
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-action gallium-flavonoid compounds for combating Pseudomonas aeruginosa infection†
Xiaojun
He‡
ad,
Bingjie
Han‡
a,
Runming
Wang‡
bc,
Yu
Guo
a,
Richard Y T
Kao
c,
Hongyan
Li
b,
Hongzhe
Sun
 *b and
Wei
Xia
*a
*b and
Wei
Xia
*a
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-sen University, Guangzhou, 510275, China. E-mail: xiawei5@mail.sysu.edu.cn
bDepartment of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China. E-mail: hsun@hku.hk
cDepartment of Microbiology and State Key Laboratory for Emerging Infectious Diseases, The University of Hong Kong, Hong Kong, P. R. China
dSchool of Ophthalmology & Optometry, School of Biomedical Engineering, Wenzhou Medical University, Wenzhou, Zhejiang 325035, China
First published on 11th August 2023
Abstract
The opportunistic pathogen Pseudomonas aeruginosa (P. aeruginosa) causes infections that are difficult to treat, which is due to the bacterial natural resistance to antibiotics. The bacterium is also able to form a biofilm that protects the bacterium from clearance by the human immune system and leads to chronic infection. Herein, we synthesized and characterized a novel gallium compound that interferes with both the iron metabolism and quorum sensing system of P. aeruginosa to achieve a significant bactericidal activity. The compound could substantially reduce the secretion of bacterial virulence factors as well as eliminate biofilm formation. Integrative omics analysis indicates that this compound can significantly disturb the gene transcription and metabolism of P. aeruginosa. The effectiveness of the gallium compound was further validated in mammalian cell and murine skin infection models. Our study offers a new strategy to design new gallium-based antimicrobials to combat P. aeruginosa infection.
Introduction
Pseudomonas aeruginosa (P. aeruginosa) is a major opportunistic human pathogen, which is a critical priority 1 pathogen as listed by the WHO in 2017.1,2 The bacterium usually causes severe and persistent infections in immune-compromised individuals and cystic fibrosis (CF) patients.3,4 The infections caused by P. aeruginosa are usually resistant to treatment of multiple antibiotics, which is resulted from the bacterium's intrinsic drug resistance and additional acquired resistance.5 The antimicrobial tolerance of P. aeruginosa is even enhanced via the formation of biofilm, which protects the bacterium from the detrimental effects of host immunity and antibiotic treatment.6–9 Moreover, P. aeruginosa secretes a series of virulence factors, including pyocyanin, elastase, and alkaline protease (AprA), which exert important pathological roles in the colonization, survival, and infection of the bacterium.10,11 The biofilm formation and virulence factor production of P. aeruginosa are coordinately regulated by a mechanism termed quorum sensing (QS).12 The bacteria produce and release small chemical signals when rich at a high population density. The accumulated signals could bind with cognate receptors to induce virulence factor expression and biofilm formation.13,14 Given the critical role of the QS system, development of small molecules that block quorum sensing circuits is considered as a promising strategy to combat bacterial pathogenicity.15,16 The flavonoids are a group of natural products, which exhibit broad biological activities ranging from anti-microbial to anti-inflammatory.17 Particularly, recent studies indicated that flavonoids specifically inhibited the P. aeruginosa QS system as antagonists of the autoinducer-binding receptors, LasR and RhlR.18–21Like the vast majority of pathogens, P. aeruginosa requires iron for its growth. Moreover, iron depletion prevents biofilm formation of the bacterium.22,23 Therefore, interfering with P. aeruginosa iron homeostasis may serve as a potential therapeutic strategy for bacterial eradication. Recent studies demonstrated that gallium (Ga3+) could be used as a “Trojan horse” to disrupt bacterial iron metabolism due to its iron-mimetic properties. It was reported that Ga3+ had anti-biofilm activity and could effectively eradicate P. aeruginosa in two animal models, indicative of a potential therapeutic option for P. aeruginosa infection.24–26
Due to the emerging and sustained resistance to currently available antibiotics, the development of new antibacterial agents is urgently needed. One promising strategy is the development of dual-action antibacterial agents, which inhibit distinct bacterial targets at the same time. The synergistic effects could enhance antibacterial activity and reduce potential for the development of drug resistance. Herein, we report the synthesis and evaluation of antimicrobial activities of a series of novel gallium-flavonoid compounds, among which gallium-naringenin (G6) exhibits the highest bactericidal activity against P. aeruginosa with relatively low cytotoxicity to mammalian cells. Importantly, the compound exhibits dual-targeted anti-bacterial activities, i.e., disrupting both the QS system and iron metabolism of P. aeruginosa, resulting in significant attenuation of bacterial virulence factor secretion and biofilm formation.
Results and discussion
Synthesis and characterization of gallium-flavonoid compounds
Six target flavonoid ligands (Fig. 1(a)) were chosen to provide structural diversity in the composition of the gallium compounds.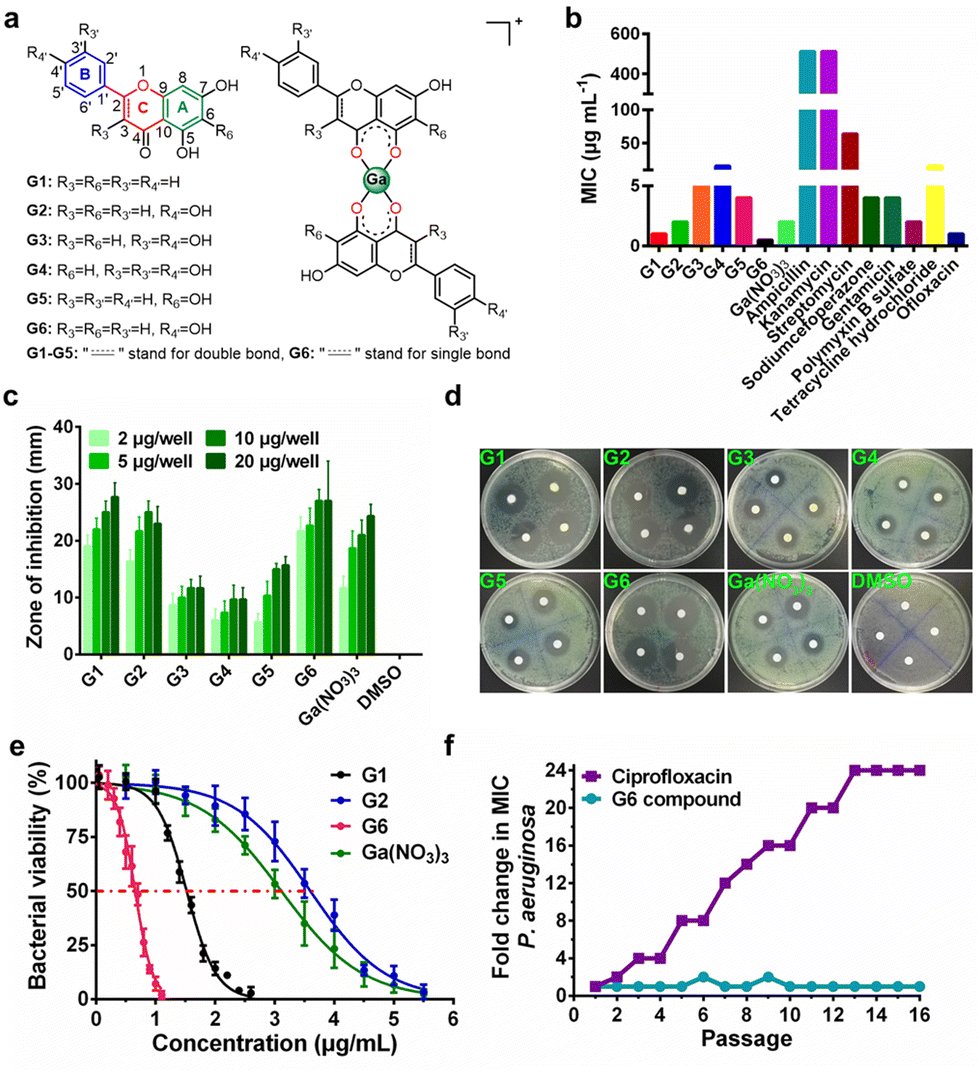 | ||
| Fig. 1 Anti-bacterial activity of gallium-flavonoid compounds in vitro. (a) The chemical structures of flavonoid ligands and gallium compounds. (b) The minimum inhibition concentration (MIC) of gallium-flavonoid compounds and antibiotics against P. aeruginosa. (c) Diameters of inhibition zones of gallium compounds, Ga(NO3)3, and DMSO. (d) Representative figures of the agar diffusion test. (e) Bacterial viability assay for gallium-flavonoid compounds and Ga(NO3)3. The IC50 values of G1, G2, G6 and Ga(NO3)3 are 1.52, 3.59, 0.65, and 3.11 μg mL−1, respectively. All experiments are performed in triplicates, and data are presented as mean ± SD. (f) Resistance development of P. aeruginosa PAO1 toward G6 and ciprofloxacin. Values are fold changes in minimal inhibitory concentration (MIC) relative to the MIC of the first passage. | ||
Chrysin (G1), baicalein (G5), and naringenin (G6) have already been identified to be antagonists against P. aeruginosa QS system.18,20 The other three analogs (apigenin, G2; luteolin, G3; quercetin, G4) have varying hydroxyl groups at different flavonoid rings. The synthesis and structural characterization of the gallium compounds are described in the ESI.† Typically, all the six flavonoids coordinate Ga3+via the 4-carbonly-5-hydroxyl site of the flavonoid (4, 5 sites) and form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 Ga3+ flavonoid compounds. These gallium compounds were stable at room temperature for at least one week as validated by UV-vis spectroscopy and HPLC analysis (ESI†).
:2 Ga3+ flavonoid compounds. These gallium compounds were stable at room temperature for at least one week as validated by UV-vis spectroscopy and HPLC analysis (ESI†).
Antibacterial activity of gallium-flavonoid compounds
To examine the anti-bacterial activities of the synthesized compounds, we first measured the minimal inhibitory concentrations (MIC) of these compounds against Gram-positive bacterium Staphylococcus aureus Newman (S. aureus), Gram-negative bacteria Escherichia coli DH5α (E. coli) and P. aeruginosa PAO1 (Table S1, ESI†). The compounds exhibit low and moderate inhibitory activities against E. coli (MIC ≥ 128 μg mL−1) and S. aureus (MIC ≥ 64 μg mL−1). In contrast, all the compounds have significantly higher inhibitory activities against P. aeruginosa with MIC values ranging from 0.5 to 16 μg mL−1, which are comparable with some commonly used antibiotics, such as Ofloxacin (Fig. 1(b)). The agar plate diffusion test was subsequently applied to further examine the antimicrobial activities of the gallium compounds. In brief, approximately 10 μL of an aqueous solution of the compounds were loaded onto filter paper discs and implanted on the surface of agar plates inoculated with P. aeruginosa. The plates were incubated at 37 °C for 24 h and the diameters of the inhibition zones were measured. In consistent with the obtained MIC results, G6 had the largest inhibition zones at all four different dilutions, followed by G1 and G2, while the negative control DMSO did not affect bacterial growth (Fig. 1(c) and (d)). As shown in Fig. 1(e), the two most potent compounds G6 and G1 had even higher anti-P. aeruginosa activity (with IC50 values of 0.65 and 1.52 μg mL−1, respectively) than the Ga3+ alone (Ga(NO3)3 with IC50 value of 3.11 μg mL−1). In addition, the bactericidal effect of the gallium-flavonoid compounds was observed using agar plate counting method (Fig. S1, ESI†).Since resistance development is a major concern for anti-bacterial agents, we subsequently assessed the ability of P. aeruginosa to develop resistance to the most potent gallium compound G6 as described previously.27 As shown in Fig. 1(f), no drug resistance was developed in P. aeruginosa after the serial passage of the bacteria in the presence of a sub-inhibitory concentration of G6 (with almost the same MIC value of 0.5 μg mL−1 at the first and 16th passages). In contrast, exposure to the antibiotic ciprofloxacin led to a rapid increase in MIC after the 16th passage (with MIC value rising from 1 to 24 μg mL−1). The results indicate that P. aeruginosa fails to develop resistance against the G6 compound.
Gallium-flavonoid compounds inhibit biofilm formation
To investigate the effects of gallium compounds on P. aeruginosa biofilm development, we first examined biofilm formation under static growth conditions in microtiter dish wells. The bacteria were cultured for 48 h and the adherent biofilms were stained with crystal violet and quantified (Fig. 2(a)). In consistence with a previous report, the presence of 0.08 μM Ga(NO3)3 reduced biofilm formation by approximately 50% compared to the negative control. Among all the gallium compounds, G6, G1, and G2 were the most potent inhibitors, which attenuated biofilm formation by approximately 90%, 70%, and 50%, respectively (Fig. 2(b)). In contrast, all the flavonoid ligands had no evident effects on biofilm formation (Fig. S2, ESI†). Furthermore, the alternation in the structure of the biofilms was also characterized by field emission scanning electron microscopy (FE-SEM). The bacterial culture conditions were the same except that a glass slide was implanted in the microtiter well. The biofilms were harvested after growing for 1 and 5 days and prepared for SEM data collection as described previously.28 Clear biofilm formation was observed in the control well, which contained dense intracellularly fiber-like structures consisting of polysaccharides and DNA.29 Incubation of compounds G3, G4, and G5 had little or no obvious effects on the biofilm formation. In contrast, compounds G1, G2, G6, and Ga(NO3)3 significantly reduced the interconnecting fiber-like structures of the biofilms. Compound G6 was particularly potent since no fiber-like structures were observed in P. aeruginosa culture even after 5 days (Fig. 2(c)). Typically, the bacterium was cultured in the absence or presence of gallium compounds with sub-inhibitory concentration at 0.08 μM, which do not affect planktonic growth of P. aeruginosa (Fig. 2(d)). The results indicate that the sub-inhibitory concentration of gallium compounds is sufficient to prevent P. aeruginosa biofilm formation.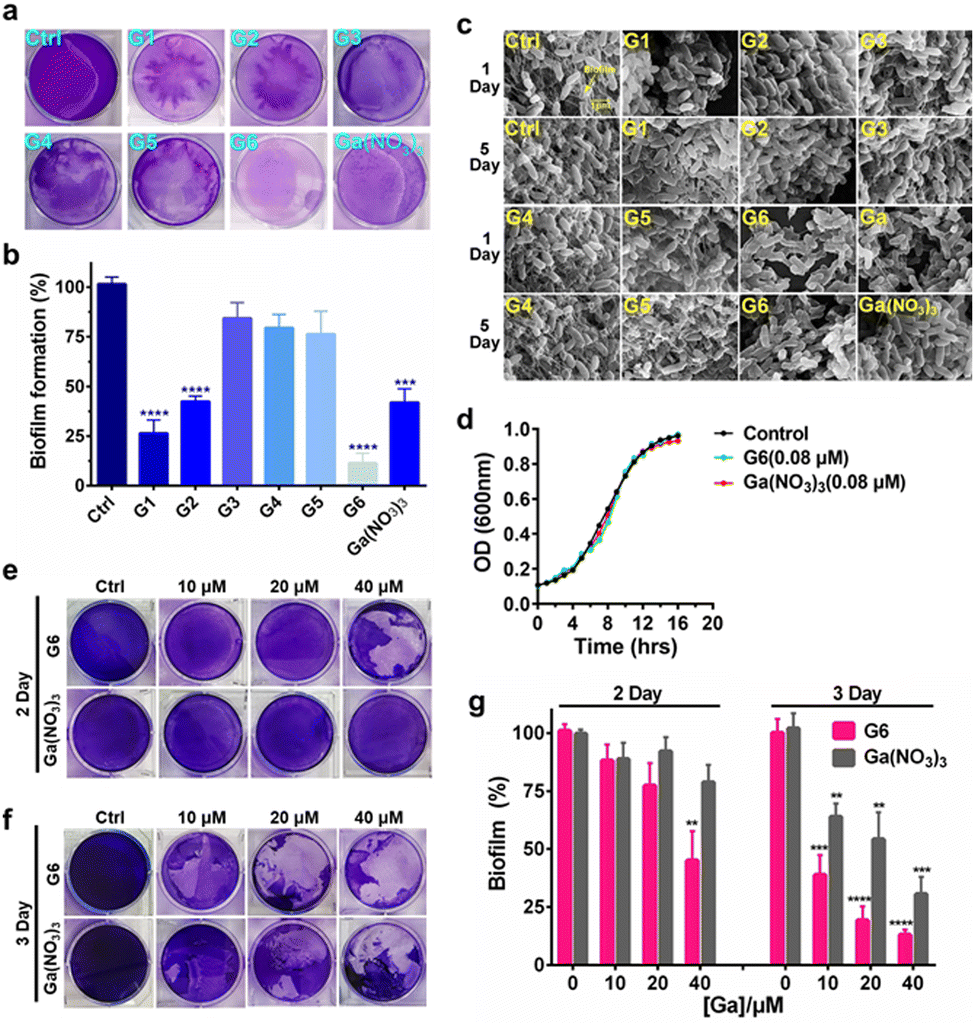 | ||
| Fig. 2 Gallium-flavonoid compounds inhibit biofilm formation and eradicate established biofilm in P. aeruginosa. (a) Representative photograph of biofilm formation of Pseudomonas aeruginosa incubated with gallium compounds and Ga(NO3)3. (b) Quantitative analysis of biofilm formation after 48 h in the absence or presence of a sub-inhibitory concentration of gallium compounds and Ga(NO3)3. (c) Scanning electron microscope (SEM) images of P. aeruginosa biofilms in the presence of a sub-lethal concentration of gallium compounds and Ga(NO3)3. (d) Growth curves of P. aeruginosa in the presence of a sub-lethal concentration of gallium compound G6 and Ga(NO3)3. Photograph of P. aeruginosa biofilm eradication by gallium compound G6 and Ga(NO3)3. The remained biofilms were stained by crystal violet after 2 days (e) and 3 days (f) incubation, respectively. (g) Biofilm eradication assay. 48 hours-old mature biofilms were incubated with gradient amounts of G6 compound and Ga(NO3)3. The biofilm was stained with crystal violet and quantified. The percentages of remaining mature biofilms in the control group are set to 100%. The biofilms in the experiment group are normalized to that in the control group. All experiments are performed in triplicates, and data are presented as mean ± SD. Two-tailed ratio paired t test was used to determine statistical significance; **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
To examine whether the gallium compounds could eradicate established biofilms, we further used the microtiter-well biofilm assay and applied the gallium compound G6 and Ga(NO3)3 after the biofilm was formed. In brief, P. aeruginosa was cultured for 48 h in a 6-well microtiter plate to allow biofilm formation. The matured biofilm was subsequently incubated with gradient amounts of G6 compound or Ga(NO3)3 (0, 10, 20, and 40 μM), respectively. The remained biofilm contents were quantified by crystal violet staining after an additional 2 or 3 day incubation (Fig. 2(e) and (f)). As shown in Fig. 2(g), 40 μM G6 compound could abrogate approximately 50% of established biofilms after 2 day incubation. On the third day, both the G6 compound and Ga(NO3)3 could significantly abolish pre-formed biofilms with G6 compound exhibiting higher activity, since only 10% of biofilms remained after treatment of 40 μM G6 compound compared to 30% for 40 μM Ga(NO3)3. Collectively, the results demonstrate that gallium compound G6 is particularly effective to prevent biofilm formation as well as eradicating established biofilms.
Transcriptomics and metabolomics analysis of P. aeruginosa
To further investigate the anti-bacterial mechanism of G6 compound, we examined the transcriptomic profiles of P. aeruginosa after exposure to the G6 compound. RNA-seq data revealed that 1527 genes in total were differentially expressed when the P. aeruginosa PAO1 strain was incubated with 4 μM G6 compound for 4 h, among which 749 genes were upregulated and 778 genes were downregulated (Fig. 3(a)). Gene ontology analysis showed that significantly altered genes were enriched in several major biological processes, including metabolic process, cellular process, cell localization, single-organism process, response to stimulus, etc. Meanwhile, 9 GO terms involved in molecular function including catalytic activity, especially for the oxidoreductase activity, nucleic acid binding transcription factor activity, transporter activity, and electron activity were significantly enriched (Fig. 3(b)), implying that the cellular redox process, protein biosynthesis, and nucleotide metabolism were disturbed after the treatment of G6 compound. According to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, the significantly altered genes could be classified into several different pathways, such as oxidative phosphorylation, TCA cycle, fatty acid biosynthesis, and biofilm formation (Fig. S3, ESI†). Further analysis demonstrated that G6 compound could significantly changed the transcription of genes involved in quorum sensing (QS) and virulence factor (VF) (Fig. 3(c)). For example, the genes involved in las (lasAB), rhl (rhlR and rhlAB) and pqs (pqsE, pqsH, pqsR, and pqsBCD) signaling circuits were significantly down-regulated, and the virulence-related genes, such as those participating in phenazine biosynthesis (phzA, phzB, phzM, phzG, and phzS), alkaline protease secretion (aprDEF) and exotoxin A (toxA) also decreased in G6 compound treated bacteria. Collectively, the results suggested that the G6 compound significantly abrogated the QS system and the virulence secretion of P. aeruginosa. The RNA-seq data were also verified by quantitative PCR (qPCR) analysis (Fig. S4, ESI†). In consistence with the RNA-seq results, only lasR gene in the QS system was significantly upregulated, whereas all other genes were downregulated after G6 treatment.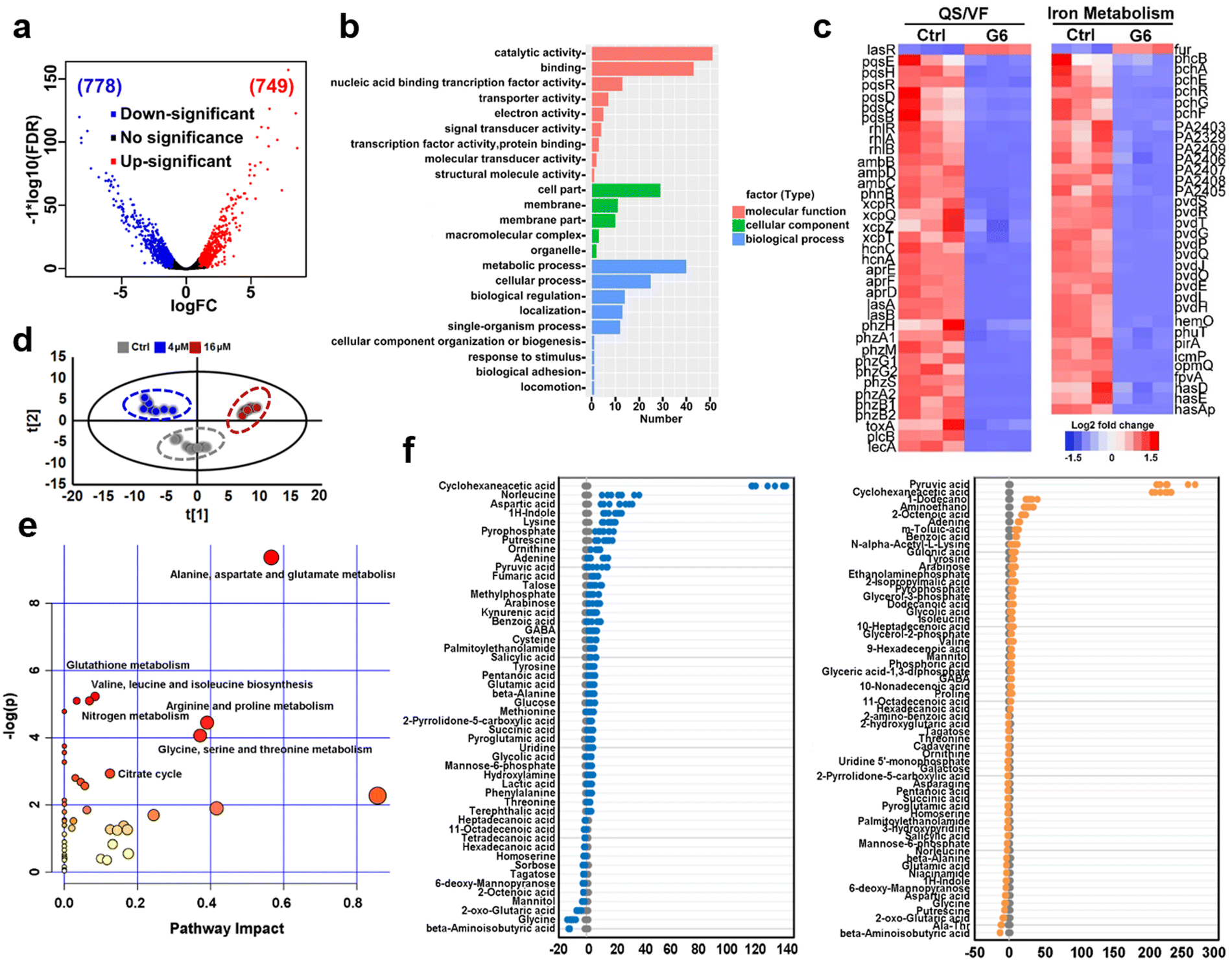 | ||
| Fig. 3 Transcriptomic and metabolic analysis of P. aeruginosa exposed to different concentrations of G6 compound. (a) Volcano plot of differentially expressed genes in transcriptomic analysis. (b) GO analysis of molecular function, cellular component, and biological process of significantly different genes. (c) Hierarchically clustered heatmaps for changes in gene transcript levels between G6 treated and control groups. Values reported as log2-fold changes over untreated control in the expression of the quorum sensing system, virulence factor, and iron metabolism. (d) PCA analysis of GC-MS profiles obtained from the control, 4 μM and 16 μM G6 treated (for 4 h) P. aeruginosa metabolite extracts. Each dot represents metabolite profiling data from one individual sample and colors indicate samples in different treatment groups. (e) KEGG pathway enrichment was analyzed of the differential P. aeruginosa metabolites after 16 μM G6 treatment. (f) Z-score plots of the differential metabolites based on control. Each point represents one metabolite in one biological repeat (grey: control; blue: 4 μM G6 treatment; orange: 16 μM G6 treatment). | ||
Since Ga3+ was reported to target P. aeruginosa iron metabolism to exert antimicrobial functionality, the transcription levels of the corresponding genes involved in iron homeostasis were also examined. Indeed, 37 among the 38 related genes were downregulated, including the genes involved in the biosynthesis of two siderophores, i.e., pyoverdine (pvd operon) and pyochelin (pch operon) and some iron transporters (has operon, fep operon). The previous study suggested that the antibacterial activity of Ga was attributed to the effect on the uptake, transport, and metabolic process of iron, especially interfering with bacteria for the synthesis of siderophores in an iron deficiency environment.30–33
The expression level of iron-sensing global regulatory protein (fur) increased by almost 2-fold, while genes involved in three siderophores, i.e., pyoverdine (pvd operon), pyochelin (pch operon), and heme acquisition protein (has operon) biosynthesis were significantly down-regulated (Fig. 3(c)). To further verify the interference of gallium on bacterial iron metabolism, inductively coupled plasma mass spectrometry (ICP-MS) was used to detect the iron content in bacteria before and after G6 treatment. Indeed, the iron content in bacteria decreased after G6 treatment (Fig. S5, ESI†), indicating that gallium has impact on the iron content in bacteria. All the data demonstrated that the G6 compound disturbed the iron metabolism of P. aeruginosa. Furthermore, we also performed metabolomic analysis to observe the changes in bacterial metabolites after G6 treatment. The results of PCA analysis showed a clear separation between the control group and the G6 treatment group (Fig. 3(d)). Among the 92 identified metabolites, the abundance of 50 metabolites in the 4 μM G6 treatment group and 61 metabolites in 16 μM G6 were significantly changed, including amino acids, carbohydrates, fatty acids, nucleotide acids, and others. Z-Score displayed variations of the differential metabolites based on the control group (Fig. 3(e)). The Z-scores for the 4 μM G6 treatment group were −12.5 to 132.2, with 37 metabolites at the higher abundance and 13 at the lower abundance. Meanwhile, the Z scores of the 16 μM G6 treatment group were −13.6 to 231.8, with the abundance of 31 metabolites up-regulated and 30 metabolites down-regulated. KEGG enrichment analysis revealed that the changed metabolites could be classified into various pathways, including alanine, aspartate, and glutamate metabolism, glutathione metabolism, TCA cycle, and nitrogen metabolism (Fig. 3(f)).
Gallium-flavonoid compounds inhibit virulence factors expression of P. aeruginosa
P. aeruginosa produces numerous virulence factors, which contribute to its ability to colonize, penetrate and survive the host's immune defense.34,35 For example, pyocyanin, a secondary metabolite secreted by P. aeruginosa can target a wide range of cellular pathways and therefore can kill mammalian lung cells upon bacterial infection.36 Given the prominent anti-biofilm activities of G1, G2, and G6 compounds, we further investigated the effects of these three gallium compounds on the expression of bacterial virulence factors, including elastase, pyocyanin, and pyoverdine (Fig. 4(a)). Phenazines are nitrogen-containing redox-active pigments produced by P. aeruginosa. The secreted pigment is recognized as an important bacterial virulence factor and plays an important role in establishing chronic and acute infections.35 As shown in Fig. 4(b), the secretion of pyocyanin was significantly suppressed in the presence of a sub-inhibitory concentration of gallium compounds (0.08 μM). In particular, G6 led to the most significant decrease (91%) in pyocyanin secretion. While compounds G1, G2, and Ga(NO3)3 reduced the pyocyanin production by 67%, 50%, and 47%, respectively. Elastase is a major virulent metalloprotease of P. aeruginosa that is believed to cause extensive tissue damage in the host during infection.37 Similarly, compound G6 exhibited the highest activity to inhibit the secretion of elastase by 89% (Fig. 4(c)). In contrast, compounds G1 and G2 were less potent, which caused 81% and 53% decrease in elastase, respectively. While siderophore pyoverdine is not only required for bacterial iron uptake but also contributes to the regulation of other virulence factors.38G6 is again the most potent compound and reduced the secretion of pyoverdine by 87%. While treatment of G1 and G2 caused 64% and 52% decrease in pyoverdine, respectively (Fig. 4(d)).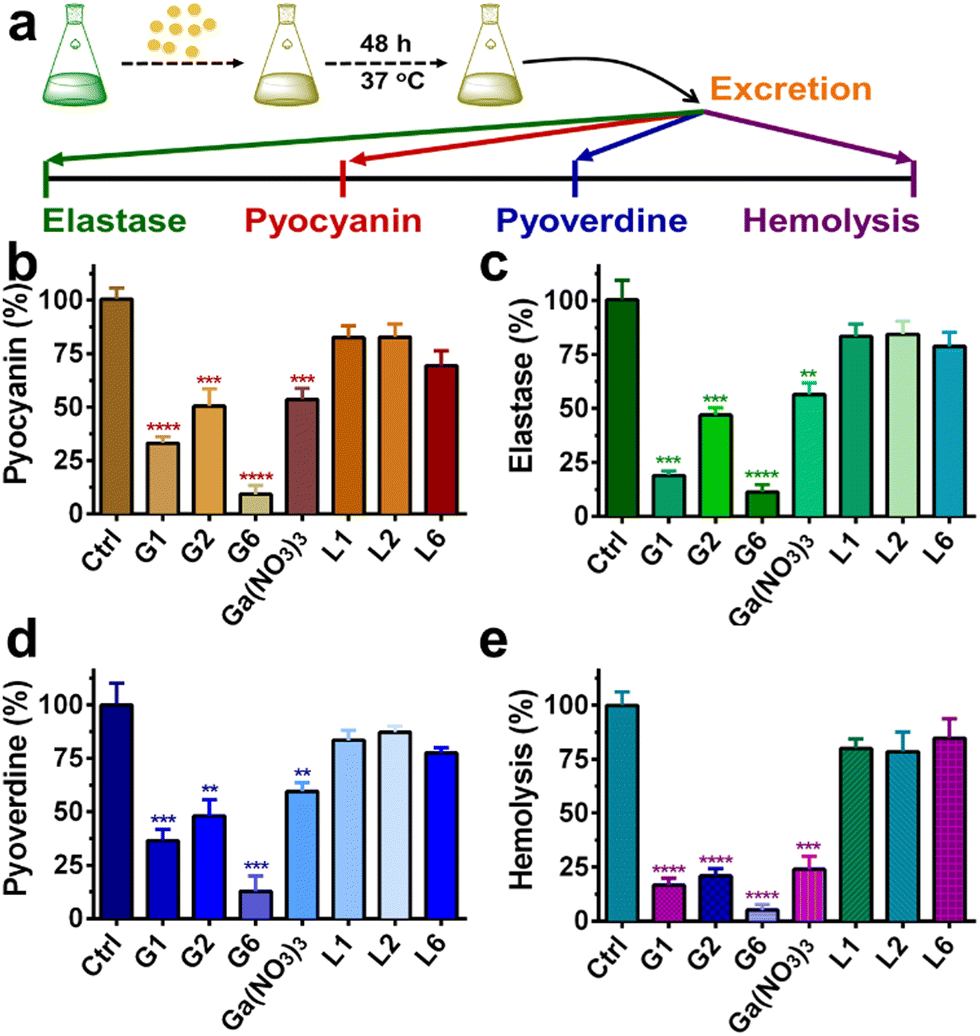 | ||
| Fig. 4 (a) Schematic illustration of virulence factors assay procedures. Quantitative analysis of secreted pyocyanin (b), elastase (c), pyoverdine (d), and hemolytic activities (e) of P. aeruginosa culture supernatant in the presence of 0.08 μM gallium compounds, Ga(NO3)3 and flavonoid ligands for 20 h. For hemolytic assay. Complete lysis of rabbit red blood cells with 0.1% SDS was used as a positive control. M9 medium was used as a negative control. All experiments are performed in triplicates, and data are presented as mean ± SD. Two-tailed ratio paired t test was used to determine statistical significance; **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
Moreover, the hemolytic activity of P. aeruginosa was also significantly suppressed in the presence of gallium compounds. The residual hemolytic activity of the bacterium was only 5% after incubation with the 0.08 μM G6 compound (Fig. 4(e)). All these results demonstrate that compound G6 has the highest inhibitory activity against virulence factor secretion among the gallium compounds. It is probably due to the different cell membrane permeability since compound G6 had the highest lipophilicity among all the compounds with a logP value of 1.86 (Table S2, ESI†). Kinetics studies also confirmed that the gallium internalization rate of P. aeruginosa was faster when incubated with G6 than that with G1, G2, or Ga(NO3)3. (Fig. S6, ESI†). It should be noted that the expression of virulence factors pyocyanin, elastase, and pyoverdine are all under the control of the P. aeruginosa QS system.39–41 Therefore, the results are consistent with the RNA-seq data that the gallium compounds could paralyze the QS system, thereby leading to the decreased expression of virulence factors. Our previous studies showed that the conserved P. aeruginosa HitABC transporter was responsible for the Ga(NO3)3 uptake.26 To further examine the internalization pathway of the gallium-flavonoid compound, both wild-type (WT) P. aeruginosa and hitAB gene knockout P. aeruginosa mutant (ΔhitAB) were treated with G6 compound and Ga(NO3)3 with the concentration of 0.16 μM and the intracellular gallium contents were measured by ICP-MS in the course of time. Consistent with previous studies, Ga(NO3)3 uptake rate of ΔhitAB-mutant was significantly abrogated (Fig. S7, ESI†). In contrast, the ΔhitAB-mutant showed a slower but still significant gallium internalization rate compared to the WT strain when incubated with the G6 compound. The results indicated that the HitABC transporter is probably involved in G6 uptake. However, P. aeruginosa might be also utilizing alternative pathways to internalize the G6 compound.
Mammalian cell and animal infection model
The cytotoxicity of these compounds to mammalian cells is subsequently investigated. The IC50 values of the gallium compounds against human hepatocyte LO2 cell lines were measured based on MTT assays (Fig. S8 and Table S3, ESI†). All the gallium compounds exhibited low cytotoxicity towards mammalian cells with IC50 values higher than 200 μM (>122.2 μg mL−1). P. aeruginosa is known to cause a cytotoxic effect on eukaryotic cells via the translocation of bacterial effectors into the host cells.42 To further examine the anti-bacterial effect of gallium compounds, the human alveolar basal epithelial cells, A549 cells were co-cultured with P. aeruginosa PAO1 strain in the absence or presence of gallium compound G6 (Fig. 5(a)). The cytotoxic effects of P. aeruginosa were examined based on A549 cell morphology after 2 h incubation. As shown in Fig. 5(b), healthy A549 cells exhibited a pebble-like shape and formed a well-spread monolayer. Upon infection with P. aeruginosa, the majority of A549 cells retracted into round shapes with drastically reduced surface area and gradually detached from the culture plate. With increasing concentrations of the G6 compound, the cytotoxic effect of P. aeruginosa on A549 cells was gradually reduced. Approximately 1.0 μg mL−1G6 compound (approximately twice the MIC value against P. aeruginosa) was sufficient to protect A549 cells from bacterial cytotoxicity, while the compound alone had no significant effects on the A549 cell morphology. In consistent, MTT assay also verified that G6 exhibited low cytotoxicity towards A549 cells with IC50 value higher than 200 μM (Fig. S9, ESI†).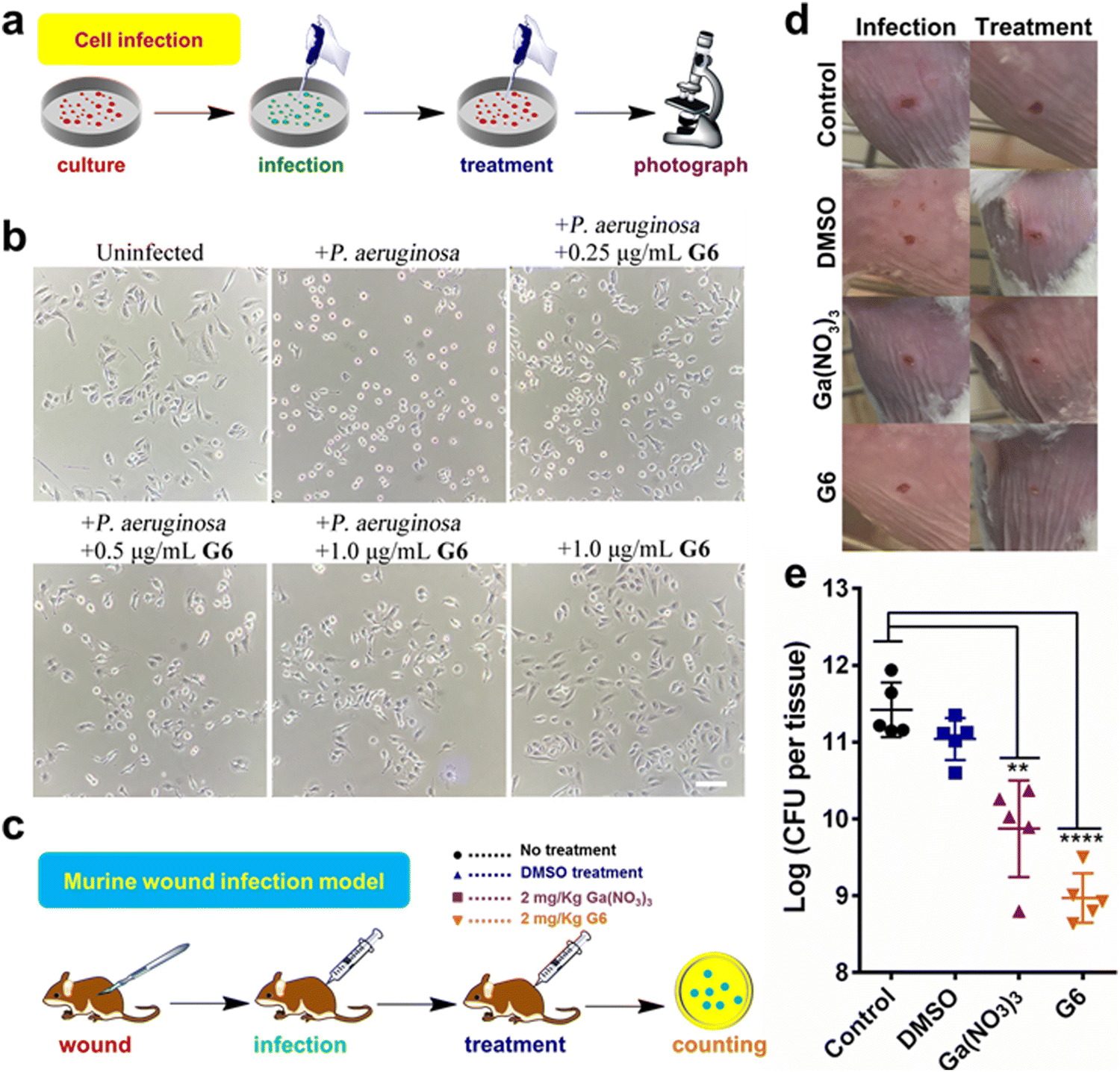 | ||
| Fig. 5 (a) Schematic illustration of bacteria-mammalian cells co-culture model to examine the anti-P. aeruginosa efficacy of G6 compound. (b) G6 compound protects the mammalian cells from the cytotoxic effect of P. aeruginosa. Human lung epithelial A549 cells were co-cultured with P. aeruginosa PAO1 in the absence and presence of different concentrations of the G6 compound. Scale bar: 50 μm. (c) Schematic illustration of mouse wound infection model to compare the anti-P. aeruginosa efficacy of Ga(NO3)3 and G6 compound. (d) Representative photographs of the lesions of the mice at 2 day and 10 days. (e) The bacterial load of local infection sites induced by P. aeruginosa PAO1 was enumerated in control, DMSO-treated, gallium nitrate-treated, and G6 compound-treated groups (20 mg kg−1 for gallium nitrate and compound G6) after 10 days. The log (CFU per tissue) values are presented as the mean ± sem. The statistical difference is determined by the Mann–Whitney U test. **p < 0.01, ****p < 0.0001. | ||
The above results prompted us to further investigate whether the gallium compound G6 could eradicate P. aeruginosa in an animal infection model (Fig. 5(c)). To this end, we used a mouse wound infection model to assess the anti-P. aeruginosa efficacy of compound G6 as described previously.43 The details were described in the ESI.† In brief, an excisional wound (∼2 mm × 2 mm) was created on the flank of each mouse. Approximately 5 × 108 CFU of P. aeruginosa in an aliquot of 10 μL was dropped into the wound to induce infection. Twice-daily treatment of Ga(NO3)3 (2 mg kg−1) or compound G6 (2 mg kg−1) was applied to the wounds. The infection sites were excised 48 h post-infection, homogenized, and serially diluted for CFU quantification. Representative close-up photographs of the lesions of the entire dorsal back are shown in Fig. 5(d), and the wound on the surface of the mouse was significantly better after 48 hours of G6 compound treatment. Furthermore, in the control group, wound models treated with vehicle had an average of 2.6 × 1011 CFU of P. aeruginosa per wound model. Treatment with Ga(NO3)3 caused a significant 2-log lower number of CFU of P. aeruginosa. Whereas, compound G6 exhibited higher activity and caused an almost 3-log lower number of bacterial CFU compared to the control group (Fig. 5(e)). The results demonstrate that compound G6 is more potent than Ga(NO3)3 to inhibit P. aeruginosa growth in a mouse wound infection model. It should be noted that we used a bacterial skin infection model in this study. However, P. aeruginosa also causes soft tissue infections, such as lung infections in patients with cystic fibrosis. Given its high anti-bacterial efficacy and low cytotoxicity, we envision that the G6 compound could also be applied to treat P. aeruginosa-caused soft tissue infections. Similar to gallium nitrate, intravenous injection of G6 might be a possible way for drug administration. Therefore, the anti-bacterial efficacy of the gallium compound G6 to treat soft tissue infections warrants further investigation.
Experimental
Reagents and instruments
All reagents were used as received without further purification. The solvents, methanol, and ethanol used in this work were obtained from Aladdin (HPLC Grade). GaCl3 was purchased from Sigma-Aldrich and handled in a glove box (Mikrouna). 1H and 13C NMR spectra were recorded on a Bruker Advance III 400 MHz or 500 MHz NMR spectrometers at ambient temperature. All chemical shifts (δ) are reported in parts per million (ppm) with tetramethylsilane (TMS) as an internal standard. ESI-MS was performed on a Thermo LTQ XL linear ion trap mass spectrometer. C and H elemental analysis was carried out on an Elementar Vario EL analyzer. Metal contents were analyzed on a Thermo iCAP RQ inductively coupled plasma mass spectrometry (ICP-MS). Fourier transforms infrared (FT-IR) spectra were recorded on a Bruker EQUINOX 55 spectrometer using the KBr pellet method. Thermogravimetric (TG) and differential thermal analysis (DTA) was performed on a Netzsch TG 209 F1 libra instrument with a sample weight between 5–10 mg over the temperature range of 25–800 °C and a heating rate of 10 °C min−1 under a nitrogen atmosphere. UV-vis spectra were recorded on a SHIMADZU UV-2600 spectrophotometry.Synthesis of gallium compounds
The gallium compounds were synthesized according to a modified procedure as described previously.44 In brief, flavonoid ligands (1.0 mmol) and GaCl3 (0.5 mmol) were added slowly into 20 mL mixed solvent of ethanol and methanol (volume ratio of 1:2). After refluxing for 6 h under a nitrogen atmosphere at 70 °C, the reaction mixture was allowed to cool to room temperature. The precipitate was collected by filtration and washed several times with cold ethanol. The yellow products were recrystallized from ethanol and dried via P4O10 overnight.
Bacterial strains and growth condition
Pseudomonas aeruginosa strain PAO1 (P. aeruginosa) was grown in M9 minimal medium (1.0 g L−1 NH4Cl, 0.5NaCl, 6.0 g L−1 Na2HPO4, 3.0 g L−1 KH2PO4) supplemented with 1.0 mM MgSO4, 1.0 mM ZnSO4, 0.2 mM CaCl2, 0.4% (w/v) glucose and 0.2% (w/v) casein hydrolysate. Gram-negative Escherichia coli strain K-12 (E. coli) was cultured in Luria–Bertani (LB) medium (5 g L−1 yeast extract, 10 g L−1 tryptone, 10 g L−1 sodium chloride). The Gram-positive Staphylococcus aureus strain Newman (S. aureus) was grown in Tryptic Soy Broth (TSB) medium (17 g L−1 Tryptone, 3 g L−1 Phytone, 5 g L−1 sodium chloride, 2.5 g L−1 dipotassium phosphate and glucose 2.5 g L−1).Antimicrobial activity
The minimal inhibitory concentration (MIC) values of the gallium compounds were determined using a standard broth dilution method as described previously.45 In brief, the compounds were two-fold serially diluted to concentrations from 512 to 0.25 μg mL−1. Approximately 50 μL of the compound solution was added to a 96-well microtiter plate and mixed with 50 μL of bacteria culture in the log phase (106 CFU mL−1). The bacterial culture was further incubated at 37 °C for 22–24 h. The OD600 value for each well was recorded on a microplate reader. The MICs value was defined as the lowest compound concentration that inhibited 95% of the bacterial growth. The IC50 values of gallium compounds G1, G2, G6, and Ga(NO3)3 against P. aeruginosa were measured similarly except that the compound concentrations were from 0.5 to 6 μg mL−1 with 0.5 μg mL−1 intervals. Each well was done in duplicates.The inhibition zone values of the gallium compounds were measured by agar plate diffusion test as described previously.46 Typically, different concentrations of the gallium compounds were prepared in a 2% DMSO solution. And approximately 10 μL of the solution was loaded onto a sterilized filter paper disk (6.0 mm in diameter). After drying, the discs were implanted on the surface of the agar plates inoculated with P. aeruginosa. The agar plates were subsequently incubated at 37 °C for 24 h for bacterial growth. The zone of inhibited bacterial growth was measured in millimeters. DMSO solution was used as a negative control and Ga(NO3)3 was used as a positive control. All experiments were performed in triplicates.
Resistance development
Development of resistance to the gallium compound G6 and the clinically relevant antibiotics ciprofloxacin were assessed as described by Habets and Brockhurst.47 Approximately 50 μL of bacterial suspension (106 CFU mL−1) in M9 medium was mixed with 50 μL gallium compound G6 or ciprofloxacin in the same medium (with the final concentration of 0.25 to 128 μg mL−1 for both G6 and ciprofloxacin) and incubated at 37 °C for 24 h. On the next day, the MIC was recorded as the minimal concentration that completely inhibited growth. Thereafter, a new passage MIC measurement assay was prepared using diluted bacterial cells grown at 0.5-fold MIC suspension. This was repeated for 16 passages and the MIC values for G6 and ciprofloxacin were recorded for each passage.P. aeruginosa biofilm assay
P. aeruginosa was cultured in a 6-well microtiter plate (Corning, NY, USA) for static biofilm formation. For biofilm inhibition assay, approximately 3 mL of bacterial overnight culture were diluted in fresh M9 medium with a final OD600 value of 0.05 and transferred into each well of a microtiter plate in the absence or presence of a sub-lethal concentration of gallium compounds (0.08 μM). The plate was further incubated at 37 °C for 48 h. For biofilm eradication assay, 3 mL of bacteria were first cultured in M9 medium in a 6-well microtiter plate at 37 °C for 48 h to allow matured biofilm formation. Gradient amounts of Ga(NO3)3 or G6 compound were then added to bacterial culture with concentrations of 10, 20, and 40 μM. The bacteria were allowed for further culture for 2 or 3 days.The P. aeruginosa biofilm was quantified according to a procedure described previously.48 In brief, weakly adherent planktonic cells were removed from the microtiter plate by washing with phosphate-buffered saline (PBS) three times. The adherent biofilms were dried and stained with 0.1% (w/v) crystal violet for 15 min. Excess dye was removed by rinsing the plate three times with PBS. Crystal violet was solubilized from the stained biofilms by adding 30% acetic acid and the biofilms were quantified by measuring the 595 nm absorbance. Each experiment was performed in triplicates.
Field emission scanning electron microscopy
Images of P. aeruginosa biofilms were recorded on a JSM-6330F (JEOL) field emission scanning electron microscopy as previously reported.49 In brief, P. aeruginosa was cultured in a 6-well microtiter plate with a sterile glass coverslip in each well. Approximately 3 mL of bacterial suspension (106 CFU mL−1) were incubated in the presence and absence of 0.08 μM gallium compounds at 37 °C for 36 h to allow biofilm formation on the coverslip. Thereafter, weakly adherent planktonic cells were removed by washing with phosphate-buffered saline (PBS) three times. The biofilm samples were fixed using 2.5% (v/v) glutaraldehyde in PBS buffer at 4 °C overnight. After rinsing twice with PBS buffer, the samples were subsequently exposed to ethanol dehydration series of 30%, 50%, 70%, 90% for 10 min, and 100% for 30 min. All the samples were finally air-dried in a vacuum desiccator and sputter-coated with palladium–gold thin film before data collection.Elastase activity assay
P. aeruginosa secreted elastase activity was examined by Elastin-Congo red assay.50 Secreted elastase could cleave the substrate and release the soluble Congo red dye, which has a typical absorption at 495 nm. In brief, P. aeruginosa overnight culture was diluted with fresh M9 medium to OD600 value of 0.05. The bacteria were then further cultured at 37 °C for 20 h in the presence of 0.08 μM gallium compounds, Ga(NO3)3, or flavonoid ligands. Thereafter, the bacterial supernatant medium was collected by centrifugation at 10000g for 20 min. The medium was further concentrated using centrifugal filter units (Ultracel-3k, Millipore), and protein concentration in the medium was determined by BCA kit. The concentrated medium was diluted with PBS buffer to a final protein concentration of 10 mg mL−1. Subsequently, 1 mL diluted medium was mixed with 1 mL of reaction buffer (100 mM tris–HCl, 10 mM CaCl2, pH 7.5) supplemented with 20 mg Elastin-Congo red elastase substrate and incubated at 37 °C for 6 h with shaking. Then 0.2 mL EDTA was added to a final concentration of 10 mM to stop the cleavage. Insoluble pellets were removed by centrifugation and the absorbance of the supernatant at 495 nm was measured.
Pyocyanin and pyoverdine assay
The secreted virulence factors pyocyanin was quantified as described previously.51P. aeruginosa was cultured in the presence of 0.08 μM gallium compounds, Ga(NO3)3, or flavonoid ligands at 37 °C for around 20 h until OD600 reached 2. Approximately 10 mL of bacterial supernatant medium was collected by centrifugation. Subsequently, 3 mL chloroform was added into medium to extract the pyocyanin and pyoverdine. The pyocyanin was reextracted into 1 mL of 0.2 M HCl to give a pink to the deep red solution after approximately 30 min. The pyocyanin concentration was determined by measuring the absorbance at 520 nm. For pyoverdine quantification, the collected bacterial culture supernatant medium was diluted in 100 mM tri–HCl (pH 8.0) and measured as OD405 normalized by the cell density of bacterial culture.52Hemolysis assay
The hemolytic activity of the P. aeruginosa secreted virulent factor was examined using rabbit erythrocytes hemolysis assay.53P. aeruginosa was cultured in the presence of 0.08 μM gallium compounds, Ga(NO3)3, or flavonoid ligands for approximately 20 h until OD600 value reached 2. The bacterial supernatant medium was collected by centrifugation. The fresh rabbit erythrocytes were washed three times with PBS buffer and diluted 50 times in M9 medium. Thereafter, approximately 1.4 mL of the prepared erythrocyte solution was mixed with 0.6 mL of the bacterial medium. Meanwhile, 0.1% SDS solution and M9 medium were used as a positive and negative control, respectively. The mixtures were incubated at 37 °C for an additional 30 min. The supernatant of the mixture was collected by centrifugation at 10000g for 10 min. The released hemoglobin in the supernatant was determined by measuring the absorbance at 595 nm. The percentage of erythrocyte lysis was calculated using the following equation: [(S − N)/(P − N)] × 100%, in which N is the OD595 value for negative control M9 medium, P is the OD595 value of 0.1% SDS positive control and S is the OD595 value of the analyzed specimen. The assays were performed twice independently and each assay was measured in duplicate.
Lipophilicity of gallium compounds
The lipophilicity of gallium compounds was examined by calculating the octanol/water partition-coefficient (LogP) value of gallium compounds using the shake-flask method.54 Octanol-saturated water (OSW) and water-saturated octanol (WSO) were prepared from PBS (pH 7.4) and octanol (250 rpm, 37 °C overnight shaking). Different concentrations of gallium compounds (0, 0.5, 1, 2, 4, 8, 16, and 32 μg mL−1) were dissolved in OSW or WSO. And the absorbance of compounds in OSW or WSO was recorded to obtain the standard curves. Compounds G1–G6 were dissolved in 5 mL of WSO to a final concentration of 5 μg mL−1, centrifuged and supernatants were collected. Approximately 2 mL of the obtained WSO solutions of gallium compounds were added to OSW (2 mL) and shaken for 12 h at 250 rpm, 37 °C. After that, the mixtures were centrifuged, and the octanol and aqueous layer were separated carefully. The absorbance of compounds in the OSW and WSO layer was recorded and the concentrations were calculated from the standard curves. The logP = log(CWSO/COSW) equation was used for the partition coefficients calculation. The experiments were performed in triplicate.
Gallium uptake assay
The amount of gallium uptake by P. aeruginosa was quantified by ICP-MS as described previously.55P. aeruginosa overnight culture was diluted to OD600 of 0.1 in the presence of 0.16 μM of gallium compounds or Ga(NO3)3. Approximately 5 mL aliquot of bacterial culture was removed and collected every hour, up to 5 h. The collected cell pellet was washed three times in PBS supplemented with 1 mM EDTA to remove surface-bound gallium. The pellet was subsequently digested by 70% nitric acid at 60 °C for 2 h and diluted with double-distilled H2O to a final HNO3 concentration of 2%. The prepared samples were analyzed on an iCAP RQ ICP-MS.Total RNA isolation and preparation for qPCR and RNA-seq
P. aeruginosa PAO1 cultures were grown to early log phase (OD600 = 0.3) and the G6 compound was added to a final concentration of 4 μM the bacteria were further cultured for 4 h at 37 °C. The G6-treated and untreated bacteria were harvested by centrifugation. And total RNA was isolated from the cultured P. aeruginosa cells using an SV Total RNA isolation kit (Promega) according to the manufacturer's instructions. And RNA was quantified by NanoDrop spectrophotometer. For qPCR analysis, cDNA was generated by reverse transcription using GoScript Reverse Transcriptase (Promega). The transcription levels of detected genes were subsequently determined by real-time PCR using the GoTaq qPCR Master Mix kit (Promega) on a StepOne Plus Real-time PCR system (Life Technologies). The primer pairs used to perform qPCR are listed in Table S4 (ESI†). In brief, the reaction mixtures consisted of 2 μL cDNA and 0.2 μM primers in a final volume of 20 μL. Each reaction cycle consisted of denaturation at 95 °C for 10 s, annealing at 50 °C for 20 s, and extension at 72 °C for 30 s, and a total of 35 cycles was performed. Melting curve analyses were performed to verify the amplification specificity. The rrsA gene (16S rRNA) was used as the internal reference gene, and the relative quantification of gene transcription was performed according to the 2−ΔΔCT method.56 For RNA-seq analysis, the quality of the isolated RNA was examined by Agilent 2100 Bioanalyzer (Agilent). Paired-end, strand-specific RNA-seq was performed using the dUTP method.57 The ribosomal RNAs were removed with Ribo-Zero rRNA Removal Kit (Illumina). Subtracted RNAs were fragmented and reverse-transcribed. The sequencing library was loaded on an Illumina HiSeq instrument according to manufacturer's instruction. RNA-seq experiments were performed in triplicates.Preparation of the GC-MS sample
20 mL of the pseudomonas aeruginosa cells (OD600 = 1.0) were collected by centrifugation at 4000g for 10 min at 4 °C for GC-MS analysis. Then, 20 mL of cold methanol were added to the samples to quench the metabolic process. Metabolites were extracted with 1 mL of cold methanol containing 10 μL of 0.1 mg mL−1 ribitol (Sigma) as an analytical internal standard through high throughput tissuelyser. After centrifugation at 12000g for 10 min, 800 μL of the supernatant was dried in a rotary vacuum centrifuge device for further analysis. Each sample had ten biological replicates.
GC-MS analysis
The dried samples need further derivation for GC-MS analysis.58 Firstly, 80 μL of 20 mg mL−1 methoxyamine hydrochloride (Sigma) in pyridine were added to the dried samples and incubated at 37 °C for 90 min. Then, the 80 μL of N-methy-N-trimethylsilyltrifluoroacetamide (MSTFA, Sigma) were added to the samples with incubation for another 30 min for acidic protons derivatization. After centrifugation at 12000g for 20 min, 140 μL of the supernatant was transferred to sample vials for chemical analysis, which was carried out by 7890A/5975C Chemstation (Agilent). 1 μL of the derivatized sample was injected into a HP-5MS column (30 m length × 250 μm i.d. × 0.25 μm thickness, Agilent J&W Scientific, Folsom, CA). The MS source temperature was maintained at 280 °C in the EI (ionized directly) mode at 70 eV ionization energy. The initial temperature of the GC oven was programmed at 85 °C for 3 min, followed by an increase to 310 °C at a rate of 20 °C min−1, and held for 7 min. Helium was used as the carrier gas at a constant flow of 1 mL min−1. The MS was operated in a range of 33–550 m/z.
Multivariate statistical analysis
The deconvolution, alignment, and identification of metabolites were performed using eRah package combined with MSD chemstation with NIST MS search 2.0 program.59 The abundance of metabolites was normalized to the mean value of the integrated peak area of ribitol. The metabolite data matrix was imported to SIMCA software (version 13.0) for principal component analysis (PCA) and orthogonal partial least squares-discriminant analysis (OPLS-DA) to reduce the high dimension of the data set for observing the difference among groups. Then, the VIP score (VIP > 1) derived from OPLS-DA models was integrated with the P value (P < 0.01) calculated by t test to screen the significantly different metabolites. Z-Score and hierarchical clustering were used to sort and visualize the differential metabolites after normalization.58 Finally, MetaboAnalyst 3.0 (https://www.metaboanalyst.ca/) was employed to analyze the enrichment pathways over the identified differential metabolites.Mammalian cell cytotoxicity
The cell cytotoxicity of gallium compounds towards mammalian cells was measured by MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide reduction assay.60 In brief, human hepatocyte LO2 cell lines were cultured in 96-well plates (8 × 103 cells per well) for 24 h at 37 °C. Thereafter, the cells were treated with gradient concentrations (0, 5, 10, 20, 40, 80, 160, 200, and 250 μM) of gallium compounds. The cells were further incubated at 37 °C for 48 h and 20 μL MTT solution (5 mg mL−1) was added to each well. The plate was incubated at 37 °C for another 4 h. Subsequently, the media were removed from the wells, and 150 μL DMSO was added to each well to dissolve the formazan crystals. The absorbance at 490 nm of each well was measured with a microplate reader. The IC50 values were determined by the percentage of cell viability versus gallium compound concentrations on a logarithmic graph. The experiments were carried out in triplicates.Mammalian cell infection by P. aeruginosa
Human alveolar basal epithelial A549 cells were cultured in 1640 medium. And gallium compound G6 was added into the medium to different final concentrations (0.25, 0.5, and 1.0 μg mL−1). P. aeruginosa overnight culture was diluted 10 times by 1640 medium and incubated at 37 °C for 2 h. Subsequently, the adapted P. aeruginosa bacterium was added to the A549 cell culture to a final concentration of 106 CFU mL−1 and the culture was further incubated at 37 °C for 2 hours. The morphology of the cells was recorded on a Nikon Eclipse TS100 inverted microscopy. A549 cells with P. aeruginosa or G6 compound alone were used as controls.Murine skin infection model
Six to eight weeks old, female BALB/c mice (18–22 g) were purchased from Charles River Laboratories, Inc. and used in the studies. Animals had free access to food and water and were randomized to cages for each experiment. In the wound infection model, static overnight cultures of P. aeruginosa PAO1 strain were harvested and washed with PBS three times before further use. Mouse hair in the infection site (of area ∼2 cm2) was removed by Veet® (according to the manufacturer's instruction) and an excisional wound (∼2 mm × 2 mm) was created on the flank of each mouse. About 5 × 108 CFU of bacteria in an aliquot of 10 μL were dropped to the wound. Groups (n = 5 for each group) of mice received monotherapy of Ga(NO3)3 (2 mg kg−1) and compound 6 (2 mg kg−1). All the compounds were mixed in cream and smeared on the infection site twice daily post-infection. Mice administrated with the vehicle were set as the control. All the mice 48 hours post-infection and the infection site were collected in an aliquot of 0.5 mL PBS and homogenized (Tissue Lyser II, QIAgen, Hilden, Germany) at a frequency of 30 Hz for 5 minutes. The bacterial loads per tissue were enumerated by agar plating.Data availability
The authors declare that all the relevant data supporting the findings of this study are available within the article and its ESI† files or from the corresponding author on request.Conclusions
In conclusion, a new class of dual-targeting gallium-flavonoid compounds, which abrogate both quorum sensing and iron metabolism pathways of P. aeruginosa, has been synthesized and characterized. In particular, a gallium-naringenin compound (G6) exhibited high antimicrobial activity and relatively low cytotoxicity to mammalian cells both in vitro and in vivo. The G6 compound could significantly attenuate P. aeruginosa virulence factor secretion, prevent biofilm formation as well as eradicate matured biofilms. Our results shed light on the development of efficient Ga-based therapies to combat P. aeruginosa infection in future.Author contributions
Xiaojun He: conceptualization, methodology, writing – original draft. Bingjie Han: conceptualization, methodology, writing – original draft, resources. Runming Wang: conceptualization, methodology. Yu Guo: methodology, investigation. Richard Y T Kao: investigation. Hongzhe Sun: writing – review & editing, supervision. Hongyan Li and Wei Xia: resources, writing – review & editing, validation, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22077142 and 22022706); Natural Science Foundation of Guangdong Province, China (2019A1515011156) and the Fundamental Research Funds for the Central Universities (23xkjc020).Notes and references
- M. Nagao, Y. Iinuma, J. Igawa, T. Saito, K. Yamashita, T. Kondo, A. Matsushima, S. Takakura, A. Takaori-Kondo and S. Ichiyama, J. Hosp. Infect., 2011, 79, 49–53 CrossRef CAS PubMed.
- A. Tsutsui, S. Suzuki, K. Yamane, M. Matsui, T. Konda, E. Marui, K. Takahashi and Y. Arakawa, J. Hosp. Infect., 2011, 78, 317–322 CrossRef CAS PubMed.
- R. N. Chernish and S. D. Aaron, Curr. Opin. Pulm. Med., 2003, 9, 509–515 CrossRef PubMed.
- R. T. Sadikot, T. S. Blackwell, J. W. Christman and A. S. Prince, Am. J. Respir. Crit. Care Med., 2005, 171, 1209–1223 CrossRef PubMed.
- K. Poole, Front. Microbiol., 2011, 2, 65 CAS.
- R. A. Bonomo and D. Szabo, Clin. Infect. Dis., 2006, 43, S49–S56 CrossRef CAS.
- J. W. Costerton, P. S. Stewart and E. P. Greenberg, Science, 1999, 284, 1318–1322 CrossRef CAS PubMed.
- D. Davies, Nat. Rev. Drug Discovery, 2003, 2, 114–122 CrossRef CAS PubMed.
- H. C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS PubMed.
- B. Rada and T. L. Leto, Trends Microbiol., 2013, 21, 73–81 CrossRef CAS PubMed.
- S. Bleves, V. Viarre, R. Salacha, G. P. Michel, A. Filloux and R. Voulhoux, Int. J. Med. Microbiol., 2010, 300, 534–543 CrossRef CAS PubMed.
- W. L. Ng and B. L. Bassler, Annu. Rev. Genet., 2009, 43, 197–222 CrossRef CAS.
- M. Schuster, C. P. Lostroh, T. Ogi and E. P. Greenberg, J. Bacteriol., 2003, 185, 2066–2079 CrossRef CAS PubMed.
- J. Lee and L. H. Zhang, Protein Cell, 2015, 6, 26–41 CrossRef CAS PubMed.
- C. T. O'Loughlin, L. C. Miller, A. Siryaporn, K. Drescher, M. F. Semmelhack and B. L. Bassler, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17981–17986 CrossRef PubMed.
- T. Defoirdt, Trends Microbiol., 2018, 26, 313–328 CrossRef CAS PubMed.
- S. Kumar and A. K. Pandey, Sci. World J., 2013, 2013, 162750 Search PubMed.
- J. Luo, B. Dong, K. Wang, S. Cai, T. Liu, X. Cheng, D. Lei, Y. Chen, Y. Li, J. Kong and Y. Chen, PLoS One, 2017, 12, e0176883 CrossRef PubMed.
- J. Luo, J.-l Kong, B. Dong, H. Huang, K. Wang, C. Hou, Y. Liang, B. Li, Y. Chen and L. Wu, Drug Des., Dev. Ther., 2016, 10, 183–203 CrossRef CAS.
- J. E. Paczkowski, S. Mukherjee, A. R. McCready, J.-P. Cong, C. J. Aquino, H. Kim, B. R. Henke, C. D. Smith and B. L. Bassler, J. Biol. Chem., 2017, 292, 4064–4076 CrossRef CAS PubMed.
- M. E. Skogman, S. Kanerva, S. Manner, P. M. Vuorela and A. Fallarero, Molecules, 2016, 21, 1211 CrossRef PubMed.
- P. K. Singh, M. R. Parsek, E. P. Greenberg and M. J. Welsh, Nature, 2002, 417, 552–555 CrossRef CAS PubMed.
- E. Banin, M. L. Vasil and E. P. Greenberg, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11076–11081 CrossRef CAS PubMed.
- Y. Kaneko, M. Thoendel, O. Olakanmi, B. E. Britigan and P. K. Singh, J. Clin. Invest., 2007, 117, 877–888 CrossRef CAS.
- E. Banin, A. Lozinski, K. M. Brady, E. Berenshtein, P. W. Butterfield, M. Moshe, M. Chevion, E. P. Greenberg and E. Banin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16761–16766 CrossRef CAS.
- Y. Guo, W. Li, H. Li and W. Xia, ACS Infect. Dis., 2019, 5, 1693–1697 CrossRef CAS.
- A. de Breij, M. Riool, R. A. Cordfunke, N. Malanovic, L. de Boer, R. I. Koning, E. Ravensbergen, M. Franken, T. van der Heijde, B. K. Boekema, P. H. S. Kwakman, N. Kamp, A. El Ghalbzouri, K. Lohner, S. A. J. Zaat, J. W. Drijfhout and P. H. Nibbering, Sci. Transl. Med., 2018, 10, eaan4044 CrossRef PubMed.
- S. Andersson, G. Dalhammar, C. J. Land and G. K. Rajarao, Appl. Microbiol. Biotechnol., 2009, 82, 535–543 CrossRef CAS PubMed.
- M. Alhede, K. N. Kragh, K. Qvortrup, M. Allesen-Holm, M. van Gennip, L. D. Christensen, P. O. Jensen, A. K. Nielsen, M. Parsek, D. Wozniak, S. Molin, T. Tolker-Nielsen, N. Hoiby, M. Givskov and T. Bjarnsholt, PLoS One, 2011, 6, e27943 CrossRef CAS.
- L. R. Bernstein, Pharmacol. Rev., 1998, 50, 665–682 CAS.
- R. García-Contreras, E. Lira-Silva, R. Jasso-Chávez, I. L. Hernández-González, T. Maeda, T. Hashimoto, F. C. Boogerd, L. Sheng, T. K. Wood and R. Moreno-Sánchez, Int. J. Med. Microbiol., 2013, 303, 574–582 CrossRef PubMed.
- J. D. Faraldo-Gomez and M. S. Sansom, Nat. Rev. Mol. Cell Biol., 2003, 4, 105–116 CrossRef CAS PubMed.
- C. Wandersman and P. Delepelaire, Annu. Rev. Microbiol., 2004, 58, 611–647 CrossRef CAS PubMed.
- P. Huber, P. Basso, E. Reboud and I. Attree, Environ. Microbiol. Rep., 2016, 8, 564–571 CrossRef PubMed.
- S. Hall, C. McDermott, S. Anoopkumar-Dukie, A. J. McFarland, A. Forbes, A. V. Perkins, A. K. Davey, R. Chess-Williams, M. J. Kiefel, D. Arora and G. D. Grant, Toxins, 2016, 8, 236 CrossRef PubMed.
- G. W. Lau, H. M. Ran, F. S. Kong, D. J. Hassett and D. Mavrodi, Infect. Immun., 2004, 72, 4275–4278 CrossRef CAS PubMed.
- B. Wretlind and O. R. Pavlovskis, Rev. Infect. Dis., 1983, 5(Suppl 5), S998–S1004 CrossRef CAS PubMed.
- I. L. Lamont, P. A. Beare, U. Ochsner, A. I. Vasil and M. L. Vasil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7072–7077 CrossRef CAS PubMed.
- T. Bjarnsholt, P. O. Jensen, T. H. Jakobsen, R. Phipps, A. K. Nielsen, M. T. Rybtke, T. Tolker-Nielsen, M. Givskov, N. Hoiby, O. Ciofu and S. C. F. Study, PLoS One, 2010, 5, e10115 CrossRef PubMed.
- J. P. Pearson, E. C. Pesci and B. H. Iglewski, J. Bacteriol., 1997, 179, 5756–5767 CrossRef CAS PubMed.
- M. Whiteley, K. M. Lee and E. P. Greenberg, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13904–13909 CrossRef CAS PubMed.
- V. T. Lee, R. S. Smith, B. Tummler and S. Lory, Infect. Immun., 2005, 73, 1695–1705 CrossRef CAS PubMed.
- S. DeLeon, A. Clinton, H. Fowler, J. Everett, A. R. Horswill and K. P. Rumbaugh, Infect. Immun., 2014, 82, 4718–4728 CrossRef PubMed.
- E. G. Ferrer, M. V. Salinas, M. J. Correa, L. Naso, D. A. Barrio, S. B. Etcheverry, L. Lezama, T. Rojo and P. A. Williams, J. Biol. Inorg. Chem., 2006, 11, 791–801 CrossRef CAS PubMed.
- I. Wiegand, K. Hilpert and R. E. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS PubMed.
- A. W. Bauer, W. M. Kirby, J. C. Sherris and M. Turck, Am. J. Clin. Pathol., 1966, 45, 493–496 CrossRef CAS PubMed.
- M. G. Habets and M. A. Brockhurst, Biol. Lett., 2012, 8, 416–418 CrossRef CAS PubMed.
- G. A. O'Toole and R. Kolter, Mol. Microbiol., 1998, 28, 449–461 CrossRef PubMed.
- C. Vuotto and G. Donelli, Methods Mol. Biol., 2014, 1147, 73–84 CrossRef PubMed.
- K. Morihara, H. Tsuzuki, T. Oka, H. Inoue and M. Ebata, J. Biol. Chem., 1965, 240, 3295–3304 CrossRef CAS PubMed.
- D. W. Essar, L. Eberly, A. Hadero and I. P. Crawford, J. Bacteriol., 1990, 172, 884–900 CrossRef CAS PubMed.
- F. Imperi, F. Tiburzi and P. Visca, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20440–20445 CrossRef CAS PubMed.
- G. Rossignol, A. Merieau, J. Guerillon, W. Veron, O. Lesouhaitier, M. G. Feuilloley and N. Orange, BMC Microbiol., 2008, 8, 189 CrossRef PubMed.
- S. Y. Han, J. Q. Qiao, Y. Y. Zhang, L. L. Yang, H. Z. Lian, X. Ge and H. Y. Chen, Chemosphere, 2011, 83, 131–136 CrossRef CAS PubMed.
- Y. Hong, Y. T. Lai, G. C. Chan and H. Sun, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3211–3216 CrossRef CAS PubMed.
- T. D. Schmittgen and K. J. Livak, Nat. Protoc., 2008, 3, 1101–1108 CrossRef CAS PubMed.
- J. Z. Levin, M. Yassour, X. A. Adiconis, C. Nusbaum, D. A. Thompson, N. Friedman, A. Gnirke and A. Regev, Nat. Methods, 2010, 7, 709–767 CrossRef CAS PubMed.
- X. Zhao, C. Wu, X. Peng and H. Li, J. Proteome Res., 2014, 13, 4155–4163 CrossRef CAS PubMed.
- X. Domingo-Almenara, J. Brezmes, M. Vinaixa, S. Samino, N. Ramirez, M. Ramon-Krauel, C. Lerin, M. Díaz, L. Ibáñez, X. Correig, A. Perera-Lluna and O. Yanes, Anal. Chem., 2016, 88, 9821–9829 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00033h |
| ‡ These authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2023 |