
Gigantic supramolecular assemblies built from dynamic hierarchical organization between inorganic nanospheres and porphyrins†
Kirill
Grzhegorzhevskii
 *a,
Mohamed
Haouas
b,
Maxence
Lion
b,
Arthur
Vashurin
c,
Andrey
Denikaev
a,
Yuriy
Marfin
c,
Grigoriy
Kim
ad,
Clément
Falaise
b and
Emmanuel
Cadot
*b
*a,
Mohamed
Haouas
b,
Maxence
Lion
b,
Arthur
Vashurin
c,
Andrey
Denikaev
a,
Yuriy
Marfin
c,
Grigoriy
Kim
ad,
Clément
Falaise
b and
Emmanuel
Cadot
*b
aUral Federal University, Institute of Natural Sciences and Mathematics, 19 Mira St., Ekaterinburg, Russia. E-mail: kirillvalentinovich@urfu.ru
bInstitut Lavoisier de Versailles CNRS, UVSQ, Université Paris-Saclay, Versailles, France
cIvanovo State University of Chemistry and Technology, Sheremetevsky str., 7, Ivanovo 153000, Russia
dInstitute of Organic Synthesis Ural Branch of the Russian Academy of Sciences, 22 Akademicheskaya St., Ekaterinburg 620990, Russia
First published on 28th November 2022
Abstract
Noncovalent ionic interactions between nanosized Keplerate-type capsules {Mo132} and tetra-cationic porphyrins have been investigated in aqueous solution using small-angle X-ray scattering, 1H NMR and photophysical methods. These complementary multiscale methods reveal the formation of large hybrid oligomers built from a short-range organization in which the cationic porphyrin is glued onto the large POM surface. The local structuring appears to be strongly dependent on the dye![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :{Mo132} ratio changing the morphology of the oligomers from linear to dense aggregates.
:{Mo132} ratio changing the morphology of the oligomers from linear to dense aggregates.
The porphyrin core represents a key part of many cofactors (e.g. vitamin B12) that are essential for living organisms.1,2 This class of dyes exhibits striking catalytic and photophysical properties, making them highly appealing components to design multi-functional systems. In context, the association of porphyrin derivatives with inorganic metal-oxo clusters such as polyoxometalates (POMs) has been used to develop catalytic and photovoltaic systems.3–5 Although covalent attachment represents a popular strategy for designing such hybrid systems,2,6 this approach has major drawbacks such as being time-consuming or the poor hydrolytic stability of covalent bonds. In contrast, the supramolecular approach could represent a straightforward synthetic route, offering both versatile and self-repairing systems, however the structures and the dynamics of such supramolecular aggregates remain largely poorly characterized or unknown.7,8
The Keplerate-type capsule [MoVI72MoV60O372(CH3COO)30 (H2O)72]42−, notated hereafter as {Mo132}, is an anionic hollow sphere of 3 nm in diameter (see Fig. 1) exhibiting unique supramolecular behaviour in aqueous solution. For instance, this giant POM has been identified as able to (i) recognize alkylammonium cations,9 (ii) form large “blackberry” structures (20–1000 nm in size) via non-bonding interactions,10–12 or (iii) encapsulate a wide variety of guest molecules.13–16 Recently, it was observed that {Mo132} is able to act as an efficient catalyst for hydrogen evolution when associated with porphyrin derivatives as photosensitizers.17 However, no structural information at the molecular or macromolecular level is available for such a hybrid assembly that remains essential to better understand the reaction mechanism of the photocatalytic tandem.
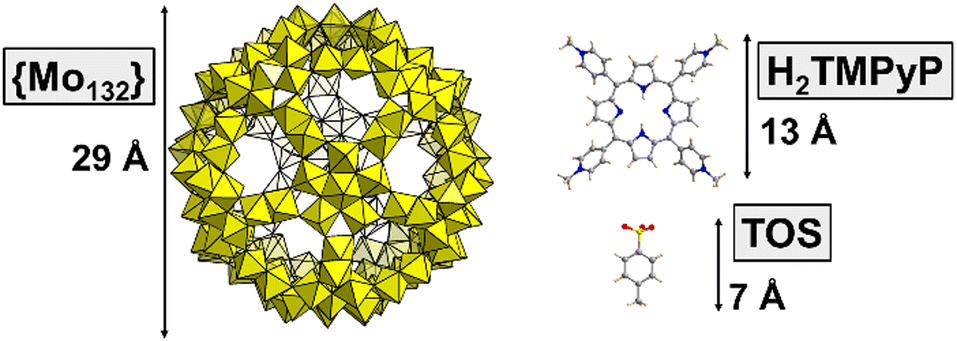 | ||
| Fig. 1 Structural representations of the molecular components involved in the formation of gigantic supramolecular assemblies. The Keplerate-type POM [Mo132O372(CH3COO)30(H2O)72]42− (or {Mo132}) is a nanosized inorganic hollow sphere. The tetra-cationic porphyrin C44H38N84+ (or H2TMPyP) is a planar dye that comprises a porphyrin core and four N-methylpyridinium side groups. This cationic dye was associated with the tosylate anion C7H7O3S− (or TOS) as a counterion. | ||
Herein, we report multiscale investigations of the supramolecular interactions between the giant polyanion {Mo132} and the tetra-cationic porphyrin 5,10,15,20-tetrakis(4-methylpyridyl)porphyrin (see Fig. 1), denoted H2TMPyP, introduced as a tosylate salt (TOS).
At first, small-angle X-ray scattering (SAXS) has been used to characterize the 0.1 mM aqueous solutions of {Mo132} containing different amounts of porphyrin (from 0.5 to 40 eq.). It is worth mentioning that SAXS has already been used successfully for studying large POMs in solution.18–20 In the absence of dye, the SAXS curve exhibits three well-defined oscillations (Fig. 2) similar to that calculated for discrete {Mo132} (Fig. S1, ESI†). The Rg value determined by using a Guinier law supported by the PDDF analysis is fully consistent with the expected values for dispersed Keplerate-type anions in aqueous solution (see Fig. S2 and S3, ESI†). In the presence of increasing amounts of H2TMPyP, no or very weak modifications are observed at high q values (>0.1 Å−1) demonstrating that the Keplerate ions remain intact in such conditions. However, an abrupt increase of the scattering intensity is observed at low q values consistent with the presence of large aggregates, the fractal dimensions of which can be anticipated from the negative slope of the scattering intensity (Fig. 2A). The SAXS curves obtained for 0.5 and 1 eq. of H2TMPyP exhibit negative slope of −0.9 and −1.2, respectively, consistent with the presence of quasi-linear aggregates built from the electrostatically-induced oligomerization involving POMs and the planar organic tetra-cations. Increasing the dye content further leads to a structural reorganization evidenced by the stronger power law dependence observed for a POM:H2TMPyP ratio larger than 2 eq. (slope = −1.7). Such a structural change could result from the formation of branched or cross-linked polymeric hybrid aggregates. For ratios larger than 5, the scattering curve shows two linear regimes (Fig. 2B) highlighted by slopes of −1.6 and −3.1 for the q ranges 0.01–0.04 and 0.05–0.15 Å−1, respectively. This shape suggests that the dye–{Mo132} aqueous mixture contains branched polymeric aggregates built from dense Keplerate-based assemblies with a diameter varying from 7 to 12 nm (Fig. S5, ESI†). For high contents of porphyrin e.g. 40 eq. the SAXS curves exhibit a similar signature to those observed for 5 eq. except for the first oscillation which becomes significantly broader and significantly shifted from 0.25 to 0.22 Å−1. Such a SAXS profile shows that the long-range organization of the hybrid aggregates is fully maintained while the short-range structural modifications occur in close vicinity of the Keplerate. To provide information about the interactions occurring at the molecular level between H2TMPyP and {Mo132}, first, a 1H NMR titration of 0.25 mM porphyrin D2O solution has been performed. Increasing the POM content from POM:dye ratio 1:100 up to 1:20 resulted in a progressive decrease in the 1H NMR signal intensities of the aromatic protons (9.3–8.8 ppm) of the porphyrin (Fig. S7, ESI†). Although quite unaffected for ratios less than 1:40, their linewidth suddenly becomes very broad at a 1:20 ratio before finally disappearing at 1:10 and larger ratios (Fig. 3). Interestingly, the signals of the benzene core in the TOS anion (7.06 and 7.46 ppm) behave differently, showing a significant gradual broadening until a POM:H2TMPyP ratio of 1:20. However, the TOS signals recover in intensity and narrow again as doublets for ratios larger than 1:10. At the same time, these protons have undergone significant deshielding (7.30 and 7.62 ppm). This means that the TOS anions are involved in a fast chemical exchange regime within the large hybrid aggregates and this process is progressively slowing down with the formation of the supramolecular assembly until POM:H2TMPyP = 1:20. For larger POM contents, the TOS anions remain mostly isolated as solvated ions as shown by the reversed NMR titration of 0.1875 mM {Mo132} solution for which the signals of TOS preserve the fine structure (Fig. 3). We also found that the NMR signal of the acetate ligands inside the Keplerate cavity at 0.77 ppm disappears gradually when the ratio POM:dye increases (Fig. S6, ESI†), due to the very low diffusion coefficient of the Keplerate ions embedded within the large supramolecular structures. Analysis of these results leads to the following statements: (i) H2TMPyP species interacting with Keplerate anions are NMR silent, whereas (ii) TOS anions are also involved in the large supramolecular hybrid aggregates as ion-pairing arrangements with H2TMPyP in the molar ratio range 1:100–1:20. In contrast, with increasing the POM content (for larger ratio than 10), the TOS anions are not involved in the supramolecular structures that are built exclusively from the ionic interaction between the POM and H2TMPyP. In such a scenario, the TOS anions were released as isolated and solvated species. In line with the 42-ionic charge of {Mo132} and the four positive charges of the H2TMPyP cation, the 1:10 ratio nearly corresponds to the stoichiometry of the electric balance. Then, the number of porphyrins interacting per Keplerate ion have also been determined by integrating the NMR data into a Langmuir model (Fig. S7, ESI†), which is consistent with a maximal capacity of about 18–19 dyes per POM. This high number suggests the formation of a double porphyrin shell around the POM capsule where the first layer involves direct contacts between the POM and dyes and the second shell should consist of porphyrins interacting with first layer through intercalated TOS anions through π–π stacking. Furthermore, the self-assembly process between the {Mo132} and H2TMPyP has also been investigated by photophysical methods, namely UV-vis spectroscopy, steady-state photoluminescence (PL) and photoluminescence excitation (PLE). Such techniques require diluted systems due to the high molar absorptivity of the components. UV-vis spectra (Fig. S8 and S9, ESI†) showed only a weak red shift of the B band (Soret) and Q-bands of the porphyrin in the presence of the POM. In addition, the fluorescence properties of pure H2TMPyP are related to the monomer and the excimer emission or to the equilibrium between the free base form and the partially deprotonated one.21 Within the 5–500 μM range, the PL spectra remain nearly unchanged (Fig. S10, ESI†). However, the Soret band diminishes in the PLE spectra (Fig. S11, ESI†) as a consequence of FRET and/or π-stacking with the TOS anions22 accompanied with PICT.23 For the molar ratio of POM:porphyrin = 1:1, the PL quenching is incomparably stronger than in the pure porphyrin solution showing a 320-fold decreased PL intensity at 711 nm. Such an effect is consistent with a charge transfer (CT) process according to the HOMO/LUMO positions at −5.9/−4.16 and −5.8/−4.19 eV for porphyrin24 and Keplerate,25 respectively. Interestingly, the CT-process was also observed26 in another macrocyclic oligopyrrole dye covalently bound to a POM {P2V3W15O62}9−.
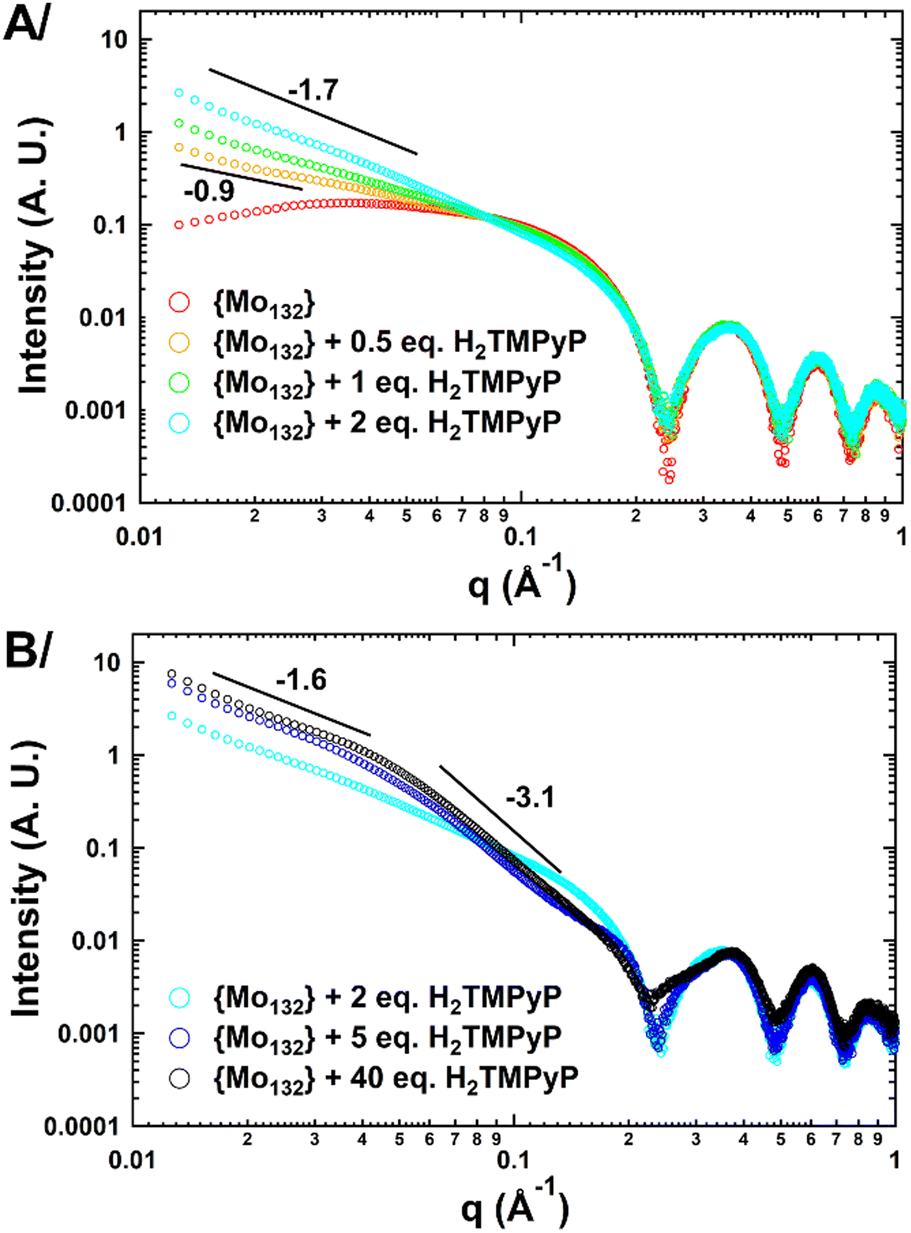 | ||
| Fig. 2 The log(q)–log(I(q)) scattering curves of aqueous solutions containing 0.1 mM {Mo132} and various amounts of H2TMPyP (A: from 0 to 2 eq.; B: from 2 to 40 eq.). | ||
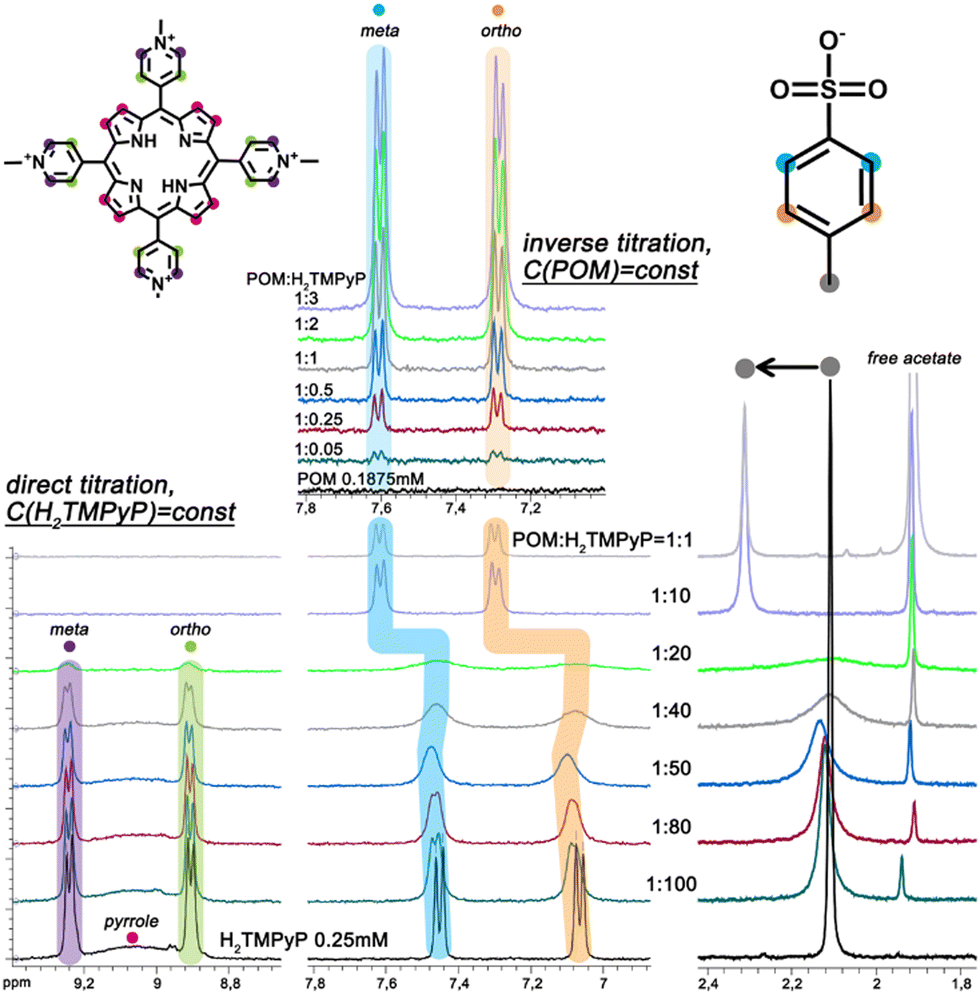 | ||
| Fig. 3
1H NMR spectra (in D2O) for different molar ratios of POM:porphyrin at a constant H2TMPyP concentration of 0.25 mM (direct titration) and at a constant {Mo132} concentration of 0.1875 mM (inverse titration). | ||
Generally, the PL spectrum loses the second and fourth components (Fig. 4, insert) and the band at 711 nm shifts to 723 nm. In the PLE spectrum the contribution of the Q1x band near 585 nm increases significantly (even after correction of POM absorption). As only free porphyrins contribute to the PL and PLE spectra, such a behavior can be simply attributed to concentration effect. In addition, the intensity sum of the emission bands at 664 and near 722 nm plotted against the POM:dye ratio is consistent with an average stoichiometry of four porphyrins per one Keplerate (Fig. 4, insert). Then, the four positive charges localized at the periphery of the porphyrin (accessible on both sides of the molecular plane), could be shared by two adjacent POMs as a ditopic linker able to associate two building blocks. Finally, the thermodynamic parameters featuring the {Mo132}–H2TMPyP interactions have been determined from Stern–Volmer titration, using the full equation for a 1:1 molar ratio after correction of the inner filter effect27 (see ESI†). The Stern–Volmer plot does not lead to a linear function (Fig. S13, ESI†) indicating complex intermolecular interactions for this system. The static quenching mechanism in the {Mo132}–H2TMPyP pair is confirmed (see Fig. S14, ESI†) since the lifetimes of the excited state are preserved: 5.4 ± 0.2 and 5.1 ± 0.2 ns for H2TMPyP and the aggregate obtained with one H2TMPyP per {Mo132}, respectively. In addition, the binding constant (KSV = 21 × 109) and related free Gibbs energy 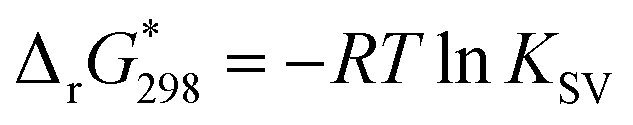
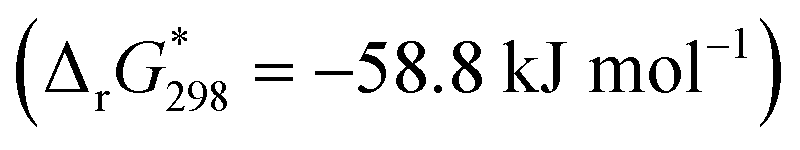 are consistent with very strong attractive interactions between the Keplerate and porphyrin ionic species. These solution studies have revealed that strong coulombic interactions between H2TMPyP and {Mo132} are probably supported by solvent effects that rely on the hydrophobic nature of the ionic species and give rise to the formation of gigantic supramolecular oligomers in which both short- and long-range organizations are controlled by the H2TMPyP:{Mo132} ratio (Fig. 5). In short, for low H2TMPyP content (1–2 eq. per POM), the porphyrins behave as ditopic linkers producing either rod-like supramolecular oligomers or branched aggregates. For a H2TMPyP content larger than 5 eq., a dense supramolecular hybrid arises from self-rearrangements into closely packed {Mo132}-porphyrin species within the oligomers. For high porphyrin concentrations (>10 eq.), the long-range structural organization remains mostly unchanged while important modifications of the molecular interactions between the primary building blocks are depicted. In such conditions, the Keplerate surface should be decorated by about 18–19 dyes that intercalate the TOS anions as guests into a double shell structure.
are consistent with very strong attractive interactions between the Keplerate and porphyrin ionic species. These solution studies have revealed that strong coulombic interactions between H2TMPyP and {Mo132} are probably supported by solvent effects that rely on the hydrophobic nature of the ionic species and give rise to the formation of gigantic supramolecular oligomers in which both short- and long-range organizations are controlled by the H2TMPyP:{Mo132} ratio (Fig. 5). In short, for low H2TMPyP content (1–2 eq. per POM), the porphyrins behave as ditopic linkers producing either rod-like supramolecular oligomers or branched aggregates. For a H2TMPyP content larger than 5 eq., a dense supramolecular hybrid arises from self-rearrangements into closely packed {Mo132}-porphyrin species within the oligomers. For high porphyrin concentrations (>10 eq.), the long-range structural organization remains mostly unchanged while important modifications of the molecular interactions between the primary building blocks are depicted. In such conditions, the Keplerate surface should be decorated by about 18–19 dyes that intercalate the TOS anions as guests into a double shell structure.
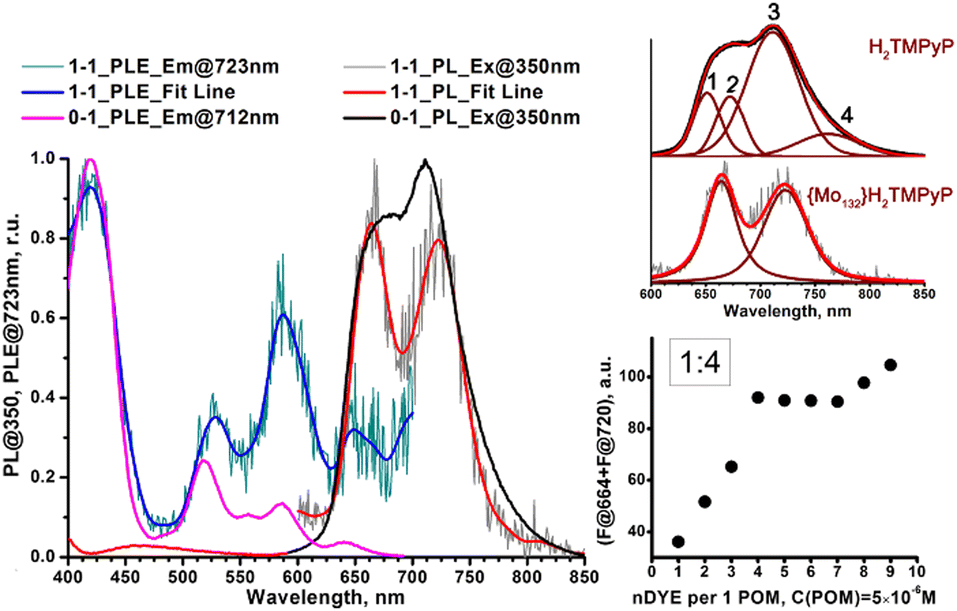 | ||
| Fig. 4 Normalized PL (excited at 350 nm) and PLE (emission at 712–723 nm) spectra of a pure porphyrin aqueous solution at 5 μM (0–1 sample) and associated {Mo132}(H2TMPyP)n at C(porphyrin) = 5 μM and 1:1 molar ratio (1–1 sample). Inserts: Black spots – sum of fluorescence peak intensity of H2TMPyP at 664 and 720 nm plotted against dye:POM molar ratio; red lines – deconvolution (with Voight function) of PL spectra (excited at 350 nm) of pure H2TMPyP and the supramolecular hybrid aggregate at a 1:1 molar ratio. | ||
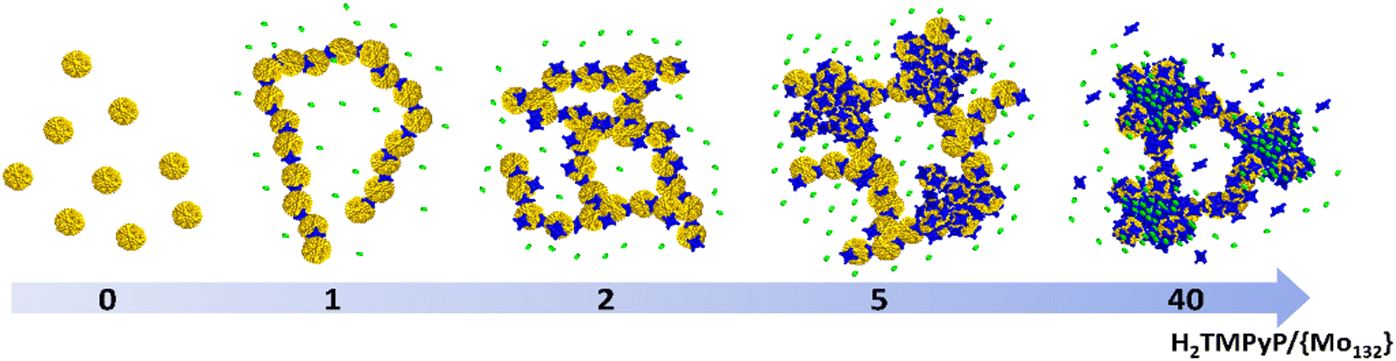 | ||
| Fig. 5 Schematic representation of hybrid supramolecular aggregates built from Keplerate (yellow), H2TMPyP (blue) and TOS (green). As the H2TMPyP:POM ratio (noted R) increases, the POM-porphyrin system gives rise to quasi-linear oligomers (R = 1), which then self-arrange into branched-type oligomers (R = 2). For 5 < R < 10, the hybrid assembly becomes denser giving rise to close-packed aggregates, while for R > 10, these packed aggregates intercalate tosylate anions that are strongly immobilized within a porphyrin-based multi-shell around the inorganic capsules. | ||
Aging the {Mo132}–H2TMPyP mixtures leads to flocculation, the rates of which vary from minutes to weeks depending on the concentration and on the POM:dye ratio. Typically, the flocculation process is faster for a POM:dye ratio close to 10, corresponding roughly to the electric balance. Infrared and Raman analysis showed that dry flocs are mostly composed of Keplerate and dye (Fig. S15–S17, ESI†). Interestingly, the Raman spectra revealed that the normal vibration modes of porphyrin shift to higher energy on the 5–12 or 1–3 cm−1 level (except for δ(pyr): 1642 → 1639 cm−1 (A1g)). Such a trend is consistent with an increase in the rigidity of the porphyrin, probably due to planar interactions with the rigid POM.
In summary, very strong associations between a Keplerate-type capsule and H2TMPyP give rise to efficient charge transfer while the structure of the resulting supramolecular oligomers can be monitored via composition of the three-component system POM/porphyrin/tosylate. This is the way for smart-design of novel hybrid materials based on porphyrins.
This work was supported by the Russian Science Foundation: Project No. 21-73-00311 for the synthesis and sample characterizations, and a public grant overseen by the French National Research Agency as part of the “Investissements d’Avenir” program (Labex Charm3at, ANR-11-LABX-0039-grant). We acknowledge SOLEIL and Dr J. Perez for synchrotron facilities and assistance in using beamline SWING.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Sugiyasu, S. Ogi and M. Takeuchi, Polym. J., 2014, 46, 674–681 CrossRef CAS.
- R. Lamare, R. Ruppert, C. Boudon, L. Ruhlmann and J. Weiss, Chem. – Eur. J., 2021, 27, 16071–16081 CrossRef CAS PubMed.
- C. Zou, Z. Zhang, X. Xu, Q. Gong, J. Li and C. Wu, J. Am. Chem. Soc., 2012, 134, 87–90 CrossRef CAS PubMed.
- Z. Huo, A. Bonnefont, Y. Liang, R. Farha, M. Goldmann, E. Saint-Aman, H. Xu, C. Bucher and L. Ruhlmann, Electrochim. Acta, 2018, 274, 177–191 CrossRef CAS.
- G. Paille, M. Gomez-Mingot, C. Roch-Marchal, B. Lassalle-Kaiser, P. Mialane, M. Fontecave, C. Mellot-Draznieks and A. Dolbecq, J. Am. Chem. Soc., 2018, 140, 3613–3618 CrossRef CAS PubMed.
- Y. Zhu, Y. Huang, Q. Li, D. Zang, J. Gu, Y. Tang and Y. Wei, Inorg. Chem., 2020, 59, 2575–2583 CrossRef CAS PubMed.
- A. Tsuda, E. Hirahara, Y. S. Kim, H. Tanaka, T. Kawai and T. Aida, Angew. Chem., Int. Ed., 2004, 43, 6327–6331 CrossRef CAS.
- A. Yokoyama, T. Kojima and S. Fukuzumi, Dalton Trans., 2011, 40, 6445–6450 RSC.
- N. Watfa, D. Melgar, M. Haouas, F. Taulelle, A. Hijazi, D. Naoufal, J. B. Avalos, S. Floquet, C. Bo and E. Cadot, J. Am. Chem. Soc., 2015, 137, 5845–5851 CrossRef CAS PubMed.
- P. P. Mishra, J. Pigga and T. Liu, J. Am. Chem. Soc., 2008, 130, 1548–1549 CrossRef CAS PubMed.
- J. Zhang, D. Li, G. Liu, K. J. Glover and T. Liu, J. Am. Chem. Soc., 2009, 131, 15152–15159 CrossRef CAS.
- F. Haso, D. Li, S. Garai, J. M. Pigga and T. Liu, Chem. – Eur. J., 2015, 21, 13234–13239 CrossRef CAS PubMed.
- A. Ziv, A. Grego, S. Kopilevich, L. Zeiri, P. Miro, C. Bo, A. Müller and I. A. Weinstock, J. Am. Chem. Soc., 2009, 131, 6380–6382 CrossRef CAS PubMed.
- R. Carr, I. A. Weinstock, A. Sivaprasadarao, A. Müller and A. Aksimentiev, Nano Lett., 2008, 8, 3916–3921 CrossRef CAS PubMed.
- R. W. Pow, W. Xuan, D.-L. Long, N. L. Bell and L. Cronin, Chem. Sci., 2020, 11, 2388–2393 RSC.
- S. Chakraborty, A. Shnaiderman Grego, S. Garai, M. Baranov, A. Müller and I. A. Weinstock, J. Am. Chem. Soc., 2019, 141, 9170–9174 CrossRef.
- A. Panagiotopoulos, A. M. Douvas, P. Argitis and A. G. Coutsolelos, ChemSusChem, 2016, 9, 3213–3219 CrossRef CAS PubMed.
- D. Sures, M. Segado, C. Bo and M. Nyman, J. Am. Chem. Soc., 2018, 140, 10803–10813 CrossRef CAS PubMed.
- C. Falaise, S. Khlifi, P. Bauduin, P. Schmid, W. Shepard, A. A. Ivanov, M. N. Sokolov, M. A. Shestopalov, P. A. Abramov, S. Cordier, J. Marrot, M. Haouas and E. Cadot, Angew. Chem., Int. Ed., 2021, 60, 14146–14153 CrossRef CAS PubMed.
- O. Renier, C. Falaise, H. Neal, K. Kozma and M. Nyman, Angew. Chem., Int. Ed., 2016, 128, 13678–13682 CrossRef.
- K. Kalyanasundaram, Inorg. Chem., 1984, 23, 2453–2459 CrossRef CAS.
- M. Makarska-Bialokoz, J. Mol. Struct., 2015, 1081, 224–232 CrossRef CAS.
- C. A. Guido, B. Mennucci, D. Jacquemin and C. Adamo, Phys. Chem. Chem. Phys., 2010, 12, 8016 RSC.
- K. Kalyanasundaram and M. Neumann-Spallart, J. Phys. Chem., 1982, 86, 5163–5169 CrossRef CAS.
- V. Fazylova, N. Shevtsev, S. Mikhailov, G. Kim, A. Ostroushko and K. Grzhegorzhevskii, Chem. – Eur. J., 2020, 26, 5685–5693 CrossRef CAS PubMed.
- R. Pütt, P. Kozłowski, I. Werner, J. Griebel, S. Schmitz, J. Warneke and K. Y. Monakhov, Inorg. Chem., 2021, 60(1), 80–86 CrossRef.
- M. van de Weert and L. Stella, J. Mol. Struct., 2011, 998, 144–150 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc05193a |
| This journal is © The Royal Society of Chemistry 2023 |