
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDerivatization of sumanenetrione through Lewis acid-mediated Suzuki–Miyaura coupling and an unprecedented ring opening†
Junyi
Han
a,
Yuta
Uetake
 ab,
Yumi
Yakiyama
*ab and
Hidehiro
Sakurai
*ab
ab,
Yumi
Yakiyama
*ab and
Hidehiro
Sakurai
*ab
aDivision of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: hsakurai@chem.eng.osaka-u.ac.jp; yakiyama@chem.eng.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 15th March 2023
Abstract
A series of sumanenetrione derivatives were synthesized through Lewis acid-mediated Suzuki–Miyaura cross-coupling. Their optical properties reflected the significantly strong electron-accepting ability of sumanenetrione. The bowl strain of the 2-hydroxyphenyl derivative brought the ring-opening response adjacent to the substituent even at room temperature without any activation, which generally requires harsh conditions.
Sumanenetrione (1) is a bowl-shaped aromatic ketone in which all three benzylic positions of sumanene are fully oxygenated (Fig. 1).1 The unique properties of 1 are represented by the strong electron-accepting ability to have the first reduction potential at −0.92 V, and the second one at −1.43 V (vs. Fc/Fc+, Fc: ferrocene).2 These values are more positive than those of boron-doped polycyclic aromatic hydrocarbons,3 fluorenone derivatives,4 and fullerene derivatives.5 Another unique feature of 1 is that the intersystem crossing (ISC) from the triplet state to S0 was faster than that of the planar triplet ketones such as triplet fluorenone. This is caused by the larger spin orbital coupling (SOC) than the planar system due to the curved structure.6 These unique electronic features make 1 a promising material for application to electron injection7 and organic phosphorescent materials.8,9
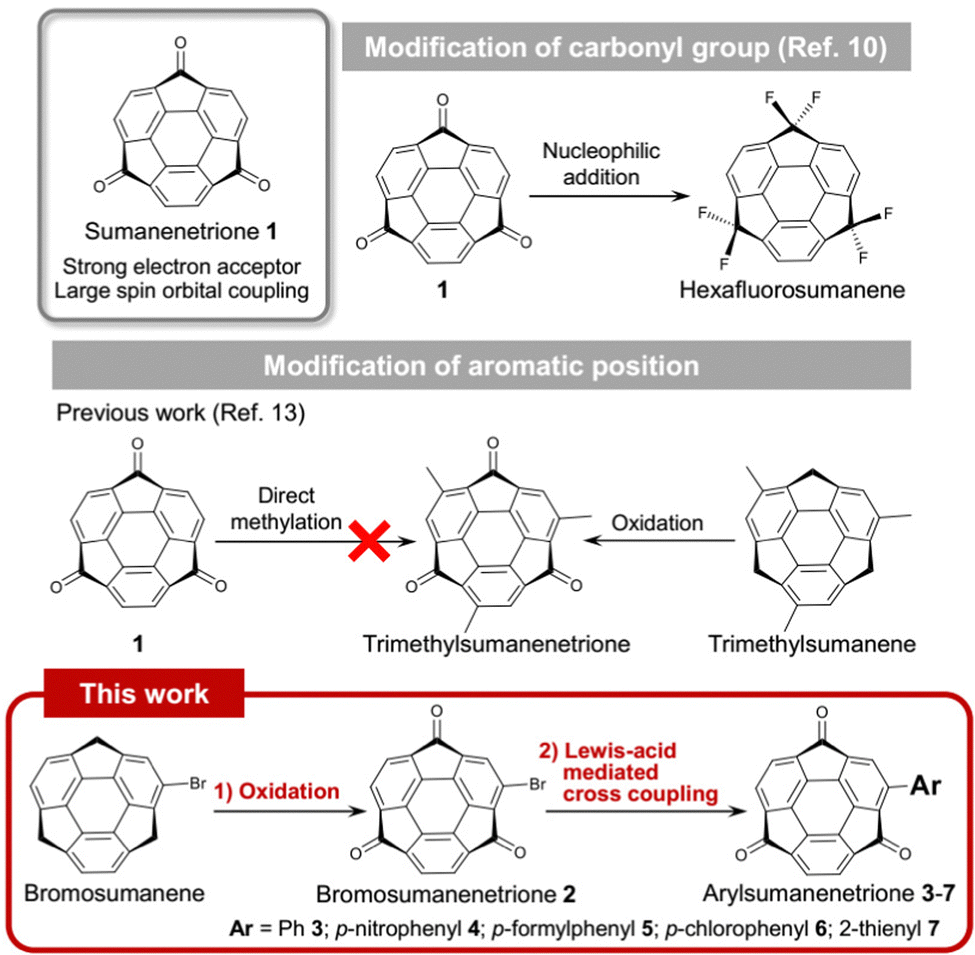 | ||
| Fig. 1 Modification of 1 at the carbonyl group and aromatic periphery. | ||
In terms of the utilization of molecular synthesis, 1 also has the potential to be a precursor for functionalized sumanenes by the modification of three carbonyl groups by nucleophilic addition reactions. Indeed, the preparation of hexafluorosumanene10 from 1 brought a new opportunity to utilize fluorosumanenes as Debye-type dielectric responsive materials.11,12 In contrast, the study of chemical modification of 1 at the aromatic periphery is scarce. Only one derivative having a sumanenetrione moiety, trimethylsumanenetrione, has been prepared by oxidation of trimethylsumanene.13 The problem of the chemical modification of 1 lies in the poor reactivity at the aromatic periphery, especially against aromatic electrophilic substitutions due to its electron-deficient character compared to the non-oxygenated sumanene. Furthermore, the susceptibility of 1 to bases significantly restricts the choice of the transformation methods (vide infra). Here, we propose a new chemical modification method of 1 employing Lewis acid-mediated Suzuki–Miyaura coupling reaction via mono-brominated derivative, bromosumanenetrione (2), to obtain various coupling products 3–7 (Fig. 1). In addition, we unexpectedly observed pentagonal ring opening in the preparation of 2-hydroxyohenyl derivative 8 even at room temperature within 1 min without any activation.
Bromosumanenetrione 2 was prepared from previously reported bromosumanene14 through Ru-mediated oxygenation in a 43% yield (Scheme 1 and Scheme S1, ESI†). First, phenylation of 2 by the Suzuki−Miyaura cross-coupling was investigated (Scheme S2 and Table S1, ESI†). When the reaction was carried out under the general Suzuki−Miyaura coupling reaction conditions using the combination of Pd(PPh3)4 and K2CO3 in a mixture of THF and water, 2 was immediately consumed to give a complicated mixture instead of the desired coupling product, indicating that 2 was unstable under basic K2CO3/H2O conditions (entry 1, ESI†). Changing the base to CsF did not solve the problem (entry 2). Therefore, we tried several representative protocols using aryl borate, which do not require the addition of an external base. In entry 3, the reaction was conducted with choline hydroxide (ChOH), an ionic liquid, which consists of a tertiary amine and hydroxide15 and potassium trifluoro(phenyl)borate instead of boronic acid. However, it also turned out to be ineffective. The trial to use a combination of sodium phenyl boronate with toluene to perform the reaction under anhydrous and base-free conditions also resulted in no reaction (entry 4).16 A further experiment using highly stable and soluble potassium phenyltriolborate17 under milder conditions still did not give a positive result (entry 5). We finally arrived at the recently reported Lewis acid-mediated Suzuki−Miyaura coupling reaction, which is applicable to base-susceptible substrates.18 When 2 was treated with phenylboronic acid in the presence of PdCl2(amphos)2 and ((tmeda)Zn(OH)(OTf))3 in THF at 80 °C for 1 h, the desired coupling product 3 was successfully obtained in 89% yield (entry 6). This success of the coupling reaction motivated us to make a variety of derivatizations with several kinds of aromatic compounds to find that all products 4–7 were obtained in good yields (Scheme 2 and Schemes S3 and S4, ESI†).19
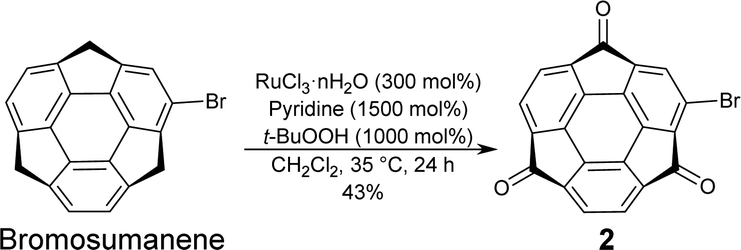 | ||
| Scheme 1 Synthetic scheme of bromosumanenetrione 2. | ||
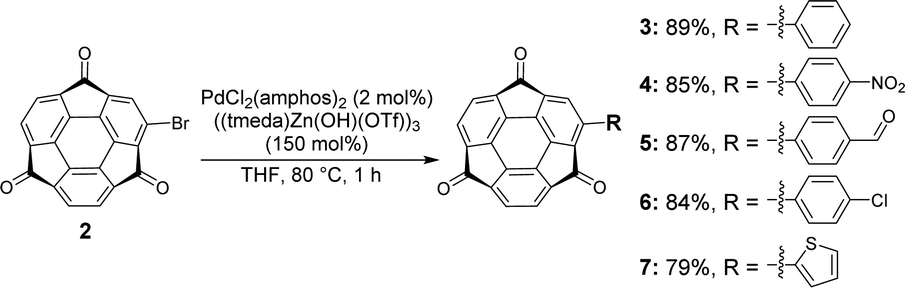 | ||
| Scheme 2 Synthesis of 4–7. | ||
The UV-vis absorption and photoluminescence (PL) spectra of 3–7 in CH2Cl2 solution (10−5 mol L−1) are shown in Fig. 2. All the compounds showed two main absorption bands, one in the UV range from 250–400 nm and the other in the visible range from 450–650 nm (Fig. 2a). The longer wavelength peak of all the phenyl derivatives 3–6 appeared at nearly the same position at 525 nm. More electron-donating thiophene-introduced 7 showed a clear red shift of the corresponding absorption peak to reach 580 nm. DFT calculations performed at the B3LYP/6-311G(d,p) level of theory revealed that, in all cases, the highest occupied molecular orbital (HOMO) was distributed over the entire molecular backbone. In contrast, the lowest unoccupied molecular orbital (LUMO) was distributed exclusively on the sumanenetrione moiety (Fig. 3), indicating that the absorption bands in the visible region were derived from the charge-transfer from the substituents to the sumanenetrione moiety. The time-dependent density functional theory (TD-DFT) calculations performed at the B3LYP/6-311G(d,p) level of theory well explained this assumption (Fig. S1 and S2, ESI†). Notably, even the nitrophenyl group, recognized as a typical strong electron-accepting unit, worked as the donor moiety in this system. This clearly demonstrated the extraordinary electron-accepting ability of 1.
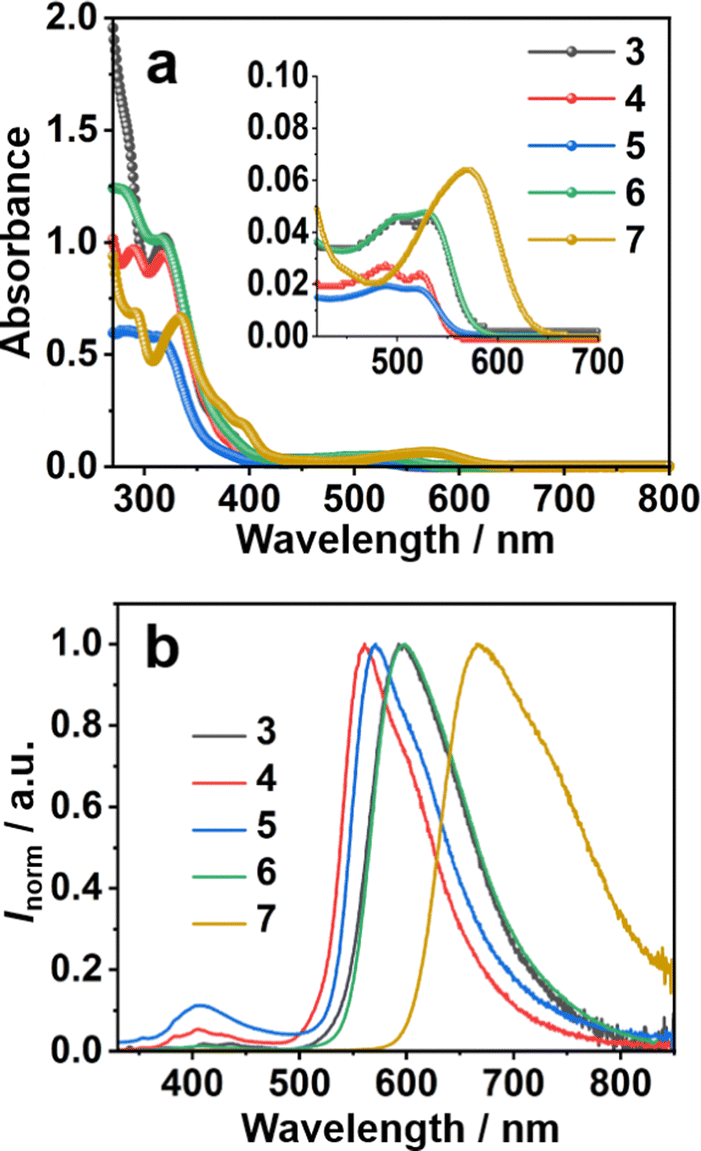 | ||
| Fig. 2 (a) UV-vis absorption spectra and (b) emission spectra (λex = 320 nm) of 3–7 in CH2Cl2 (1.0 × 10−5 mol L−1). | ||
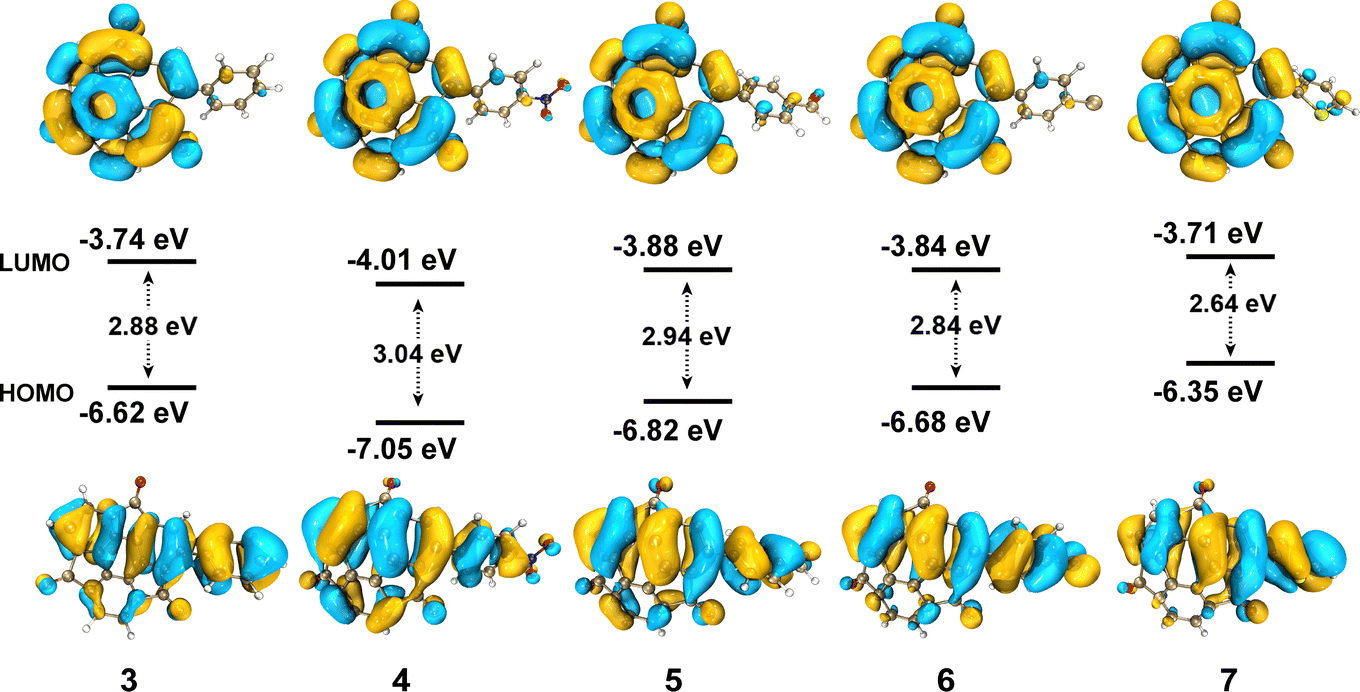 | ||
| Fig. 3 The HOMO and LUMO of 3–7 obtained by DFT calculations performed at the B3LYP/6-311G(d,p) level of theory. Isovalue = 0.02. | ||
In the case of the emission spectra, two peaks were observed in 3–7, one at 410 nm with weak intensity and the other ranging from 560 to 668 nm (Fig. 2b). The latter band showed a red shift as the electron-donating ability of the substituent increased. Such HOMO and LUMO orientation indicated that the luminescence at 410 nm came from the locally-excited (LE) state of the sumanenetrione moiety.20 In comparison, the luminescence from 560 to 668 nm was thought to be from a charge-transfer state. The HOMO and LUMO energy gap decreased with the electron-donating thiophene group in 7 through the increment of the HOMO level, which is consistent with the experimental UV absorption and PL spectra.
Among the coupling products, 7 showed significant positive solvatoluminochromism as a function of the permittivity of the organic solvent (Fig. 4). Changing the solvent from hexane to acetonitrile caused almost a 100 nm red shift of the emission from the visible to NIR region, indicating a significant intermolecular charge-transfer (ICT) nature in the excited state. The Lippert–Mataga plot21,22 of 7 referring to the Stokes shift Δν and the solvent polarizability Δf revealed that 7 shows two-section lines with an inflection point. In low polarity solvents, 7 exhibited a significant dipole moment change (Δμ is 11.22 D). However, in high polarity solvents, the dipole moment changes of 7 were much smaller than in the case of less polar solvents (Δμ is 1.21 D, Table S2, ESI†). This phenomenon of 7 exhibiting both charge transfer and local excited properties is probably caused by the hybridization of the LE and CT states.23,24
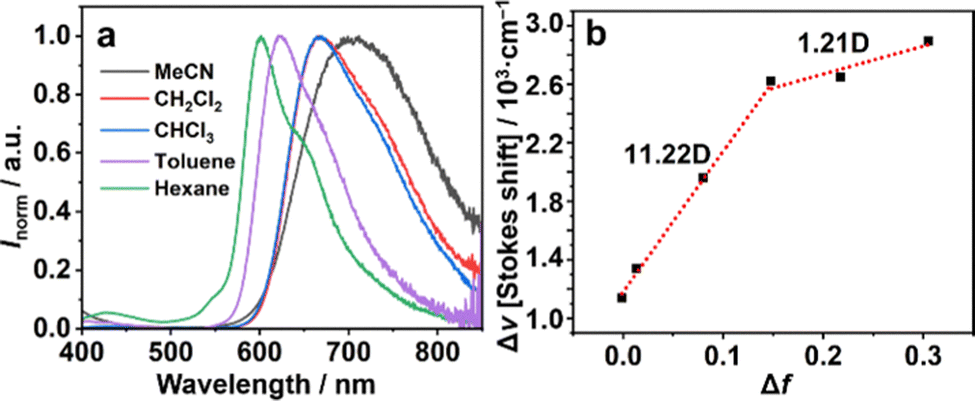 | ||
| Fig. 4 (a) PL spectra (λex = 320 nm) of 7 in four types of solvents (c = 10−5 M). (b) Linear fitting of Lipert–Mataga plots of 7. | ||
Following the optical measurement results of electron-donating thiophene-introduced 7, further preparation of 2-hydroxyphenyl derivative 8, which is expected to be a valuable ligand for complexation with metal ions, was investigated.25–28 The reaction was performed under the same conditions using 2-hydroxyphenylboronic acid. The crude 1H NMR spectrum proved the formation of the desired product 8 (see ESI†). However, it was found that 8 was unexpectedly unstable and converted to another product during the purification process. In particular, the use of MeOH gave an identifiable and isolable product 9. When the percentage of MeOH to THF was increased to 90%, the reaction took place even within one minute to give 9 in 47% yield (Scheme 3). Although the data quality was insufficient, single crystal X-ray structure analysis results also supported the characterized structure, in which the adjacent pentagonal ring of the substituent opened to produce a more planar molecular skeleton (Scheme 3 and Fig. S3, ESI†). Generally, the corresponding ring-opening reaction from the planar fluorenone to biphenyl carboxylic acid requires much harsh conditions with a strong base (i.e., 270–300 °C under a molten state of sodium hydroxide and potassium hydroxide conditions).29 In contrast, the ring-opening of 8 proceeds even at room temperature without the addition of acid or base, indicating that the strain release effect probably worked as a driving force for the ring-opening reaction.30,31 Since the C−C bond cleavage proceeded exclusively at the position adjacent to the 2-hydroxyphenyl group, its participation in the reaction mechanism was strongly expected. A possible reaction passway of the ring-opening with a supportive proposition of DFT calculations is initiated by the formation of hemiketal intermediates generated by the nucleophilic addition of MeOH to the carbonyl carbon (Fig. S4, ESI†). Further C−C bond cleavage was probably caused via an intramolecular proto-deacylation-like mechanism, which was assisted by forming a hydrogen-bonded six-membered ring structure.
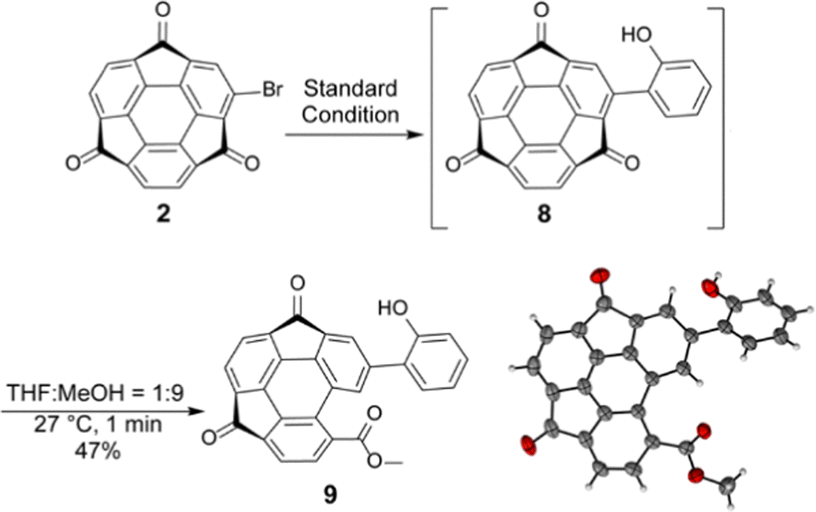 | ||
| Scheme 3 Unexpected ring opening from 8 and the thermal displacement ellipsoid plot of 9 in a crystal structure with 50% probability. | ||
In summary, we overcame the inherent difficulty of chemical modification of 1 using the Lewis-acid mediated Suzuki−Miyaura coupling reaction via bromosumanenetrione to obtain various coupling products. The optical properties of the products revealed that the sumanenetrione moiety has significant electron-accepting ability. Even at room temperature, the high strain of the bowl caused the ring-opening reaction on 2-hydroxyphenyl-substituted sumanenetrione at the adjacent pentagonal ring to occur without activation. This study opens a new way for the rational use of sumanenetrione as a new type of curved-π electron-acceptor. Further investigation via fine energy gap tuning by appropriate electron-donor introduction will afford a great opportunity for the potential application of sumanenetrione in organic optoelectronic devices.
We thank Prof. Takashi Niwa and Prof. Takamitsu Hosoya (Tokyo Medical and Dental University) for providing ((tmeda)Zn(OH)(OTf))3. We also thank Prof. Mao Minoura (Rikkyo University) for his help in the single crystal X-ray diffraction data collection of 9. This study was supported by a Grant-in-Aid for Transformative Research Areas “Science of 2.5 Dimensional Materials” (No. JP21H05233) and JSPS KAKENHI (JP19H00912, JP20H00400 and JP20K15279), a Mitsubishi Gas Chemical Award in Synthetic Organic Chemistry, Japan, and RIKEN-Osaka University Science and Technology Hub Collaborative Research Program from RIKEN and Osaka University. The theoretical calculations were performed at the Research Centre for Computational Science, Okazaki, Japan (Project: 22-IMS-C068). H. J. thanks the Otsuka Toshimi Scholarship Foundation for kindly providing scholarships (19-S5 and 20-S4).
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Amaya, M. Hifumi, M. Okada, Y. Shimizu, T. Moriuchi, K. Segawa, Y. Ando and T. Hirao, J. Org. Chem., 2011, 76, 8049–8052 CrossRef CAS PubMed.
- S. Higashibayashi, B. B. Shrestha, Y. Morita, M. Ehara, K. Ohkubo, S. Fukuzumi and H. Sakurai, Chem. Lett., 2014, 43, 1297–1299 CrossRef CAS.
- C. Mutzel, J. M. Farrell, K. Shoyama and F. Wurthner, Angew. Chem., Int. Ed., 2022, 61, e202115746 CrossRef PubMed.
- N. A. Kukhta, D. Volyniuk, J. V. Grazulevicius and G. Sini, J. Mater. Chem. C, 2018, 6, 1679–1692 RSC.
- D. A. Heredia, A. M. Durantini, J. E. Durantini and E. N. Durantini, J. Photochem. Photobiol., C, 2022, 51, 100471 CrossRef CAS.
- K. Kanahara, M. D. M. R. Badal, S. Hatano, M. Abe, S. Higashibayashi, N. Takashina and H. Sakurai, Bull. Chem. Soc. Jpn., 2015, 88, 1612–1617 CrossRef CAS.
- S. E. Koops, B. C. O’Regan, P. R. Barnes and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 4808–4818 CrossRef CAS PubMed.
- H. F. Higginbotham, M. Okazaki, P. de Silva, S. Minakata, Y. Takeda and P. Data, ACS Appl. Mater. Interfaces, 2021, 13, 2899–2907 CrossRef CAS.
- T. Hosono, N. O. Decarli, P. Z. Crocomo, T. Goya, L. E. de Sousa, N. Tohnai, S. Minakata, P. de Silva, P. Data and Y. Takeda, J. Mater. Chem. C, 2022, 10, 4905–4913 RSC.
- B. M. Schmidt, B. Topolinski, S. Higashibayashi, T. Kojima, M. Kawano, D. Lentz and H. Sakurai, Chem. – Eur. J., 2013, 19, 3282–3286 CrossRef CAS PubMed.
- M. Li, J. Wu, K. Sambe, Y. Yakiyama, T. Akutagawa, T. Kajitani, T. Fukushima, K. Matsuda and H. Sakurai, Mater. Chem. Front., 2022, 6, 1752–1758 RSC.
- M. Li, X. Chen, Y. Yakiyama, J. Wu, T. Akutagawa and H. Sakurai, Chem. Commun., 2022, 58, 8950–8953 RSC.
- R. Tsuruoka, S. Higashibayashi, T. Ishikawa, S. Toyota and H. Sakurai, Chem. Lett., 2010, 39, 646–647 CrossRef CAS.
- T. Amaya, S. Seki, T. Moriuchi, K. Nakamoto, T. Nakata, H. Sakane, A. Saeki, S. Tagawa and T. Hirao, J. Am. Chem. Soc., 2009, 131, 408–409 CrossRef CAS PubMed.
- S. R. Joo, G. T. Kwon and S. H. Kim, Asian J. Org. Chem., 2020, 9, 584–587 CrossRef CAS.
- A. N. Cammidge, V. H. M. Goddard, H. Gopee, N. L. Harrison, D. L. Hughes, C. J. Schubert, B. M. Sutton, G. L. Watts and A. J. Whitehead, Org. Lett., 2006, 8, 4071–4074 CrossRef CAS.
- Y. Yamamoto, M. Takizawa, X.-Q. Yu and N. Miyaura, Angew. Chem., Int. Ed., 2008, 120, 942–945 CrossRef.
- T. Niwa, Y. Uetake, M. Isoda, T. Takimoto, M. Nakaoka, D. Hashizume, H. Sakurai and T. Hosoya, Nat. Catal., 2021, 4, 1080–1088 CrossRef CAS.
- The synthesis of phenylsumanenetrione was also achievable by Negishi coupling under the base-free conditions despite the lower yield than under the present Suzuki–Miyaura coupling conditions. See the details in ESI†.
- Y. Morita, S. Nakao, S. Haesuwannakij, S. Higashibayashi and H. Sakurai, Chem. Commun., 2012, 48, 9050–9052 RSC.
- E. v Lippert, Z. Elektrochem., Ber. Bunsenges. Phys. Chem., 1957, 61, 962–975 CAS.
- N. Mataga, Y. Kaifu and M. Kozumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- Y. Gao, S. Zhang, Y. Pan, L. Yao, H. Liu, Y. Guo, Q. Gu, B. Yang and Y. Ma, Phys. Chem. Chem. Phys., 2016, 18, 24176–24184 RSC.
- S. Zhang, L. Yao, Q. Peng, W. Li, Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. Wang, P. Lu, F. Li, S. Su, B. Yang and Y. Ma, Adv. Funct. Mater., 2015, 25, 1755–1762 CrossRef CAS.
- A. Kasprzak, A. Kowalczyk, A. Jagielska, B. Wagner, A. M. Nowicka and H. Sakurai, Dalton Trans., 2020, 49, 9965–9971 RSC.
- A. Kasprzak and H. Sakurai, Dalton Trans., 2019, 48, 17147–17152 RSC.
- A. Kasprzak and H. Sakurai, Chem. Commun., 2021, 57, 343–346 RSC.
- A. Kasprzak, A. Tobolska, H. Sakurai and W. Wroblewski, Dalton Trans., 2022, 51, 468–472 RSC.
- E. R. Andrews, R. W. Fleming, J. M. Grisar, J. C. Kihm, D. L. Wenstrup and G. D. Mayer, J. Med. Chem., 1974, 17, 882–886 CrossRef CAS PubMed.
- L. Ge, C. Zhang, C. Pan, D. X. Wang, D. Y. Liu, Z. Q. Li, P. Shen, L. Tian and C. Feng, Nat. Commun., 2022, 13, 5938 CrossRef CAS PubMed.
- J. Turkowska, J. Durka and D. Gryko, Chem. Commun., 2020, 56, 5718–5734 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed methods for the experiments and computational analyses, supporting figures and tables, and single crystal X-ray analysis results. See DOI: https://doi.org/10.1039/d3cc00394a |
| This journal is © The Royal Society of Chemistry 2023 |