
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsoform-specific optical activation of kinase function reveals p38-ERK signaling crosstalk†
Wenyuan
Zhou
a,
Amy
Ryan
a,
Chasity P.
Janosko
a,
Karsen E.
Shoger
bc,
Jason M.
Haugh
d,
Rachel A.
Gottschalk
bc and
Alexander
Deiters
 *ac
*ac
aDepartment of Chemistry, University of Pittsburgh, Pittsburgh, PA 15260, USA. E-mail: deiters@pitt.edu
bDepartment of Immunology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15260, USA
cCenter for Systems Immunology, University of Pittsburgh, Pittsburgh, PA 15261, USA
dDepartment of Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, NC 27606, USA
First published on 25th August 2023
Abstract
Evolution has diversified the mammalian proteome by the generation of protein isoforms that originate from identical genes, e.g., through alternative gene splicing or post-translational modifications, or very similar genes found in gene families. Protein isoforms can have either overlapping or unique functions and traditional chemical, biochemical, and genetic techniques are often limited in their ability to differentiate between isoforms due to their high similarity. This is particularly true in the context of highly dynamic cell signaling cascades, which often require acute spatiotemporal perturbation to assess mechanistic details. To that end, we describe a method for the selective perturbation of the individual protein isoforms of the mitogen-activated protein kinase (MAPK) p38. The genetic installation of a photocaging group at a conserved active site lysine enables the precise light-controlled initiation of kinase signaling, followed by investigation of downstream events. Through optical control, we have identified a novel point of crosstalk between two major signaling cascades: the p38/MAPK pathway and the extracellular signal-regulated kinase (ERK)/MAPK pathway. Specifically, using the photoactivated p38 isoforms, we have found the p38γ and p38δ variants to be positive regulators of the ERK signaling cascade, while confirming the p38α and p38β variants as negative regulators.
Introduction
The various substrates and cellular functions associated with the p38 MAPK are notably diverse.1 The p38/MAPK pathway is important for the stress-induced cellular response as it has been shown to promote not only apoptosis, but also survival, cell growth, and differentiation in response to extracellular stimuli such as UV damage, heat, inflammatory cytokines, and growth factors.2–5 This signaling pathway has been demonstrated to suppress tumorigenesis through oncogene-induced senescence, replicative senescence, DNA-damage responses, and contact inhibition through its impact on several cell-cycle regulators.6–8 The p38 signaling pathway has also been linked to inflammation and immune (innate and adaptive) responses through the p38-dependent transcriptional activation of immune regulators such as cytokines and receptors.9–11Four isoforms of p38 – p38α, p38β, p38γ, and p38δ – have been identified. The contradictory nature of protein isoforms with redundant or specific functions has long been a topic of research. The evolution of protein isoforms through alternative splicing and the extension of multigene families has corresponded with the evolution of multicellularity, supporting their role in providing biological diversity.12 While some isoforms have demonstrated identical roles in a functional context, research has revealed isoform differentiation in a specialized context. This has been attributed to varying cell type- and tissue-dependent expression patterns, as well as alternative cellular spatiotemporal dynamics and divergent regulatory mechanisms.12 All three major MAPK signaling proteins (ERK, JNK, and p38) have multiple isoforms, displaying varying degrees of redundancy in the regulation of major cellular processes such as differentiation, apoptosis, and proliferation.13 For example, while the p38 isoforms share overlapping upstream mitogen-activated protein kinase kinase (MAPKK) activators, they differ in tissue distribution, regulatory mechanisms (external stimuli, feedback control via kinases or phosphatases), and downstream protein targets (Fig. 1(A)).14 The p38α/β isoforms are expressed in most tissues, while p38γ is primarily found in the skeletal muscle,15–18 and p38δ is expressed in the lung, pancreas, kidney, and small intestine.19 They also express differently in various inflammatory cell lineages, including neutrophiles and macrophages.20
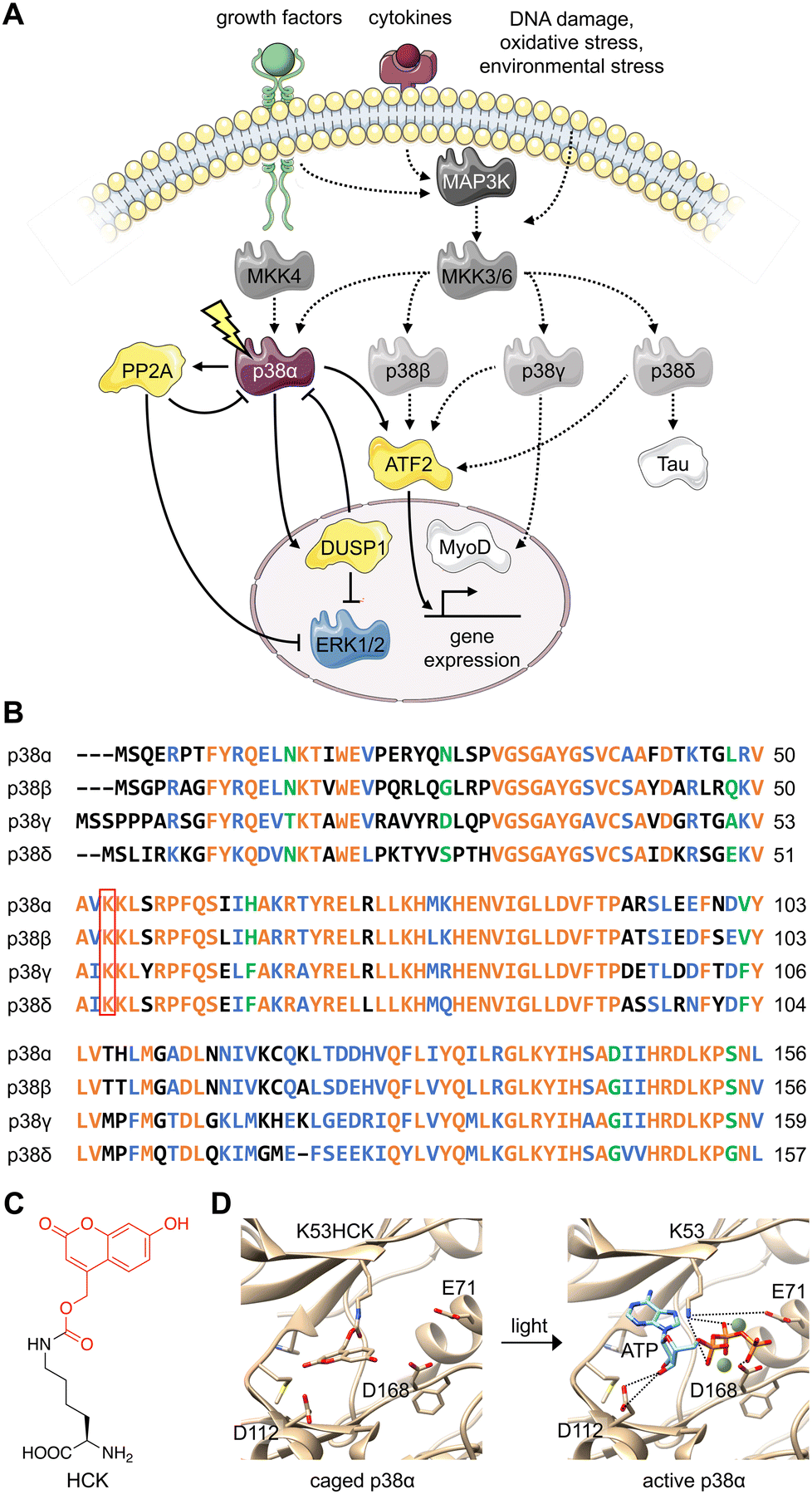 | ||
| Fig. 1 Design of an optically controlled p38 isoform. (A) Light activation of constitutively active p38α leads to isoform-specific perturbation of downstream targets. Solid arrows represent light-mediated p38 signaling. Dotted arrows represent endogenous p38 signaling, stimulated through cellular stress. (B) Alignment of p38 isoform sequences in the conserved N-terminal ATP binding pocket, with the catalytically critical lysine outlined in red. Alignment was completed by Clustal Omega. Orange = identical residues; blue = residues of strong similarity; green = residues of weak similarity. (C) Chemical structure of hydroxycoumarin lysine (HCK) depicting the caging group in red. (D) Light activation of photocaged p38α. Light exposure (365 nm or 405 nm) restores the native active site and ATP binding (PDB: 2BAQ).62 To generate the model on the left (caged p38), ATP was superimposed into the position of an ATP-analogue inhibitor. | ||
Various levels of isoform redundancy have been observed depending on the cellular context, and this redundancy is often correlated with protein sequence homology.13 The four p38 isoforms share a certain level of homology, including a common threonine-glycine-tyrosine (TGY) motif for phosphorylation by MAPKKs. They can be further divided into two subsets: p38α/β share 75% sequence homology, p38γ/δ share 70% sequence homology, and p38γ/δ share about 60% homology to p38α/β (Fig. 1(B)).14 Even though p38α, the most broadly expressed isoform, has been comprehensively studied, the characterization of the β, γ, and δ isoforms is underdeveloped due to a lack of tools (e.g., small molecule modifiers) that acutely (on a minute timescale) activate or deactivate their function with complete isoform specificity. These confounding factors also blur our understanding of the relationship between p38 signaling and other signal transduction pathways. Optogenetic approaches combine the specificity of genetic perturbations with the spatial and temporal precision of light as an external control element.21,22 Acute and selective activation of kinase and phosphatase activities have been explored. Small molecule-induced activation of signaling pathways has been demonstrated23 but obviously does not offer spatial control. Importantly, photoactivatable dimerization inducers24 and photoswitchable kinase inhibitors25 have also been developed. These small molecules achieved reversibility either through re-dosing or light-induced isomerization, which is not shared by caged UAAs. It should be noted, however, that background activity and a lack of spatial precision were observed for these compounds. Previously, kinases coupled with light-responsive protein domains have also allowed light-induced perturbation of signaling pathways,26–29 which required extensive screening for the desirable configurations of fusion proteins. These efforts may not be easily transferrable from one kinase of interest to another, hindering easy application of light-responsive protein domains across different targets. Different UAAs, activatable with specific optogenetic and chemogenetic inducers have been applied to optical control of kinase and phosphatase function.23,30–32 They have been shown to enable activation with ultra-low background and high spatiotemporal specificity with general applicability to different targets with identified catalytic active-site residues, but do not allow for reversibility. All in all, these different approaches complement each other and should be selected based on the biological question(s) asked.
Several open questions regarding p38 signaling remain, including if the isoforms perform redundant or non-redundant physiological functions, and if potential crosstalk of each isoform with other closely related MAPK pathways occurs. To that end, we set out to demonstrate the conditional control of each individual p38 isoform, enabled by a unique combination of genetic and photochemical approaches, and triggered by irradiation. We33–39 and others40–43 have previously demonstrated a generally applicable strategy to create light-activated kinases through replacement of a critical lysine residue within the ATP binding domain with a photocaged lysine. With this methodology, proteins with extremely similar sequence and structure can be independently and acutely controlled, enabling spatial and temporal dissection of isoform function. While we target p38 here, our approach is applicable to any set of isoforms in the human kinome.
Results and discussion
To parse the functions of the p38 isoforms, it is essential that each isoform can be activated without cell surface stimulation of the entire MKK3(6)/p38 signaling pathway. This precludes the possibility that any upstream MAPKKK or MAPKK would simultaneously activate multiple p38 isoforms or perturb the others MAPK signaling pathways, such as the MEK/ERK pathway.44The p38 isoforms were therefore rendered constitutively active by introducing the following mutations: p38α D176A F327S, p38β D176A, p38γ F324S, and p38δ D179A.45 These mutations induce conformational changes within the activation loop (residues 171–183), stimulate autophosphorylation of the TGY motif, and result in constitutive activation without the need for upstream activators.46–51 The constitutively active mutants were used for all experiments to decouple the resulting activity from parallel growth factor-induced signaling. To achieve light activation of p38, a conserved lysine residue critical for catalytic function (p38α K53, p38β K53, p38γ K56, and p38δ K54) in the N-terminal ATP-binding pocket52,53 was mutated to hydroxycoumarin lysine (HCK)54 through unnatural amino acid mutagenesis in cells with an expanded genetic code (Fig. 1(C)).37,55 The hydroxycoumarin protection group removes the positive charge from the lysine side chain, disrupting an electrostatic interaction with the α-phosphate of ATP, as well as a stabilizing salt bridge interaction with the conserved glutamate E71.56,57 The introduction of HCK thus renders the p38 kinase inactive until a brief 365–405 nm light exposure removes the caging group, restoring the native active site. Through a decaging study using an HCK synthetic intermediate, we showed a dose–response relationship for the release of the caging group with respect to the duration of irradiation (Fig. S1, ESI†). ATP docking is then restored and stabilized through electrostatic interactions with the catalytic lysine and acidic residues D112 and D168,57 initiating a phosphoryl transfer reaction toward the cognate substrates (Fig. 1(D)). One advantage for the use of HCK is that hydroxycoumarin and its photolysis byproduct, hydroxycoumarin alcohol (Fig. S2, ESI†), are of low cytotoxicity and do not generate reactive oxygen species nor activate the p38 signaling pathway.58–61
NIH3T3 cells were selected for analyzing kinase activity since they grow as a monolayer, facilitating fluorescence imaging and imaging cytometry.63 The uniform and efficient expression of photocaged p38 kinases is important for the assessment of light-induced kinase activity through western blot analysis, since a low population of cells expressing the kinase would consequently yield a low concentration of phosphorylated substrate. A positive signal can thus be easily diluted by the greater population of cells lacking the caged protein. We therefore endeavored to increase the pool of caged p38 (and its downstream targets) through the generation of a stable cell line expressing a chimeric synthetase (HCKRS) specific for the caged lysine and four copies of the tRNACUA. The HCKRS was engineered for accepting HCK as a substrate and combines the N-terminus of PylRS from Methanosarcina barkeri and the C-terminus of PylRS from Methanosarcina mazei since this variant provides higher amber codon suppression efficiency than other previously tested by us.64 In brief, the genetic elements expressing these components were integrated in the NIH3T3 genome by the PiggyBac transposase via flanking inverted terminal repeat sequences. The monoclonal cell line with the highest UAA incorporation efficiency and PylRS/PylT expression level was selected and cryopreserved, designated from here as the NIH3T3 stable cell line (NIH3T3HCK). The p38 isoform genes carrying the discussed activation mutations, the amber codon mutation, and a C-terminal monomeric tagRFP-HA fluorophore attached via a flexible GGGS linker65 were cloned into a pcDNA backbone. An EF1α promoter was chosen for protein expression, as it provided the most efficient amber codon suppression compared to other plasmid-based systems (Fig. 2(A)).66 This construct allowed for confirmation of HCK-containing, full-length p38 isoform expression through fluorescence microscopy. HCK-dependent expression of each of the caged isoforms in NIH3T3HCK cells was further confirmed by western blot (Fig. 2(B)).
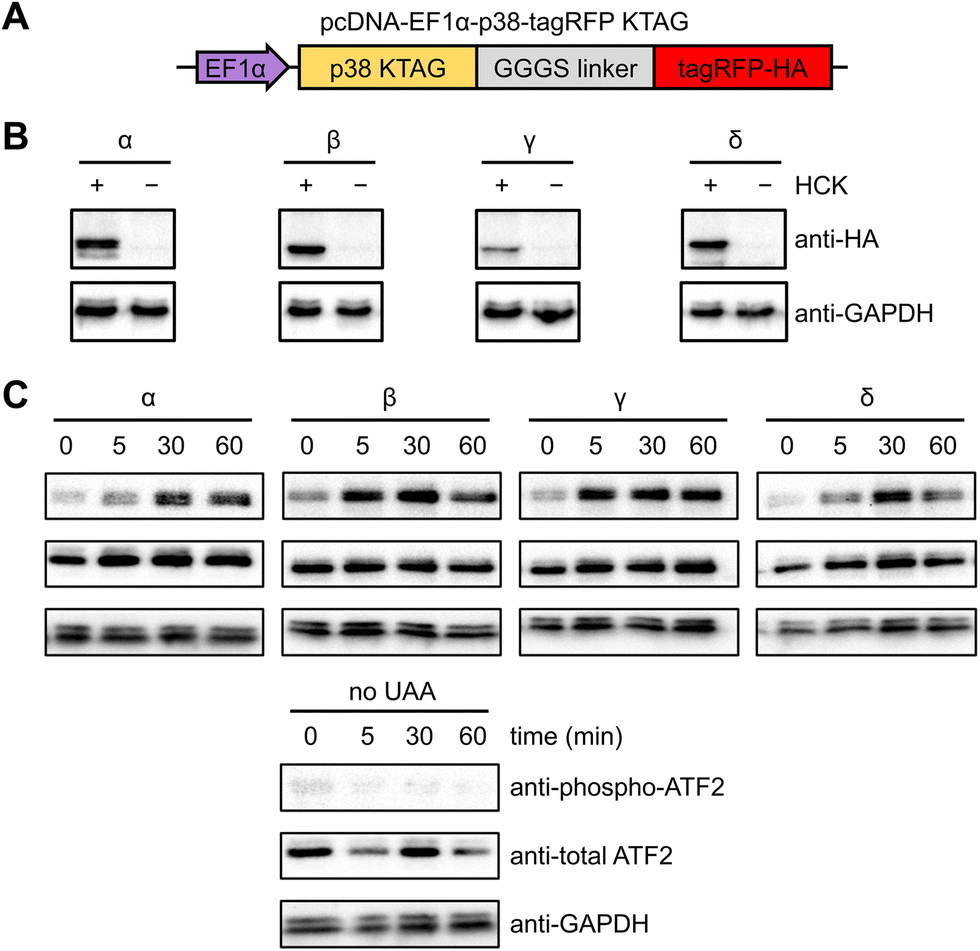 | ||
| Fig. 2 Expression and light activation of photocaged p38 isoforms in NIH3T3HCK cells. (A) Construct for the expression of photocaged p38 isoforms in the NIH3T3 cell line stably expressing the PylRS/PylT pair. (B) Western blot analysis of photocaged p38-tagRFP isoform expression in the absence or presence of HCK (0.25 mM). (C) Western blot analysis of p38 light activation, leading to phosphorylation of ATF2, a pan-p38 substrate. | ||
To test the ability of the photocaged p38 isoforms to efficiently catalyze substrate phosphorylation in a light-dependent manner, we chose to assess a pan-p38 substrate, the activating transcriptional factor 2 (ATF2). All four p38 enzymes, once activated, lead to the transcriptional activation and phosphorylation of the T69/71 “phosphoswitch” sites of ATF2.19,67–69 The T69/71 dual phosphorylation occurs in a two-step manner: T71 is predominantly phosphorylated by the Ras/MEK/ERK pathway, while T69 phosphorylation is carried out by p38 MAPK.70 It is important to note that according to an in vitro kinetics study, phosphorylation of mono-phosphorylated ATF2 differs depending on which threonine is phosphorylated first. Phosphorylation of T71 had no significant effect on the rate of T69 phosphorylation, but T69 phosphorylation reduced the efficiency of T71 phosphorylation to 2.5% of unphosphorylated ATF2.71 Thus, we set out to validate in vivo light activation of caged p38 isoforms by probing ATF2 pT69 levels in the presence of a constitutively active MEK1 (MEK1 S222/224D) for the prerequisite phosphorylation of ATF2 T71. NIH3T3HCK cells expressing each of the photocaged p38 isoforms and caMEK1 were serum-starved for 4 hours before irradiation (365 nm, transilluminator, 3 min). Cells were harvested and lysed at 0 min (−UV), 5 min, 30 min, and 60 min after light activation. Phospho-ATF2 levels increased rapidly in response to light-activated p38, but not in NIH3T3HCK cells grown in the absence of HCK (i.e., no caged kinase expression), confirming optical activation of p38 kinase activity in a temporally controlled manner (Fig. 2(C)). The kinetics of ATF2 T71 phosphorylation (the phosphorylation site detected in Fig. 2(C)) also indicates that the p38β and γ isoforms have a preference for this site, while the p38α and δ isoforms do not. These results from our cell-based studies are supported by earlier, biochemical assays.67 The reduced phospho-ATF2 levels (Fig. 2(C)) could be explained by the negative feedback mechanism for p38 regulation through the activation of phosphatases, such as DUSP1. These phosphatases interact with multiple downstream targets, including p38 and ATF2, which resulted in the dephosphorylation of the autophosphorylated TGY motif of decaged cap38 as well as phospho-ATF2.72 The observation of changes in phosphor-ATF2 levels demonstrates that photoactivation of p38 is a valuable tool to study direct and transient cellular responses.
A set of kinase translocation reporters (KTR) fused to Clover, a bright monomeric variant of EGFP, were reported to monitor kinase activity with high signal-to-noise ratio.73 By combining a MAPK-specific distant docking region, a nuclear export signal (NES), a bipartite nuclear localization sequence (bNLS), and a fluorescent protein, these MAPK activity reporters reside mainly in the nucleus when the cognate MAPK is not active. The reporter is excluded from the nucleus after the activated cognate MAPK enters the nucleus and phosphorylates the bNLS and NES sequences (Fig. S3, ESI†). Translocation of KTR Clover is represented by the ratio between Clover fluorescence intensity in the cytoplasm and the nucleus (C/N ratio) of individual cells. We aimed to use the both the ERK and p38 KTR Clover constructs for real-time analysis of p38-mediated regulation of MAPK signaling.
Serum starved NIH3T3 cells expressing both caged p38-tagRFP and MAPK KTR Clover (Fig. 3(A)) were imaged following 365 nm irradiation (2 s, DAPI filter). Prior to stimulation, both ERK KTR and p38 KTR Clover localized in the nucleus due to suppressed MAPK signaling of the serum starved cells. As expected, light activation of p38α triggered p38 KTR Clover translocation (Fig. 3(B)).73 A similar response was observed upon activation of p38α's close analogue, p38β (Fig. 3(C)). Light activation of the p38α/β isoforms was shown to have no effect on the ERK/MAPK pathway, a result that is supported by a previous report demonstrating the failure of p38α and β to trigger ERK KTR Clover translocation upon stimulation with anisomycin,73 a chemical inducer of stress pathways.74 Alternatively, light activation of p38γ (Fig. 3(D)) and p38δ (Fig. 3(E)) failed to trigger the translocation of p38 KTR Clover. This can be attributed to the MAPK docking site of p38 KTR Clover based on the p38α-specific substrate, Mef2C, which is not shared by the p38γ/δ isoforms.1 Intriguingly, light activation of caged p38γ/δ resulted in rapid cytoplasmic translocation of ERK KTR Clover, plateauing within 20 minutes of irradiation.
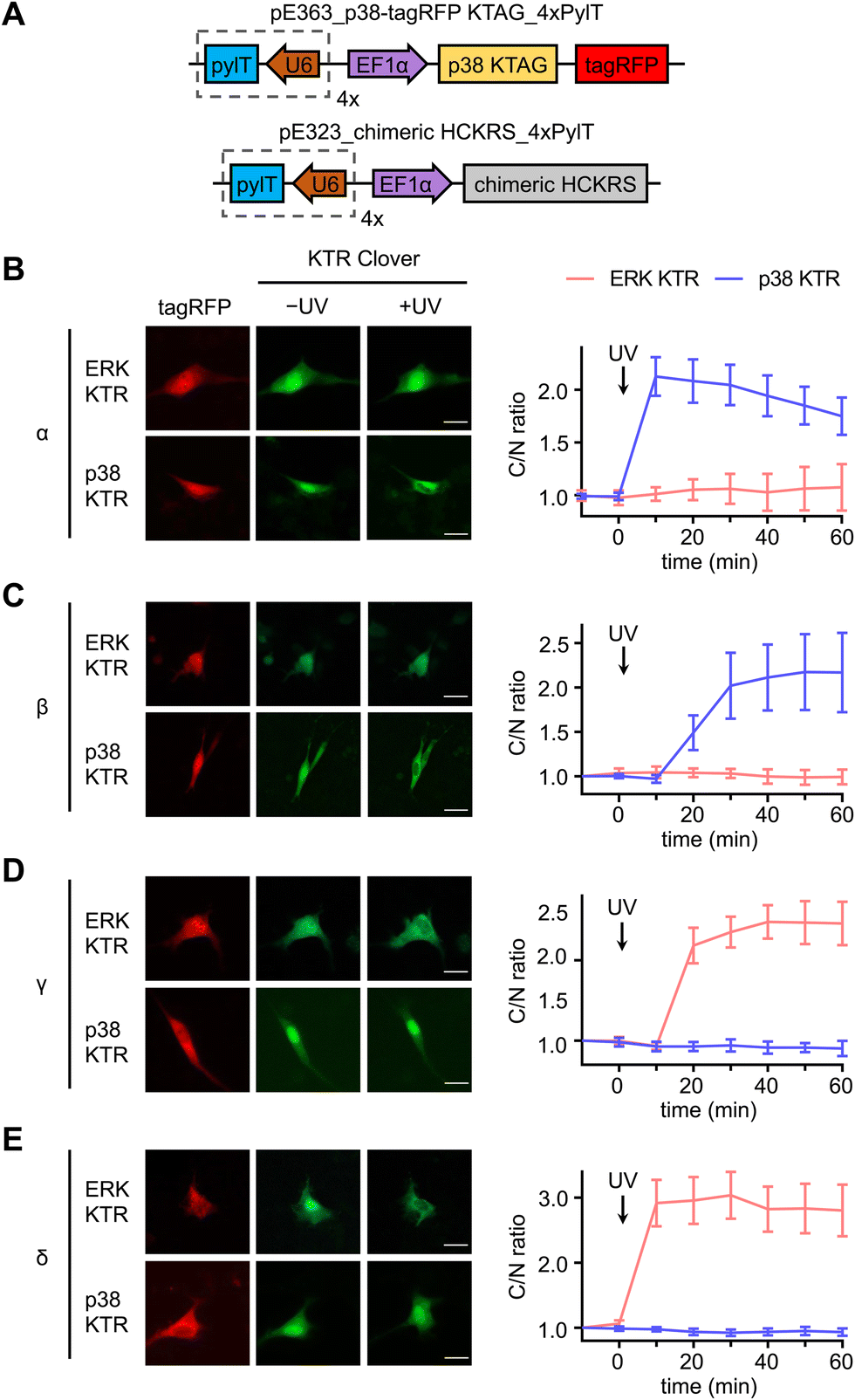 | ||
| Fig. 3 Divergent functions of p38α/β and p38γ/δ subsets in NIH3T3 cells. (A) Plasmid maps for the expression of photocaged p38 in cells: pE363_p38-tagRFP KTAG_4xPylT and pE323_chimeric HCKRS_4xPylT. Representative fluorescence microscopy images and time-course quantification of C/N ratio of Clover fluorescence before and after stimulation of irradiated cells expressing ERK KTR Clover or p38 KTR Clover and, (B) caged p38α (ERK: n = 4; p38: n = 33), (C) caged p38β (ERK: n = 11; p38: n = 10), (D) caged p38γ (ERK: n = 25; p38: n = 12), or (E) caged p38δ (ERK: n = 20; p38: n = 25). Scale bars = 20 μm. Error bars represent 95% confidence intervals. | ||
In a similar fashion, serum starved NIH3T3HCK cells expressing both the caged p38-tagRFP and ERK KTR Clover (Fig. 4(A)) were imaged following sequential 365 nm irradiation (2 s, DAPI filter) and surface stimulation with FGF. Importantly, light activation of caged p38α (Fig. 4(B)) or p38β (Fig. 4(C)) was sufficient for suppression of FGF-induced ERK/MAPK signaling in cells, as indicated by the reduced nuclear exportation of ERK KTR Clover (Fig. S5, ESI†). While FGF alone resulted in a nearly 2-fold change in C/N fluorescence intensity, light-activated p38α or p38β effectively reduced the level of reporter translocation back to baseline. This indicated negative crosstalk between the active p38 and ERK signaling pathways, a mechanism which may be mediated by the p38-dependent activation of the ERK1/2-specific phosphatase PP2A.75 As anticipated from the previous result, light activation of caged p38γ (Fig. 4(D)) and p38δ (Fig. 4(E)) has no effect on FGF-induced ERK/MAPK signaling, as cell surface stimulation induces ERK/MAPK signaling.
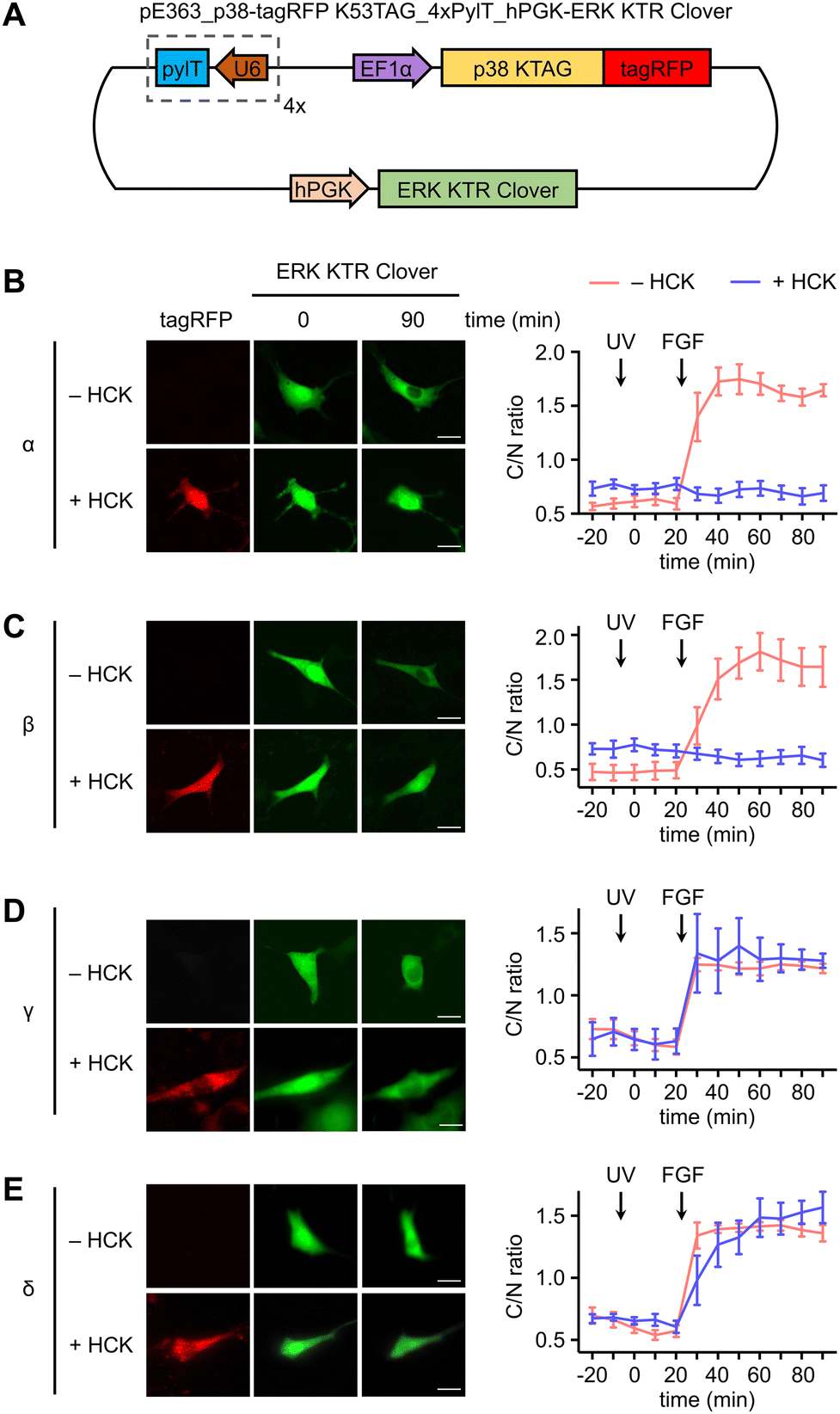 | ||
| Fig. 4 Investigation of crosstalk between p38 isoforms and the ERK signaling pathway. (A) Plasmid map for the expression of photocaged p38 in the NIH3T3HCKcell line: pE363_p38-tagRFP K53TAG_4xPylT_hPGK-ERK KTR Clover. hPGK = humanized PGK promoter. Representative fluorescence images of NIH3T3HCK cells expressing each p38-tagRFP KTAG in the absence or presence of HCK (0.25 mM) and time-course quantification of C/N ratio of Clover fluorescence before and after stimulation of irradiated cells in the absence or presence of caged (B) caged p38α (n = 5), (C) caged p38β (n = 5), (D) caged p38γ (n = 5), or (E) caged p38δ (n = 5). UV treatment at 0 min timepoint; FGF treatment at 25 min timepoint. Scale bars = 20 μm. Error bars represent 95% confidence intervals. | ||
These results establish a precedent for opposing roles of the p38 isoform subset in the crosstalk attenuation of a parallel MAPK signaling pathway, with broader implications as to the isoform-specific role of the p38γ/δ subset in cellular development in response to stress. It is possible that positive regulation of ERK/MAPK signaling through the p38γ/δ subset is responsible for dampening the p38α/β-mediated ERK inhibition. The predominant form of feedback regulation would be dependent on the cell type-dependent isoform expression levels. Changes in substrate specificity may be attributed to differences in isoform structure and cellular context. Notably, ERK2 and p38γ share almost identical structures within the activation loop,76 and ERK and p38δ recognize a similar FXF (Phe-Xaa-Phe-(Pro)) motif, indicating possible overlap in substrate identity and supporting a role in promotion of the same pathways.77 Our results may also provide mechanistic insight as to the seemingly contradictory roles of p38 in certain disease states. For example, similar to the function of ERK1/2, the p38γ/δ isoforms confer resistance to drug-induced apoptosis in renal cell carcinoma, contradicting the pro-apoptotic function of p38α/β.78 Given these parallels, it is not entirely surprising that p38α/β and p38γ/δ subsets play opposite roles in association with the ERK signaling pathway.
To further validate the results observed using the ERK KTR reporter assay, we utilized immunofluorescence for the direct analysis of ERK phosphorylation in response to light-induced p38γ/δ. Serum-starved NIH3T3 cells expressing photocaged p38γ or p38δ were stimulated through exposure to 405 nm light (1 min, LED), and following a 60 minutes incubation, cells were fixed and stained using antibodies against phospho-ERK1/2 (T202/Y204 phosphosites, pERK1/2) and HA, followed by fluorophore-labeled secondary antibodies. Fluorescence intensity levels were quantified for individual cells using a high content imager, and p38 expressing transfected cells were gated based on HA expression (Fig. S6A and B, ESI†). Relative to the nonirradiated control, nuclear ERK phosphorylation was significantly higher in cells expressing p38γ (Fig. 5(A)) and p38δ (Fig. 5(B)) post-irradiation. This is consistent with the translocation mechanism of phosphorylated ERK,79 and supports positive crosstalk between p38γ/δ and the ERK/MAPK signaling pathway. Additionally, this data further validates our results demonstrating rapid activation of ERK KTR upon stimulation of the caged p38γ and p38δ proteins through positive regulation of ERK. Importantly, in the absence of p38δ or p38γ expression, cells exposed to light showed no significant increase in pERK1/2 levels relative to the nonirradiated control (Fig. S6C, ESI†) confirming that irradiation is not responsible for ERK activation.
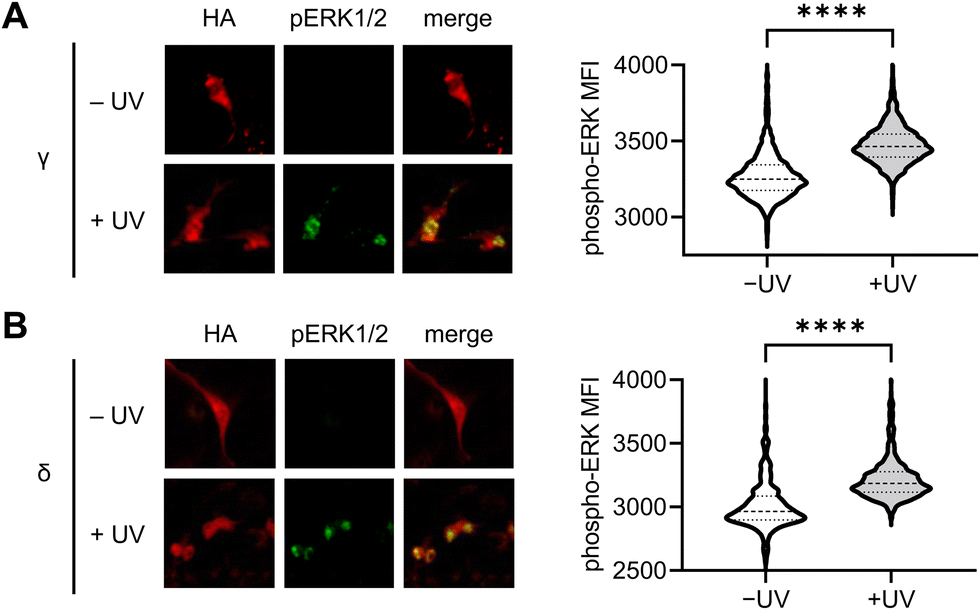 | ||
| Fig. 5 Immunofluorescence analysis of p38γ/δ-induced ERK phosphorylation in NIH3T3 cells. Representative fluorescence microscopy images and quantification of pERK1/2 immunofluorescence intensity in HA expressing cells, before and after photoactivation (405 nm LED, 1 min) of cells expressing (A) caged p38γ (−UV, n = 1957; +UV, n = 1956) and (B) caged p38δ (−UV, n = 569; +UV, n = 704). p < 0.0001 for both isoforms from unpaired two-tailed Mann–Whitney test. | ||
To delineate the interaction between the p38γ/δ isoforms and the ERK signaling pathway, select nodes at different levels of the ERK signaling cascade were blocked by utilizing small molecule inhibitors for Raf (LY3009120),80 MEK (U0126)81 and ERK (SCH772984)82 (Fig. 6(A)). NIH3T3 cells expressing ERK KTR Clover and photocaged p38δ or p38γ were pre-treated with LY3009120 (1 μM), U0126 (10 μM), SCH772984 (1 μM), or DMSO for 30 min. Following inhibition, the cells were irradiated with 365 nm light (2 s, DAPI filter), and p38/ERK crosstalk was monitored through the extent of ERK KTR Clover translocation. While DMSO had no significant effect on KTR activity upon activation of p38δ and p38γ, all three inhibitors suppressed the crosstalk between the ERK pathway and either p38γ (Fig. 6(B)) or p38δ (Fig. 6(C)). This may indicate that the crosstalk is mediated by activating upstream regulators in the Ras/Raf/MEK/ERK signaling pathway, likely Ras. While p38 has been identified as a negative regulator of ERK signaling both directly (through the phosphorylation of fibroblast growth factor receptor substrate-2 (FRS2)83) and indirectly (through the activation of the phosphatase PP2A75), this is the first reported example of positive ERK regulation in response to p38 activation. Notably, the previous observations of downregulation of ERK signaling were not obtained through p38 isoform-specific perturbation.
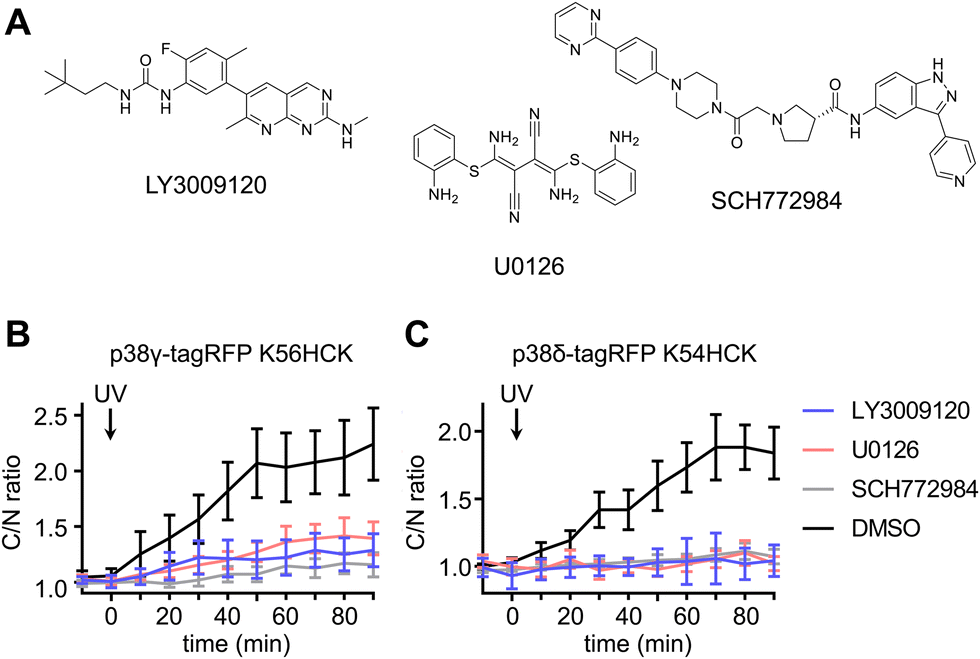 | ||
| Fig. 6 Light activation of photocaged p38γ/δ isoforms in the presence of ERK pathway inhibitors. (A) Structures of the Raf inhibitor LY3009120, the MEK inhibitor U0126, and the ERK inhibitor SCH772984. Time-course quantification of C/N ratio of Clover fluorescence before and after irradiation of cells expressing ERK KTR Clover and (B) caged p38γ in the presence of inhibitors LY3009120 (n = 6), U0126 (n = 23), SCH772984 (n = 20), or DMSO (n = 21), or (C) caged p38δ in the presence of LY3009120 (n = 12), U0126 (n = 20), SCH772984 (n = 15), or DMSO (n = 21). p = 0.0012, p = 0.0032, and p = 0.0002 for comparison of DMSO and LY3009120, U0126, or SCH772984, respectively, in cells expressing caged p38γ. p = 0.0003, p = 0.0004, and p = 0.0005 for comparison of DMSO and LY3009120, U0126, or SCH772984, respectively, in cells expressing caged p38δ. Significance was calculated from unpaired two-tailed student's t test. Error bars represent 95% confidence intervals. | ||
Conclusions
In conclusion, we have demonstrated the usefulness of engineering individual light-responsive protein isoforms to dissect their unique contributions to cell signaling mechanisms through precise control of kinase activity. Through mutagenesis with photocaged lysine, we have combined the exquisite specificity of genetic manipulation with the precise temporal regulation of optical activation. Installation of the hydroxycoumarin photocaging group on the ε-amino group of a critical active site lysine in kinases ensures disruption of ATP cofactor binding. This can be attributed to two factors: (1) the steric displacement of ATP by the hydroxycoumarin caging group, effectively blocking the active site, and (2) the removal of the electrostatic interactions between the critical lysine side chain and the alpha phosphate on ATP.56,57 Light-induced decaging restores the native active site, leading to ATP binding, and enzymatic function of the constitutively active kinase.Using this approach, we established diverging functions of the four isoforms of p38: α, β, γ, and δ. Western blot analysis of a pan-p38 substrate, ATF2, revealed light-induced ATF2 phosphorylation by all four isoforms, confirming the endogenous activity of the light-triggered p38 isoforms. We next set out to establish the specificity of all four isoforms for regulation of the ERK/MAPK and p38/MAPK signaling pathways. As expected, KTR translocation indicated the ability of the decaged p38α/β subset to activate the p38/MAPK pathway, while the ERK/MAPK signaling pathway remained inactive. Interestingly, the decaged p38γ/δ isoform subset had the opposite result: while p38 KTR Clover remained inactive and nuclear sequestered, ERK KTR Clover was rapidly and extensively activated. This was further supported by the increase of ERK1/2 phosphorylation in response to light-mediated p38γ/δ activation, as assessed through immunofluorescence measurements of cellular pERK1/2 levels. Ras-dependent activation of the p38 signaling pathway84 has been demonstrated and is well established, however, evidence for crosstalk in the reverse direction – from p38 to ERK – has been insufficient. In one incidence, p38 was shown to be indispensable for ERK activation in a specific, GMP-dependent protein kinase-dependent manner when induced by thrombin.85 The results presented in this study not only revealed unexpected crosstalk between the ERK/MAPK pathway and the p38γ/δ isoforms without upstream induction, but also demonstrated the effectiveness of the optically triggered kinase strategy for selective light activation of kinase isoforms.
To determine the signaling node at which p38 crosstalk occurs within the ERK/MAPK signaling cascade, we tested p38-dependent ERK signaling activity in the presence of three inhibitors. Crosstalk was completely suppressed through application of a Raf (LY3009120), MEK (U0126) and ERK (SCH772984) inhibitor compared to the DMSO control, suggesting the point of ERK/MAPK signaling perturbation occurs at or above the Raf level. The crosstalk interaction requires further biochemical analysis to better understand the specific mechanism of ERK/MAPK attenuation by the decaged p38γ/δ isoforms. The p38-dependent negative attenuation of ERK signal transduction is hypothesized to occur through three different mechanisms: p38-mediated deactivation of scaffolding protein FRS2,83 PP2A-mediated deactivation of kinase suppressor of Ras-1 (KSR1), and PP2A-mediated deactivation of Raf1.75 However, our results represent the first demonstration of positive ERK signaling regulation upon p38 activation.
Collectively, these results support the efficiency and usefulness of photocaging for the acute control of kinase activity, leading to the dissection of complex mechanisms of protein signaling with complete isoform specificity. Given the level of sequence homology and functional redundancy often found between protein isoforms, this level of specificity is a major advantage of our technique. The small size of the hydroxycoumarin caging group, paired with the restoration of the native active site upon decaging minimizes the possibility of structural disruption. This has the benefit of reducing the need for protein engineering and diminishing the possibility of background activity in the dark state, both of which are common challenges of optogenetic technologies.86
Future applications of this caging technique may be useful as an alternative to traditional biochemical analysis in determining the mechanistic function of individual isoforms with spatiotemporal accuracy and complete genetic specificity. For example, it could be utilized to investigate the extensive substrate landscape for each of the p38 isoforms, including how cell type and status (e.g., cell cycle) may contextualize isoform function, both of which are currently underrepresented areas of p38 research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (MCB-1330746) and the National Institutes of Health (R35GM146896, R01AI175067). We would like to thank the Chin Lab for kindly providing the pE323 and pE363 plasmid backbones. We would also like to thank Dr Kevin Janes (no. 47575), Dr Roger Vais (no. 20355 and 20353), Dr Kirill Alexandrov (no. 67077), Dr Silvia Corvera (no. 42635), and Dr Markus Covert (no. 59150 and 59152) for depositing the indicated Addgene plasmids. We thank Dr Toufiqur Rahman for help with imaging protocols.References
- A. Cuenda and S. Rousseau, Biochim. Biophys. Acta, Mol. Cell Res., 2007, 1773, 1358–1375 CrossRef CAS PubMed.
- A. Mansouri, L. D. Ridgway, A. L. Korapati, Q. Zhang, L. Tian, Y. Wang, Z. H. Siddik, G. B. Mills and F. X. Claret, J. Biol. Chem., 2003, 278, 19245–19256 CrossRef CAS.
- X. Sui, N. Kong, L. Ye, W. Han, J. Zhou, Q. Zhang, C. He and H. Pan, Cancer Lett., 2014, 344, 174–179 CrossRef CAS.
- T. Wada and J. M. Penninger, Oncogene, 2004, 23, 2838–2849 CrossRef CAS.
- T. Zarubin and J. Han, Cell Res., 2005, 15, 11–18 CrossRef CAS PubMed.
- J. Han and P. Sun, Trends Biochem. Sci., 2007, 32, 364–371 CrossRef CAS PubMed.
- D. V. Bulavin and A. J. Fornace, Adv. Cancer Res., 2004, 92, 95–118 CrossRef CAS PubMed.
- Y. Xu, N. Li, R. Xiang and P. Sun, Trends Biochem. Sci., 2014, 39, 268–276 CrossRef CAS PubMed.
- F. Dodeller and H. Schulze-Koops, Arthritis Res. Ther., 2006, 8, 205 CrossRef PubMed.
- J. D. Ashwell, Nat. Rev. Immunol., 2006, 6, 532–540 CrossRef CAS PubMed.
- G. Huang, L. Z. Shi and H. Chi, Cytokine, 2009, 48, 161–169 CrossRef CAS PubMed.
- P. W. Gunning, eLS, John Wiley & Sons, Ltd, 2006> Search PubMed.
- M. K. Saba-El-Leil, C. Frémin and S. Meloche, Front. Cell Dev. Biol., 2016, 4(67), 1–9 Search PubMed.
- A. Risco and A. Cuenda, J. Signal Transduction, 2012, 2011, e520289 Search PubMed.
- Z. Li, Y. Jiang, R. J. Ulevitch and J. Han, Biochem. Biophys. Res. Commun., 1996, 228, 334–340 CrossRef CAS PubMed.
- Y. Jiang, C. Chen, Z. Li, W. Guo, J. A. Gegner, S. Lin and J. Han, J. Biol. Chem., 1996, 271, 17920–17926 CrossRef CAS PubMed.
- Y. Jiang, H. Gram, M. Zhao, L. New, J. Gu, L. Feng, F. D. Padova, R. J. Ulevitch and J. Han, J. Biol. Chem., 1997, 272, 30122–30128 CrossRef CAS PubMed.
- A. Cuenda and D. S. Dorow, Biochem. J., 1998, 333(Pt 1), 11–15 CrossRef CAS PubMed.
- S. Kumar, P. C. McDonnell, R. J. Gum, A. T. Hand, J. C. Lee and P. R. Young, Biochem. Biophys. Res. Commun., 1997, 235, 533–538 CrossRef CAS PubMed.
- K. K. Hale, D. Trollinger, M. Rihanek and C. L. Manthey, J. Immunol., 1999, 162, 4246–4252 CrossRef CAS.
- D. Tischer and O. D. Weiner, Nat. Rev. Mol. Cell Biol., 2014, 15, 551–558 CrossRef CAS PubMed.
- P. A. Gagliardi and O. Pertz, Dev. Cell, 2019, 48, 289–290 CrossRef CAS PubMed.
- Y. Liu, R. Zeng, R. Wang, Y. Weng, R. Wang, P. Zou and P. R. Chen, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2025299118 CrossRef CAS PubMed.
- A. V. Karginov, F. Ding, P. Kota, N. V. Dokholyan and K. M. Hahn, Nat. Biotechnol., 2010, 28, 743–747 CrossRef CAS PubMed.
- Y.-H. Tsai, S. Essig, J. R. James, K. Lang and J. W. Chin, Nat. Chem., 2015, 7, 554–561 CrossRef CAS PubMed.
- J. E. Toettcher, O. D. Weiner and W. A. Lim, Cell, 2013, 155, 1422–1434 CrossRef CAS PubMed.
- H. E. Johnson, Y. Goyal, N. L. Pannucci, T. Schüpbach, S. Y. Shvartsman and J. E. Toettcher, Dev. Cell, 2017, 40, 185–192 CrossRef CAS PubMed.
- A. Levskaya, O. D. Weiner, W. A. Lim and C. A. Voigt, Nature, 2009, 461, 997–1001 CrossRef CAS PubMed.
- H. Wang, M. Vilela, A. Winkler, M. Tarnawski, I. Schlichting, H. Yumerefendi, B. Kuhlman, R. Liu, G. Danuser and K. M. Hahn, Nat. Methods, 2016, 13, 755–758 CrossRef CAS.
- W. Zhou and A. Deiters, Curr. Opin. Chem. Biol., 2021, 63, 123–131 CrossRef CAS PubMed.
- A. Gautier, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2011, 133, 2124–2127 CrossRef CAS PubMed.
- A. Liaunardy-Jopeace, B. L. Murton, M. Mahesh, J. W. Chin and J. R. James, Nat. Struct. Mol. Biol., 2017, 24, 1155–1163 CrossRef CAS PubMed.
- A. Gautier, D. P. Nguyen, H. Lusic, W. An, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2010, 132, 4086–4088 CrossRef CAS PubMed.
- N. Ankenbruck, T. Courtney, Y. Naro and A. Deiters, Angew. Chem., Int. Ed., 2018, 57, 2768–2798 CrossRef CAS PubMed.
- A. Deiters, Curr. Opin. Chem. Biol., 2009, 13, 678–686 CrossRef CAS PubMed.
- L. Gardner and A. Deiters, Curr. Opin. Chem. Biol., 2012, 16, 292–299 CrossRef CAS PubMed.
- T. Courtney and A. Deiters, Curr. Opin. Chem. Biol., 2018, 46, 99–107 CrossRef CAS PubMed.
- D. D. Young and A. Deiters, Org. Biomol. Chem., 2007, 5, 999–1005 RSC.
- A. S. Baker and A. Deiters, ACS Chem. Biol., 2014, 9, 1398–1407 CrossRef CAS PubMed.
- L. Fenno, O. Yizhar and K. Deisseroth, Annu. Rev. Neurosci., 2011, 34, 389–412 CrossRef CAS PubMed.
- H.-M. Lee, D. R. Larson and D. S. Lawrence, ACS Chem. Biol., 2009, 4, 409–427 CrossRef CAS PubMed.
- G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900–4921 CrossRef CAS PubMed.
- D. S. Lawrence, Curr. Opin. Chem. Biol., 2005, 9, 570–575 CrossRef CAS PubMed.
- S. Md. T. Rahman, W. Zhou, A. Deiters and J. M. Haugh, J. Biol. Chem., 2020, 295, 8494–8504 CrossRef CAS PubMed.
- M. Avitzour, R. Diskin, B. Raboy, N. Askari, D. Engelberg and O. Livnah, FEBS J., 2007, 274, 963–975 CrossRef CAS PubMed.
- R. Diskin, N. Askari, R. Capone, D. Engelberg and O. Livnah, J. Biol. Chem., 2004, 279, 47040–47049 CrossRef CAS PubMed.
- L. Kim, L. D. Rio, B. A. Butcher, T. H. Mogensen, S. R. Paludan, R. A. Flavell and E. Y. Denkers, J. Immunol., 2005, 174, 4178–4184 CrossRef CAS PubMed.
- J. Beenstock, S. Ben-Yehuda, D. Melamed, A. Admon, O. Livnah, N. G. Ahn and D. Engelberg, J. Biol. Chem., 2014, 289, 23546–23556 CrossRef CAS PubMed.
- J. Beenstock, D. Melamed, N. Mooshayef, D. Mordechay, B. P. Garfinkel, N. G. Ahn, A. Admon and D. Engelberg, Mol. Cell. Biol., 2016, 36(10), 1540–1554 CrossRef CAS PubMed.
- N. Askari, R. Diskin, M. Avitzour, R. Capone, O. Livnah and D. Engelberg, J. Biol. Chem., 2007, 282, 91–99 CrossRef CAS PubMed.
- N. Trempolec, N. Dave-Coll and A. R. Nebreda, Cell, 2013, 152(3), 656 CrossRef CAS PubMed.
- V. B. Pillai, N. R. Sundaresan, S. A. Samant, D. Wolfgeher, C. M. Trivedi and M. P. Gupta, Mol. Cell. Biol., 2011, 31(11), 2349–2363 CrossRef CAS PubMed.
- S. Kumar, M. M. McLaughlin, P. C. McDonnell, J. C. Lee, G. P. Livi and P. R. Young, J. Biol. Chem., 1995, 270, 29043–29046 CrossRef CAS PubMed.
- J. Luo, R. Uprety, Y. Naro, C. Chou, D. P. Nguyen, J. W. Chin and A. Deiters, J. Am. Chem. Soc., 2014, 136, 15551–15558 CrossRef CAS PubMed.
- M. A. Shandell, Z. Tan and V. W. Cornish, Biochemistry, 2021, 60, 3455–3469 CrossRef CAS PubMed.
- S. S. Taylor and A. P. Kornev, Trends Biochem. Sci., 2011, 36, 65–77 CrossRef CAS PubMed.
- A. Kuzmanic, L. Sutto, G. Saladino, A. R. Nebreda, F. L. Gervasio and M. Orozco, eLife, 2017, 6, e22175 CrossRef PubMed.
- G. J. Finn, B. Creaven and D. A. Egan, Melanoma Res., 2001, 11, 461–467 CrossRef CAS PubMed.
- L. F. Cruz, G. F. de Figueiredo, L. P. Pedro, Y. M. Amorin, J. T. Andrade, T. F. Passos, F. F. Rodrigues, I. L. A. Souza, T. P. R. Gonçalves, L. A. R. dos Santos Lima, J. M. S. Ferreira and M. G. de F. Araújo, Biomed. Pharmacother., 2020, 129, 110432 CrossRef CAS PubMed.
- R. S. Givens, M. Rubina and J. Wirz, Photochem. Photobiol. Sci., 2012, 11, 472–488 CrossRef CAS PubMed.
- Q. Zheng, S. Jockusch, Z. Zhou and S. C. Blanchard, Photochem. Photobiol., 2014, 90, 448–454 CrossRef CAS PubMed.
- J. E. Sullivan, G. A. Holdgate, D. Campbell, D. Timms, S. Gerhardt, J. Breed, A. L. Breeze, A. Bermingham, R. A. Pauptit, R. A. Norman, K. J. Embrey, J. Read, W. S. VanScyoc and W. H. J. Ward, Biochemistry, 2005, 44, 16475–16490 CrossRef CAS PubMed.
- R. W. Holley and J. A. Kiernan, Proc. Natl. Acad. Sci. U. S. A., 1968, 60, 300–304 CrossRef CAS PubMed.
- D. I. Bryson, C. Fan, L.-T. Guo, C. Miller, D. Söll and D. R. Liu, Nat. Chem. Biol., 2017, 13, 1253–1260 CrossRef CAS PubMed.
- E. M. Merzlyak, J. Goedhart, D. Shcherbo, M. E. Bulina, A. S. Shcheglov, A. F. Fradkov, A. Gaintzeva, K. A. Lukyanov, S. Lukyanov, T. W. J. Gadella and D. M. Chudakov, Nat. Methods, 2007, 4, 555–557 CrossRef CAS PubMed.
- J. W. Chin, Nature, 2017, 550, 53–60 CrossRef CAS PubMed.
- H. Enslen, J. Raingeaud and R. J. Davis, J. Biol. Chem., 1998, 273, 1741–1748 CrossRef CAS PubMed.
- G. A. Keesler, J. Bray, J. Hunt, D. A. Johnson, T. Gleason, Z. Yao, S.-W. Wang, C. Parker, H. Yamane, C. Cole and H. S. Lichenstein, Protein Expression Purif., 1998, 14, 221–228 CrossRef CAS PubMed.
- D. L. Long and R. F. Loeser, Osteoarthr. Cartil., 2010, 18, 1203–1210 CrossRef CAS PubMed.
- D. M. Ouwens, N. D. de Ruiter, G. C. M. van der Zon, A. P. Carter, J. Schouten, C. van der Burgt, K. Kooistra, J. L. Bos, J. A. Maassen and H. van Dam, EMBO J., 2002, 21, 3782–3793 CrossRef CAS PubMed.
- W. F. Waas, H.-H. Lo and K. N. Dalby, J. Biol. Chem., 2001, 276, 5676–5684 CrossRef CAS PubMed.
- W. Breitwieser, S. Lyons, A. M. Flenniken, G. Ashton, G. Bruder, M. Willington, G. Lacaud, V. Kouskoff and N. Jones, Genes Dev., 2007, 21, 2069–2082 CrossRef CAS PubMed.
- S. Regot, J. J. Hughey, B. T. Bajar, S. Carrasco and M. W. Covert, Cell, 2014, 157, 1724–1734 CrossRef CAS PubMed.
- D. D. Young and P. G. Schultz, ACS Chem. Biol., 2018, 13, 854–870 CrossRef CAS PubMed.
- S. Grethe and M. I. Pörn-Ares, Cell. Signalling, 2006, 18, 531–540 CrossRef CAS PubMed.
- S. Bellon, M. J. Fitzgibbon, T. Fox, H.-M. Hsiao and K. P. Wilson, Structure, 1999, 7, 1057–1065 CrossRef CAS PubMed.
- Handbook of cell signaling, ed. R. A. Bradshaw and E. A. Dennis, Academic Press, Amsterdam, San Diego, Calif, 2004, vol. 1 Search PubMed.
- M. Ambrose, A. Ryan, G. C. O’Sullivan, C. Dunne and O. P. Barry, Mol. Pharmacol., 2006, 69, 1879–1890 CrossRef CAS PubMed.
- A. Plotnikov, K. Flores, G. Maik-Rachline, E. Zehorai, E. Kapri-Pardes, D. A. Berti, T. Hanoch, M. J. Besser and R. Seger, Nat. Commun., 2015, 6, 6685 CrossRef CAS PubMed.
- J. R. Henry, M. D. Kaufman, S.-B. Peng, Y. M. Ahn, T. M. Caldwell, L. Vogeti, H. Telikepalli, W.-P. Lu, M. M. Hood, T. J. Rutkoski, B. D. Smith, S. Vogeti, D. Miller, S. C. Wise, L. Chun, X. Zhang, Y. Zhang, L. Kays, P. A. Hipskind, A. D. Wrobleski, K. L. Lobb, J. M. Clay, J. D. Cohen, J. L. Walgren, D. McCann, P. Patel, D. K. Clawson, S. Guo, D. Manglicmot, C. Groshong, C. Logan, J. J. Starling and D. L. Flynn, J. Med. Chem., 2015, 58, 4165–4179 CrossRef CAS PubMed.
- M. F. Favata, K. Y. Horiuchi, E. J. Manos, A. J. Daulerio, D. A. Stradley, W. S. Feeser, D. E. Van Dyk, W. J. Pitts, R. A. Earl, F. Hobbs, R. A. Copeland, R. L. Magolda, P. A. Scherle and J. M. Trzaskos, J. Biol. Chem., 1998, 273, 18623–18632 CrossRef CAS PubMed.
- E. J. Morris, S. Jha, C. R. Restaino, P. Dayananth, H. Zhu, A. Cooper, D. Carr, Y. Deng, W. Jin, S. Black, B. Long, J. Liu, E. DiNunzio, W. Windsor, R. Zhang, S. Zhao, M. H. Angagaw, E. M. Pinheiro, J. Desai, L. Xiao, G. Shipps, A. Hruza, J. Wang, J. Kelly, S. Paliwal, X. Gao, B. S. Babu, L. Zhu, P. Daublain, L. Zhang, B. A. Lutterbach, M. R. Pelletier, U. Philippar, P. Siliphaivanh, D. Witter, P. Kirschmeier, W. R. Bishop, D. Hicklin, D. G. Gilliland, L. Jayaraman, L. Zawel, S. Fawell and A. A. Samatar, Cancer Discovery, 2013, 3, 742–750 CrossRef CAS PubMed.
- M. Zakrzewska, L. Opalinski, E. M. Haugsten, J. Otlewski and A. Wiedlocha, Int. J. Mol. Sci., 2019, 20, 1826 CrossRef CAS PubMed.
- T. Matsumoto, K. Yokote, K. Tamura, M. Takemoto, H. Ueno, Y. Saito and S. Mori, J. Biol. Chem., 1999, 274, 13954–13960 CrossRef CAS PubMed.
- Z. Li, G. Zhang, R. Feil, J. Han and X. Du, Blood, 2006, 107, 965–972 CrossRef CAS PubMed.
- C. P. O’Banion and D. S. Lawrence, ChemBioChem, 2018, 19, 1201–1216 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC decaging study of coumarin lysine, cell viability, schematic of the MAPK KTR reporter mechanism, micrograph and time course data for p38-mediated suppression of ERK/MAPK signaling, full time course micrographs for Fig. 3 and 4, and detailed biological protocols, primer sequences, and plasmid maps. See DOI: https://doi.org/10.1039/d2cb00157h |
| This journal is © The Royal Society of Chemistry 2023 |