-Lor, J. V. Sancho and F. Hernández, J. Chromatogr. A, 2010, 1217, 622–632 CrossRef PubMed.
- V. d. J. Gaffney, V. V. Cardoso, A. Rodrigues, E. Ferreira, M. J. Benoliel and C. M. M. Almeida, Quim. Nova, 2014, 37, 138–149 CrossRef.
- R. Salgado, J. Noronha, A. Oehmen, G. Carvalho and M. Reis, Water Sci. Technol., 2010, 62, 2862–2871 CrossRef CAS PubMed.
- I. Baranowska and B. Kowalski, Water Sci. Technol., 2009, 60, 449–458 CrossRef CAS PubMed.
- C. Almeida, Separations, 2021, 8, 16 CrossRef CAS.
- M. Borecka, A. Białk-Bielińska, G. Siedlewicz, K. Kornowska, J. Kumirska, P. Stepnowski and K. Pazdro, J. Chromatogr. A, 2013, 1304, 138–146 CrossRef CAS.
- P. Konieczka and J. Namieśnik, J. Chromatogr. A, 2010, 1217, 882–891 CrossRef CAS PubMed.
- ISO 8466-1, International Standard Organization, 1990.
- J. van Trijp and A. H. Roos, RIKILT-DLO, model for the calculation of calibration curves, RIKILT report 91.02, Wageningen, The Netherlands, 1991 Search PubMed.
- ISO/IEC 17025, International Organization for Standardization, 2017.
- ISO 11352, International Standard Organization, 2012.
- Infarmed, Monitorization of the market, available at: https://www.infarmed.pt/portal/page/portal/infarmed/monitorizacao_do_mercado/observatorio/estatistica_do_medicamento/estmed-2011.pdf, accessed on 05.10.12.
- Infarmed, Prontuário Terapêutico, 2010.
- P. Verlicchi and E. Zambello, Sci. Total Environ., 2015, 538, 750–767 CrossRef CAS.
- J. Brozinski, M. Lahti, A. Meierjohann, A. Oikari and L. Kronberg, Environ. Sci. Technol., 2013, 47, 342–348 CrossRef CAS.
- M. Lahti and A. Oikari, Chemosphere, 2011, 85, 826–831 CrossRef CAS PubMed.
- U. Memmert, A. Peither, R. Burri, K. Weber, T. Schmidt, J. Sumpter and A. Hartmann, Environ. Toxicol. Chem., 2013, 32, 442–452 CrossRef CAS PubMed.
- J. Sumpter, R. Donnachie and A. Johnson, Aquat. Toxicol., 2014, 151, 57–60 CrossRef CAS PubMed.
- Directive 2013/39/EU, Official Journal of the European Union L226, 2013.
- A. Cravo, S. Silva, J. Rodrigues, V. V. Cardoso, M. J. Benoliel, C. Correia, M. R. Coelho, M. J. Rosa and C. M. M. Almeida, Environ. Res., 2022, 208, 112632 CrossRef PubMed.
- M. Henriques, V. Cardoso, A. Rodrigues, E. Ferreira, M. Benoliel and C. Almeida, J. Water Resour. Prot., 2010, 2, 818–829 CrossRef.
- V. Gaffney, C. M. M. Almeida, A. Rodrigues, E. Ferreira, M. J. Benoliel and V. V. Cardoso, Water Res., 2015, 72, 199–208 CrossRef.
- J. Rodrigues, S. Albino, S. Silva, A. Cravo, V. Cardoso, M. Benoliel and C. Almeida, Food Analytical Methods, 2019, 12, 838–851 CrossRef.
- EURACHEM/CITAC, Quantifying Uncertainty in Analytical Measurement, Guide CG 4, 2nd edn, 2000 Search PubMed.
- S. Ellison and A. E. Williams, Eurachem/CITAC Guide: Quantifying Uncertainty in Analytical Measurement, 3rd edn, 2012, ISBN 978-0-948926-30-3, available from: https://www.eurachem.org Search PubMed.
- V. Barwick and S. Ellison, VAM Project 3.2.1, Development and Harmonization of Measurement Uncertainty Principles, Part (d): Protocol for Uncertainty Evaluation from Validation Data, 2000.
- EA, European co-operation for accreditation, EA guidelines on the expression of uncertainty in quantitative testing, EA-4/16, 2003 Search PubMed.
- B. Magnusson, T. Näykki, H. Hovind and M. Krysell, Handbook for calculation of measurement uncertainty in environmental laboratories, Oslo, Norway, 2012 Search PubMed.
- Guia RELACRE 31, Associação de Laboratórios Acreditados de Portugal, 2018.
- M. Stuber and T. Reemtsma, Anal. Bioanal. Chem., 2004, 378, 910–916 CrossRef PubMed.
- SANTE/11312/2021, available at: https://www.eurl-pesticides.eu/docs/public/tmplt_article.asp?CntID=727, accessed on 22.11.22.
- A. Kot-Wasik, J. Debska, A. Wasik and J. Namiesnik, Chromatographia, 2006, 64, 13–21 CrossRef.
- L. Vera-Candioti, M. D. Gil García, M. Martínez Galera and H. C. Goicoechea, J. Chromatogr. A, 2008, 1211, 22–32 CrossRef PubMed.
- M. D. Gil García, F. Cañada Cañada, M. J. Culzoni, L. Vera-Candioti, G. G. Siano, H. C. Goicoechea and M. Martínez Galera, J. Chromatogr. A, 2009, 1216, 5489–5496 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ay01676a |
| This journal is © The Royal Society of Chemistry 2023 |