
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparison of click-capable oxaliplatin and cisplatin derivatives to better understand Pt(II)-induced nucleolar stress†
Andres S.
Guerrero‡
 a,
Paul D.
O’Dowd‡
bc,
Hannah C.
Pigg
a,
Katelyn R.
Alley
a,
Darren M.
Griffith
bc and
Victoria J.
DeRose
*a
a,
Paul D.
O’Dowd‡
bc,
Hannah C.
Pigg
a,
Katelyn R.
Alley
a,
Darren M.
Griffith
bc and
Victoria J.
DeRose
*a
aDepartment of Chemistry and Biochemistry, University of Oregon, Eugene, OR, USA. E-mail: derose@uoregon.edu
bDepartment of Chemistry, RCSI, Dublin, Ireland
cSSPC, the Science Foundation Ireland Research Centre for Pharmaceuticals, Ireland
First published on 16th August 2023
Abstract
Pt(II) chemotherapeutic complexes have been used as predominant anticancer drugs for nearly fifty years. Currently there are three FDA-approved chemotherapeutic Pt(II) complexes: cisplatin, carboplatin, and oxaliplatin. Until recently, it was believed that all three complexes induced cellular apoptosis through the DNA damage response pathway. Studies within the last decade, however, suggest that oxaliplatin may instead induce cell death through a unique nucleolar stress pathway. Pt(II)-induced nucleolar stress is not well understood and further investigation of this pathway may provide both basic knowledge about nucleolar stress as well as insight for more tunable Pt(II) chemotherapeutics. Through a previous structure-function analysis, it was determined that nucleolar stress induction is highly sensitive to modifications at the 4-position of the 1,2-diaminocyclohexane (DACH) ring of oxaliplatin. Specifically, more flexible and less rigid substituents (methyl, ethyl, propyl) induce nucleolar stress, while more rigid and bulkier substituents (isopropyl, acetamide) do not. These findings suggest that a click-capable functional group can be installed at the 4-position of the DACH ring while still inducing nucleolar stress. Herein, we report novel click-capable azide-modified oxaliplatin mimics that cause nucleolar stress. Through NPM1 relocalization, fibrillarin redistribution, and γH2AX studies, key differences have been identified between previously studied click-capable cisplatin mimics and these novel click-capable oxaliplatin mimics. These complexes provide new tools to identify cellular targets and localization through post-treatment Cu-catalyzed azide–alkyne cycloaddition and may help to better understand Pt(II)-induced nucleolar stress. To our knowledge, these are the first reported oxaliplatin mimics to include an azide handle, and cis-[(1R,2R,4S) 4-methylazido-1,2-cyclohexanediamine]dichlorido platinum(II) is the first azide-functionalized oxaliplatin derivative to induce nucleolar stress.
Introduction
Pt(II)-based chemotherapeutic complexes are widely used in clinical settings and include three FDA-approved complexes: cisplatin, carboplatin, and oxaliplatin.1,2 The mechanism of action of cisplatin has been extensively researched and attributed to cell death via the DNA Damage Response (DDR) pathway. The mechanism of action for oxaliplatin, however, is less well-characterized.3–5 Both cisplatin and oxaliplatin can create intra- and inter-strand Pt(II)–DNA adducts.3,6 Differences in DNA binding capacity were an early indicator that oxaliplatin may work through a mechanism of action different to that of cisplatin.3 Studies have shown for example that oxaliplatin treatment results in lower cellular DNA Pt adduct levels than found following cisplatin treatment, while still eliciting higher cell cytotoxicity in some cases.6 Recent findings suggest that unlike cisplatin and carboplatin, oxaliplatin along with phenanthriplatin7 and a recent ruthenium complex8 may induce cell death via ribosome biogenesis stress.9–13 The mechanisms by which Pt(II) complexes interact with the nucleolus to cause this specific inhibition of ribosome biogenesis and subsequent cell death are not well understood.Through limited structure-function analyses, we previously determined that a Pt(II) complex with the 1,2-diaminocyclohexane (DACH) ring of oxaliplatin, dichloro(1,2-diaminocyclohexane)Pt(II) (DACH-Pt), as well as dichloro Pt(II) complexes with 1,2-diaminocyclopentane (DACP-Pt) and 1,2-phenylenediamine, are capable of nucleolar stress induction.11 To further investigate the influence of ligand ring structure on nucleolar stress, the impact of substituents at the 4-position of the DACH ring was investigated. It was identified that complexes with axial or equatorial methyl, ethyl, and propyl substituents induced nucleolar stress, while the installation of more rigid or bulky substituents such as an isopropyl (axial or equatorial) and acetamide (axial) resulted in complexes that no longer induce nucleolar stress.14 Finally, it was found that the 1R,2R isomers of DACH-Pt and DACP-Pt were more effective in nucleolar stress induction than 1S,2S isomers or the racemic mixtures.13 These studies lay groundwork for azide installments onto the DACH ring that may enable target identification of oxaliplatin mimics via Cu-catalyzed azide–alkyne cycloaddition (CuAAC).
Molecular target identification using Pt(II) complexes modified for azide–alkyne cycloaddition “click” chemistry has been previously reported by us and others.15–21 These complexes, such as azido-Pt (Fig. 1), have been used in vitro to identify nucleotide and protein binding,15 and in vivo for in-cell localization through fluorescence microscopy16,20 and chemical proteomics utilizing streptavidin/biotin enrichment.16 However, like cisplatin, azido-Pt and 1,3-Pt do not induce nucleolar stress.11 Thus, new azide-modified reagents are needed to pursue target identification relevant to Pt(II)-induced nucleolar stress.
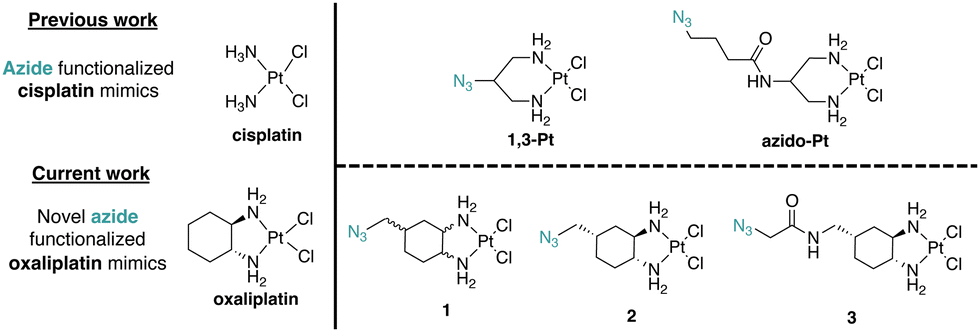 | ||
| Fig. 1 Azide-functionalized cisplatin and oxaliplatin mimics used in this study. | ||
Herein, we report the synthesis of novel azide click-capable oxaliplatin mimics (1, 2, 3) and find key differences in their influence on nucleolar properties in comparison to the previously synthesized azido-Pt and 1,3-Pt (Fig. 1). We have identified that azido-Pt, 1,3-Pt, and 3 do not induce nucleolar stress based on NPM1 relocalization and fibrillarin assays. The enantiomerically pure (1R,2R,4S) complex 2 induces a robust nucleolar stress response at regular treatment concentrations (10 μM), whereas the stereoisomeric mixture 1 induces a lower nucleolar stress response at the same treatment concentration. To further examine the relationship between NPM1 relocalization caused by complex 2 and the DDR pathway,12 γH2AX assays were performed. Unlike the case for cisplatin, H2AX activation is not significant for oxaliplatin or azide-modified complexes 1 and 2.
To our knowledge, these are the first reported oxaliplatin mimics to include an azide handle. Complex 2 has potential to be a crucial tool in understanding Pt(II)-induced nucleolar stress as it is, to our knowledge, the first azide-functionalized oxaliplatin derivative demonstrated to induce nucleolar stress.
Results
Synthesis of 1, 2, and 3
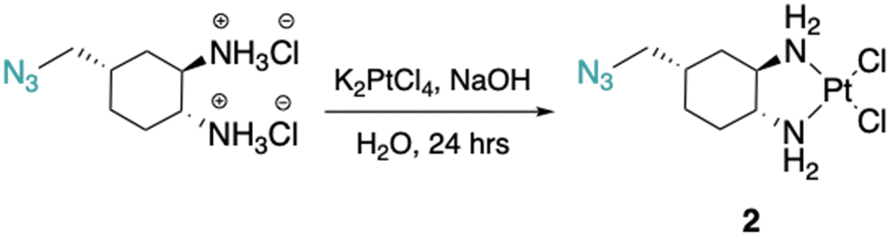
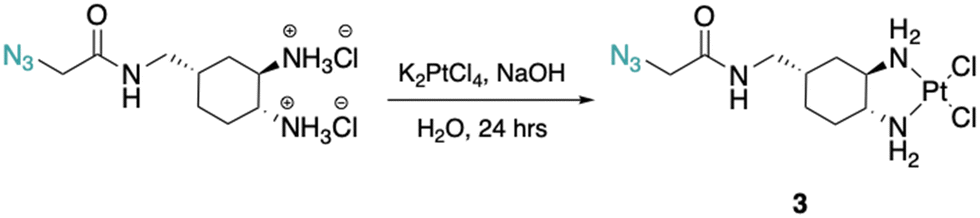
The DACH ligand in 1 is a mixture of four trans-isomers, as two enantiomeric pairs ((1R,2R,4S)/(1S,2S,4R) and (1R,2R,4R)/(1S,2S,4S)), whereas the DACH ligand in 2 is enantiomerically pure trans-(1R,2R,4S). Furthermore, 2 possesses only (1R,2R)-configuration with respect to the two amine groups, as is the case for oxaliplatin. The DACH ligand used in the synthesis of 1 was synthesized from a racemic starting material while the DACH ligand used for 2 was synthesized from an enantiomerically pure starting material (Fig. 2). With regards to 2, the 4S-conformation is retained throughout each step of the synthesis and the (1R,2R,4S) isomer is solely isolated by column chromatography following the diazidation step. Synthesis of the DACH ligand for 3 follows a similar synthetic approach to 2, but after a reduction of the terminal azide, chloroacetyl chloride was linked through an amide bond and a terminal azide was installed (see ESI†). Complexes 1–3 were synthesized upon deprotonation of the hydrochloride salts of the respective DACH ligand with base, and subsequent reaction with K2PtCl4.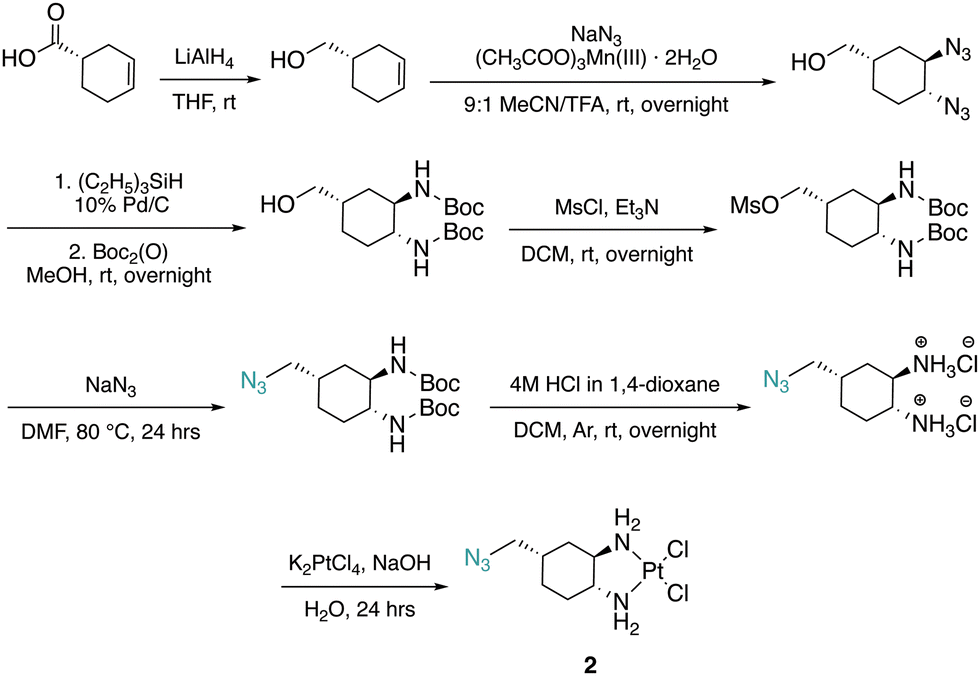 | ||
| Fig. 2 Synthesis of 2. | ||
X-ray crystal structures for the tartrate salt of the enantiomerically pure DACH ligand found in 2 and for the diiodo analogue of 2, [Pt(DACH-CH2N3)I2], were determined (Fig. 3). In both structures, the CH2N3 group at position 4-occupies an axial position on the cyclohexane ring, while the two amine groups lie in equatorial positions with trans-configuration. The presence of the CH2N3 group in an axial position results in ring flattening with reduced dihedral angles, ranging from 164.0° to 167.1°, between the group and its neighboring axial protons across both structures.22 The DACH ligand acts as a bidentate ligand coordinating to the platinum center through the two nitrogen atoms of the amines. Overall the N–Pt–N bond angles and Pt–N bond lengths are similar to those previously reported for oxaliplatin and a 4-methyl oxaliplatin derivative.23 Crystallographic data, CCDC 2256792 and 2256793.†.
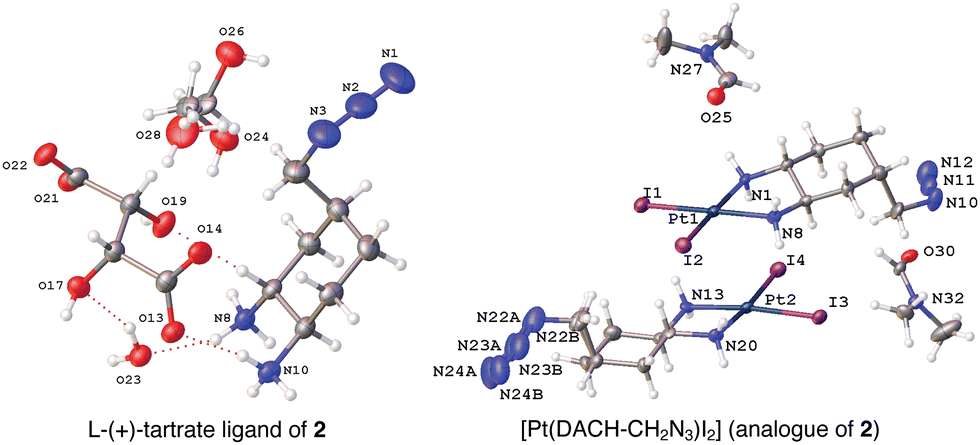 | ||
| Fig. 3 X-ray structures of the L-(+)-tartrate salt of the enantiomerically pure DACH ligand found in 2 and [Pt(DACH-CH2N3)I2], the diiodo analogue of 2. | ||
In vitro binding of azide-modified Pt(II) complexes to hairpin DNA and fluorophore conjugation
To measure DNA binding and click reactivity, azido-Pt, 1,3-Pt, 1, 2, and 3 were incubated with a 20-nucleotide DNA hairpin (HP) containing a single GG site. A subsequent Cu-free click reaction was performed using Alexa-647 DBCO24 and conjugation of the fluorophore was analyzed by dPAGE and fluorescence imaging. All complexes tested were able to readily bind to DNA after a total of 48 hours of incubation. Cisplatin and cisplatin mimics (azido-Pt and 1,3-Pt) appear to have a higher DNA binding capacity than oxaliplatin and complexes 1 and 2. Interestingly, complex 3, which acts as an oxaliplatin mimic, shows DNA binding results similar to cisplatin and cisplatin mimics. All azide-containing complexes (1,3-Pt, azido-Pt, 1, 2, and 3) show conjugation to the Alexa-647 fluorophore, confirming their ability to bind to DNA and subsequently undergo click reactions (Fig. 4). | ||
| Fig. 4 dPAGE analysis of Pt(II) complexes incubated with HP DNA. Complexes labeled as “click” were first incubated with HP for 24 h followed by a click reaction with Alexa-647 DBCO for an additional 24 h, giving a total DNA incubation time of 48 h. Control complexes were incubated for 48 h with HP with no click reaction. All samples were purified by spin column before analysis by dPAGE. Grey: DNA stained with SYBR gold (539 nm emission wavelength), red: DNA-Pt-647 DBCO complex (671 emission wavelength). | ||
In-cell localization through fluorescent tagging of 1,3-Pt, azido-Pt, 1, 2, and 3
To visualize localization of the click-capable cisplatin and oxaliplatin mimics in mammalian cell culture, fluorescence imaging was used (Fig. 5). A549 cells were treated for 24 hours with the click-capable complexes and subsequently treated with Alexa-488-alkyne for post-treatment fluorescent labeling via the CuAAC reaction. In agreement with previous results in HeLa cells,16 both 1,3-Pt and azido-Pt localized to the nucleoli of cells and around the cellular nucleus. Interestingly, the oxaliplatin click-capable mimics (1, 2, 3) showed cellular localization similar to that of the cisplatin mimics. These initial cell imaging studies required a higher concentration of Pt(II) complexes (50 μM) than used for nucleolar stress assays (10 μM, vide infra). Under these conditions, cellular localization via post-treatment fluorescent labeling is not sufficient to distinguish possible variations that could differentiate the mechanism of action of oxaliplatin and cisplatin. Future higher sensitivity imaging may allow visualization of cellular localization at lower Pt concentrations.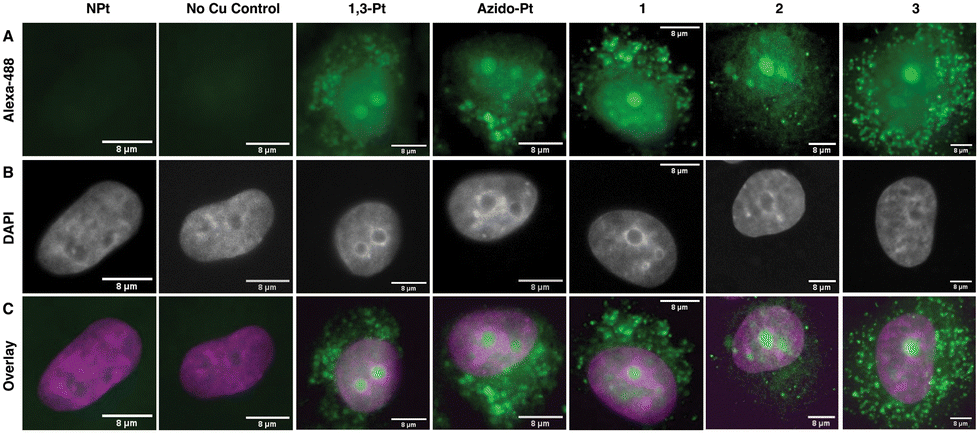 | ||
| Fig. 5 Confocal images of fluorescent cellular localization of azide-modified Pt(II) complexes. Panel (A) shows fluorescent labeling of A549 cells treated with 50 μM of azide-modified Pt(II) for 24 h and subsequent CuAAC with alkyne-tagged Alexa-488. The Pt(II)-free and Cu-free controls show little to no signal indicating low background fluorescence. Panel (B) shows the nuclei labeled via DAPI stain. Panel (C) shows the overlay of Alexa-488 (green) labeling and DAPI (magenta). | ||
NPM1 relocalization occurs for oxaliplatin-mimic 2 but not for cisplatin-mimics 1,3-Pt and azidoplatin
Azido-Pt has been previously reported to not induce nucleolar stress as assayed by NPM1 relocalization.11 To confirm and further investigate properties of azido-Pt and 1,3-Pt, NPM1, fibrillarin and γH2AX assays were performed in A549 cells at 24 h drug treatment with either 10 or 20 μM treatment concentrations. Relocalization of the nucleolar protein NPM1 is one hallmark of nucleolar stress induction, as shown following treatment with ActD, a known nucleolar stress inducer used as a positive control in this study.12,25 Distribution of NPM1 can be observed through immunofluorescence and quantified via the coefficient of variation (CV, see Methods), where cells exhibiting nucleolar stress show CV values of ∼0.6 and those not exhibiting stress have CV values of ∼1.0.11 In Fig. 6, cells treated with azido-Pt and 1,3-Pt at both 10 and 20 μM drug concentrations appear to be unstressed as the nucleoli are amorphous in shape and NPM1 is not relocalized throughout the nucleoplasm. This is further supported by the CV values ∼1.0.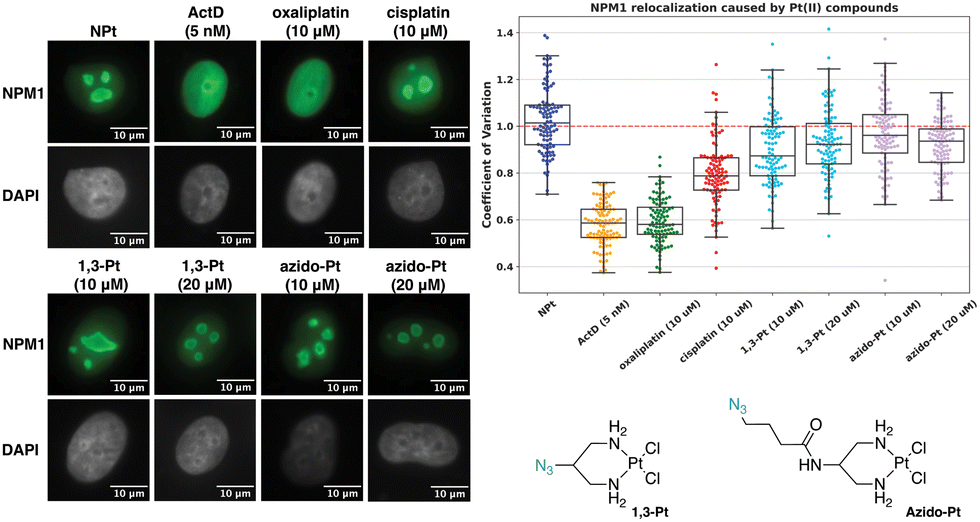 | ||
| Fig. 6 Visualization and quantification of NPM1 relocalization (NPM1: green, DAPI: gray) induced by 1,3-Pt and azido-Pt. Treatment conditions indicated (10 or 20 μM for Pt(II) complexes, 5 nM for ActD) in A549 cells at 24 h treatment. Representative images and CV calculations based on 3 trials are shown. Full cell images found in ESI,† Fig. S2. For each treatment data set, boxes represent median, first and third quartiles, and vertical lines are the range of data with outliers (see Experimental Section). Scale bar = 10 μm. | ||
The NPM1 relocalization assay was next performed with azide-modified oxaliplatin mimics 1, 2, and 3. For these studies, the stereochemistry at the 1- and 2-position of the DACH ring is important, making 1R,2R oxaliplatin and 1R,2R DACH-Pt appropriate complexes to compare to 1, 2, and 3.13 As observed in Fig. 7, complex 1 induces a slight nucleolar stress response at 10 μM and more significant nucleolar stress at 20 μM. For the enantiomerically pure complex 2, nucleolar stress is observed at both 10 and 20 μM treatment concentrations. These results highlight the importance of stereochemistry about the 1- and 2-position of the DACH ring and agree with previous work demonstrating that the 1R,2R enantiomers for DACH-Pt and DACP-Pt show higher degrees of nucleolar stress than the 1S,2S enantiomers.13 Similar trends have been observed for other Pt(II) complexes in different cell lines.26 Complex 3 does not appear to induce nucleolar stress at 10 or 20 μM, which may be attributed to the amido group installed at the 4-position of the DACH ring. A similar effect was observed in a previous study, in which complexes with a bulky acetamide modification directly off the 4-position of the DACH ring did not induce nucleolar stress.14
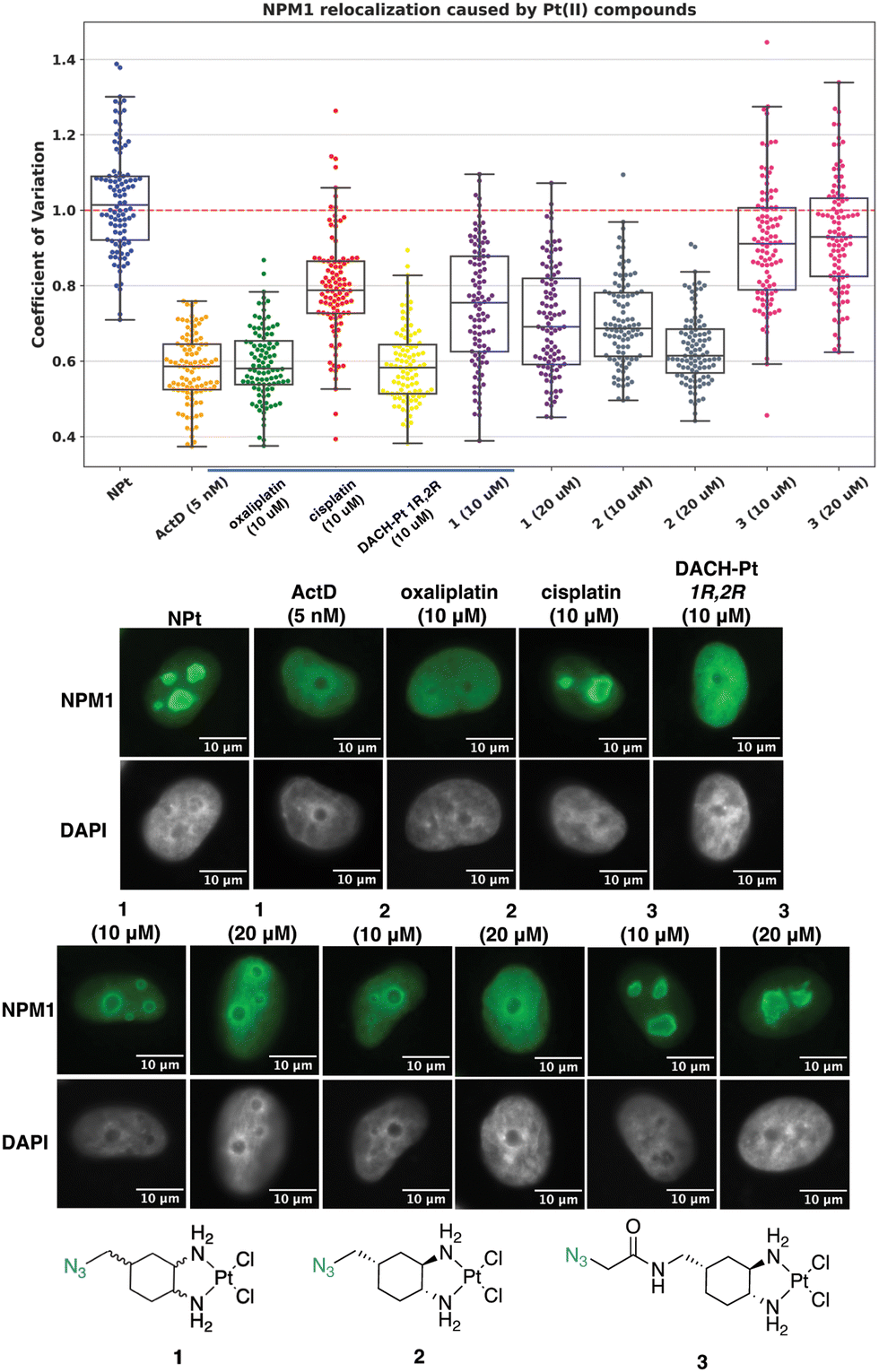 | ||
| Fig. 7 Visualization and quantification of NPM1 relocalization (NPM1: green, DAPI: gray) induced by 1, 2, and 3. Treatment conditions indicated (10 or 20 μM for Pt(II) complexes, 5 nM for ActD) in A549 cells at 24 h treatment. Representative images and CV calculations based on 3 trials are shown. Full cell images found in ESI,† Fig. S4. For each treatment data set, boxes represent median, first and third quartiles, and vertical lines are the range of data with outliers (see Experimental Section). Scale bar = 10 μm. | ||
Fibrillarin condensation occurs for oxaliplatin-mimic 2 but not for cisplatin-mimics 1,3-Pt and azido-Pt
Fibrillarin, a nucleolar protein found in the dense fibrillar component of the nucleolus, was used to further demonstrate the effects of the Pt(II) complexes on the nucleolus. Fibrillarin is thought to be a key participant in the first step of processing pre-ribosomal RNA. Upon inhibition of nucleolar processes such as rRNA synthesis, fibrillarin demonstrates nucleolar segregation in which fibrillarin phase-separates from the granular component of the nucleolus and forms nucleolar caps. This morphological change is a downstream effect of NPM1 relocalization.12,27,28In Fig. 8, treatment with the known stress inducers oxaliplatin and ActD results in the formation of fibrillarin-containing nucleolar caps. Upon treatment with azido-Pt and 1,3-Pt, and cisplatin, fibrillarin shows an even distribution throughout the nucleolus. Nucleolar cap formation upon treatment with complex 2 at 10 μM is observed. However, with complex 3 nucleolar cap formation is not observed and the fibrillarin resembles that of the untreated cells. The results of nucleolar cap formation are in agreement with NPM1 redistribution studies, demonstrating that novel complex 2 induces nucleolar stress at 10 μM.
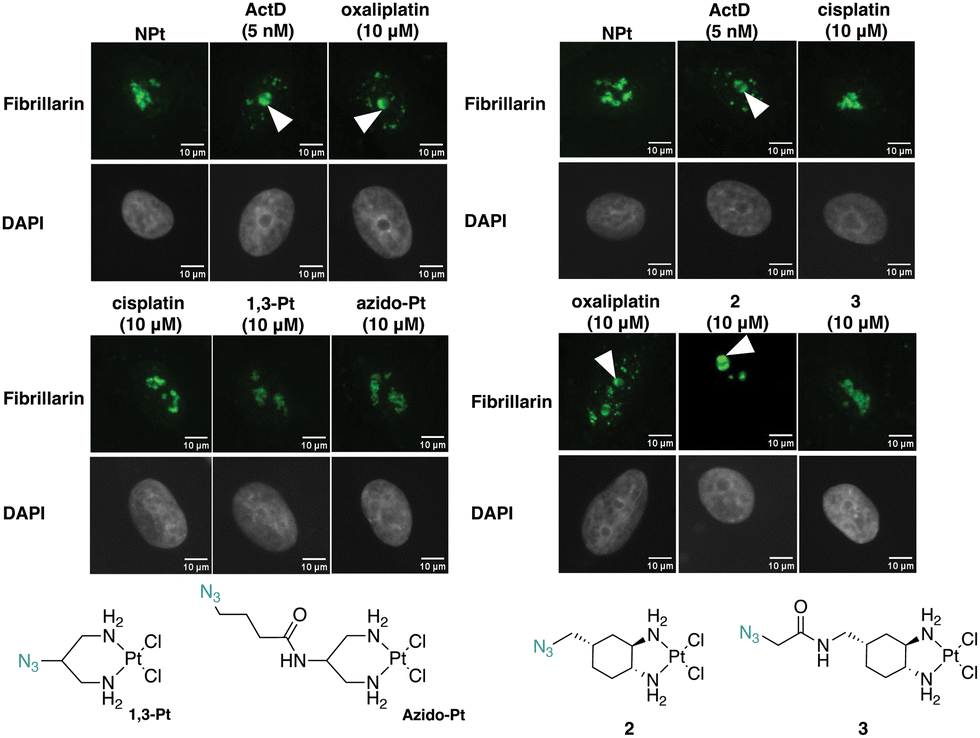 | ||
| Fig. 8 Nucleolar morphological changes monitored by fibrillarin (fibrillarin: green, DAPI: gray). Treatment condition at 10 μM for azido-Pt, 1,3-Pt, 2, and 3 and 5 nM for ActD in A549 cells at 24 h treatment. Nucleolar cap formation is indicated by a white arrow. Scale bar = 10 μm. | ||
Oxaliplatin-mimics 2 and 3 do not induce significant γH2AX
Previous studies have found that in certain instances, nucleolar stress can occur downstream of the DDR pathway.12,29 The phosphorylated protein γH2AX is a biomarker of DDR as it attracts factors and proteins involved in repairing DNA damage induced by environmental agents or chemotherapeutics, such as cisplatin.30–32 This allows γH2AX to be used as one method to quantify DDR activation upon treatment with platinum complexes.12 In Fig. 9, the levels of γH2AX following treatment with 1,3-Pt and azido-Pt compared to cisplatin levels have a statistically significant difference (p < 0.05). Although different, γH2AX levels with 1,3-Pt are higher (statistically significant, p < 0.05) compared to oxaliplatin, while azido-Pt γH2AX levels are very similar to that of oxaliplatin (N.S., p > 0.05). Lower γH2AX levels caused by the more highly functionalized azido-Pt matches previous studies with functionalized cisplatin mimics that found lower cytotoxicity compared to cisplatin, and required higher concentrations to produce similar levels of γH2AX.20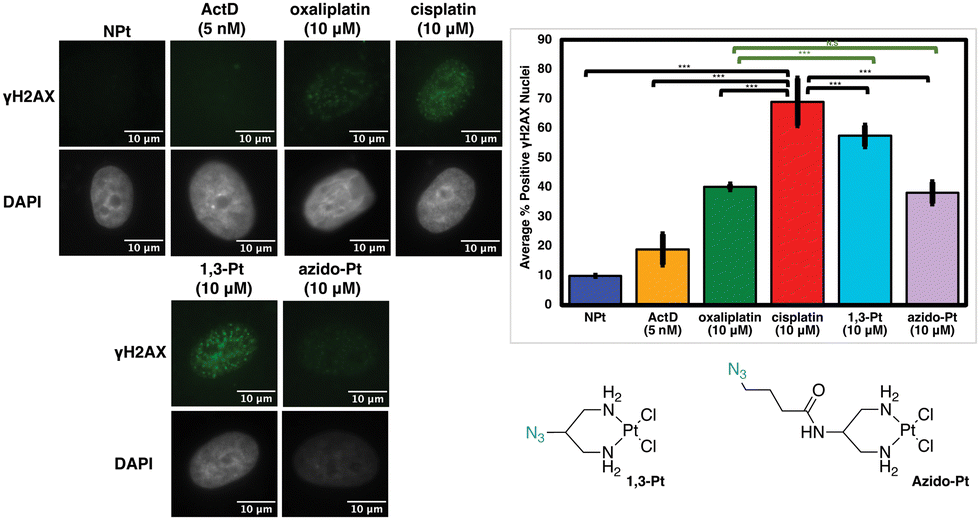 | ||
| Fig. 9 Visualization and quantification of γH2AX (γH2AX: green, DAPI: gray) as an indicator of DDR induced by azido-Pt and 1,3-Pt. Treatment conditions indicated (10 μM for Pt(II) complexes, 5 nM for ActD) in A549 cells at 24 h treatment; average of 3 trials and std; quantifications performed described in Experimental Section. Scale bar = 10 μm. ***p < 0.05, N.S. p > 0.05. | ||
Although complex 2 is inducing nucleolar stress, this could potentially be activated as a downstream effect of the DDR pathway as shown previously with cisplatin.12 As shown in Fig. 10, however, the level of γH2AX after treatment with complex 2 is similar to that of oxaliplatin (N.S., p > 0.05) and significantly lower than the extent of cisplatin-induced γH2AX (p < 0.05). Complex 3 follows the same trend as 2. This could be explained by the extensive modification of the bulky substituents at the 4-position of the DACH ring and the DACH ring itself when compared to cisplatin, which could be preventing recruitment of proteins involved in the DDR pathway.
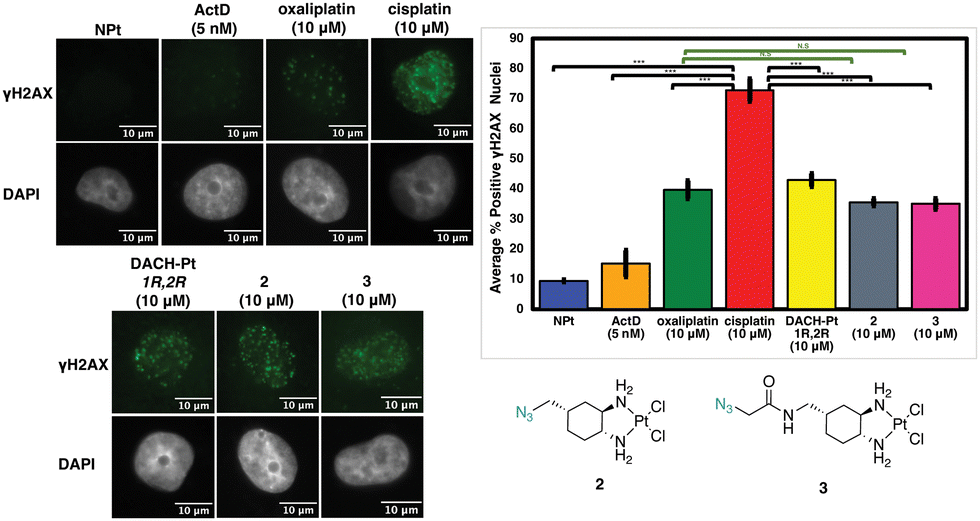 | ||
| Fig. 10 Visualization and quantification of γH2AX (γH2AX: green, DAPI: gray) as an indicator of DDR induced by 2 and 3. Treatment conditions indicated (10 μM for Pt(II) complexes, 5nM for ActD) in A549 cells at 24 h treatment; average of 3 trials and std; quantification performed described in Experimental Section. Scale bar = 10 μm. ***p < 0.05, N.S. p > 0.05. | ||
Discussion
Investigating nucleolar stress induction through Pt(II) complexes is of great importance in the field of medicine and biology as the nucleolus displays abnormalities and over-activation in many types of cancer. This over-activation allows for over-expression of essential proteins that cause uncontrollable cellular replication. Understanding how small molecules influence the functions of the nucleolus may provide insight on how to trigger pathways, such as nucleolar stress, or further highjack regulatory proteins with targeted therapeutics and inhibit cell proliferation.33In this work we sought to explore novel azide-functionalized oxaliplatin mimics in comparison with previously synthesized azide-functionalized cisplatin mimics.15–21 A bona-fide azide-modified oxaliplatin mimic would allow further bioanalytical studies to identify key differences leading to nucleolar stress induction by Pt(II) complexes, and gain insight into how nucleolar stress is being induced by small molecules in general. We utilized dPAGE analysis to demonstrate that these complexes can bind to DNA and undergo ring-strained cycloaddition for fluorescent labeling in vitro. In addition, we demonstrated that these complexes enter cells and undergo CuACC for fluorescent labeling in fixed mammalian cell culture. This in-cell localization confirmed that Pt(II) complexes concentrate in the nucleolus,16,20,34 but at the resolution of current experiments did not differentiate localization between click-functionalized cisplatin and oxaliplatin mimics.
We used NPM1 relocalization and fibrillarin cap formation to identify nucleolar stress induction in A549 cells following 24 hours treatment with each complex. We find that oxaliplatin-mimic 2, which has a DACH ligand with (1R,2R) configuration, induces nucleolar stress and fibrillarin cap formation. Complex 1, which has a mixed (1R,2R)/(1S,2S) configuration, requires higher treatment concentrations for similar NPM1 redistribution, and the more highly functionalized 3 does not induce nucleolar stress at up to 20 μM treatment concentrations. These results reinforce the conclusion that stereochemistry about the amine ligands is a vital factor in the ability of Pt(II) complexes to cause nucleolar stress, as was observed in a previous study,13 and that nucleolar stress may not be afforded with bulkier amido groups at the 4-position of the DACH ring,14 as in complex 3.
We observe nucleolar disruption in the form of NPM1 relocalization and fibrillarin cap formation in A549 cells treated with oxaliplatin and azide-modified 1 and 2, but not with cisplatin or related 1,3-platin and azido-Pt. The characteristics of these markers of nucleolar disruption are in some contrast with a recent report35 that shows more broad distribution of fibrillarin, and less broad non-nucleolar distribution of NPM1, with oxaliplatin treatment. These differences may be due to detection method (endogenously tagged proteins35vs. immunofluorescence) and/or differences in cell lines. An interesting hypothesis was raised that selective nucleolar partitioning and preferential binding of oxaliplatin to fibrillarin causes nucleolar disruption.35 The preliminary cell imaging studies in Fig. 5 suggest that both oxaliplatin and cisplatin-type Pt(II) compounds localize to the nucleolus, which does not support a theory of differential partitioning, but as noted above these studies require higher resolution for more definitive conclusions. The click-capable oxaliplatin mimic reported here provides a tool for detecting fibrillarin and other possible in-cell Pt-protein interactions related to nucleolar stress.
To further examine the relationships between nucleolar stress and DDR, we utilized a γH2AX detection assay. Prior studies show that in general, oxaliplatin induces significantly lower γH2AX levels than does cisplatin.10,12,36 Consistent with this, we find that azide-modified, nucleolar stress-inducing complex 2 induces low levels of γH2AX, similar to observations with oxaliplatin, and 1,3-Pt causes higher levels of γH2AX, consistent with being a cisplatin mimic and acting through DDR. While these results support the observation that nucleolar stress induction by oxaliplatin-like Pt(II) complexes is accompanied by less observable DNA damage as monitored by γH2AX, a recent study reports partial protection from oxaliplatin-induced nucleolar stress via inhibition of DDR signaling kinases ATM and ATR.36 Moreover, the more highly modified azido-Pt and 3 do not cause nucleolar stress, but also do not induce high levels of γH2AX. Thus, further studies are warranted to confirm relationships between different DDR pathway markers and nucleolar stress induction. Similarly, further studies are needed to investigate the relationships between nucleolar stress and cytotoxicity caused by these Pt(II) compounds.11
Conclusion
Here we report novel azide-functionalized oxaliplatin mimics and compare their properties to previously synthesized azide-functionalized cisplatin mimics to identify key differences in Pt(II)-induced nucleolar stress. We identify novel complex 2 as a potential tool to study nucleolar stress as it induces key markers including relocalization of NPM1 and the formation of fibrillarin caps. We find that stereochemistry is crucial to induce a significant degree of nucleolar stress, specifically when comparing complex 1, which possesses a DACH ligand as a mixture of trans-isomers, and complex 2, which possesses an enantiomerically pure trans-(1R,2R,4S)-DACH ligand. Finally, we find that further modification through an amido linkage at the 4-position of the DACH ring prevents nucleolar stress from being induced. With complex 2 being a tool to study Pt(II)-induced nucleolar stress, a more in-depth analysis of the molecular targets (DNA, RNA, proteins) can be pursued in order to further understand the complex mechanism of nucleolar stress induction by small molecule platinum complexes.Experimental section
Images were taken using a HC PL Fluotar 63×/1.3 oil objective mounted on a Leica DMi8 fluorescence microscope or HCX PL APO 63×/1.40–0.6 objective mounted on Leica DFC365 FX with Leica Application Suite X software.Cell culture and treatment
A549 human lung carcinoma cells (#CCL-185, American Type Culture Collection) were cultured in 5% CO2 at 37 °C in Dulbecco's Modified Eagle Medium (DMEM) with 10% Fetal Bovine Serum (FBS) and 1% antibiotic–antimycotic. Treatments were performed on cells that had been grown for 11–26 passages to 70% confluency. All treatments were performed for 24 hours at 10 or 20 μM concentrations of the platinum complexes. 1,3-Pt, azido-Pt, 1, and 3 were made in 5 mM stocks, while 2 was made in a 2.5 mM stock due to difference in solubilities. The complex stock solutions were made with DMF (1,3-Pt, azido-Pt, 1, 2, and 3), water (oxaliplatin, cisplatin), or DMSO (ActD) Stock solutions were diluted into media immediately prior to drug treatment. Treatments were performed in triplicate and additional replicates are available from the corresponding author upon reasonable request.Immunofluorescence
Cells were grown on coverslips (Ted Pella product no. 260368, round glass coverslips, 10 mm diam. 0.16–0.19 mm thick) as described above. After treatment was complete, cells were washed with phosphate buffered saline (PBS) and fixed with 4% paraformaldehyde (PFA) in PBS for 20 minutes at room temperature. PFA was removed using aspiration and cells were permeabilized with 0.5% Triton-X in PBS for 20 minutes at room temperature. Two ten-minute blocking steps were then performed with 1% bovine serum albumin (BSA) in PBST (PBS with 0.1% Tween-20). Cells were incubated for one hour in primary antibody NPM1 or γH2AX for DDR and for an hour and a half with the primary antibody Fibrillarin for nucleolar stress response (NPM1 monoclonal antibody, FC-61991, ThermoFisher, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :800 dilution in PBST with 1%BSA) (Phospho-Histone H2A.X) (Ser139) Monoclonal Antibody (CR55T33, ThermoFisher, 2.5 μg in PBST with 1% BSA), (anti-Fibrillarin antibody ab4566 from Abcam, 1:500 dilution) and 1 hour in secondary antibody for NPM1 or γH2AX and an 1.5 h for fibrillarin (Goat Anti-Mouse IGG H&L Alexa Fluor® 488, ab150113, Abcam, 1:1000 dilution in PBST with 1% BSA), with three 5 minute wash steps using PBST between antibody incubations. Washed again in the same manner before mounting the slides. Coverslips were then mounted on slides with ProLong™ Diamond Antifade Mountant with DAPI (ThermoFisher) according to manufacturer's instructions.
:800 dilution in PBST with 1%BSA) (Phospho-Histone H2A.X) (Ser139) Monoclonal Antibody (CR55T33, ThermoFisher, 2.5 μg in PBST with 1% BSA), (anti-Fibrillarin antibody ab4566 from Abcam, 1:500 dilution) and 1 hour in secondary antibody for NPM1 or γH2AX and an 1.5 h for fibrillarin (Goat Anti-Mouse IGG H&L Alexa Fluor® 488, ab150113, Abcam, 1:1000 dilution in PBST with 1% BSA), with three 5 minute wash steps using PBST between antibody incubations. Washed again in the same manner before mounting the slides. Coverslips were then mounted on slides with ProLong™ Diamond Antifade Mountant with DAPI (ThermoFisher) according to manufacturer's instructions.
Image processing and quantification
The quantification of NPM1 relocalization was performed in an automated fashion using a Python 3 script. Images were preprocessed in ImageJ,37,38 to convert the DAPI and NPM1 channels into separate 16-bit grayscale images. Between 50–250 cells were analyzed for each treatment group. Nuclei were segmented using the DAPI images using Li thresholding function in the Scikit-Image Python package.39 The coefficient of variation (CV) for individual nuclei, which is defined as the standard deviation in pixel intensity divided by the mean pixel intensity, was calculated from the NPM1 images using the SciPy Python package. All the data was normalized to the no-treatment in each experiment. NPM1 imaging results for each complex were observed in triplicate. Data are represented as boxplots generated using Seaborn within Python.Quantification of γH2AX intensity and foci was performed with CellProfiler 4.2.1 software.40 In one analysis method, a “percent positive” value was calculated for each treatment condition relative to the untreated control. A threshold was determined for a positive γH2AX result based on the 90th percentile intensity value of the untreated control for each time point. Nuclei in the experimental samples with integrated intensity levels higher than this were counted as positive for γH2AX. Significance testing was done via t-test to obtain a p value.
In vitro DNA gel binding and fluorophore clicking
Hairpin DNA sequence (TATGGTATTTTTATACCATA) (280 μM) was folded by rapid heating to 90 °C and slow cooling to 4 °C in 10 mM Na2HPO4/NaH2PO4 buffer (pH 7.1), 0.1 M NaNO3, and 10 mM Mg(NO3)2. The platinum complex (830 μM) was then added and the solution was incubated at 37 °C for 24 h. For click complexes, 195 μM of Alexa-657 DBCO (AZDye) was add and incubated 37 °C for an additional 24 h. Non-click controls complexes were incubated for an additional 24 h at 37 °C. All complexes were then purified with Sephadex G-25 Medium size exclusion resin (GE Healthcare) on laboratory-prepared spin columns (BioRad) to remove unbound platinum and fluorophore. Purified samples were added at a DNA concentration of 200 ng on dPAGE (19:1 20% acrylamide in 8 M urea) and ran at 180 V for 2 hours. Gels were then stained with SYBR gold for 5 minutes and imaged using a GE Amersham Typhoon gel imager.
In-cell fluorescent clicking and localization
Cells were grown on coverslips (Ted Pella product no. 260368, round glass coverslips, 10 mm diam. 0.16–0.19 mm thick) as described above. Cells were then treated with 50 μM of azide-modified Pt(II) complexes for 24 hours. Following treatment cells were washed with PBS 3X for 5 minutes each and fixed with 4% PFA in PBS for 20 minutes. Cells were then permeabilized with 2% Triton-X in PBS for 1 hour. Two ten-minute blocking steps were then performed with 5% bovine serum albumin (BSA) in PBST (PBS with 0.1% Tween-20). Cells were then incubated with 750 μM iodoacetamide in PBS for 1 hour. Following this, a click cocktail containing 25 mM sodium ascorbate, 10 mM THPTA, 5 μM Alexa-488 alkyne (AZDye), and 1 mM CuSO4 in PBS was added to cells and incubated for 1 hour. Control treatments for no Cu had all components of the click cocktail added excluding the CuSO4. After the click reaction, cells were washed 3X with PBST for 5 minutes each. Cells were then incubated with 1.66 mM Hoechst 33342 in 2% Triton-X in PBS for 30 minutes. Cells were then washed with 2% Triton-X in PBS 2X for 15 minutes each and mounted on slides with antifade fluorescence mounting media (Abcam) and left to cure overnight before imaging. All the above steps were done at room temperature.Synthesis
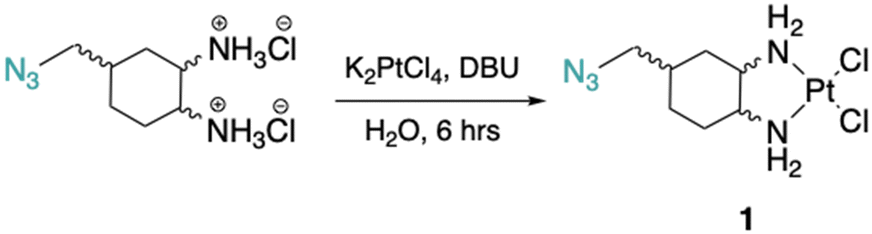
Cis-[((1R,2R,4S)/(1S,2S,4R), (1R,2R,4R)/(1S,2S,4S)) 4-methylazido-1,2-cyclohexanediamine]dichlorido platinum(II) (1). ((1R,2R,4S)/(1S,2S,4R), (1R,2R,4R)/(1S,2S,4S)) 4-methylazido-1,2-cyclohexanediammonium dihydrochloride (0.1 g, 0.3 mmol) was dissolved in 1.5 mL of dI water, 5 drops of DBU (or until basic) was added to deprotonate the amines, and K2PtCl4 (0.1 g, 0.3 mmol) was added and stirred at rt for ∼6 hours. Precipitate was washed with dI water (3×), cold ethanol (3×), diethyl ether (3×), and dried to produce 1 as a light-yellow solid (0.04 g, 33% yield). 1H NMR (600 MHz, DMF-d7) ((1R,2R,4S), (1S,2S,4R)): δ 5.74–5.51 (m, 2H), 5.16–4.86 (m, 2H), 3.47–3.40 (m, 2H), 2.70–2.51 (dd, J = 60.7, 11.5 Hz, 2H), 2.18–2.11 (d, J = 13.6 Hz, 1H), 1.99–1.90 (s, 2H), 1.72–1.60 (m, 3H), 1.39–1.26 (m, 1H). 13C NMR (151 MHz, DMF-d7): δ 64.6, 64.4, 63.9, 59.7, 56.9, 53.4, 37.9, 34.3, 34.2, 31.4, 29.3, 28.0, 27.1. 195Pt NMR (129 MHz, DMF-d7) ((1R,2R,4S), (1S,2S,4R)): δ −2271.84; ((1R,2R,4R),(1S,2S,4S)): δ −2259.07. HRMS (ESI+): m/z calcd for C7H16N5Cl2Pt: 435.0430 [M + H]+; found: 435.0439.
Cis-[(1R,2R,4S) 4-methylazido-1,2-cyclohexanediamine]dichlorido platinum(II) (2).
(1R,2R,4S) 4-methylazido-1,2-cyclohexanediammonium dihydrochloride (0.2 g, 0.8 mmol) was dissolved in dI water (5 mL) and two equiv. of NaOH was added to form the free-amine form of the ligand. This solution was added dropwise to a solution of K2PtCl4 (0.3 g, 0.8 mmol) in dI water (10 mL). The reaction mixture was stirred in the dark at room temperature for 24 hours resulting in the formation of a light-yellow precipitate which was isolated by vacuum filtration and washed with dI water. This solid was dried under vacuum overnight to yield 2 as a light-yellow solid (0.4 g, 68% yield). 1H NMR (500 MHz, DMF-d7): δ: 5.63–5.51 (m, 2H), 5.05–4.84 (m, 2H), 3.43 (d, 2H, 7.2 Hz), 2.72–2.69 (m, 1H), 2.57–2.53 (m, 1H), 2.14 (d, 1H, 13.0 Hz), 1.97–1.90 (m, 2H), 1.75–1.59 (m, 3H), 1.49-1.43 (m, 1H). 13C NMR (126 MHz, DMF-d7): δ 64.6, 59.8, 53.4, 34.3, 34.3, 28.0, 27.1. 195Pt NMR (107 MHz, DMF-d7): δ −2269.25. FT-IR νmax (cm−1): (N–H) 3244, (N–H) 3183, (C–H) 2929, (C–H) 2863, (N
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NN) 2091, (N–H) 1553, 742. MS (ESI+) (DMSO/MeOH) [M–Cl + DMSO]+: m/z calcd for C9H21ClN5OPtS: 477.1, found: 477.0. HRMS (ESI+) (DMF/MeOH) [M–Cl + DMF]+: m/z calcd for C10H22ClN6OPt: 472.1191, found: 472.1190.
NN) 2091, (N–H) 1553, 742. MS (ESI+) (DMSO/MeOH) [M–Cl + DMSO]+: m/z calcd for C9H21ClN5OPtS: 477.1, found: 477.0. HRMS (ESI+) (DMF/MeOH) [M–Cl + DMF]+: m/z calcd for C10H22ClN6OPt: 472.1191, found: 472.1190.
Cis-[(1R,2R,4S) 4-methyl(1-azidomethylamide)-1,2-cyclohexanediamine]dichlorido platinum(II) (3).
(1R,2R,4S) 4-Methyl(1-azidomethylamido)-1,2-cyclohexaneammonium dihydrochloride (0.3 g, 1.0 mmol) was dissolved in dI water (5 mL) and two equiv. of NaOH was added to form the free-amine form of the complex. This solution was added dropwise to a solution of K2PtCl4 (0.4 g, 1.0 mmol) in dI water (10 mL). The reaction mixture was stirred in the dark at room temperature for 24 hours resulting in the formation of a light-yellow precipitate which was isolated by vacuum filtration and washed with dI water. This solid was dried under vacuum overnight to yield 3 as a light-yellow solid (0.3 g, 57%). 1H NMR (500 MHz, DMF-d7): δ 8.16 (t, 1H, 5.6 Hz), 5.67–5.47 (m, 2H), 5.16–5.05 (m, 2H), 3.94 (s, 2H), 3.25 (t, 2H, 6.8 Hz), 2.88–2.85 (m, 1H), 2.72–2.67 (m, 1H), 2.09 (d, 1H, 12.5 Hz), 1.92–1.89 (m, 2H), 1.79 (d, 1H, 14.8 Hz), 1.70 (td, 1H, 12.5, 5.0 Hz), 1.55 (d, 1H, 13.6 Hz), 1.44–1.35 (m, 1H). 13C NMR (126 MHz, DMF-d7): δ 168.6, 64.7, 60.1, 52.3, 41.1, 34.8, 34.4, 28.1, 26.9. 195Pt NMR (107 MHz, DMF-d7): δ −2271.09. FT-IR νmax (cm−1): (N–H) 3256, (N–H) 3186, (C–H) 2930, (N
 NN) 2109, 1674, 1633, 1552, 1060, 1034, 762. MS (ESI+) (DMSO/MeOH) [M–Cl + DMSO]+: m/z calcd for C11H24ClN6O2PtS: 534.1, found: 534.1. HRMS (ESI+) (DMF/MeOH) [M–Cl + DMF]+: m/z calcd for C12H25ClN7O2Pt: 529.1406, found: 529.1409.
NN) 2109, 1674, 1633, 1552, 1060, 1034, 762. MS (ESI+) (DMSO/MeOH) [M–Cl + DMSO]+: m/z calcd for C11H24ClN6O2PtS: 534.1, found: 534.1. HRMS (ESI+) (DMF/MeOH) [M–Cl + DMF]+: m/z calcd for C12H25ClN7O2Pt: 529.1406, found: 529.1409.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation [CHE2109255 to V. J. D., NSF DGE-2022168 to ASG and KRA]. DMG and POD gratefully acknowledge funding received from the Synthesis and Solid State Pharmaceutical Centre (SSPC), financed by a research grant from Science Foundation Ireland (SFI) and co-funded under the European Regional Development Fund under [grant number 12/RC/2275_P2]. This work is also supported by the Department of Chemistry and Biochemistry and the Material Science Institute at the University of Oregon. We gratefully acknowledge Dr Brendan Twamley, (School of Chemistry, Trinity College Dublin, University of Dublin, Dublin 2, Ireland) for determining the X-ray crystal structures described within.Notes and references
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS PubMed.
- G. B. Kauffman, Platinum Met. Rev., 2010, 54, 250–256 CrossRef CAS.
- R. K. Mehmood, Oncol. Rev., 2014, 8, 256 Search PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- S. Rottenberg, C. Disler and P. Perego, Nat. Rev. Cancer, 2021, 21, 37–50 CrossRef CAS PubMed.
- F. Biommaert, H. C. M. van Dijk-Knijnenburg, F. J. Dijit, L. den Engelse, R. A. Baan, F. Berends and A. M. J. Fichtinger-Schepman, Biochem., 1995, 34, 8474–8480 CrossRef PubMed.
- C. E. McDevitt, M. V. Yglesias, A. M. Mroz, E. C. Sutton, M. C. Yang, C. H. Hendon and V. J. DeRose, J. Inorg. Chem., 2019, 24, 899–908 CAS.
- R. J. Mitchell, S. M. Kriger, A. D. Fenton, D. Havrylyuk, A. Pandeya, Y. Sun, T. Smith, J. E. DeRouchey, J. M. Unrine, V. Oza, J. S. Blackburn, Y. Wei, D. K. Heidary and E. C. Glazer, RSC Chem. Biol., 2023, 4, 344–353 RSC.
- K. Burger, B. Muhl, T. Harasim, M. Rohrmoser, A. Malamoussi, M. Orban, M. Kellner, A. Gruber-Ebert, E. Kremmer, M. Holzel and D. Eick, J. Biol. Chem., 2010, 285, 12416–12425 CrossRef CAS PubMed.
- P. M. Bruno, Y. Liu, G. Y. Park, J. Murai, C. E. Koch, T. J. Eisen, J. R. Pritchard, Y. Pommier, S. J. Lippard and M. T. Hemann, Nat. Med., 2017, 23, 461–471 CrossRef CAS PubMed.
- E. C. Sutton, C. E. McDevitt, J. Y. Prochnau, M. V. Yglesias, A. M. Mroz, M. C. Yang, R. M. Cunningham, C. H. Hendon and V. J. DeRose, J. Am. Chem. Soc., 2019, 141, 18411–18415 CrossRef CAS PubMed.
- E. C. Sutton and V. J. DeRose, J. Biol. Chem., 2021, 296, 100633, DOI:10.1016/j.jbc.2021.100633.
- H. C. Pigg, M. V. Yglesias, E. C. Sutton, C. E. McDevitt, M. Shaw and V. J. DeRose, ACS Chem. Biol., 2022, 17, 2262–2271 CrossRef CAS PubMed.
- C. E. McDevitt, A. S. Guerrero, H. M. Smith and V. J. DeRose, ChemBioChem, 2022, 23, e202200130, DOI:10.1002/cbic.202200130.
- R. M. Cunningham and V. J. DeRose, ACS Chem. Biol., 2017, 12, 2737–2745 CrossRef CAS PubMed.
- R. Wirth, J. D. White, A. D. Moghaddam, A. L. Ginzburg, L. N. Zakharov, M. M. Haley and V. J. DeRose, J. Am. Chem. Soc., 2015, 137, 15169–15175 CrossRef CAS PubMed.
- J. D. White, M. F. Osborn, A. D. Moghaddam, L. E. Guzman, M. M. Haley and V. J. DeRose, J. Am. Chem. Soc., 2013, 135, 11680–11683 CrossRef CAS PubMed.
- A. D. Moghaddam, J. D. White, R. M. Cunningham, A. N. Loes, M. M. Haley and V. J. DeRose, Dalton Trans., 2015, 44, 3536–3539 RSC.
- J. D. White, L. E. Guzman, L. N. Zakharov, M. M. Haley and V. J. DeRose, Angew. Chem., Int. Ed., 2015, 54, 1032–1035 CrossRef CAS PubMed.
- A. Moretton, J. Slyskova, M. E. Simaan, E. A. Arasa-Verge, M. Meyenberg, D. A. Cerron-Infantes, M. M. Unterlass and J. I. Loizou, Front. Oncol., 2022, 12, 874201 CrossRef CAS PubMed.
- N. Farrer and D. M. Griffith, Curr. Opin. Chem. Biol., 2020, 55, 59–68 CrossRef CAS PubMed.
- C. Vanasschen, E. Molnár, G. Tircsó, F. K. Kálmán, É. Tóth, M. Brandt, H. H. Coenen and B. Neumaier, Inorg. Chem., 2017, 56, 7746–7760 CrossRef CAS PubMed.
- M. S. Galanski, A. Yasemi, S. Slaby, M. A. Jakupec, V. B. Arion, M. Rausch, A. A. Nazarov and B. K. Keppler, Eur. J. Med. Chem., 2004, 39, 707–714 CrossRef CAS PubMed.
- J. M. Eeftens, J. V. Torre and D. R. Burnham, BMC Biophys., 2015, 8, 9 CrossRef PubMed.
- K. Yang, J. Yang and J. Yi, Cell Stress, 2018, 2, 125–140 CrossRef PubMed.
- P. Papadia, A. Barbanente, N. Ditaranto, J. D. Hoeschele, G. Natile, C. Marzano, V. Gandin and N. Margiotta, Dalton Trans., 2021, 50, 15655–15668 RSC.
- D. L. J. Lafontaine, J. A. Riback, R. Bascetin and C. P. Brangwynne, Nat. Rev. Mol. Cell Biol., 2021, 22, 165–182 CrossRef CAS PubMed.
- M. van Sluis and B. McStay, Curr. Opin. Cell Biol., 2017, 46, 81–86 CrossRef CAS PubMed.
- A. IKoike, H. Nishikawa, W. Wu, Y. Okada, A. R. Venkitaraman and T. Ohta, Cancer Res., 2010, 70, 6746–6756 CrossRef PubMed.
- M. Podhorecka, A. Skladanowski and P. Bozko, J. Nucleic Acids, 2010, 920161 Search PubMed.
- O. Surova and B. Zhivotovsky, Oncogene, 2013, 32, 3789–3797 CrossRef CAS PubMed.
- S. P. Jackson and J. Bartek, Nature, 2009, 461, 1071–1078 CrossRef CAS PubMed.
- P. Carotenuto, A. Pecoraro, G. Palma, G. Russo and A. Russo, Cells, 2019, 8, 1090 CrossRef CAS PubMed.
- A. A. Legin, A. Schintlmeister, N. S. Sommerfeld, M. Eckhard, S. Theiner, S. Reipert, D. Strohhofer, M. A. Jakupec, M. S. Galanski, M. Wagner and B. K. Keppler, Nanoscale Adv., 2021, 3, 249–262 RSC.
- H. B. Schmidt, Z. A. Jaafar, B. E. Wulff, J. J. Rodencal, K. Hong, M. O. Aziz-Zanjani, P. K. Jackson, M. D. Leonetti, S. J. Dixon, R. Rohatgi and O. Brandman, Cell Rep., 2022, 41, 111629 CrossRef CAS PubMed.
- M. Nechey, D. Wang and R. E. Kleiner, Cell Chem. Biol., 2023, 30, 1–14 CrossRef PubMed.
- I. Schindelin, E. Arganda-Carreras, V. Frise, M. Kaynig, S. Longair, T. Pietzsch, C. Preibisch, S. Rueden, B. Saalfeld, J. Schmid, D. Tinevez, V. White, K. Hartenstein, P. Eliceiri, A. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef PubMed.
- C. T. Rueden, J. Schindelin, M. C. Hiner, B. E. DeZonia, A. E. Walter, E. T. Arena and K. W. Eliceiri, BMC Bioinf., 2017, 18, 529 CrossRef PubMed.
- S. Van der Walt, J. L. Schonberger, J. Nunez-Iglesias, F. Boulogne, J. D. Warner, N. Yager, E. Gouilart, T. Yu and the scikit-image contributors, PeerJ, 2014, 2, e453 CrossRef PubMed.
- A. E. Carpenter, T. R. Jones, M. R. Lamprecht, C. Clarke, I. H. Kang, O. Friman, D. A. Guertin, J. H. Chang, R. A. Lindquist, J. Moffat, P. Golland and D. M. Sabatini, Genome Biol., 2006, 7, R100 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2256792 and 2256793. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cb00055a |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2023 |