
Palladium-catalyzed B7–11 penta-arylation of the {CB11} monocarborane cluster†
Yujie
Jin
a,
Jizeng
Sun
a,
Kang
Zhang‡
a,
Jiyong
Liu
a,
Michael
Wörle
b and
Simon
Duttwyler
 *a
*a
aDepartment of Chemistry, Zhejiang University, 38 Zheda Road, Hangzhou 310027, China. E-mail: duttwyler@zju.edu.cn
bDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1, Zürich 8093, Switzerland
First published on 28th November 2022
Abstract
Regioselective, five-fold B–H activation of the monocarborane cluster [CB11H12]− at the positions B7–11 has been accomplished. Selective substitution of these positions by B–H activation has not been reported before. Our protocol involves directing group assistance by the carboxylic acid functionality and is based on palladium catalysis using iodoarene coupling partners. Penta-arylated products are obtained in a single step with yields ranging from 42% to 89% and with good functional group tolerance. X-Ray crystal structures for five new compounds confirm the selective substitution of the lower belt of the monocarborane cage.
The unique steric and electronic features of carboranes have fascinated chemists since their discovery.1 {CB11}- and {C2B10}-based boranes possess high thermodynamic stability and lead to unique inorganic–organic hybrid molecules upon covalent modification of B and/or C vertices. Applications of such compounds have been reported in the fields of coordination chemistry, fluorescence/phosphorescence, materials science and medicinal chemistry.1–5 Monocarboranes, owing to their single negative charge and exceptional inertness, are superior weakly coordinating anions (WCA) and have therefore been used in the isolation of reactive cations and in the catalysis mediated by cationic catalysts.6,7 Furthermore, a range of applications in the areas of ligand design, liquid crystals and energy storage have emerged in the past years.8,9 Several strategies for the B-vertex functionalization of the monocarborane [CB11H12]− (1, Fig. 1a) have been established. Direct electrophilic substitution is used to access halogenated derivatives for WCA applications,6 B-iodo derivatives can undergo Pd-catalyzed cross coupling.10 Furthermore, procedures via “onium zwitterions” have been developed.8 Transition metal-catalyzed B–H activation of 1 has emerged as an alternative synthetic tool and has been a central focus of our group's research.11,12 Its power lies in its step-economy and ability to afford derivatives that are otherwise inaccessible.
 | ||
| Fig. 1 (a) Structure and numbering of the [CB11H12]− cage; (b) selected recent poly-arylation reactions of boron clusters; (c) summary of this work. | ||
B–H activation/arylation of boron cages has primarily been reported for {C2B10} clusters.13 Pioneering work on ortho-dicarbaboranes stems from the groups of Xie and Cao.13q,r In a study from 2021, Yan, Zhang and Shi achieved poly-arylation of ortho-, meta- and para-dicarbaboranes (one selected example is depicted in Fig. 1b); similar results using iron catalysis have appeared very recently.13a,b As far as B–H arylation of {CB11} cages is concerned, we are aware of only three reports. Lavallo found that 1 reacts with in situ-generated trityl cation to give the product {CB11}–(C6H4)–CH(Ph)2 by electrophilic substitution at the B12 position.14 Furthermore, sequential C1/B2 annulation was reported by Uchiyama, and five-fold B2–6 arylation was developed in our group (Fig. 1b).12d,15
Selective catalytic B–H functionalization of the lower belt (B7–11) of 1 is an unsolved challenge. Weller reported on the poly-ethylation of 1 by ethenylation–hydrogenation cycles starting from (PPh3)2Rh[CB11H12], ultimately yielding a product with two ethyl groups on the upper belt, two ethyl groups on the lower belt and a B12-ethyl group.16 Installing a C1-methyl or C1-silyl group resulted in lower degrees of substitution, such as [1-(i-Pr3Si)-CB11H8-7,9,12-Et3]−. All of these reactions required a full equivalent of rhodium and could not be rendered catalytic.
Here we describe for the first time catalytic lower-belt B–H activation/functionalization of the {CB11} cage. The transformation is enabled by B12-COOH directing group assistance and occurs with iodoarene coupling partners under palladium catalysis. It affords penta-arylated products in moderate to high yields in a single step.
At the outset of our studies, we evaluated conditions for the arylation of [CB11H11-12-COOH]− (2). This starting material was reported earlier17 and contains the simple carboxylic acid directing group, which can act as a functional group handle for further modification. Optimization details are provided in the ESI† (p. S12, Table S1). The degree of substitution and bulk purity after purification could reliably be verified by full-range (–)-ESI-mass spectrometry (m/z = 100–1200) because the products have a single negative charge and exhibit very little fragmentation. Successful coupling was achieved using iodoarene (6 equiv.), 10% Pd(OAc)2, Ag2CO3 (6 equiv.)/NaOAc (5 equiv.) as additives and dimethylformamide as the solvent. At slightly elevated temperatures of 40–60 °C, arylation at the B7–11 positions was reached within 18–48 hours.
Various penta-arylated compounds 3 with electron-donating and electron-withdrawing groups were prepared according to the established conditions (Table 1). They were fully characterized by NMR spectroscopy (1H, 1H{11B}, 11B, 11B{1H}, 13C{1H}) and mass spectrometry. Phenyl, para-alkylaryl and para-haloaryl substitution took place in yields of 54% to 87% (3a–i). Furthermore, the heteroatom-containing groups –CN, –NO2, –CO2Me and –CHO were well tolerated (3j–m). Yields tended to be higher for electron-deficient coupling partners because of faster conversion and cleaner product formation; 3l was isolated in a reduced yield of 54% due to a more difficult chromatographic separation. Electron-rich iodoarenes, such as in the case of 3b–d, required a longer reaction time and higher temperature (48 h/60 °C), resulting in the formation of larger amounts of by-products. Extension of the π system and meta-substitution gave products 3n–s in yields of 42% to 85%. Unsatisfactory conversion was observed for ortho-substituted coupling partners. In the 11B NMR spectra, the resonances of B2–6 and B7–11 were uniformly observed as broad signals at ca. −16 ppm and −4 ppm, respectively. For comparison, the values for 2 are −16.6 ppm and −13.0 ppm. The B12 position experienced a slight shielding from 4.3 ppm in 2 to ca. 6 ppm in the penta-arylated 3.

|
Crystals suitable for X-ray diffraction were obtained for 3a, 3b, 3h and 3q (Fig. 2). In 3a and 3b, two and three rings are almost perfectly aligned with the B12–C1 axis, while the other rings adopt a propeller-like geometry. In 3h and 3q, all rings prefer the propeller-like arrangement. Generally speaking, the penta-aryl substitution imposes no significant strain on the carborane cage, as evidenced by cluster B–B distances similar to those of the parent carborane 1 and starting material 2.
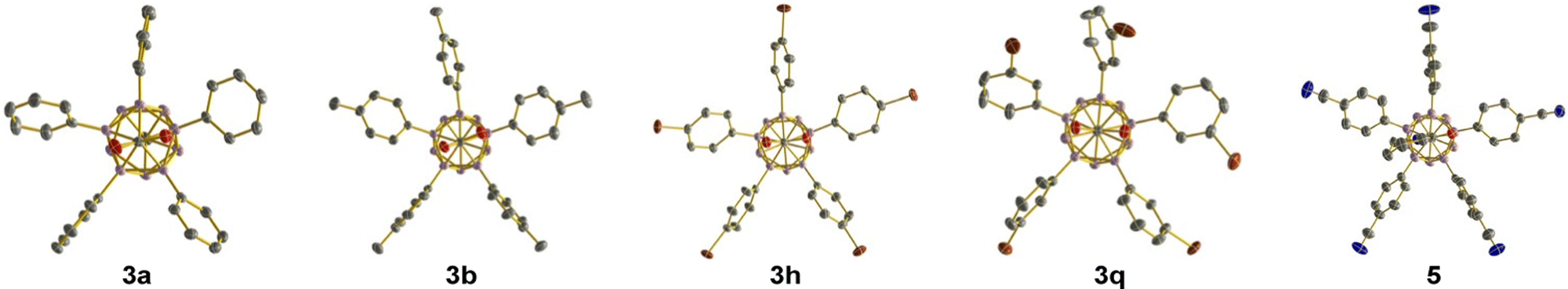 | ||
| Fig. 2 Anionic parts of the X-ray crystal structures of 3a, 3b, 3h, 3q and 5 (50% displacement ellipsoids, H atoms omitted for clarity). For 3b, only one of the four anions in the asymmetric unit is shown. | ||
The utility of the carboxylic acid functionality for synthetic modification was demonstrated by transformation of 3j into ester 4 and amide 5 (Scheme 1a). Treatment of 3j with K2CO3 and iodomethane in dimethylformamide gave the desired methyl ester 4 in almost quantitative yield. Amide formation was achieved in two steps via the acid chloride. Oxalyl chloride in combination with catalytic dimethylformamide was used to activate the carboxylic acid moiety, converting it to the acid chloride. The formation of this intermediate was confirmed by NMR spectroscopy of the crude solid after removal of the volatiles. Subsequent treatment with pyrrolidine and triethylamine as an additional base furnished amide 5 in 84% overall yield. The identity of 5 was further confirmed by X-ray diffraction (Fig. 2). In this structure, two of the rings are aligned perpendicular to the lower belt, while the other three show a propeller-like arrangement. Finally, scalability of the methodology was exemplified by penta-arylation of 1.5 mmol of 2 with para-cyanoiodobenzene (Scheme 1b). 1.02 g of 3j was isolated in 82% yield, identical to that in Table 1. In our previous study with COOH at the C1 position, the directing group could be removed in DMF at elevated temperatures.12d We tested its removability for products 3a and 3j under similar conditions (DMF/100 °C, neutral conditions or with 10 equiv. HOAc or with 10 equiv. KOAc), but obtained unchanged products. The use of Na in THF did not lead to COOH cleavage, either.
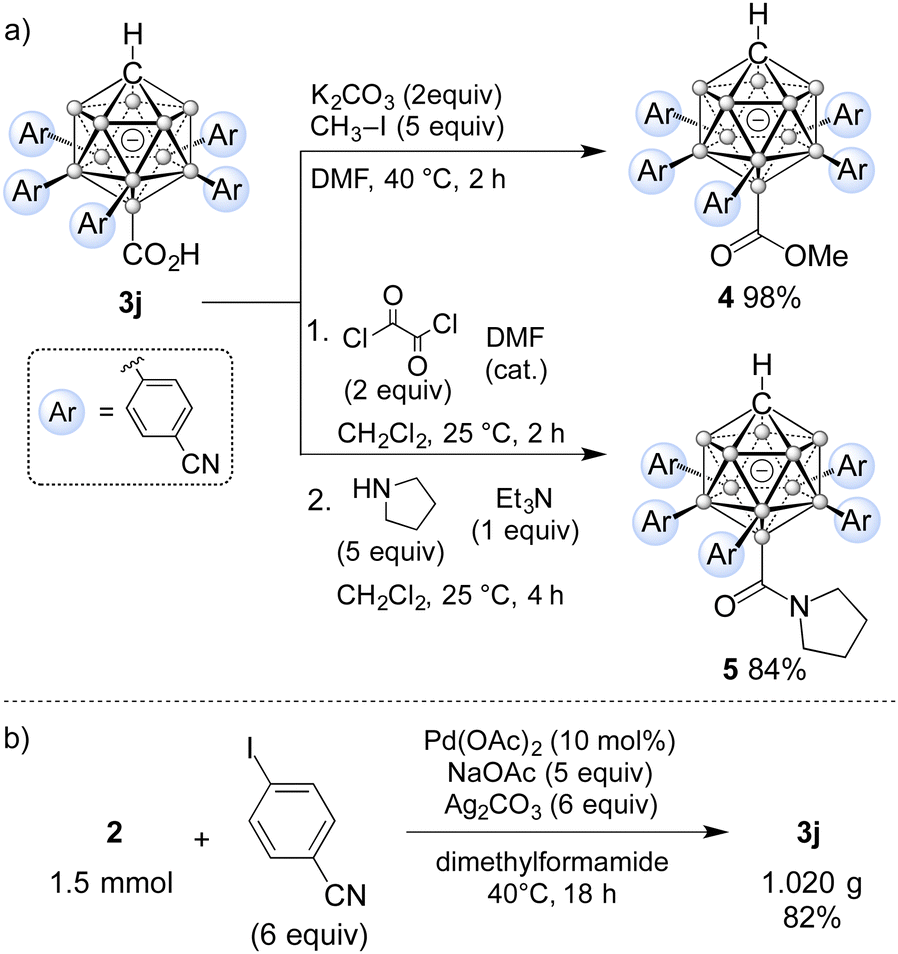 | ||
| Scheme 1 (a) Additional transformations of 3j to give ester 4 and amide 5; (b) gram-scale synthesis of 3j. | ||
In conclusion, we have developed a method to penta-arylate the monocarborane cluster at the B7–11 positions. For the first time, catalytic regioselective B–H activation/B–C coupling of these vertices was achieved, and substrate screening revealed good functional group tolerance. Supplementary versatility of the carboxylic acid moiety was verified by ester and amide formation without affecting the remaining C–H and B–H bonds. Thus, these findings enable the synthesis of a new class of C5-symmetrical monocarborane derivatives.
Y. J. synthesized and characterized the new compounds. J. S. and K. Z. synthesized some of the starting materials. J. L. and M. W. carried out the crystallographic analysis. S. D. designed and supervised the study. S. D. and Y. J. wrote the manuscript.
Financial support by the National Natural Science Foundation of China (No. 21871231) and the Special Funds for Basic Scientific Research of Zhejiang University (No. 2019QNA3010 and K20210335) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Boron Science: New Technologies and Applications, ed. N. S. Hosmane, Taylor & Francis Books/CRC, Boca Raton, FL, 2011 Search PubMed; (b) R. N. Grimes, Carboranes, 3rd edn, Elsevier, London, 2016 Search PubMed; (c) In Handbook of Boron Science: With Applications in Organometallics Catalysis, Materials and Medicine, ed. N. S. Hosmane and R. Eagling, World Scientific Publishing, London, 2018 Search PubMed; (d) Review: C. Douvris and J. Michl, Chem. Rev., 2013, 113, PR179–PR233 CrossRef.
- I. B. Sivaev, M. Y. Stogniy and V. I. Bregadze, Coord. Chem. Rev., 2021, 436, 213795–213889 CrossRef CAS.
- R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef.
- (a) J. Yan, W. Yang and Q. Zhang, Chem. Commun., 2020, 56, 11720–11734 RSC; (b) Materials: R. Fernandez-Alvarez, V. Ďordovič, M. Uchman and P. Matějíček, Langmuir, 2018, 34, 3541–3554 CrossRef CAS; (c) R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC; (d) B. P. Dash, R. Satapathy, J. A. Maguire and N. S. Hosmane, New J. Chem., 2011, 35, 1955–1972 RSC.
- (a) P. Stockmann, M. Gozzi, R. Kuhnert, M. B. Sárosi and E. Hey-Hawkins, Chem. Soc. Rev., 2019, 48, 3497–3512 RSC; (b) In Boron-Based Compounds: Potential and Emerging Applications in Medicine, ed. E. Hey-Hawkins and C. Viñas Teixidor, John Wiley & Sons, Oxford, 2018 Search PubMed; (c) Z. J. Leśnikowski, J. Med. Chem., 2016, 59, 7738–7758 CrossRef; (d) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS; (e) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS.
- (a) I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982–14024 CrossRef CAS; (b) C. Knapp, Weakly Coordinating Anions: Halogenated Borates and Dodecaborates. Comprehensive Inorganic Chemistry II, Elsevier, Amsterdam, 2013, vol. 1, pp. 651–679 Search PubMed; (c) C. A. Reed, Acc. Chem. Res., 2010, 43, 121–128 CrossRef CAS PubMed; (d) C. A. Reed, Acc. Chem. Res., 1998, 31, 133–139 CrossRef CAS.
- Recent selected example: Catalysis: H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2021, 143, 15490–15507 CrossRef CAS.
- (a) For reviews on applications of monocarboranes including the field of energy storage see: S. P. Fisher, A. W. Tomich, S. O. Lovera, J. F. Kleinsasser, J. Guo, M. J. Asay, H. M. Nelson and V. Lavallo, Chem. Rev., 2019, 119, 8262–8290 CrossRef CAS; (b) S. P. Fisher, A. W. Tomich, J. Guo. and V. Lavallo, Chem. Commun., 2019, 55, 1684–1701 RSC.
- (a) R. Jakubowski, A. Pietrzak, A. C. Friedli and P. Kaszyński, Chem. – Eur. J., 2020, 26, 17481–17494 CrossRef CAS; (b) M. O. Ali, J. C. Lasseter, R. Żurawiński, A. Pietrzak, J. Pecyna, J. Wojciechowski, A. C. Friedli, D. Pociecha and P. Kaszyński, Chem. – Eur. J., 2019, 25, 2616–2630 CrossRef CAS PubMed; (c) J. Pecyna, R. Żurawiński, P. Kaszyński, D. Pociecha, P. Zagórski and S. Pakhomov, Eur. J. Inorg. Chem., 2016, 2923–2931 CrossRef CAS; (d) J. Pecyna, P. Kaszyński, B. Ringstrand, D. Pociecha, S. Pakhomov, A. G. Douglass and V. G. Young, Inorg. Chem., 2016, 55, 4016–4025 CrossRef CAS; (e) P. Kaszyński and B. Ringstrand, Angew. Chem., Int. Ed., 2015, 54, 6576–6581 CrossRef.
- (a) R. M. Dziedzic and A. M. Spokoyny, Chem. Commun., 2019, 55, 430–442 RSC; (b) For a recent report on Pd coupling to the {B12} cage, see: M. K. Al-Joumhawy, T. Marei, A. Shmalko, P. Cendoya, J. La Borde and D. Gabel, Chem. Commun., 2021, 57, 10007–10010 RSC.
- For reviews on B–H activation of monocarboranes, see: (a) I. B. Sivaev, Russ. J. Inorg. Chem., 2021, 66, 1289–1342 CrossRef CAS; (b) M. Hamdaoui, R. Varkhedkar, J. Sun, F. Liu and S. Duttwyler, Synthetic Inorganic Chemistry, Elsevier, Amsterdam, 2021, pp. 343–389 Search PubMed; (c) J. Kanazawa, Y. Kitazawa and M. Uchiyama, Chem. – Eur. J., 2019, 25, 9123–9132 CrossRef CAS PubMed; (d) S. Duttwyler, Pure Appl. Chem., 2018, 90, 733–744 CrossRef CAS ; for recent reviews on B–H activation of neutral {C2B10} clusters, see: ; (e) Z. Qiu and Z. Xie, Acc. Chem. Res., 2021, 54(21), 4065–4079 CrossRef CAS PubMed; (f) X. Zhang and H. Yan, Coord. Chem. Rev., 2019, 378, 466–482 CrossRef CAS; (g) Y. Quan and Z. Xie, Chem. Soc. Rev., 2019, 48, 3660–3673 RSC.
- Selected examples of monocarborane B–H activation: (a) R. Varkhedkar, F. Yang, R. Dontha, J. Zhang, J. Liu, B. Spingler, S. van der Veen and S. Duttwyler, ACS Cent. Sci., 2022, 8, 322–331 CrossRef CAS PubMed; (b) Y. Shen, K. Zhang, X. Liang, R. Dontha and S. Duttwyler, Chem. Sci., 2019, 10, 4177–4184 RSC; (c) F. Lin, Y. Shen, Y. Zhang, Y. Sun, J. Liu and S. Duttwyler, Chem. – Eur. J., 2018, 24, 551–555 CrossRef CAS PubMed; (d) F. Lin, J. L. Yu, Y. Shen, S. Q. Zhang, B. Spingler, J. Liu, X. Hong and S. Duttwyler, J. Am. Chem. Soc., 2018, 140, 13798–13807 CrossRef CAS PubMed; (e) Y. Shen, J. Liu, T. Sattasatchuchana, K. K. Baldridge and S. Duttwyler, Eur. J. Inorg. Chem., 2017, 4420–4424 CrossRef CAS; (f) Y. Shen, Y. Pan, J. Liu, T. Sattasathuchana, K. K. Baldridge and S. Duttwyler, Chem. Commun., 2017, 53, 176–179 RSC; (g) Y. Shen, Y. Pan, K. Zhang, X. Liang, J. Liu, B. Spingler and S. Duttwyler, Dalton Trans., 2017, 46, 3135–3140 RSC.
- (a) P. Zhou, Y. Chen and Z. Xie, ACS Catal., 2022, 12, 8761–8767 CrossRef CAS; (b) H. Cao, M. Chen, F. Sun, Y. Zhao, C. Lu, X. Zhang, Z. Shi and H. Yan, ACS Catal., 2021, 11, 14047–14057 CrossRef CAS; (c) Y. Chen, H. Lyu, Y. Quan and Z. Xie, Org. Lett., 2021, 23, 4163–4167 CrossRef CAS PubMed; (d) K. Cao, J. Wu, C. Zhang, L. Ding and J. Yang, ChemistrySelect, 2021, 6, 10178–10181 CrossRef CAS; (e) Y. Baek, K. Cheong, D. Kim and P. H. Lee, Org. Lett., 2021, 23, 1188–1193 CrossRef CAS PubMed; (f) C. Li, Q. Ning, W. Zhao, H. Cao, Y. Wang, Y. Hong, C. Lu and Y. Liang, Chem. – Eur. J., 2021, 27, 2699–2706 CrossRef CAS PubMed; (g) Y. Chen, Y. Quan and Z. Xie, Chem. Commun., 2020, 56, 7001–7004 RSC; (h) Y. Au, H. Lyu, Y. Quan and Z. Xie, Chin. J. Chem., 2020, 38, 383–388 CrossRef CAS; (i) Z. Zhang, X. Zhang, J. Yuan, C. Yue, S. Meng, J. Chen, G. Yu and C. Che, Chem. – Eur. J., 2020, 26, 5037–5050 CrossRef CAS PubMed; (j) Z. Zhang, X. Zhang, J. Yuan, C. Yue, S. Meng, J. Chen, G. Yu and C. Che, Chem. – Eur. J., 2020, 26, 5037–5050 CrossRef CAS PubMed; (k) Y. Liang, L. Yang, B. B. Jei, R. Kuniyil and L. Ackermann, Chem. Sci., 2020, 26, 10764–10769 RSC; (l) H. Lyu, J. Zhang, J. Yang, Y. Quan and Z. Xie, J. Am. Chem. Soc., 2019, 141, 4219–4224 CrossRef CAS PubMed; (m) W. Mu, W. Liu, R. Cheng and D. Fang, ACS Omega, 2019, 4, 465–474 CrossRef CAS PubMed; (n) T. Xu, K. Cao, C. Zhang, J. Wu, L. Jiang and J. Yang, Chem. Commun., 2018, 54, 13603–13606 RSC; (o) X. Zhang and H. Yan, Chem. Sci., 2018, 9, 3964–3969 RSC; (p) X. Zhang, H. Zheng, J. Li, F. Xu, J. Zhao and H. Yan, J. Am. Chem. Soc., 2017, 139, 14511–14517 CrossRef CAS PubMed; (q) D. Zhao and Z. Xie, Angew. Chem., Int. Ed., 2016, 55, 3166–3170 CrossRef CAS; (r) Y. Quan and Z. Xie, Angew. Chem., Int. Ed., 2015, 55, 1295–1298 CrossRef PubMed; (s) K. Cao, Y. Huang, J. Yang and J. Wu, Chem. Commun., 2015, 51, 7257–7260 RSC.
- J. F. Kleinsasser, S. P. Fisher, F. S. Tham and V. Lavallo, Eur. J. Inorg. Chem., 2017, 4417–4419 CrossRef CAS.
- G. Akimoto, M. Otsuka, K. Miyamoto, A. Muranaka, D. Hashizume, R. Takita and M. Uchiyama, Chem. – Asian. J., 2018, 13, 913–917 CrossRef CAS PubMed.
- (a) E. Molinos, S. K. Brayshaw, G. Kociok-Köhn and A. S. Weller, Dalton Trans., 2007, 4829–4844 RSC; (b) E. Molinos, S. K. Brayshaw, G. Kociok-Köhn and A. S. Weller, Organometallics, 2007, 26, 2370–2382 CrossRef CAS; (c) E. Molinos, G. Kociok-Köhn and A. S. Weller, Chem. Commun., 2005, 3609–3611 RSC.
- A. J. Rosenbaum, D. H. Juers and M. A. Juhasz, Inorg. Chem., 2013, 52, 10717–10719 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of synthesis and characterization of compounds (PDF); CIF files for the X-ray crystal structures of 3a, 3b, 3h, 3q and 5. CCDC 2206555–2206559. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc05422a |
| ‡ Present address: Yuanpei College, Shaoxing University, 2799 Qunxian Road, Shaoxing 312000, China. |
| This journal is © The Royal Society of Chemistry 2023 |