
Light-switched selective catalysis with NADH mimic functionalized metal–organic capsules†
Jianwei
Wei
,
Liang
Zhao
 *,
Yu
Zhang
,
Peng
Zhou
,
Guangzhou
Liu
and
Chunying
Duan
*,
Yu
Zhang
,
Peng
Zhou
,
Guangzhou
Liu
and
Chunying
Duan
State Key Laboratory of Fine Chemicals, Zhang Dayu School of Chemistry, Dalian University of Technology, Dalian 116024, P. R. China. E-mail: zhaol@dlut.edu.cn
First published on 2nd December 2022
Abstract
By incorporating an active site model of nicotinamide adenine dinucleotide (NADH) as an electron regulator, a redox-active metal–organic capsule as an efficient photocatalyst was obtained for the light switchable synthesis of a series of aromatic azoxy and amino compounds from their corresponding nitroaromatics under either purple (420 nm) or blue (455 nm) LED light irradiation.
Controllable selective synthesis using photocatalysts is an intriguing but challenging research topic in chemistry,1 especially for light switching methods.2,3 Generally, the capability of accessing different catalytic pathways upon simple tuning of the irradiation wavelength could be attributed to the different excited states of the forming intermediates, photosensitizers, etc.4 Enzymes exhibit excellent selectivity benefitting from the specific cavity and properties of electron transfer, including hydride transfer,5 electron bifurcation,6 multiple optical excitations,7etc. Analogous to the reaction specificity achieved in enzyme pockets, metal–organic capsules attract considerable interest due to their high symmetry, stability and rich chemical properties, which are also powerful platforms for regulating the selectivity via engineering their catalytic cavities.8–10 However, modifying the electron transfer pathways and kinetics to regulate reaction selectivity remains largely unexplored, and a few visible light switchable catalysis methods were reported previously.11–13 One of the important relevant methods is the switchable selective reduction of nitroarenes into different products, due to the inherent complexity of nitro reduction networks.14,15
Recently, inspired by the ability of natural cofactor NADH to be used as the efficient electron regulator, we utilized NADH mimics to modify the electron donating behaviour and balance the inner and outer electron transfer kinetics to switch the group selectivity of bifunctional substrates.16 We envisioned that incorporating different photoredox processes into internal and external cavities of supramolecular catalysts could facilitate different catalytic manifolds by changing the irradiation wavelengths, providing the possibility to switch the selectivity of products. Herein, we constructed a new redox-active metal–organic capsule containing NADH mimics for the light switchable selective synthesis of aromatic azoxy and amino compounds from nitroaromatics under mild conditions (Scheme 1). Under 420 nm LED irradiation, the capsule could convert the photoinduced single electron transfer of ruthenium(II) trisbipyridine (Ru(bpy)32+) into a double electron reduction inside the cavity, quickly reducing the in situ formed nitroso intermediate to provide hydroxylamine species, followed by encapsulating these two molecules to produce azoxy compounds via a condensation pathway. Alternatively, irradiating with a 455 nm LED, utilizing Ru(bpy)32+ to trigger consecutive multistep single electron processes outside the cavity, was proposed to give amine products. By simply changing the irradiation wavelength, switchable selectivity has been achieved to efficiently tune the photosynthesis of aromatic azoxy and amine compounds from nitroaromatics.
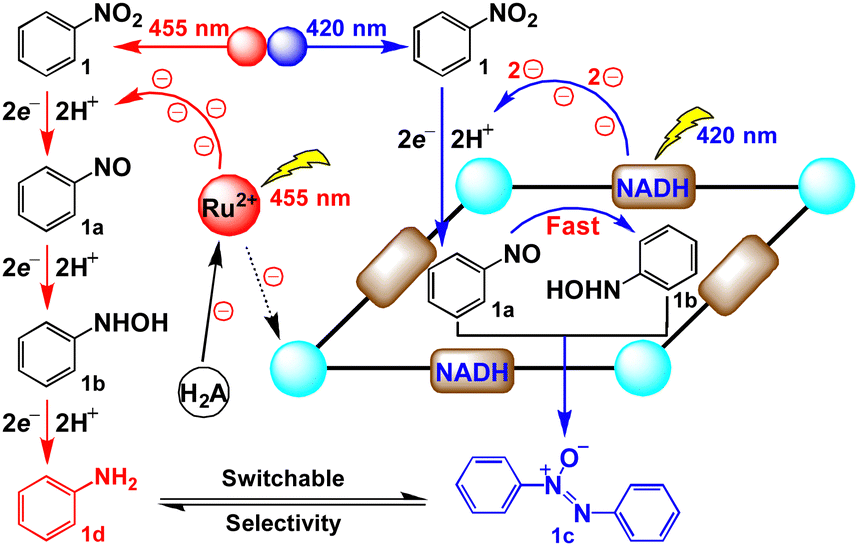 | ||
| Scheme 1 Selective reduction of nitroarenes inside and outside the capsule for switching the selectivity under different irradiation wavelengths. | ||
Ligand H2TPB containing NADH mimics was synthesized by a Schiff-base reaction of 4-phenyl-1-(thiophen-2-ylmethyl)-1,4-dihydropyridine-3,5-dicarbohydrazide and 2-pyridylaldehyde in ethanol. Metal–organic capsules were assembled by reacting H2TPB with the corresponding metal salts in CH3CN. Diffraction-grade single crystals of Zn–TPB with redox-inactive zinc ions and Ni–TPB with redox-active nickel ions were obtained by diffusion of diethyl ether into capsule solutions. Single crystal structure analysis revealed that both capsules Zn–TPB and Ni–TPB exhibit similar square configurations via the connection of four ligands and four metal ions (Fig. 1a and Fig. S1, S2, ESI†), alternatively. Each metal ion is chelated in an octahedral geometry by a pair of widely delocalized N2O chelators from two ligands positioned in a mer configuration. The coordinated amide groups located in the square provide a confined cavity for suitable substrate inclusion. The outwards thiophene rings help the capsule to form a large opening window (about 9.0 Å for one edge) in favour of the ingress and egress of guests.17 Four NADH mimics are located on the parallel side of the square with the active H–atoms orientated in the interior, endowing the possibility to regulate a double electron reduction from reactions outside the cavity.
 | ||
| Fig. 1 (a) Procedure for the synthesis of the capsule Zn–TPB and its crystal structure showing the resulting confined space of the capsule. Zn, cyan; S, yellow; N, blue; O, red and C, grey. ESI-MS spectra of Zn–TPB (0.1 mM) (b) and of Zn–TPB (c) with 5 equiv. of 1 in CH3CN. The insets show the isotopic patterns at m/z = 1223.1879 and 1346.1938. | ||
The stability of Zn–TPB in solution was characterized by the ESI-MS spectrum that exhibited an intense peak at 1223.1879 corresponding to [H2Zn4(TPB)4]2+ (Fig. 1b).18 Guest binding within the capsule was evident by ESI-MS analysis of a mixture of Zn–TPB and nitrobenzene (1), which showed a new peak corresponding to [H2Zn4(TPB)4⊃(1)2]2+ species at 1346.1938 (Fig. 1c). This was also determined from the DOSY spectra with the same diffusion coefficient of Zn–TPB and 1 (D = 7.08 × 10−11 m2 s−1) (Fig. S51, ESI†).19 The UV-Vis spectra of Zn–TPB exhibit an absorption band at 420 nm (Fig. 2a). When excited at 420 nm, Zn–TPB exhibited a bright emission at 490 nm (Fig. 2b). Upon addition of 1, only 7% emission was quenched, whereas the intermediate nitrosobenzene (1a) exhibited the strongest quenching (24%) over 1 and intermediate N-phenylhydroxylamine (1b) (Fig. S30, ESI†). Meanwhile, UV-Vis titration confirmed that Zn–TPB could form a stable host–guest complex with 1, 1a or 1b (Fig. S12–S14, ESI†). The host–guest complex was further confirmed by fluorescence titration of Zn–TPB upon the addition of 1, 1a or 1b (Fig. S22–S24, ESI†). Furthermore, isothermal titration calorimetry (ITC) of Zn–TPB upon the addition of 1, 1a or 1b revealed an appropriate n value of 2.0 (Fig. 2c).20 We also recognized that 1b exhibited the largest negative free energy change with Zn–TPB over 1 and 1a (Fig. S32–S34, ESI†), and the corresponding nitroso and nitro species hardly squeezed the hydroxylamine species from the cavity of Zn–TPB. We assumed that the potential double electron reduction from 1a to 1b was the most efficient step in the tandem reductions.21 The in situ formed 1b has the potential to interact with the unreduced 1a species, forming azoxybenzene (1c) when the cavity is large enough to encapsulate more than one substrate.
 | ||
Fig. 2 (a) UV-Vis spectra of H2TPB (40 µM), Zn–TPB (10 µM), Ru(bpy)32+ (0.1 mM) and Zn–TPB/Ru(bpy)32+ (10 µM/0.1 mM) in CH3CN/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, pH 5.0). (b) Luminescence spectra of Zn–TPB (10.0 µM), Ru(bpy)32+ (0.1 mM) and a mixture of Zn–TPB and Ru(bpy)32+ in CH3CN/H2O (1:1, pH 5.0), excited at 420 nm and 455 nm, respectively. The fitting curves of the ITC test in CH3CN/H2O (1:1, pH 5.0) show the effect of Zn–TPB with 1, 1a and 1b (c) and 2, 3 and ATP (d), respectively. :1, pH 5.0). (b) Luminescence spectra of Zn–TPB (10.0 µM), Ru(bpy)32+ (0.1 mM) and a mixture of Zn–TPB and Ru(bpy)32+ in CH3CN/H2O (1:1, pH 5.0), excited at 420 nm and 455 nm, respectively. The fitting curves of the ITC test in CH3CN/H2O (1:1, pH 5.0) show the effect of Zn–TPB with 1, 1a and 1b (c) and 2, 3 and ATP (d), respectively. | ||
Under standard conditions, irradiation of a CH3CN/H2O (1:1, 5 mL) solution containing Ru(bpy)32+ (0.5 mM), Zn–TPB (50 µM), 1 (10.0 mM), HCOOH (0.05 M) and ascorbic acid (H2A, 0.1 M) with a blue LED light (455 nm) at room temperature for 6 h afforded aniline (1d) in 81% yield and 1c in 17% yield (Table 1, entry 1). Altering the light sources from 455 nm to 420 nm, the yield of 1c increased to 54% and the yield of 1d decreased to 35% (entry 2). Without Zn–TPB, the system with either 455 nm or 420 nm LED irradiation afforded product 1d over 90% yield under the same conditions (entries 3 and 4), suggesting that simple Ru(bpy)32+ could form the amino compound by photoinduced single electron transfer through the direct route.22,23 Control experiments revealed that the absence of any of these individual components or light led to a failure in the reduction (entries 5 and 6). Notably, the ligand H2TPB was not an efficient alternative for Zn–TPB (entries 7 and 8). Therefore, the new photocatalytic system composed of photosensitizers and capsules can effectively reduce the nitro groups, and more importantly, the selectivity can be switched by changing the wavelength of light.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Substrate | Irradiation (nm) | Yield/% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Azoxy | Amine | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
a Standard conditions: substrate (10.0 mM), Ru(bpy)32+ (0.5 mM), catalyst (0.05 mM for Zn–TPB/Ni–TPB; 0.20 mM for H2TPB), and HCOOH/H2A (0.05/0.1 M) in CH3CN/H2O (1:1, pH 5.0) under LED irradiation for 6 h.
b Without HCOOH/H2A.
c Without Ru(bpy)32+.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Zn–TPB | 1 | 455 | 17 | 81 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Zn–TPB | 1 | 420 | 54 | 35 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | — | 1 | 455 | Trace | 93 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | — | 1 | 420 | Trace | 91 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5b | — | 1 | 455 | 0 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | — | 1 | — | 0 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | H2TPB | 1 | 455 | Trace | 92 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | H2TPB | 1 | 420 | Trace | 91 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | Ni–TPB | 1 | 455 | 15 | 80 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | Ni–TPB | 1 | 420 | 90 | 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11c | Zn–TPB | 1 | 420 | 43 | 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12c | Ni–TPB | 1 | 420 | Trace | Trace | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | Zn–TPB | 2 | 455 | 0 | 95 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | Zn–TPB | 2 | 420 | 0 | 92 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | Zn–TPB | 3 | 455 | 0 | 95 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16 | Zn–TPB | 3 | 420 | 0 | 93 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

When 4,4″-di-tert-butyl-5′-nitro-1,1′:3′,1″-terphenyl (2), which is larger than the capsule cavity, was introduced into the system, no azoxy compound could be formed, but amino product 2d was detected, regardless of changing the reaction light (420 nm or 455 nm) (entries 13 and 14). Outside the cavity, only amine compounds could be formed through the direct route. In the case of 1-nitropyrene (3), it has a medium size, larger than 1 but smaller than 2. The ITC test of Zn–TPB upon the addition of 3 reveals a good n value of 1.0 (Fig. 2d and Fig. S36, ESI†). When substrate 3 was introduced into the system, azoxy compounds also could not be formed, but amino product 3d was detected under either 420 nm or 455 nm LED irradiation (entries 15 and 16), suggesting that the inclusion of more than one substrate was essential for producing azoxy compounds.
To examine the influence of the host–guest effect on the switchable selective reduction of nitroarenes, we conducted kinetic experiments using the initial rate method. For substrate 1, the initial formation rate of 1c was 1.3 mM h−1 and the formation rate of 1d was 0.8 mM h−1 with Zn–TPB under 420 nm (Fig. S54, ESI†), consistent with the aforementioned reaction yields and selectivities. Meanwhile, the system resulted in the initial formation rate of 2d as 1.2 mM h−1 and the formation rate of 3d as 1.6 mM h−1 with no detection of azoxy compounds under the same conditions (Fig. S54, ESI†). When dislodging Zn–TPB, the formation rate of 1d increased to 1.8 mM h−1 with no formation of 1c, and the formation rate of 2d and 3d was maintained under 420 nm (Fig. S55, ESI†). Altering the irradiation wavelength from 420 nm to 455 nm, all the formation rates of amine increased (Fig. S55, ESI†), due to the stronger fluorescence efficiency of Ru(bpy)32+ excited at 455 nm (Fig. 2b). Moreover, the formation rate ratio of the amine product (R420/R455) is proportional to fluorescence ratio of Ru(bpy)32+ (F420/F455) excited by different wavelengths, and the absorption ratio at different wavelengths (A420/A455) (Fig. S56, ESI†). Therefore, we speculated that the selectivity of the system was caused by the different responses of Zn–TPB and Ru(bpy)32+ under different wavelengths, respectively (Fig. 2b). As a result, the ratios of both fluorescence (FZn–TPB/FRu) and absorption (AZn–TPB/ARu) of Zn–TPB and Ru(bpy)32+ in the photocatalytic system exhibit a positive correlation with the yield and the rate ratio of 1c/1d under two different light sources (Fig. S57, ESI†).
To further improve the selectivity, an analogous Ni–TPB with redox-active nickel ions was designed and prepared because the presence of nickel ions could enhance the electron transfer between the capsule and Ru(bpy)32+.24 The ESI-MS spectrum of Ni–TPB exhibits a peak of [H2Ni4(TPB)4]2+ at 1209.1890, suggesting a stable M4L4 structure in solution (Fig. S7, ESI†). The properties of the host–guest complex between Ni–TPB and the substrate and their intermediates were consistent with those of Zn–TPB, confirmed by the ESI-MS spectrum, UV-Vis absorption spectra and ITC assay (Fig. S8, S9, S15–S17 and S38–S40, ESI†). As expected, under standard conditions, Ni–TPB could maintain not only the yield (80%) and selectivity (84%) of 1d under 455 nm irradiation (Table 1, entry 9), but could also sharply increase the yield and selectivity of 1c to 90% and 96% under 420 nm irradiation, respectively (entry 10), indicating that localizing double and single electron transfer inside and outside the pocket could comfortably tune the reaction selectivity.
It should be noted that the formation rate of 1c by Ni–TPB was faster than that of Zn–TPB under 420 nm irradiation, whereas the formation rate of 1d was opposite (Fig. S58, ESI†). The luminescence quenching of Ru(bpy)32+ by Ni–TPB was stronger than that of Zn–TPB, and Ni–TPB could reduce the lifetime of Ru(bpy)32+ by 69 ns higher than that of Zn–TPB (Fig. S31, ESI†), revealing the greater ability of Ni–TPB to regulate the electron transfer between the capsule and Ru(bpy)32+. Without Ru(bpy)32+, Zn–TPB could only yield 43% of 1c with 1d in 4% yield, and both products were traced for Ni–TPB under standard conditions (entries 11 and 12), suggesting that Ru(bpy)32+ has a significant effect on the capsule catalysis. Meanwhile, to further gain insights into the light switchable catalysis, 1a and 1b as reaction substrates, respectively, were introduced into the photocatalytic system. The results showed that using 1a (10.0 mM) as a substrate under standard conditions, Ni–TPB system resulted in an initial formation rate k1 of up to 6.9 mM h−1 for product 1c and produced traces of 1d under 420 nm (Fig. S59, ESI†). Alternatively, 1b as a substrate led to an initial formation rate k2 of 2.6 mM h−1 for 1d, and 1c was not observed under the same conditions. However, when changing to 455 nm irradiation, k1 decreased to 3.6 mM h−1, and k2 increased to 3.2 mM h−1 (Fig. S59, ESI†). The ratio of k1 and k2 under 420 nm irradiation was calculated as 2.7, which is larger than that under 455 nm irradiation as 1.1, further suggesting that the introduction of nickel ions regulated the electron transfer between the capsule and Ru(bpy)32+ to increase the reaction rate inside the capsule and decrease the rate outside.16 This is beneficial to enhance the reaction selectivity.
To further determine whether the condensation pathway occurred inside or outside the capsule, a nonreactive species, adenosine triphosphate (ATP),25 was introduced to carry out the inhibition experiment.26 The ITC test of Ni–TPB upon addition of ATP revealed the formation of the host–guest complex with a dissociation constant of 7.08 µM (Fig. 2d and Fig. S44, ESI†). This was further confirmed using ESI-MS, UV-Vis, and DOSY spectra (Fig. S10, S19 and S53, ESI†), suggesting that ATP could be encapsulated in the capsule. As expected, the addition of ATP (20.0 mM) into the Ni–TPB system resulted in an effective quenching with only 32% yield of 1c and 60% yield of 1d (Fig. S60, ESI†). Another non-ionic inhibitor 2,6-dimethylpyridine also quenched the condensation pathway well (Fig. S45 and S60, ESI†).
From a mechanistic viewpoint, the NADH mimic-modified capsule captures two substrate molecules to form a stable host–guest complex that enforces close contact between the NADH model and substrate to produce a nitroso compound. And then one nitroso molecule is reduced to hydroxylamine under 420 nm LED irradiation through double electron reduction process inside the cavity. Finally, the condensation of hydroxylamine molecule with another nitroso molecule proceeds quickly to give an azoxy compound in the cavity of capsule. The negative redox potential of the azoxy compound prevented the further reduction of 1c (Fig. S62, ESI†).27 And it could leave the cavity due to the large dissociation constant (Fig. S39, ESI†), which was consistent with the DOSY spectrum (Fig. S52, ESI†). Therefore, azoxy compounds could be selectively obtained by the condensation pathway. When changing the irradiation to 455 nm, the single electron transfer from excited Ru(bpy)32+ to substrates23 selectively produced amine compounds along the direct pathway outside the capsule. Furthermore, Ni–TPB could improve the switchable selectivity, by enhancing the electron transfer between the capsule and photosensitizer. The superiority of such supramolecular photocatalysis systems that promote the light switchable selective reduction could be extended to a wide range of nitroarenes, forming azoxy compounds under purple light and producing amine compounds under blue light, respectively (Table 2). Moreover, this photocatalytic system can synthesize the esterification of azoxy natural products (12c), azoxymycins C (Table 2).28

This work was supported by the NSFC (grant no. 22171034, 21820102001, and 21890381).
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341 RSC.
- Y. Sakakibara and K. Murakami, ACS Catal., 2022, 12, 1857 CrossRef CAS.
- D. Lunic, E. Bergamaschi and C. J. Teskey, Angew. Chem., Int. Ed., 2021, 60, 20594 CrossRef CAS.
- S. Ilic, A. Alherz, C. B. Musgrave and K. D. Glusac, Chem. Soc. Rev., 2018, 47, 2809 RSC.
- W. Buckel and R. K. Thauer, Chem. Rev., 2018, 118, 3862 CrossRef CAS PubMed.
- X. Fang, S. Kalathil and E. Reisner, Chem. Soc. Rev., 2020, 49, 4926 RSC.
- Z. Dong, Q. Luo and J. Liu, Chem. Soc. Rev., 2012, 41, 7890 RSC.
- M. D. Ward, C. A. Hunter and N. H. Williams, Acc. Chem. Res., 2018, 51, 2073 CrossRef CAS.
- M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Catal., 2020, 3, 969 CrossRef CAS.
- I. Ghosh and B. König, Angew. Chem., Int. Ed., 2016, 55, 7676 CrossRef CAS PubMed.
- Y. Markushyna, C. M. Schusslbauer, T. Ullrich, D. M. Guldi, M. Antonietti and A. Savateev, Angew. Chem., Int. Ed., 2021, 60, 20543 CrossRef CAS PubMed.
- Y. Dai, C. Li, Y. Shen, T. Lim, J. Xu, Y. Li, H. Niemantsverdriet, F. Besenbacher, N. Lock and R. Su, Nat. Commun., 2018, 9, 60 CrossRef PubMed.
- A. Corma and P. Serna, Science, 2006, 313, 332 CrossRef CAS.
- D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611 CrossRef CAS PubMed.
- J. Wei, L. Zhao, C. He, S. Zheng, J. N. H. Reek and C. Duan, J. Am. Chem. Soc., 2019, 141, 12707 CrossRef CAS PubMed.
- F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204 CrossRef.
- H. Wang, C. Guo and X. Li, CCS Chem., 2021, 3, 3161 Search PubMed.
- N. Geue, R. E. P. Winpenny and P. E. Barran, Chem. Soc. Rev., 2022, 51, 8 RSC.
- J. Tellinghuisen, Anal. Biochem., 2003, 321, 79 CrossRef CAS PubMed.
- C. Bryant and M. DeLuca, J. Bio. Chem., 1991, 266, 4119 CAS.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803 RSC.
- A. R. Todorov, S. Aikonen, M. Muuronen and J. Helaja, Org. Lett., 2019, 21, 3764 CrossRef CAS.
- D. Hong, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2017, 139, 6538 CrossRef CAS PubMed.
- P. A. Gale, N. Busschaert, C. J. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205 RSC.
- L. J. Jongkind, J. Elemans and J. N. H. Reek, Angew. Chem., Int. Ed., 2019, 58, 2696 CrossRef CAS.
- X. Chong, C. Liu, Y. Huang, C. Huang and B. Zhang, Natl. Sci. Rev., 2020, 7, 285 CrossRef CAS PubMed.
- Y. Y. Guo, Z. H. Li, T. Y. Xia, Y. L. Du, X. M. Mao and Y. Q. Li, Nat. Commun., 2019, 10, 4420 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystal structure and additional spectroscopic data. CCDC 1961723 and 1961724. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04530c |
| This journal is © The Royal Society of Chemistry 2023 |