
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intimate relationship between C–I reductive elimination, aryl scrambling and isomerization processes in Au(III) complexes†
Sara
Fernández-Moyano
 ,
Guillermo
Marcos-Ayuso
,
Marconi N.
Peñas-Defrutos
*,
Camino
Bartolomé
and
Pablo
Espinet
*
,
Guillermo
Marcos-Ayuso
,
Marconi N.
Peñas-Defrutos
*,
Camino
Bartolomé
and
Pablo
Espinet
*
IU CINQUIMA/Química Inorgánica, Facultad de Ciencias, Universidad de Valladolid, Valladolid 47071, Spain. E-mail: marconi_44@hotmail.com; espinet@qi.uva.es
First published on 23rd January 2023
Abstract
19F NMR monitoring shows that heating trans-[AuIIIRf2I2]− solutions (Rf = C6F3Cl2-3,5) leads to formation of cis-[AuRf2I2]−, [AuRf3I]− and [AuRfI3]−via kinetic competition between isomerization and Rf/I scrambling. The system evolution is driven by the easy Rf–I reductive elimination from [AuRfI3]− (forming also [AuI2]−), which is faster than any of the Rf–Rf couplings from the coexisting species, hindering the commonly desired and thermodynamically preferred C–C coupling. A kinetic model where I− dissociation triggers both isomerization and transmetalation steps is proposed, which fits well the experimental data. DFT calculations support that the lower bond strength of AuIII–I compared to other halides produces a pathway switch that makes C–I coupling kinetically preferred. Consequently, it is better avoided in reactions looking for C–C coupling.
In the recent years, C–C cross coupling based on the AuIII/AuI pair has become a hot topic as a potential alternative to PdII/Pd0 chemistry.1,2 Efficient aryl–aryl coupling processes mediated by gold systems,3 including one example of the more challenging ArF–ArF (ArF = perhaloaryl group) reductive elimination (RE) from well-defined Au(III) complexes have been reported.4,5 In this context we planned to prepare cis-AuIIIRf2 adducts (Rf = C6F3Cl2-3,5) to study the C–C coupling possibilities, as we did previously with cis-[PdIIPf2Ln] (Pf = C6F5) species.6 For this mechanistic study we use Rf aryl complexes, which display simple and clean 19F NMR spectra.
We recently reported that, trying to synthesize (μ-Cl)2[AuRf2]2, an unexpected Rf/Cl scrambling process was observed (Scheme 1, above).7 There are just a few reported cases of aryl scrambling phenomena involving Au(III) complexes. Those reported by Luzuriaga8 (Scheme 1, middle) and Nevado9 groups (Scheme 1, below), have in common that they are only detected when strong oxidants come into play, and are lacking any mechanistic investigation.
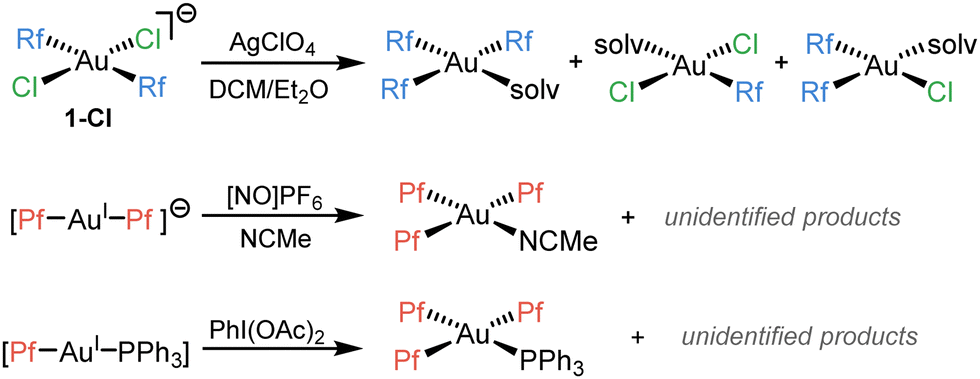 | ||
| Scheme 1 Reported cases of aryl scrambling involving AuIII complexes. | ||
In the case of our previous study (Scheme 1, above), the scrambling occurs in more conventional conditions, and is triggered by halide abstraction from trans-[AuRf2Cl2]− (1-Cl) with 1 equiv. of AgClO4. Evaporation of the solvent (specifically Et2O) and dissolution in the non-coordinating CHCl3 reverted the scrambling forming the intended dimer (μ-Cl)2[AuRf2]2.7 The reaction conditions precluded to obtain kinetic information, but the drastically different outcome observed when using cis-[AuRf2Cl2]− (2-Cl) (no scrambling detected) supported satisfactorily our mechanistic proposal of kinetic competition between isomerization and scrambling from the unstable trans-[AuRf2Cl(solv)].7 Moreover, it is worth noting that, upon heating, solutions of 1-Cl led selectively to the cis complex 2-Cl, which is reluctant to undergo coupling. On the contrary, accessible Rf–Rf reductive elimination (RE) barriers were observed for cis-[AuRf2Cl(L)] (L = μ-Cl, OEt2) derivatives, while no traces of Rf–Cl (hypothetically resulting from a C–Cl coupling) were detected in any case.7
In this context, here we study the surprising reactivity displayed by solutions of the iodo-complex (NBu4)trans-[AuRf2I2] (1) in poorly coordinating solvents. Complex 1 was obtained in excellent yield by oxidation of (NBu4)[AuRf2],10 with I2 (see details in ESI†).
The synthesis of cis-[AuPf2I2]− (Pf = C6F5), by heating the trans complex in CH2Cl2, was reported long ago (without 19F NMR data), but we confirmed that a clean isomerization does not occur in those conditions (see Fig. S1, ESI†).11 Similarly, the behaviour of 1 in tetrachloroethane (TCE) solution is not simple.12 Instead of leading selectively to (NBu4)cis-[AuRf2I2] (2), as it might be expected by analogy with the Cl complexes, a mixture of several species bearing Rf groups was observed by 19F NMR (Fig. 1 and Fig. S3, ESI†).
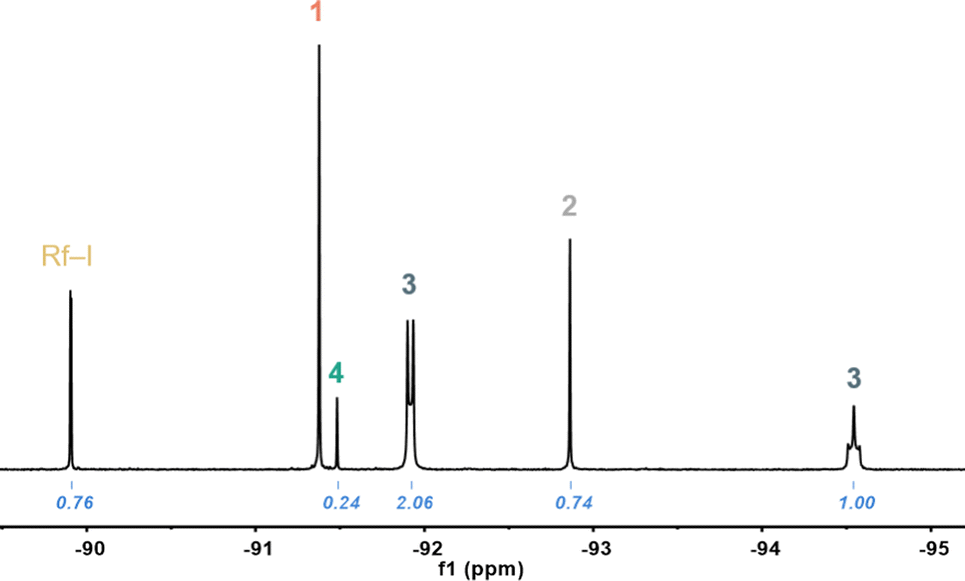 | ||
| Fig. 1 Fortho region of the 19F NMR spectrum recorded from solutions of 1 in TCE-d2 after heating at 323 K for 1 day. Assignment of the signals and corresponding integrals. | ||
The two chemically inequivalent Fortho signals in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and displaying multiplicity are consistent with (NBu4)[AuRf3I] (3), a square-planar species formed presumably by Rf/I scrambling. The pseudo doublet and triplet multiplicities of the Fortho signals reveal hindered rotation of the three Rf groups,13 imposed by the bulky iodo group, since when fast rotation is allowed, as reported for [AuRf3(OH2)],7 triplet and quintet signals with relative integrals 2:1 are found. Complex 3 could be selectively synthesized by reaction of [AuRf3(OH2)] with (NBu4)I and was fully characterized (details in ESI,† see Fig. S7 for its X-ray structure).
:1 ratio and displaying multiplicity are consistent with (NBu4)[AuRf3I] (3), a square-planar species formed presumably by Rf/I scrambling. The pseudo doublet and triplet multiplicities of the Fortho signals reveal hindered rotation of the three Rf groups,13 imposed by the bulky iodo group, since when fast rotation is allowed, as reported for [AuRf3(OH2)],7 triplet and quintet signals with relative integrals 2:1 are found. Complex 3 could be selectively synthesized by reaction of [AuRf3(OH2)] with (NBu4)I and was fully characterized (details in ESI,† see Fig. S7 for its X-ray structure).
The Fortho resonance at −89.9 ppm (doublet, 2F) and the Fpara signal at −108.1 ppm (triplet, 1F), correspond to the organic Rf–I, which must be the result of C–I reductive elimination, while no traces of Rf–Rf were observed. Interestingly, this C–I coupling seems to be fast at 323 K in contrast with the remarkably slow C–C RE observed for 2-Cl at 393 K in TCE. For stoichiometry reasons, the concentrations of tris-aryl and mono-aryl species must be identical, and consequently we assign the signal at −91.5 ppm to (NBu4)[AuRfI3] (4). Finally, the signal at −92.9 ppm corresponds to (NBu4)cis-[AuRf2I2] (2). The identity of the latter was confirmed by reaction of 2-Cl with excess of KI (details in ESI,† see also Fig. S4).
After prolonged heating of 1 for 2 days at 353 K in TCE, the final products of the reaction were 3 + Rf–I, in 1:1 ratio, by 19F NMR. Obviously, some gold (specifically half of it) was missing, but we were able to crystallize from the reaction mixture the AuI complex (NBu4)[AuI2] (5) blind to 19F NMR. This adjusts the chemical balance (Scheme 2). It is worth noting that signals of both bis-aryl complexes 1 and 2 disappear, confirming the scrambling completeness (Fig. S5, ESI†).
 | ||
| Scheme 2 Final outcome of the reaction of 1 in TCE. Products are formed after heating 2 days at 353 K. | ||
For better understanding of this remarkable reactivity, the evolution of 1 in TCE-d2 at 338 K was monitored by 19F NMR. The concentration vs. time data plot obtained for the different species is shown in Fig. 2 (dots are the experimental data). The kinetics of the competitive processes occurring is not trivial. Clearly, an induction period is observed in the disappearance of 1, meaning that either an intermediate or a product is catalysing the transformation. The addition of complexes 3, 5 or the organic molecule Rf–I did not affect the rate of the reaction and the curve of complex (NBu4)[AuRfI3] (4) confirms that it is thermodynamically unstable and behaves as the intermediate that gives rise to Rf–I + (NBu4)[AuI2] (5) by C–I reductive elimination (Scheme 2).
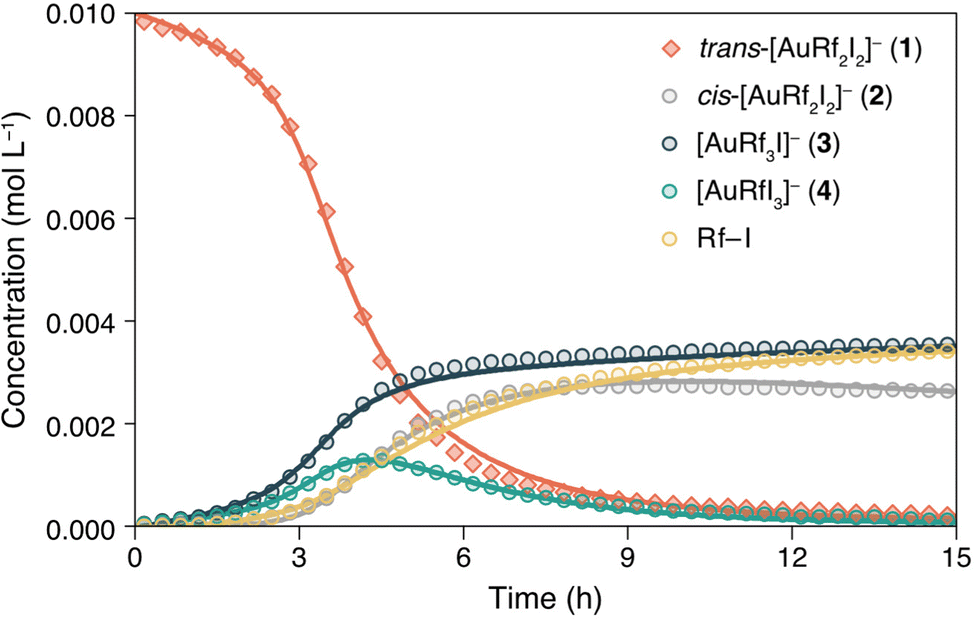 | ||
| Fig. 2 Concentration vs. time plot of experimental data (dots) obtained by 19FNMR monitoring and COPASI-fitted values (continuous lines) of the fluorinated-species observed when heating complex 1 in TCE-d2 at 338 K. | ||
The isomerization in gold(III) systems has been proposed to follow a dissociative + topomerization mechanism.14 Transmetalation reactions (scrambling is one of them) typically require ligand dissociation in square-planar complexes.15 Interestingly, addition of substoichiometric amounts of (NBu4)I to solutions of 1, specifically 20 mol% in identical conditions to the experiment in Fig. 2 (TCE-d2, 338 K, 15 h), suppresses the reactivity. This supports a dissociative mechanism, where I− coordination sequesters low concentration intermediates.16
The reaction evolution was satisfactorily fitted using COPASI software (see microkinetic details in ESI†) and the simplified kinetic model depicted in Scheme 3, which involves dissociative steps. Only six elemental reactions were needed to reproduce the experimental data precisely (see continuous lines in Fig. 2).
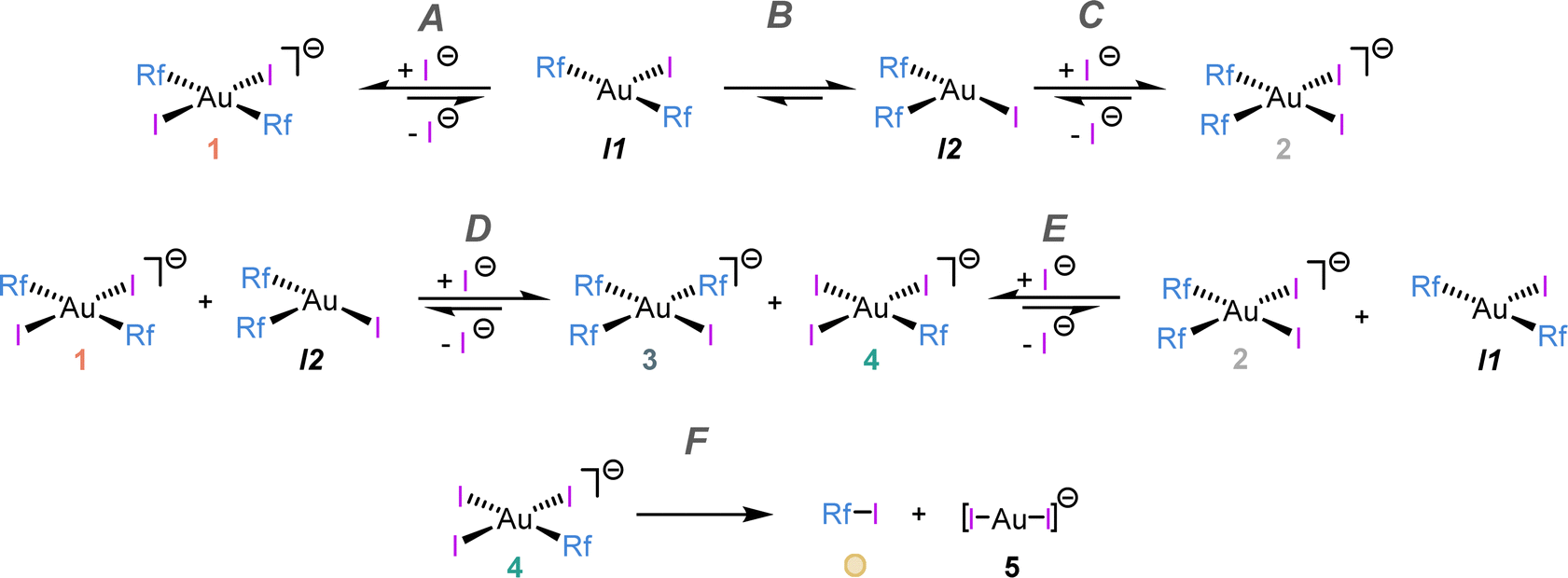 | ||
| Scheme 3 Kinetic model used for the non-linear fitting shown in Fig. 2. Elemental reactions are labelled with letters (A–F). | ||
The pair of reactions A/B in Scheme 3 accounts for the induction period observed in the disappearance of 1 (orange trace, Fig. 2). We propose that iodide dissociation from the reactant, forming an unobservable tricoordinate intermediate I1, triggers the process.17 Obviously, the first equilibrium A is very disfavoured thermodynamically. On the contrary, isomerization from the trans-intermediate I1 leading to a more stable cis tricoordinate species labelled as I2 must be exergonic, attending to trans influence reasons (step B). The latter, I2, reacts with the initial complex 1 catalysing its consumption forming the scrambling species 3 and 4 by Rf/I transmetalation (Scheme 3D, dark and light green lines in Fig. 2).18 It is worth remarking that the preferred reactivity combining trans with cis 3- and 4-coordinated species was also proposed in our previous report.7
Scheme 3C shows the capture of I2 by iodide coordination forming the cis anionic complex 2, which kinetically competes with the scrambling shown in reaction D. At long reaction times, complex 2 slowly disappears (grey line in Fig. 2) by reaction with the minute amount of I1 present in solution (reaction E). This plausible scrambling process leads as well to 3 + 4, similarly to the 1 + I2 reaction.
The last reaction of the kinetic model (step F) is the irreversible C–I coupling from 4, producing Rf–I (yellow line in Fig. 2) + 5. This reductive elimination step is the driving force bringing the reaction, at longer times or higher temperature, to the final products shown in Scheme 2, which is satisfactorily reproduced by our kinetic model (see Fig. S12, ESI†). The key role of iodide dissociation in the model is fully supported by the lethal effect of addition of substoichiometric (NBu4)I on the reactivity.
Although there are several other potential sources of Rf–I coupling in the mixture (1, 2, 3), the good COPASI fitting of the model supports that the only Rf–I reductive elimination energetically accessible in the experimental conditions (338 K) occurs on [AuRfI3]− (4). On the other hand, complexes 2 and 3 have Rf groups in cis arrangement that could undergo Rf–Rf coupling, but this biphenyl was not observed in any case.
For further support of the kinetic conclusions, we performed DFT calculations (see ESI† for details). The thermodynamic (ΔG) and kinetic (ΔG‡) computational results of the possible C–I and C–C RE processes from complexes 1–4 and selected data from the Cl analogues 2-Cl and 4-Cl for comparison, are given in Table 1.
| Entry | Comp. | RE type | Products | ΔG | ΔG‡ |
|---|---|---|---|---|---|
| 1 | 1 | C–I | Rf–I + [AuRfI]− | −3.0 | +37.3 |
| 2 | 2 | C–I | Rf–I + [AuRfI]− | −1.6 | +35.4 |
| 3 | 3 | C–I | Rf–I + [AuRf2]− | +6.4 | +48.3 |
| 4 | 4 | C–I | Rf–I + [AuI2]− | −9.6 | +22.8 |
| 5 | 2 | C–C | Rf–Rf + [AuI2]− | −35.0 | +26.6 |
| 6 | 3 | C–C | Rf–Rf + [AuRfI]− | −29.9 | +35.0 |
| 7 | 4-Cl | C–Cl | Rf–Cl + [AuCl2]− | −12.0 | +31.5 |
| 8 | 2-Cl | C–C | Rf–Rf + [AuCl2]− | −31.1 | +29.0 |
Entries 1–4 contain the computed data for the possible C–I RE processes from the mixture of species in Fig. 1, revealing that the coupling from complex 3 is thermodynamically disfavoured (ΔG = +6.4 kcal mol−1).19 Besides, Rf–I extrusion is only scarcely exergonic from 1 and 2 (entries 1 and 2). Remarkably, the concerted C–I RE from the electron poorest species,20 namely [AuRfI3]− (4), is both the most thermodynamically favoured Rf–I coupling (i.e −9.6 kcal mol−1, it may indeed be considered irreversible) and, more importantly, the fastest process in Table 1 (ΔG‡ = +22.8 kcal mol−1, entry 4). The stabilizing effect of [AuI2]− (5) + Rf–I formation drives the equilibria in Scheme 3 to scrambling completeness. The remaining non-used groups make up [AuRf3I]− (3), and the outcome shown in Scheme 2.
The Rf–Rf coupling, which is by far the most thermodynamically favoured (entries 5 and 6) is absent of the reaction for kinetic reasons: the ΔG‡ values computed for the Rf–Rf coupling processes from 2 and 3 (the only species with Rf groups arranged cis) are clearly higher than C–I from 4 (i.e +26.6 kcal mol−1 in entry 5), or inaccessible (entry 6).21 Fig. S8 (ESI†) shows the associated transition states optimized for both competing processes.
Replacing I by Cl (entries 7 and 8), the significant kinetic preference for Rf–I vs. Rf–Rf coupling is reverted. While both ΔG and ΔG‡ values for Rf–Rf formation from cis-[AuRf2Cl2]− (2-Cl) are similar to those found from the I-analogue 2, the activation barrier for the C–Cl coupling from a hypothetical [AuRfCl3]− (4-Cl) is clearly higher (ΔG‡ = +29.0 vs. +31.5 kcal mol−1) highlighting the key role of the halide. Besides, the deep stability of complex 2-Cl makes the eventual scrambling thermodynamically highly disfavoured. The AuIII–X elongation required to reach the C–X coupling TS is more demanding for the stronger AuIII–Cl bond (8.7 kcal mol−1 ΔG‡ difference between entries 4 and 7).19a,22
We also confirmed experimentally that the behaviour of 1-Br is analogue to 1-Cl, and isomerizes to 2-Br (that eventually undergoes Rf–Rf coupling under harsh conditions), while no traces of Rf/Br scrambling are detected (see ESI† for details).
Finally, the narrow energy difference between trans-[AuRf2I2]− (1) and cis-[AuRf2I2]− (2) isomers (i.e −1.4 kcal mol−1) contrasts with the selective isomerization observed for the Cl analogue (ΔG for 1-Cl/2-Cl conversion is −10.5 kcal mol−1). The latter is a reminder of the participation of opposite influences in simple isomeric structures: attending to the transphobia concept a cis arrangement should be preferred for both halides,23 but the bulkiness of the iodo ligand plays an important destabilizing role on the cis isomer 2,24 making the Rf/I scrambling thermodynamically accessible in our case. This analysis warns that both crowding repulsive effects and electronic aspects need to be taken into account to rationalize some complex reactivity patterns.
In conclusion, the nature of the halide in [AuRf2X2]− complexes (X = Cl, Br, I) plays a decisive influence on the evolution of these species in solution. While the iodide compound undergoes Rf/I scrambling and feasible Rf–I coupling (pathway favoured by the comparative weakness of the AuIII–I bond), selective trans/cis isomerization and challenging Rf–Rf coupling is found for Cl and Br derivatives. This potentially problematic kinetic competition should be considered in the development of new Au catalysed C–C cross-coupling processes.
We thank the Spanish MCINN (Project PID2020-118547GB-I00) and the JCyL (Project VA224P20) for the funding provided. We acknowledge Miguel Claros and Eric Mates-Torres for help. M. N. P-D. thanks the UVa for a Margarita Salas fellowship (ref. CONVREC-2021-221).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews see: (a) V. W. Bhoyare, A. G. Tathe, A. Das, C. C. Chintawar and N. T. Patil, Chem. Soc. Rev., 2021, 50, 10422–10450 RSC; (b) A. Nijamudheen and A. Datta, Chem. – Eur. J., 2020, 26, 1442–1487 CrossRef CAS PubMed; (c) C. Fricke, W. B. Reid and F. Schoenebeck, Eur. J. Org. Chem., 2020, 7119–7130 CrossRef CAS.
- For general gold catalysed organic transformations see: (a) M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022–15045 CrossRef CAS PubMed; (b) L. Rocchigiani and M. Bochmann, Chem. Rev., 2021, 121, 8364–8451 CrossRef CAS PubMed.
- (a) W. J. Wolf, M. S. Winston and F. D. Toste, Nat. Chem., 2014, 6, 159–164 CrossRef CAS PubMed; (b) S. Kramer, Synthesis, 2020, 2017–2030 CrossRef CAS.
- K. Kang, S. Liu, T. Xu, D. Wang, X. Leng, R. Bai, Y. Lan and Q. Shen, Organometallics, 2017, 36, 4727–4740 CrossRef CAS.
- For other cross-coupling reactions using stoichiometric perhaloroaryl gold complexes, see: (a) X. C. Cambeiro, T. C. Boorman, P. Lu and I. Larrosa, Angew. Chem., Int. Ed., 2013, 52, 1781–1784 CrossRef CAS PubMed; (b) M. Hofer, E. Gomez-Bengoa and C. Nevado, Organometallics, 2014, 33, 1328–1332 CrossRef CAS.
- E. Gioria, J. del Pozo, J. M. Martínez-Ilarduya and P. Espinet, Angew. Chem., Int. Ed., 2016, 55, 13276–13280 CrossRef CAS PubMed.
- S. Fernández-Moyano, M. N. Peñas-Defrutos, C. Bartolomé and P. Espinet, Chem. Commun., 2021, 57, 125–128 RSC.
- A. T. Overton, J. M. López-de-Luzuriaga, M. E. Olmos and A. A. Mohamed, Organometallics, 2012, 31, 3460–3462 CrossRef CAS.
- M. Hofer and C. Nevado, Eur. J. Inorg. Chem., 2012, 1338–1341 CrossRef CAS.
- E. J. Fernández, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, M. Montiel, M. E. Olmos, J. Pérez, R. C. Puelles and J. C. Sáenz, Dalton Trans., 2005, 1162–1164 RSC.
- R. Usón, A. Laguna, J. García and M. Laguna, Inorg. Chim. Acta, 1979, 37, 201–207 CrossRef.
- The behaviour in CDCl3 is analogous (see Fig. S2, ESI†) but it does not allow to heat above 323 K as needed for kinetic monitoring. Aryl scrambling was also observed in other poorly coordinating solvents, such as toluene, but 1 is scarcely soluble in aromatic solvents.
- P. Espinet, A. C. Albéniz, J. A. Casares and J. M. Martínez-Ilarduya, Coord. Chem. Rev., 2008, 252, 2180–2208 CrossRef CAS.
- (a) S. Komiya, T. A. Albright, R. Hoffmann and J. K. Kochi, J. Am. Chem. Soc., 1976, 98, 7255–7265 CrossRef CAS; (b) A. Tamaki, S. A. Magennis and J. K. Kochi, J. Am. Chem. Soc., 1974, 96, 6140–6148 CrossRef CAS.
- (a) M. Gazvoda, M. Virant, B. Pinter and J. Kosmrlj, Nat. Commun., 2018, 9, 4814–4822 CrossRef PubMed; (b) M. H. Pérez-Temprano, J. A. Casares, A. R. de Lera, R. Álvarez and P. Espinet, Angew. Chem., Int. Ed., 2012, 51, 4917–4920 CrossRef PubMed.
- C. Bartolomé, Z. Ramiro, M. N. Peñas-Defrutos and P. Espinet, ACS Catal., 2016, 6, 6537–6545 CrossRef.
- Naked tricoordinate species are likely just fugacious. [AuRf2I(w)] intermediates (w = any adventitious labile ligand, such as the solvent), formed after I− dissociation, are more plausible.
- The kinetic model simplifies the Rf/I transmetalation as elemental reactions (D, E). However, it probably proceeds via a multi-step mechanism featuring dimers with Rf and/or I bridges.
- For oxidative addition of Ar–I to AuI see: (a) J. A. Cadge, J. F. Bower and C. A. Russell, Angew. Chem., Int. Ed., 2021, 60, 24976–24983 CrossRef CAS PubMed; (b) M. Joost, A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654–14657 CrossRef CAS PubMed; (c) Y. Yang, L. Eberle, F. F. Mulks, J. F. Wunsch, M. Zimmer, F. Rominger, M. Rudolph and A. S. K. Hashmi, J. Am. Chem. Soc., 2019, 141, 17414–17420 CrossRef CAS PubMed; (d) I. Fernández, L. P. Wolters and F. M. Bickelhaupt, J. Comput. Chem., 2014, 35, 2140–2145 CrossRef PubMed.
- K. Kang, S. Liu, C. Xu, Z. Lu, S. Liu, X. Leng, Y. Lan and Q. Shen, Organometallics, 2021, 40, 2231–2239 CrossRef CAS.
- For C–C vs. C–X coupling competition see: (a) M. S. Winston, W. J. Wolf and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 7921–7928 CrossRef CAS PubMed; (b) R. Bhattacharjee, A. Nijamudheen and A. Datta, Chem. – Eur. J., 2017, 23, 4169–4179 CrossRef CAS PubMed.
- M. Baya, A. Pérez-Bitrián, S. Martínez-Salvador, A. Martín, J. M. Casa, B. Menjón and J. Orduna, Chem. – Eur. J., 2018, 24, 1514–1517 CrossRef CAS PubMed.
- J. Vicente, A. Arcas, D. Bautista and P. G. Jones, Organometallics, 1997, 16, 2127–2138 CrossRef CAS.
- S. Coco, F. Díez-Expósito, P. Espinet, C. Fernández-Mayordomo, J. M. Martín-Álvarez and A. M. Levelut, Chem. Mater., 1998, 10, 3666–3671 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of the complexes including NMR spectra, microkinetic model, computational details and X-ray data. CCDC 2221886–2221889. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc06415d |
| This journal is © The Royal Society of Chemistry 2023 |