
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolybdenum(IV) β-diketonate complexes as highly active catalysts for allylic substitution reactions†
Fabio
Masero
 and
Victor
Mougel
*
and
Victor
Mougel
*
Department of Chemistry and Applied Biosciences, Laboratory of Inorganic Chemistry, ETH Zurich, 8093 Zurich, Switzerland. E-mail: mougel@inorg.chem.ethz.ch
First published on 14th March 2023
Abstract
The synthesis and characterization of a series of Mo(IV) bis-β-diketonate (Rdiket, R = Me, tBu, Ph) dichloride (RMoIVCl2) and bistriflate (RMoIV(OTf)2) complexes are reported. All complexes are characterized in solid and solution state by XRD and 1H NMR spectroscopy. We demonstrate that the bistriflate complexes constitute highly active catalysts for allylic substitution reactions.
Understanding the chemical and reactivity properties of six-coordinate Mo(IV) complexes is key to a better understanding of a wide number of Mo-based enzymatic systems.1–4 However, the vast majority of the reported six-coordinate Mo(IV) complexes are supported by neutral, strongly π-accepting donor ligands such as CO, NO or phosphines,5 and only a handful of mononuclear six-coordinate Mo(IV) compounds bearing anionic oxygen or sulfur donating ligands have been reported in literature,6–10 at the difference of much more common Mo(VI) counterparts. A case in point is the octahedral Mo(IV) bis-acetylacetonate (Mediket) dichloride complex, [MoIV(Mediket)2Cl2] (MeMoIVCl2), early reported11–13 but not structurally characterized. It was proposed to display cis coordination of the chloride ligands based on the analysis of IR stretches12,13 and suggested to be a coordination polymer type structure11 owing to its very low solubility.12,13 Interestingly, MeMoIVCl2 was proposed to act as a pre-catalyst for allylic substitution reactions when combined in situ with silver salts, such as AgOTf (TfO = trifluoromethylsulfonate) or AgSbF6, (vide infra).14–17 Nevertheless, the identity of the catalytically active species as well as the one generated upon addition of the silver salts had never been investigated. Here, we report the systematic preparation and characterization of a series of Mo(IV) complexes supported by β-diketonate ligands as well as the investigation of their catalytic activity for allylic substitution reactions. We found that while the parent Mo(IV) complexes are the catalytically most active species, in situ generated higher valent Mo oxo and nitrido complexes also yield catalytically active species, yet performing at much lower rates. We show that the careful design of catalytic reaction conditions based on this mechanistic analysis enabled efficient and fast catalysis at low catalyst loading.
By analogy with the reported synthesis of MeMoIVCl2,12,13 we explored here the preparation of a series of Mo(IV) dichloride bis-β-diketonate (Rdiket, R = Me, tBu, Ph, CF3) complexes by protonolysis of in situ generated [MoCl4(CH3CN)2] from [MoCl5] in refluxing acetonitrile (MeCN), with the corresponding β-diketone ligand. The resulting RMoIVCl2 complexes were isolated in pure form in 49%, 47% and 67% yields for R = Me, Ph and tBu respectively (Scheme 1). Nevertheless, the complexes displayed significant differences in solubility: while for R = Me and Ph, highly insoluble materials were obtained, the use of tBu groups as terminal substituents on the diketonate ligand enabled significantly improving the solubility of the complex in common solvents such as toluene or tetrahydrofuran (THF), facilitating its purification, isolation and characterization. We did not succeed in isolating the targeted complex for R = CF3 but instead identified products of a disproportionation reaction by single crystal X-ray diffraction (XRD, Fig. S3, ESI†). We hypothesize that the strong electron withdrawing properties of the CF3-substituted diketonate ligand do not allow stabilizing the electron-poor Mo(IV) complex.
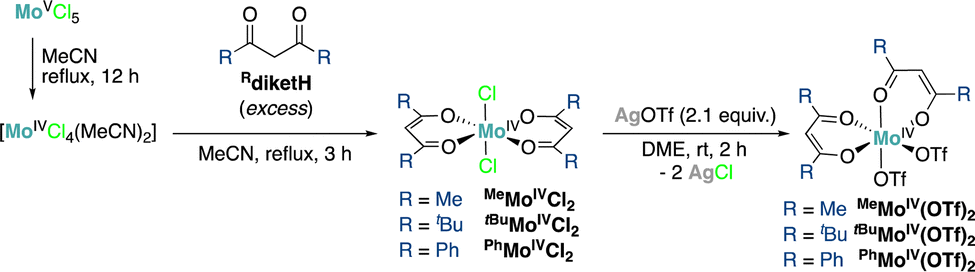 | ||
| Scheme 1 Synthesis of Mo(IV) β-diketonate complexes. | ||
Exchange of the chloride ligands with −OTf anions was carried out via salt metathesis: addition of two equivalents of solid AgOTf to a solution (R = tBu) or a suspension (R = Me, Ph) of RMoIVCl2 in 1,2-dimethoxyethane resulted in the precipitation of AgCl as a colorless solid and the formation of red-orange solutions.‡ Filtration followed by removal of volatiles under reduced pressure afforded the desired bistriflate (RMoIV(OTf)2) complexes (R = tBu, Me, Ph) as bright red-orange solids in 71%, 86% and 76% yields, respectively. Reactions with AgSbF6, previously used for the in situ generation of active catalysts for allylic substitution reactions,14 yielded complex mixtures. We attempted to determine the identity of these reaction byproducts in the case of R = tBu, revealing the occurrence of ligand scrambling and oxidation of the complex (Fig. S4, ESI†).
The solid state structures of RMoIVCl2 (R = Me, tBu, Ph) and RMoIV(OTf)2 (R = Me, tBu, Ph) complexes were characterized by single crystal XRD (Fig. 1). All compounds were found to crystallize as mononuclear coordination complexes, enabling us to unambiguously disclose the initially proposed formulation of MeMoIVCl2 as a coordination polymer.11 We observed that all the Mo(IV) dichloride complexes crystallized with the two chloride ligands trans to each other. Note that while both MeMoIVCl2 and tBuMoIVCl2 can be crystallized directly from the reaction mixture as distorted octahedral complexes with the two chloride ligands in apical positions with nearly identical bond metrics,PhMoIVCl2 precipitates from the reaction mixture as a dark black amorphous powder. We only achieved growing suitable crystals for XRD analysis by slow diffusion of npentane into a saturated pyridine solution. The solid state structure yet revealed the presence of an additional equatorial pyridine ligand, giving rise to a seven-coordinate complex.
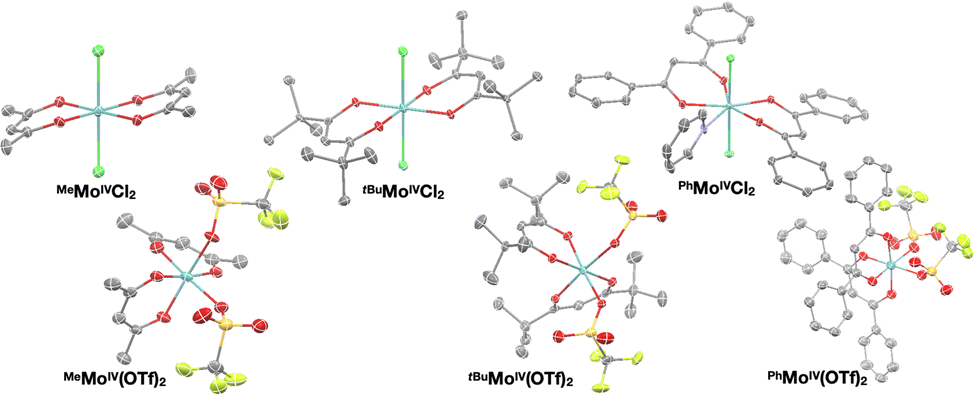 | ||
| Fig. 1 Crystal structures of dichloride- (top) and bistriflate complexes (bottom). Ellipsoids are drawn at the 50% probability level and all hydrogen atoms were omitted for clarity. | ||
XRD analysis of the bis-triflate complexes (R = Me, tBu) showed that both complexes display distorted octahedral geometries where the two triflate ligands are positioned cis to each other, in contrast to the trans coordination geometry observed for their dichloride counterparts. Despite this change in coordination geometry, the MoIV–O(Rdiket) average bond lengths are essentially identical when moving from the dichloride (dMo–O = 2.01 Å) to the bistriflate (dMo–O = 1.97 Å) complexes, and in the same range than in previously reported solid state structures of diketonate Mo complexes such as [MoIII(Mediket)3],18,19 [{MoVO(tBudiket)2}2-μ-O]20 and [MoVIO2(Mediket)2]21 (ΔdMo–O(diket) < 0.1 Å, see Table S6, ESI†). Worthy of note is the fact that, in the case of MeMoIVCl2, the chelate formed by the diketonate ligand and the Mo center significantly deviates from planarity (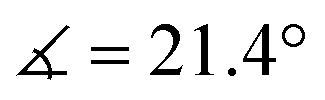 ), breaking up the complexes’ overall D4h symmetry (Fig. 2a). We further identified that the chelate is nearly planar (
), breaking up the complexes’ overall D4h symmetry (Fig. 2a). We further identified that the chelate is nearly planar (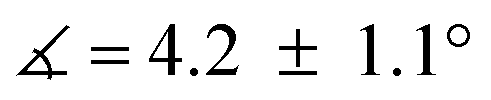 ) in lower-valent [MoIII(Mediket)3]18,19 while high-valent [MoVIO2(Mediket)2]21 displays a similar bending of the diketonate ligand (
) in lower-valent [MoIII(Mediket)3]18,19 while high-valent [MoVIO2(Mediket)2]21 displays a similar bending of the diketonate ligand (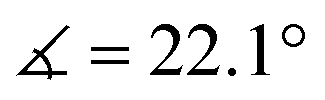 ). We propose that this deviation results from a non-zero orbital overlap between the HOMO of the anionic β-diketonate ligand22 and an empty orbital of appropriate symmetry of the metal fragment (an example with the dz2 orbital is highlighted in Fig. 2b). Population of these empty orbitals upon reduction of the Mo center will disfavor this interaction, resulting in a more planar coordination mode of the β-diketonate ligand. The larger angle observed in the case of tBuMoIVCl2 (
). We propose that this deviation results from a non-zero orbital overlap between the HOMO of the anionic β-diketonate ligand22 and an empty orbital of appropriate symmetry of the metal fragment (an example with the dz2 orbital is highlighted in Fig. 2b). Population of these empty orbitals upon reduction of the Mo center will disfavor this interaction, resulting in a more planar coordination mode of the β-diketonate ligand. The larger angle observed in the case of tBuMoIVCl2 (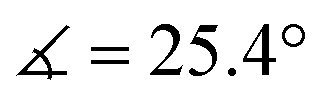 ) is in line with the stronger electron-donating properties of the tBu groups of its supporting ligand, increasing its π-donation capabilities. DFT calculations further support this hypothesis, reproducing well the trend, yet at a smaller magnitude (see Sections S4 and S7, and Fig. S44, ESI†).
) is in line with the stronger electron-donating properties of the tBu groups of its supporting ligand, increasing its π-donation capabilities. DFT calculations further support this hypothesis, reproducing well the trend, yet at a smaller magnitude (see Sections S4 and S7, and Fig. S44, ESI†).
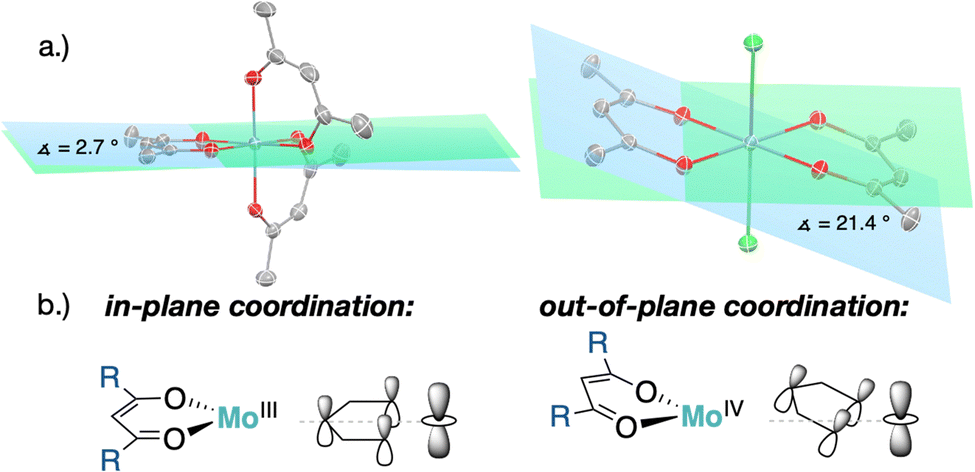 | ||
| Fig. 2 (a) Solid state structures of [MoIII(Mediket)3] (left)18 and MeMoIVCl2 (right) with highlighted bending angle (ellipsoids at the 50% probability level, all hydrogens omitted for clarity), (b) qualitative molecular orbital interaction diagram for bent vs. linear β-diketonate coordination to Mo. | ||
Both the Mo(IV) dichloride and bistriflate complexes are paramagnetic compounds, as highlighted by magnetic susceptibility measurements in both the solid and liquid state, suggesting a high spin d2 electronic configuration (Section S2, ESI†). Yet, this paramagnetic character did not impede the characterisation of the complexes by 1H and 19F (for bis-triflate complexes) NMR (Section S5, ESI†). In solution, both cis- and trans-isomers are observed for all the complexes synthesized, independently of the present X− ligand (X− = Cl− or TfO−) (Fig. S20–S36, ESI†). As an exception, in MeCN, the most polar solvent studied here, tBuMoIV(OTf)2 was found to be uniquely present as its cis isomer.
Owing to its increased solubility, the following detailed solution studies were only performed for the tBu-substituted diketonate complexes but the close similitudes between the 1H and 19F NMR spectra of all three complex families let us infer that the same conclusions can be drawn for the whole series. The ratio between the cis- and trans-isomers determined by 1H NMR and summarized for tBuMoIVCl2 and tBuMoIV(OTf)2 in Table 1 appeared to be both dependent of the anionic X− ligand and the solvent used. A significant influence of the polarity of the media on the cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :trans ratio was observed, with more polar solvents favoring the cis isomer. The behavior at different temperatures was studied using variable temperature NMR (Fig. S39–S41, ESI†). The Cl− and TfO− complexes displayed significantly different behavior: for tBuMoIVCl2 the cis:
:trans ratio was observed, with more polar solvents favoring the cis isomer. The behavior at different temperatures was studied using variable temperature NMR (Fig. S39–S41, ESI†). The Cl− and TfO− complexes displayed significantly different behavior: for tBuMoIVCl2 the cis:![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) trans ratio is nearly independent on temperature, while for tBuMoIV(OTf)2 the cis isomer is more favored at low temperature, in agreement with the cis-configuration observed in the single crystal structure. In case of the triflate complexes, structural information could be extracted from 19F NMR spectroscopy. Even in a coordinating solvent such as MeCN, the triflate ligands retain a characteristic 19F chemical shift (e.g. δ ≈ −40 ppm for tBuMoIV(OTf)2), very distinct from free, non-coordinated −OTf anions (δ ≈ −80 ppm). This result suggests that the triflate ligands remain bound to Mo, in contrast to previous literature examples,23 and highlights the strong Lewis acidity of the Mo center in this series.
trans ratio is nearly independent on temperature, while for tBuMoIV(OTf)2 the cis isomer is more favored at low temperature, in agreement with the cis-configuration observed in the single crystal structure. In case of the triflate complexes, structural information could be extracted from 19F NMR spectroscopy. Even in a coordinating solvent such as MeCN, the triflate ligands retain a characteristic 19F chemical shift (e.g. δ ≈ −40 ppm for tBuMoIV(OTf)2), very distinct from free, non-coordinated −OTf anions (δ ≈ −80 ppm). This result suggests that the triflate ligands remain bound to Mo, in contrast to previous literature examples,23 and highlights the strong Lewis acidity of the Mo center in this series.
:trans) ratios at 298 K determined by 1H NMR
| C6D6 | CD3Cl | CD2Cl2 | CD3CN | |
|---|---|---|---|---|
| tBuMoIVCl2 | 1:2.1 |
1.2:1 |
2.7:1 |
4.9:1 |
| tBuMoIV(OTf)2 | 1.2:1 |
2.5:1 |
10.7:1 |
cis only |
We investigated the catalytic activity for allylic substitution reactions of the in situ generated mixtures in the presence of RMoIVCl2/AgOTf (R = Me, Ph, tBu) in a 1:2 ratio, using the coupling of cinnamyl alcohol (1) with electron-rich aromatics as carbon nucleophiles (e.g. phenol (2), anisole or p-cresol) as prototypical reactions. All three complexes proved efficient to promote the coupling (Section S3, ESI†). As no significant difference in catalytic activity could be detected with Mo(IV) complexes supported by the other β-diketonate ligands investigated here, we decided to restrict the more detailed reactivity and mechanistic studies to the most soluble complexes of the series, supported by tBudiket ligands.
Using tBuMoIV(OTf)2 as a catalyst, full conversion was observed at a catalyst loading of 2 mol% within less than one hour at ambient temperature and the expected coupling products were isolated as mixtures of ortho- and para regio-isomers in good yields (92% for the coupling of 1 and 2, Scheme 2a). The catalytic activity of tBuMoIV(OTf)2 was further tested in a 3,3-cyclocoupling reaction of allylic alcohol 3 with p-cresol. This reaction – of interest to generate chroman moieties found notably in several pharmaceutical drugs24–26 – may be described as a domino reaction, where an allylic substitution is followed by a Lewis acid-catalyzed cyclization step. This reaction was found challenging in the original report using the in situ generated catalyst obtained with RMoIVCl2/AgSbF6 (1:2 ratio) mixtures, with a reported yield of only 27%.141H NMR analysis of the reaction using 2 mol% of tBuMoIV(OTf)2 as a catalyst revealed the formation of the initial C–C coupling product 4 and its full conversion to the cyclized product 5 over prolonged reaction times (Scheme 2b and Fig. S5, S6, ESI†). Chroman 5 could be isolated in 62% yield, a significant improvement with respect to the previously reported yield obtained using in situ generated catalysts (vide supra). This reaction was previously reported to be catalyzed by Lewis-27,28 or Brønsted29 acids under comparably harsher conditions, typically requiring elevated temperature (up to 150 °C), high catalyst loadings (5–15 mol%) or a large excess of p-cresol (up to 30 equiv.). To the best of our knowledge, the conditions for the preparation of chroman 5 described here (2 mol% catalyst loading, p-cresol (1.3 equiv.), 25 °C, no additives) are among the mildest reported to date.
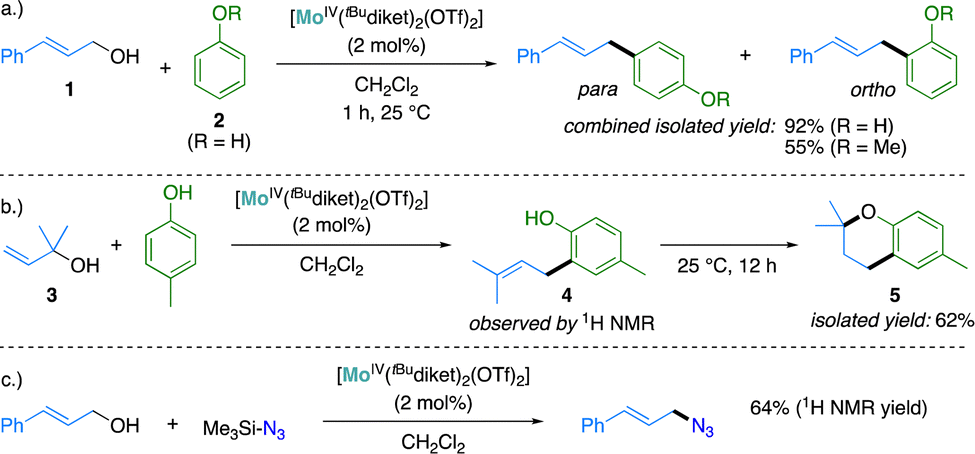 | ||
| Scheme 2 Substitution of allylic alcohols with aromatic nucloephiles (a and b) and with trimethylsilyl azide (c) catalyzed by tBuMoIV(OTf)2. | ||
Intrigued by the change of color of the reaction mixture from red to green under catalytic conditions, we attempted identifying the new species generated in situ and responsible of this color change. During the catalytic substitution of allylic alcohols, water (H2O) is expected as the main by-product. Here, we were able to show that [MoV(O)(tBudiket)2(OTf)] (tBuMoV(O)OTf) is formed upon hydrolysis of tBuMoIV(OTf)2 (Scheme 3). To probe this assumption, we independently investigated the reactivity of tBuMoIV(OTf)2 with stoichiometric amounts of H2O. When a slight excess of H2O (5 equiv.) was added to a THF solution of tBuMoIV(OTf)2, a broad signal at 2.6 ppm appeared in the 1H NMR spectrum. Cooling this reaction mixture in acetonitrile at −35 °C afforded bright green crystals of tBuMoV(O)OTf (Fig. S19, ESI†). Interestingly, while the NMR and UV-Vis spectra of tBuMoIV(OTf)2 do not change upon addition of (excess) 2, addition of stoichiometric amounts of allylic alcohol 1 to tBuMoIV(OTf)2 led to the fast appearance of the same broad signal at δ = 2.6 ppm in the 1H NMR spectrum of the mixture, pointing towards the in situ generation of tBuMoV(O)OTf (Fig. S18, ESI†). Relatedly, 1H NMR and UV-Vis spectra recorded in situ in the conditions used for the catalytic allylic substitution reaction of 1 with 2 (1 mol% tBuMoIV(OTf)2) confirm the fast formation of tBuMoV(O)OTf under catalytic conditions (Fig. S14 and S17, ESI†). Further, the conversion of the substrates is still observed when tBuMoV(O)OTf is the main Mo species, suggesting that it may also be catalytically active. To test this hypothesis, tBuMoV(O)OTf (1 mol%) was used as a catalyst for the allylic substitution of 1 with 2. Full conversion was observed after 4 h at room temperature. However, in situ1H NMR monitoring of the reaction revealed a much lower product formation rate, as if using tBuMoIV(OTf)2 as the catalyst (Fig. S13, ESI†). A long induction period of ca. 40 min was observed, suggesting that tBuMoV(O)OTf is not the actual catalytically active species, but a resting state of the catalyst. In addition, time-dependent UV-Vis spectroscopic studies carried out under catalytic conditions (Fig. S15, ESI†) did not exhibit significant changes over the course of the reaction, ruling out the in situ regeneration of the original Mo(IV) catalyst, and suggesting the formation of a new catalytically competent species. Assuming that the generation of the Mo(V) resting state is mainly driven by the accumulation of H2O released during the catalytic run, we attempted to optimize reaction conditions to prevent its formation. The presence of 4 Å molecular sieves (MS) as an additive to standard catalytic conditions using the Mo(IV) complex, led to much faster catalysis (98% conversion after 7 minutes, Fig. S12, ESI†). This suggests that the H2O by-product can be efficiently displaced by the MS, to significantly increase the lifetime of the Mo(IV) catalyst and disfavor the formation of Mo(V) oxo resting states. Based on these findings, we propose that under standard catalytic conditions, two separated catalytic cycles involving either the Mo(IV) or the Mo(V) complex as active species are operative (Scheme S2, ESI†), even though for catalyst loadings >1 mol%, the latter can be neglected due to its comparably slow product formation rate.
 | ||
| Scheme 3 Reactivity of tBuMoIV(OTf)2 with allylic alcohol or H2O and TMSN3. | ||
tBuMoIV(OTf)2 is also an active catalyst promoting C–N bond formation reactions, enabling the formation of allylic azides (Scheme 2c and Fig. S7, ESI†). Interestingly, the catalytic reaction mixture did not display the characteristic change of color to green observed in C–C coupling reaction, but instead turned dark brown, suggesting the formation of at least one additional new species. We explored the reactivity of the second reactant, TMSN3 (TMS = SiMe3) with tBuMoIV(OTf)2. At the difference of 2, which did not display any significant reactivity with tBuMoIV(OTf)2, a strong change of color to dark brown was observed immediately after addition of stoichiometric amounts of TMSN3 (1.2 equiv.) to tBuMoIV(OTf)2. Single crystal XRD measurements pointed to the formation of a new Mo(VI) species, namely [MoVI(N)(tBudiket)2(OTf)] (tBuMoVI(N)OTf, Fig. S19, ESI†). The fact, that under catalytic conditions, TMSN3 is present in large excess with respect to tBuMoIV(OTf)2, suggests that the above-described reaction is likely to be present in significant amounts in catalytic conditions. Similarly to what observed with tBuMoV(O)OTf, tBuMoVI(N)OTf can catalyze allylic substitutions but is much more sluggish (ca. 25% conversion after 12 h, Fig. S8, ESI†) than the parent Mo(IV) complex.
The in-depth investigation of the coordination chemistry properties of a series of β-diketonate Mo(IV) complexes enabled to identify that the strongly Lewis acidic complexes RMoIV(OTf)2 are highly active catalysts for the substitution of allylic alcohols. Reactivity tests combined with in situ spectroscopic studies revealed that high-valent Mo(V) oxo or Mo(VI) nitrido complexes are quickly formed in situ under catalytic conditions. These species can also act as pre-catalysts for the targeted reactions, yet at the expense of much slower rates. This observation enabled to further rationally optimize reaction conditions to lower the rate of formation of these species and respectively improve reaction rates and conversion.
We acknowledge funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 853064). We deeply thank Dr Michael Wörle, Dr René Verel, and Dr Christopher Gordon for their help with XRD, NMR, and DFT studies, respectively.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Hille, J. Hall and P. Basu, Chem. Rev., 2014, 114, 3963–4038 CrossRef CAS PubMed.
- G. Schwarz, R. R. Mendel and M. W. Ribbe, Nature, 2009, 460, 839–847 CrossRef CAS PubMed.
- L. B. Maia and J. J. Moura, J. Biol. Inorg. Chem., 2015, 20, 403–433 CrossRef CAS.
- L. B. Maia and J. J. Moura, J. Biol. Inorg. Chem., 2011, 16, 443–460 CrossRef CAS PubMed.
- C. G. Young, Comprehensive Coordination Chemistry II, Pergamon, 2003, pp. 415–527 Search PubMed.
- F. A. Cotton and G. Schmid, Inorg. Chem., 1997, 36, 2267–2278 CrossRef CAS PubMed.
- J. Bendix and A. Bogevig, Inorg. Chem., 1998, 37, 5992–6001 CrossRef CAS PubMed.
- E. Solari, C. Maltese, M. Latronico, C. Floriani, A. Chiesi-Villa and C. Rizzoli, J. Chem. Soc., Dalton Trans., 1998, 2395–2400 RSC.
- M. J. Bezdek and P. J. Chirik, Dalton Trans., 2016, 45, 15922–15930 RSC.
- J. Song, Q. Liao, X. Hong, L. Jin and N. Mezailles, Angew. Chem., Int. Ed., 2021, 60, 12242–12247 CrossRef CAS PubMed.
- M. L. Larson and F. W. Moore, Inorg. Chem., 1964, 3, 285–286 CrossRef CAS.
- G. Doyle, Inorg. Chem., 1971, 10, 2348–2350 CrossRef CAS.
- A. van den Bergen, K. S. Murray and B. O. West, Aust. J. Chem., 1972, 25, 705–713 CrossRef CAS.
- A. V. Malkov, P. Spoor, V. Vinader and P. Kocovsky, J. Org. Chem., 1999, 64, 5308–5311 CrossRef CAS PubMed.
- A. V. Malkov, B. P. Farn, N. Hussain and P. Kočovský, Collect. Czech. Chem. Commun., 2001, 66, 1735–1745 CrossRef CAS.
- A. V. Malkov and P. Kočovský, Collect. Czech. Chem. Commun., 2001, 66, 1257–1268 CrossRef CAS.
- B. Nay, M. Collet, M. Lebon, C. Chèze and J. Vercauteren, Tetrahedron Lett., 2002, 43, 2675–2678 CrossRef CAS.
- A. Y. Ledneva, S. B. Artemkina, D. A. Piryazev and V. E. Fedorov, J. Struct. Chem., 2015, 56, 1021–1023 CrossRef CAS.
- C. L. Raston and A. H. White, Aust. J. Chem., 1979, 32, 507–512 CrossRef CAS.
- A. Y. Ledneva, S. B. Artemkina, P. A. Stabnikov, L. V. Yanshole and V. E. Fedorov, J. Struct. Chem., 2017, 58, 758–762 CrossRef CAS.
- D. H. Johnston, C. King, A. Seitz and M. Sethi, IUCrData, 2021, 6, 210778 CrossRef PubMed.
- S. Evans, A. Hamnett, A. F. Orchard and D. R. Lloyd, Faraday Discuss. Chem. Soc., 1972, 54, 227–250 RSC.
- S. J. Smith, C. M. Whaley, T. B. Rauchfuss and S. R. Wilson, Inorg. Chem., 2006, 45, 679–687 CrossRef CAS.
- S. Salamone, C. Colin, I. Grillier-Vuissoz, S. Kuntz, S. Mazerbourg, S. Flament, H. Martin, L. Richert, Y. Chapleur and M. Boisbrun, Eur. J. Med. Chem., 2012, 51, 206–215 CrossRef CAS PubMed.
- M. Sawa and H. Harada, Curr. Med. Chem., 2006, 13, 25–37 CrossRef CAS PubMed.
- M. M. Singh, Med. Res. Rev., 2001, 21, 302–347 CrossRef CAS PubMed.
- Y. Yamamoto and K. Itonaga, Org. Lett., 2009, 11, 717–720 CrossRef CAS PubMed.
- S. Madabhushi, R. Jillella, K. R. Godala, K. K. R. Mallu, C. R. Beeram and N. Chinthala, Tetrahedron Lett., 2012, 53, 5275–5279 CrossRef CAS.
- Y. Ishino, M. Mihara, N. Hayakawa, T. Miyata, Y. Kaneko and T. Miyata, Synth. Commun., 2001, 31, 439–448 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2239585, 2240098–2240102. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc00572k |
| ‡ When chloride triflate exchange reactions were performed in THF, rapid polymerization of the solution occurred, highlighting the high Lewis-acidity of the formed Mo(IV) triflate complexes. |
| This journal is © The Royal Society of Chemistry 2023 |