
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChloroacetyl boronate N-tosylhydrazone as a versatile synthetic building block†
Ryuya
Miyazaki
a,
Kei
Muto
 *b and
Junichiro
Yamaguchi
*a
*b and
Junichiro
Yamaguchi
*a
aDepartment of Applied Chemistry, Waseda University, 513 Wasedatsurumakicho, Shinjuku, Tokyo 162-0041, Japan. E-mail: junyamaguchi@waseda.jp
bWaseda Institute for Advanced Study, Waseda University, 513 Wasedatsurumakicho, Shinjuku, Tokyo 162-0041, Japan. E-mail: keimuto@aoni.waseda.jp
First published on 23rd May 2023
Abstract
Chloroacetyl boronate N-tosylhydrazone (CABT) has been synthesized as a reagent to generate a diverse range of molecular skeletons. CABT can undergo a series of transformations involving nucleophilic substitution of the chloride and coupling of the N-tosylhydrazones, followed by boryl group functionalization. We further demonstrated that this CABT reagent could enable a diversity-oriented synthesis.
Given the importance of diversity-oriented synthesis, the development of synthetic methods and reagents capable of generating a diverse range of molecular skeletons is highly valuable not only in organic synthesis, but also in medicinal chemistry.1 In order to support this growing field, building blocks allowing for multi-directional transformations would be highly desirable.
The utility of organoborons in organic synthesis is undoubtedly high, exemplified by known C–B bond transformations such as Suzuki–Miyaura coupling (C–C bond formation),2 Chan–Lam coupling (C–N bond formation),3 oxidation (C–O bond formation), and Matteson homologation.4 Meanwhile, the chemistry of N-tosylhydrazones has rapidly evolved in recent years.5–10 Beside the classic reactivity of N-tosylhydrazone as a dinitrogen source, they can also generate highly reactive diazo species through a base-promoted decomposition process known as Bamford–Stevens reaction.11 By capturing these diazo species with transition-metal catalysts, N-tosylhydrazones have been multi-directionally transformed in Z–H (Z = O, NR, S) insertion,12 cross-coupling,13,14 and other transformations.15,16 Given the reactivities of organoborons and N-tosylhydrazones, we envisioned that combining these two functional groups into a single molecule17 could yield a versatile molecular building block for organic synthesis. In this context, two related examples have recently appeared (Fig. 1A). Liu developed azaborine-diazomethane and demonstrated that the diazo moiety of this chemical was amenable to C–C bond formations.18 Another example is boryl formyl-N-tosylhydrazone, which reacted under Pd-catalyzed cross-coupling with benzyl bromides to furnish valuable alkenylborons.19,20 Despite these pioneering works, the structural diversity of the obtained compounds is far from satisfactory. To build a wider range of molecular skeletons, we herein report the development of chloroacetyl boronate N-tosylhydrazone (CABT; Fig. 1B). Our design of this reagent is rather straightforward: (a) the chlorine atom could be substituted with nucleophiles, leading to various substituted boryl-N-tosylhydrazones; (b) the N-tosylhydrazone moiety of the generated synthetic intermediate could then be converted by transition-metal-catalyzed21 reactions and dinitrogen-containing heterocycle synthesis; (c) the remaining boryl group could be used to assemble with other building blocks. As such, the designed CABT reagent is expected to function as a versatile two-carbon unit in organic synthesis.
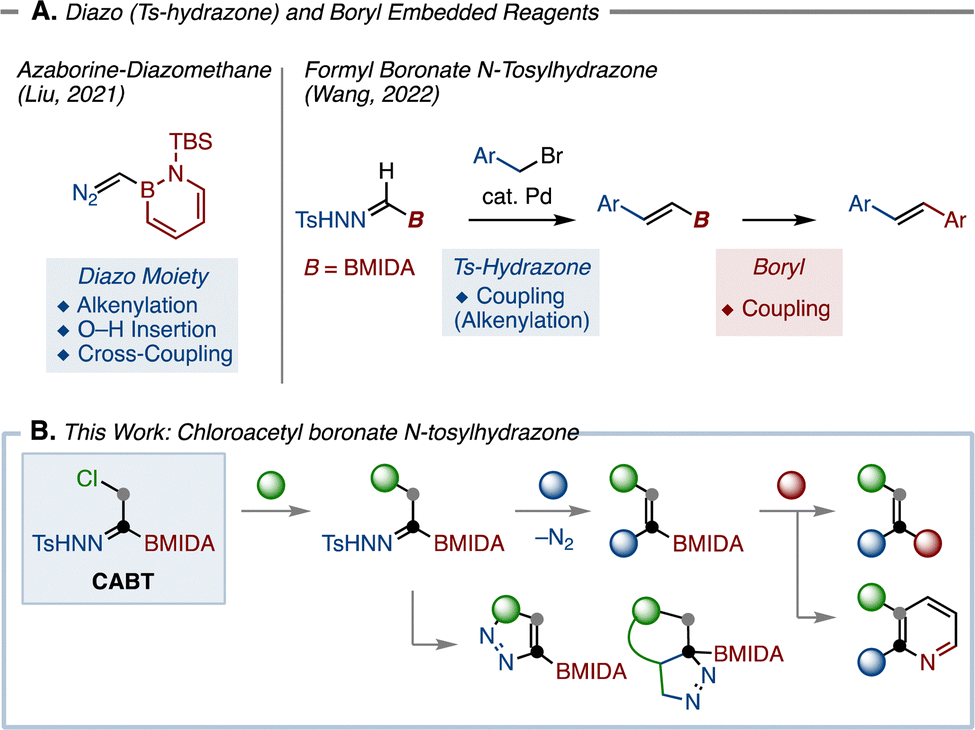 | ||
| Fig. 1 (A) Borylated diazo species (B) Chloroacetyl boronate N-tosylhydrazone. | ||
Our study commenced with the synthesis of CABT (Scheme 1). At the outset, it was expected that CABT can be synthesized from an acylboron species.22 Considering the stability of acylborons, we decided to include the well-known MIDA boronate (MIDA = methyliminodiacetic acid) into the structure of CABT. Following Yudin's α-bromoacetyl boronate synthesis,23 ring-opening chlorination of commercially available BMIDA-ethylene oxide produced chlorohydrin 2. This chlorohydrin was then oxidized by Dess–Martin periodinane (DMP) to form acylboron 3. Finally, the reaction of 3 with tosylhydrazide produced the desired CABT in a good yield. Of note, this procedure is highly reproducible and scalable, and does not require any silica-gel column purification. The obtained CABT was a white solid that exhibited high stability at room temperature.
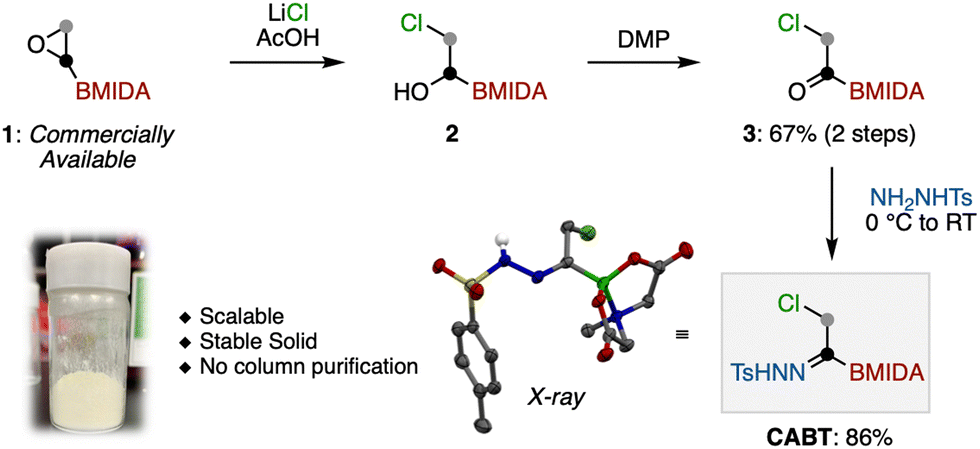 | ||
| Scheme 1 The synthesis of CABT. | ||
The reaction of CABT was next investigated. First, substitution of the C–Cl bond was examined (Scheme 2). Because CABT has three reactive functional groups on only two carbons, it was imperative to find reaction conditions that are highly chemo-selective. Fortunately, we found that Coltart's copper-catalyzed substitution reaction using Grignard reagents matched well with this purpose.24 With a slight modification of this protocol, primary alkyl (4a–4e) and secondary alkyl (4f, 4g) group substituted boryl-N-tosylhydrazones were synthesized in moderate to good yields by using the corresponding Grignard reagents. The introduction of aryl and alkenyl groups was also possible, furnishing 4h and 4i in 86% and 77% yield, respectively. Of note, this substitution afforded 4 as a E/Z mixture, which can be isomerized to Z-isomer by treating with a catalytic amount of Ts-hydrazide (see the Supplementary Information). Additionally, the substitution reaction with thiol and malonate in the presence of base led to the formation of 4j and 4k, respectively.
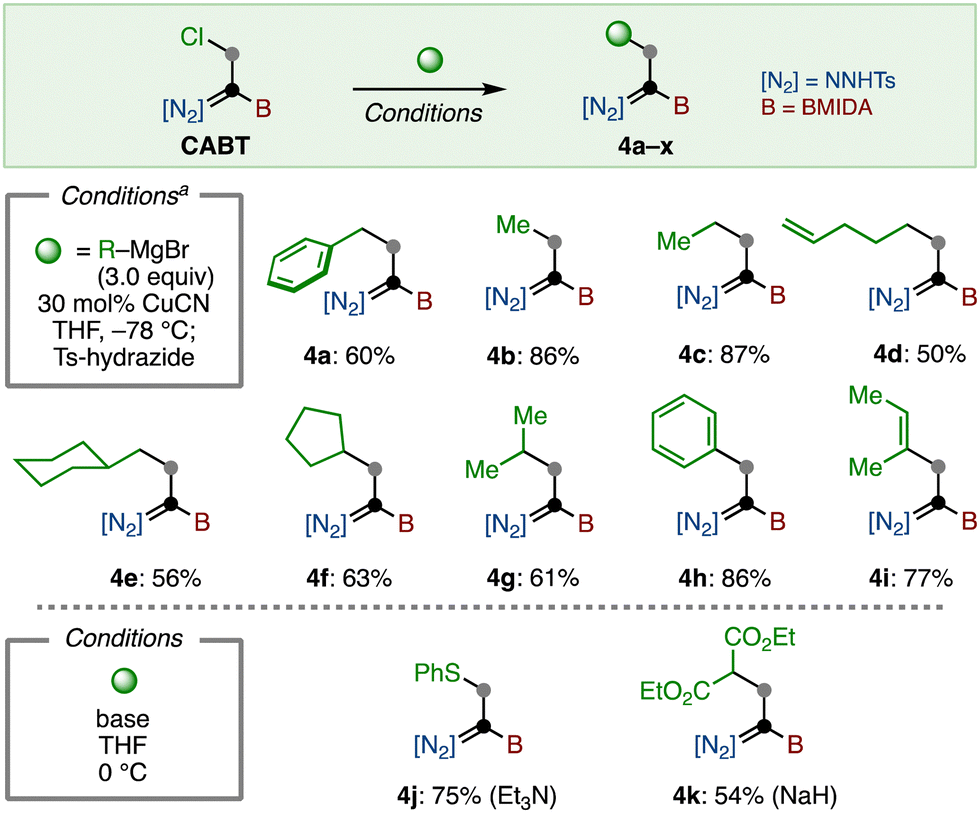 | ||
| Scheme 2 Substitution of C–Cl bond. a Obtained as a Z-isomer. | ||
Next, we pursued the transformation of the N-tosylhydrazone group (Scheme 3). The Wang group reported a coupling reaction of boryl-N-tosylhydrazones with benzyl bromides under the influence of a palladium catalyst.20,25 However, these conditions were not operative for our boryl-N-tosylhydrazone 4. After exploration of reaction conditions, we found that Barluenga's coupling reaction of 4a with p-tolyl bromide in the presence of Pd2(dba)3, XPhos, and LiOtBu in 1,4-dioxane15 produced desired coupling product 5a in 9% as a mixture of E/Z isomers. In this case, alkyl cyanide 6 was generated as a main product in 79% yield. Undesired 6 was presumably generated through the formation of borate, which was subsequently transformed to alkyl cyanide with the elimination of tosylamide. To suppress this unexpected side reaction, we next employed NaH as a less nucleophilic base than LiOtBu. Delightfully, we obtained desired product 5 in an improved 55% yield. Finally, changing the solvent from dioxane to 1,2-dimethoxyethane (DME) led to 85% yield of 5a. Using these identified conditions (with NaH in DME), we evaluated the substrate scope of this procedure (Scheme 3). It was found that this coupling reaction was not influenced by the substituent at the α-position of the hydrazone. For example, both primary and secondary alkyl-substituted alkenyl borons (5a–5e) were synthesized in good to moderate yields, albeit with poor E/Z selectivity. α-Phenylated product 5f was also obtained in 73% yield. Next, various aryl bromides were reacted with 4a under the same conditions. Ortho- and meta-tolyl bromides were suitable substrates, generating the corresponding alkenyl borons 5g and 5h in good yields. This reaction was not significantly affected by the electronic nature of the aryl bromides, as para-alkoxy- and trifluoromethyl-phenyl bromides gave products 5i and 5j in good yields. Ester (5k) and carbamate (5l) functional groups were tolerated under these conditions. Introduction of thiophene (5m) was also successful. In addition to aryl bromide, aryl triflates reacted under these conditions. Furthermore, estrone triflate coupled to boryl-N-tosylhydrazone 4a to furnish 5n in 63% yield (E/Z = 69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :31).
:31).
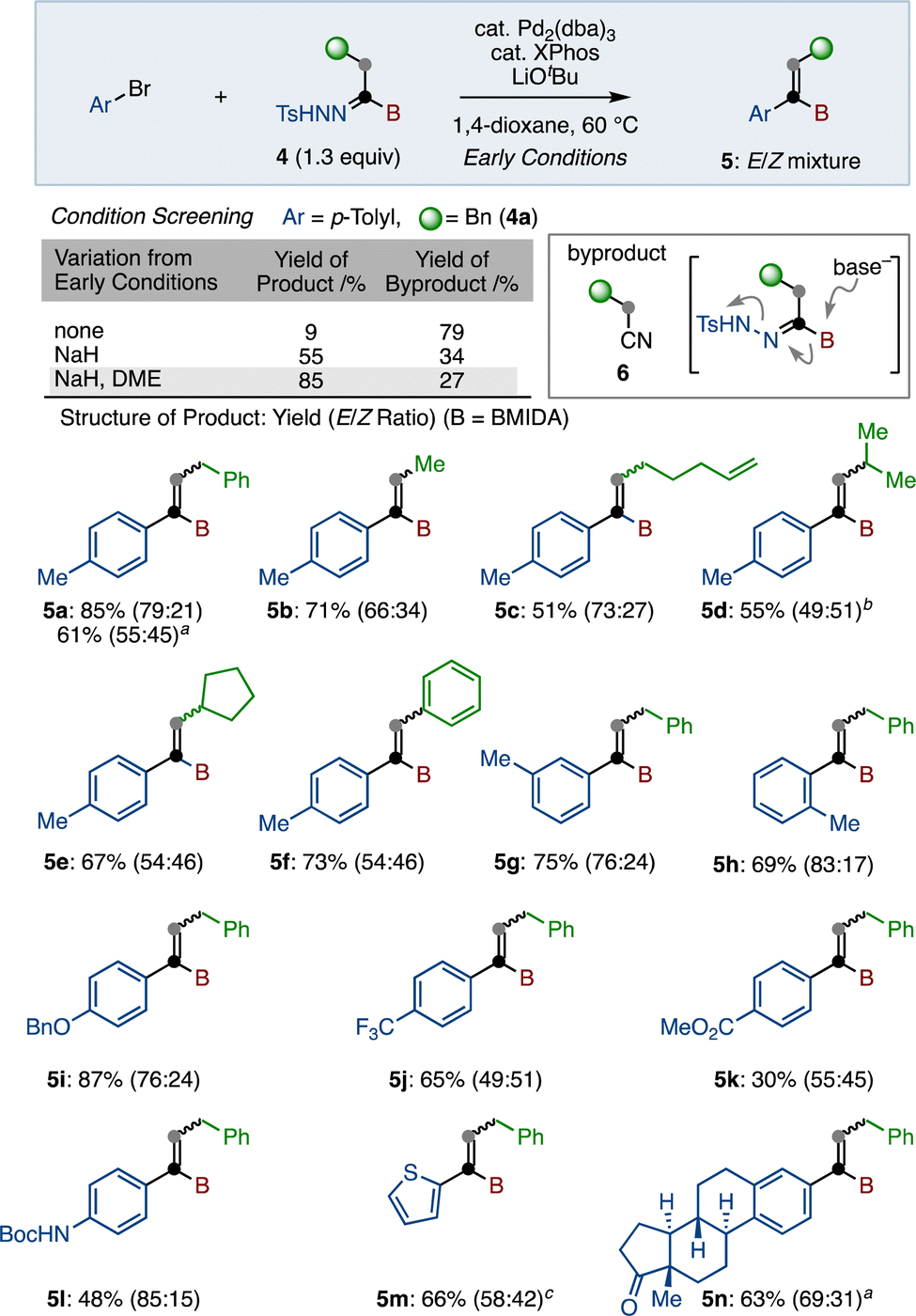 | ||
| Scheme 3 Cross-coupling of N-tosylhydrazones. a Aryl–OTf was used instead of Aryl–Br. b Reaction conducted in 1,4-dioxane. c Reaction conducted at 70 °C. | ||
Finally, we demonstrated that the remaining boryl functional group on alkenyl MIDA-boronate 5a can be engaged in chemical reaction (Scheme 4). Suzuki–Miyaura coupling of 5a and p-anisyliodide furnished tri-substituted alkene 7 in 87% yield. Similarly, Rh-catalyzed conjugate addition of 5a to cyclohexenone was possible, giving β-substituted ketone 8 in a good yield.26 After the conversion of the MIDA moiety to pinacolate 9, a copper-mediated azidation as well as a copper-catalyzed formal [4+2] cycloaddition27 generated alkenyl azide 10 and multiply substituted quinoline 12, respectively.
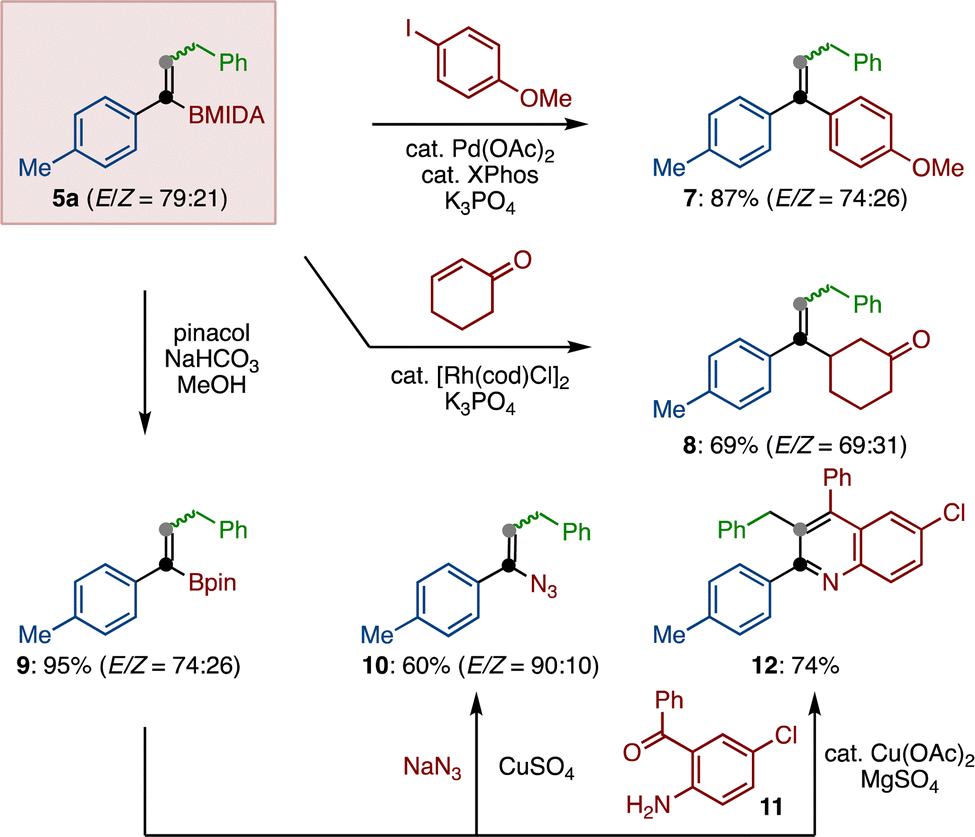 | ||
| Scheme 4 Transformation of the boron moiety. | ||
The CABT reagent can also be used to prepare a wide range of organoboron building blocks (Scheme 5). Following sequential transformations of 1 through chloride substitution and cross-coupling, alkenylboron 5a was prepared. This alkenylboron 5a was subjected to mCPBA epoxidation to form borylated epoxide 13 in 92% yield. The obtained 13 was further converted to aldehyde 14 in 44% yield through a Lewis acid-mediated ring-opening followed by a Meerwein rearrangement.28 On the other hand, the chloride substitution of CABT with p-toluidine under air provided borylated 1,2,3-triazole 15, albeit in 26% yield.29 After the chloride substitution of CABT with Grignard reagent, the boryl-N-tosylhydrazones bearing an alkenyl chain were found to cyclize under the thermal conditions in the presence of catalytic amount of rhodium, giving borylated diazo-cycloalkanes 16–18.30
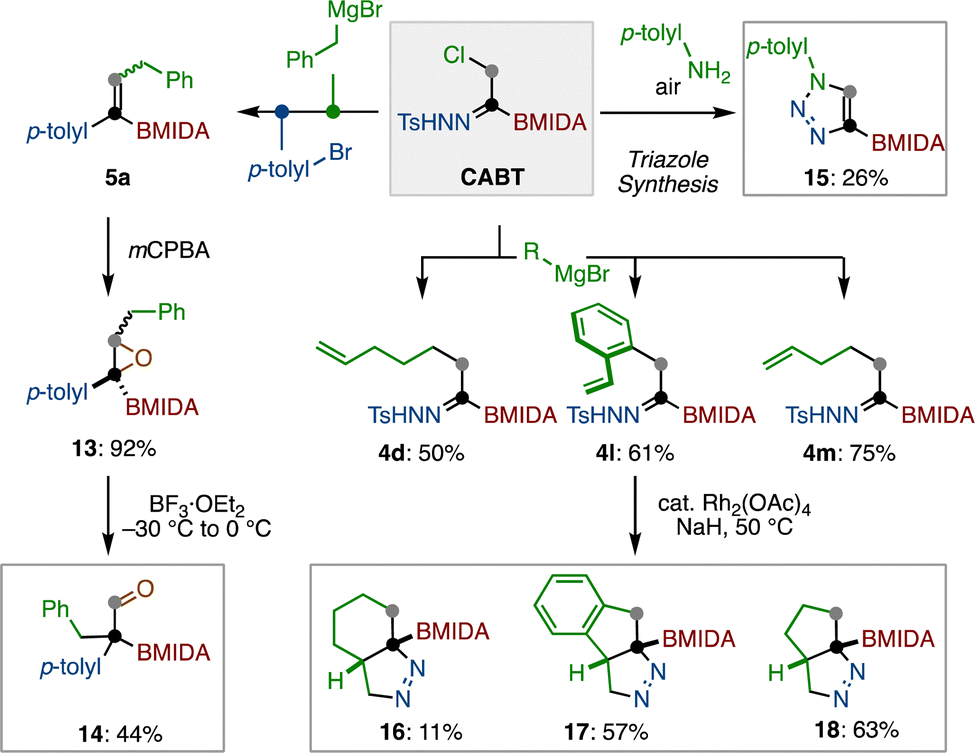 | ||
| Scheme 5 Synthesis of various boron-containing compounds. | ||
In summary, we have developed a versatile synthetic building block, chloroacetyl boronate N-tosylhydrazone (CABT). Practical preparation and facile handling of CABT are valuable traits of this new reagent. Sequential transformations of CABT (chloride substitution, N-tosylhydrazone coupling and [3+2] cyclization, as well as post-boryl transformations) were established, allowing producing a diverse range of molecular skeletons. Given the importance of diversity-oriented synthesis in medicinal chemistry this reagent could help accelerate scaffold explorations in hit-to-led efforts in early drug discovery.
This work was supported by JSPS KAKENHI Grant Number JP21H05213 (Digi-TOS) (to J.Y.), and JP21K05079 (to K.M.). This work was partly supported by JST ERATO Grant Number JPMJER1901 (to J.Y.). Dr Kenta Kato is acknowledged for helping X-ray crystallographic analysis. The Materials Characterization Central Laboratory in Waseda University is acknowledged for the support of HRMS measurement.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46–58 CrossRef PubMed; (b) W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 CrossRef PubMed; (c) C. J. Gerry and S. L. Schreiber, Nat. Rev. Drug Discovery, 2018, 17, 333–352 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- M. J. West, J. W. B. Fyfe, J. C. Vantourout and A. J. B. Watson, Chem. Rev., 2019, 119, 12491–12523 CrossRef CAS PubMed.
- J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31–55 CAS.
- J. Barluenga and C. Valdés, Angew. Chem., Int. Ed., 2011, 50, 7486–7500 CrossRef CAS PubMed.
- Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560–572 RSC.
- Y. Xia and J. Wang, Chem. Soc. Rev., 2017, 46, 2306–2362 RSC.
- M. Jia and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 9134–9166 CrossRef CAS PubMed.
- Y. Xia and J. Wang, J. Am. Chem. Soc., 2020, 142, 10592–10605 CrossRef CAS PubMed.
- J. Radolko, P. Ehlers and P. Langer, Adv. Synth. Catal., 2021, 363, 3616–3654 CrossRef CAS.
- J. R. Fulton, V. K. Aggarwal and J. de Vicente, Eur. J. Org. Chem., 2005, 1479–1492 CrossRef CAS.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Angew. Chem., Int. Ed., 2010, 49, 4993–4996 CrossRef CAS PubMed.
- For a review, see: (a) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810–13889 CrossRef CAS PubMed ; For selected examples, see: ; (b) J. Barluenga, P. Moriel, C. Valdés and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 5587–5590 CrossRef CAS PubMed; (c) J. Barluenga, M. Tomás-Gamasa, P. Moriel, F. Aznar and C. Valdés, Chem. – Eur. J., 2008, 14, 4792–4795 CrossRef CAS PubMed; (d) E. Brachet, A. Hamze, J.-F. Peyrat, J.-D. Brion and M. Alami, Org. Lett., 2010, 12, 4042–4045 CrossRef CAS PubMed; (e) M. Roche, A. Hamze, O. Provot, J.-D. Brion and M. Alami, J. Org. Chem., 2013, 78, 445–454 CrossRef CAS PubMed; (f) D. P. Ojha and K. R. Prabhu, J. Org. Chem., 2012, 77, 11027–11033 CrossRef CAS PubMed; (g) J. Barluenga, M. Escribano, P. Moriel, F. Aznar and C. Valdés, Chem. – Eur. J., 2009, 15, 13291–13294 CrossRef CAS PubMed.
- K. Ishitobi, K. Muto and J. Yamaguchi, ACS Catal., 2019, 9, 11685–11690 CrossRef CAS.
- (a) H. Kato, I. Musha, M. Komatsuda, K. Muto and J. Yamaguchi, Chem. Sci., 2020, 11, 8779–8784 RSC; (b) A. Yanagimoto, Y. Uwabe, Q. Wu, K. Muto and J. Yamaguchi, ACS Catal., 2021, 11, 10429–10435 CrossRef CAS.
- N. F. C. Ritchie, A. J. Zahara and S. M. Wilkerson-Hill, J. Am. Chem. Soc., 2022, 144, 2101–2106 CrossRef CAS PubMed.
- Borylated diazo compounds have been synthesized, but have never been investigated for their reactivity. Examples, see: (a) U. Schöllkopf, B. Bánhidai, H. Frasnelli, R. Meyer and H. Beckhaus, Justus Liebigs Ann. Chem., 1974, 1767–1783 CrossRef; (b) M. P. Arthur, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1991, 113, 5856–5857 CrossRef CAS; (c) J.-M. Sotiropoulos, A. Baceiredo, K. H. von Locquenghien, F. Dahan and G. Bertrand, Angew. Chem., Int. Ed. Engl., 1991, 30, 1154–1156 CrossRef; (d) A. Ansorge, D. J. Brauer, H. Bürger, T. Hagen and G. Pawelke, Angew. Chem., Int. Ed. Engl., 1993, 32, 384–385 CrossRef; (e) L. Weber, H. B. Wartig, H.-G. Stammler and B. Neumann, Organometallics, 2001, 20, 5248–5250 CrossRef CAS.
- Y. Liu, R. Puig de la Bellacasa, B. Li, A. B. Cuenca and S.-Y. Liu, J. Am. Chem. Soc., 2021, 143, 14059–14064 CrossRef CAS PubMed.
- Y. M. Ivon, I. V. Mazurenko, Y. O. Kuchkovska, Z. V. Voitenko and O. O. Grygorenko, Angew. Chem., Int. Ed., 2020, 59, 18016–18022 CrossRef CAS PubMed.
- S. Li, M. Li, S.-S. Li and J. Wang, Chem. Commun., 2022, 58, 399–402 RSC.
- Very recently, a catalytic reaction of in situ-generated boryl Au-carbene was reported, see: Y. Zheng, J. Jiang, Y. Li, Y. Wei, J. Zhang, J. Hu, Z. Ke, X. Xu and L. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202218175 CrossRef CAS PubMed.
- D. Wu, J. Taguchi, M. Tanriver and J. W. Bode, Angew. Chem., Int. Ed., 2020, 59, 16847–16858 CrossRef CAS PubMed.
- S. Adachi, S. K. Liew, C. F. Lee, A. Lough, Z. He, J. D. S. Denis, G. Poda and A. K. Yudin, Org. Lett., 2015, 17, 5594–5597 CrossRef CAS PubMed.
- J. M. Hatcher and D. M. Coltart, J. Am. Chem. Soc., 2010, 132, 4546–4547 CrossRef CAS PubMed.
- Q. Xiao, J. Ma, Y. Yang, Y. Zhang and J. Wang, Org. Lett., 2009, 11, 4732–4735 CrossRef CAS PubMed.
- F. Albrecht, O. Sowada, M. Fistikci and M. M. K. Boysen, Org. Lett., 2014, 16, 5212–5215 CrossRef CAS PubMed.
- Y. Li, Z. Cao, Z. Wang, L. Xu and Y. Wei, Org. Lett., 2022, 24, 6554–6559 CrossRef CAS PubMed.
- Z. He and A. K. Yudin, J. Am. Chem. Soc., 2011, 133, 13770–13773 CrossRef CAS PubMed.
- H.-W. Bai, Z.-J. Cai, S.-Y. Wang and S.-J. Ji, Org. Lett., 2015, 17, 2898–2901 CrossRef CAS PubMed.
- This reaction proceeded under thermal conditions without catalyst, but the yields of product were lower than the catalytic reactions. See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: For experimental procedures, spectroscopic data for compounds, and crystallographic data in CIF. CCDC 2257615 (for CABT) and 2257609 (for 4a). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc02086j |
| This journal is © The Royal Society of Chemistry 2023 |