
The effect of substituents and molecular aggregation on the room temperature phosphorescence of a twisted π-system†
Cristian A. M.
Salla
a,
Giliandro
Farias
b,
Ludmilla
Sturm
c,
Pierre
Dechambenoit
c,
Fabien
Durola
 c,
Aydemir
Murat
de,
Bernardo
de Souza
*b,
Harald
Bock
*c,
Andrew P.
Monkman
*d and
Ivan H.
Bechtold
*a
c,
Aydemir
Murat
de,
Bernardo
de Souza
*b,
Harald
Bock
*c,
Andrew P.
Monkman
*d and
Ivan H.
Bechtold
*a
aDepartment of Physics, Universidade Federal de Santa Catarina, 88040-900 Florianópolis, SC, Brazil. E-mail: ivan.bechtold@ufsc.br
bDepartment of Chemistry, Universidade Federal de Santa Catarina, 88040-900 Florianópolis, SC, Brazil. E-mail: bernadsz@gmail.com
cCentre de Recherche Paul Pascal, CNRS & Université de Bordeaux, 115, av. Schweitzer, 33600 Pessac, France. E-mail: harald.bock@crpp.cnrs.fr
dDepartment of Physics, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: a.p.monkman@durham.ac.uk
eErzurum Technical University, Department of Fundamental Sciences, Erzurum, Turkey
First published on 7th December 2022
Abstract
Considering the relevance of room temperature phosphorescent (RTP) materials, we discuss the influence of donor and acceptor groups substituted on to a twisted three-fold symmetric hydrocarbon homotruxene, which presents a persistent RTP, even in the absence of donor or acceptor moieties, under ambient conditions as a result of the twisted π-system. Compared to a fluorine acceptor, a donor methoxy group increases the phosphorescence decay rate in solution, while in the solid-state, molecular aggregation and packing yield a very persistent phosphorescence visible by the eye. The RTP of the intrinsically apolar homotruxene is found to be modulated by polar substituents, whose main impact on the solid-state emission is due to altered packing in the crystal.
Introduction
Persistent RTP materials have been explored for applications in bioimaging,1,2 organic optoelectronics,3 sensors,4 anti-counterfeiting,5,6 and encryption7,8 due to their significant Stokes shift, long lifetime, stronger afterglow, simple operation, and stability.9,10 Although most persistent RTP compounds are heavy metal-containing complexes, metal-free organic materials are an environmentally friendly, low-cost, and easy-to-process alternative.9,11In phosphors containing heavy metals, such as Ir and Pt, the charge transfer between metal and ligand usually has a large associated spin–orbit coupling (SOC), which enhances both the intersystem crossing (ISC) and phosphorescence (Ph) rates.12,13 On the other hand, with metal-free organic phosphors, the weak SOC slows the triplet state population formation through ISC and limits its radiative decay.14 Moreover, the longer excited-state lifetime of the triplet state increases the probability of quenching and non-radiative decay under ambient conditions.7 Thus, several groups have investigated molecular designs and the control of intermolecular interactions to promote ISC and suppress non-radiative decay to obtain organic RTP materials with high quantum yield and long lifetime.15 The most common strategies are based on the incorporation of heteroatoms with lone pairs, such as O and N.16 Approaches involving molecular aggregation,17,18 co-crystallization,19,20 polymeric systems,21 host–guest doping systems,22,23 energy-gap narrowing,24,25 twisted π systems,16,26 and structures without lone pairs27 have also been explored. The inclusion of functional groups has proved to be an important tool for taking full advantage of the El-Sayed rule, promoting an efficient ISC between states with different orbital configurations, populating the triplet states, and adjusting the RTP.10,28,29 The presence of functional groups also stabilizes the excited triplet state by improved spatial packing.15,30–32 Furthermore, introducing electron-donating or withdrawing substituents could affect the π–π interactions of aromatic systems.10,15,28
Recently, we reported the hydrocarbon homotruxene (HTX) with a twisted π-system that allows for pronounced SOC leading to persistent RTP in ambient conditions and a quantum yield of 5.6%, clearly visible by the eye after switching off the excitation.26 Its halogenation with the lightest halogen fluorine mainly impacts the prompt fluorescence (PF), while the heavy atom effect (HAE) enhances the ISC and Ph mechanisms with chlorine and bromine. The higher excited triplet states that dominate in ISC are less centered on the heavy atoms than the lower excited triplet states that dominate Ph, thus the HAE impacts Ph much more than ISC.16
Here, we explored the effect of the new methoxy homolog (HTX-MeO) compared with its unsubstituted homolog (HTX) and its homolog substituted with withdrawing fluorine (HTX-F) (Fig. 1), both already reported. Time-dependent measurements and density functional theory (DFT) calculations indicated that the methoxy donor group does not significantly affect the spin–orbit coupling matrix elements (SOCME), but the Ph rate was increased in solution. The molecular packing and aggregation in powder led to a long-lived emission, which allows the Ph to be visible by the eye after switching off the excitation (see the Movie of the ESI†).
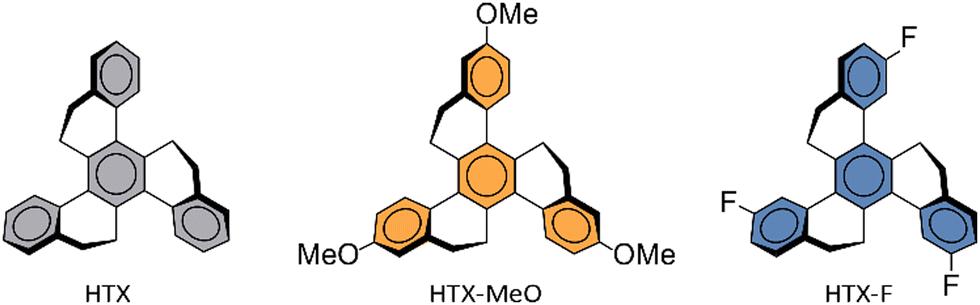 | ||
| Fig. 1 Molecular structure of HTX, HTX-F and HTX-MeO. | ||
Experimental
Single crystal X-ray diffraction
The crystallographic data were collected with a Bruker APEX II diffractometer equipped with a graphite monochromator centered on the path of MoKα radiation. A single crystal, made by slow diffusion of ethanol in chloroform solution, was coated with CargilleTM NHV immersion oil and mounted on a fiber loop, followed by data collection at 120 K. The program SAINT was used to integrate the data, which was thereafter corrected using SADABS.33 The data were cut at a resolution of 0.95 Å as there is no diffraction above, and Rint becomes larger. The result is a low θmax value as well as poor resolution and data/parameters ratio. As the crystal is a very thin needle, the diffraction is very low, and the anisotropic shape makes the diffraction strongly dependent on the orientation of the crystal. Hence, several reflections at “high” angles were not present. The structure was refined by a full-matrix least-squares method on F2 using SHELXL-2018.34 All non-hydrogens were refined with anisotropic displacement parameters. Hydrogen atoms were assigned to ideal positions and refined isotropically using a riding model. CCDC 2191273.† C33H30O3, monoclinic, P21/c (no. 14), a = 21.3546(13), b = 8.2429(6), c = 27.6061(18) Å, β = 102.563(3)°, V = 4743.0(5) Å3, Z = 8, T = 120 K, 192![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 119 reflections measured, 4558 unique (Rint = 0.1463), GoF = 1.037, R1 = 0.0436, wR2 (all reflections) = 0.1153.
119 reflections measured, 4558 unique (Rint = 0.1463), GoF = 1.037, R1 = 0.0436, wR2 (all reflections) = 0.1153.
Photophysical measurements
Absorption and fluorescence spectra were collected using a UV-3600 double beam spectrometer (Shimadzu) and Fluoromax fluorescence spectrometer (Jobin Yvon), respectively. The solutions were degassed in a long-necked quartz cuvette using three freeze–thaw cycles and then mounted in a liquid nitrogen cryostat (Janis Research) for the measurements at low temperatures. Time-resolved photoluminescence spectra (including Ph) decays were measured using either a time-correlated single photon counting set-up (TCSPC, Horiba Deltaflex) with a range of nanoLED (357 nm) and laser diode (330 nm) excitation sources, or a nanosecond gated spectrograph-coupled iCCD (Stanford, 4Picos) and a high energy pulsed Nd:YAG laser emitting at 355 nm (SL312, EKSPLA); the pulse duration was approximately 150 ps and the energy of per pulse was chosen around 100 μJ. Emission was focused onto a spectrograph and detected on a sensitive gated iCCD camera (Stanford Computer Optics) having sub-nanosecond resolution. The quantum yields were determined using a Hamamatsu Photonics Absolute Quantum Yield Measurement System model c9920–02G with an integrating sphere.Theoretical modeling
Geometries of the molecules were optimized in vacuum, using the Orca 4.2.135 software package within ZORA-PBE0/ZORA-Def2-TZVP(-f) level of theory. Dispersion effects were included using Grimme's D3 correction with Becke-Johnson (BJ) damping.36 The evaluation of the four-center integrals was accelerated with the RIJCOSX algorithm.37,38 RIJ requires the specification of an auxiliary basis set for the Coulomb part (Def2/J) and a numerical integration grid for the exchange part (GRID4 and GRIDX4), as discussed elsewhere.35 TD-DFT/TDA was employed to obtain the first 30 singlet and spin-adapted triplet excited states. The same calculation protocol was used to optimize the geometry and calculate the Hessian of the first excited state. The first triplet was optimized from the ground state of a UKS calculation. To include solvent effects in the excited state energies, a conductor-like polarizable continuum model (CPCM) was used, using 2-methyltetrahydrofuran (2-MeTHF) where the refractive index and dielectric constant were used as published by Aycook.39 SOC on top of the TD-DFT results was included by using quasi-degenerate perturbation theory.39 The SOC integrals used here are calculated using a mean-field named RI-SOMF(1X), described elsewhere.40 Images of the complex geometries were obtained using the Chemcraft program.Results & discussion
Synthesis and photophysical properties
2,8,14-Trimethoxy-homotruxene HTX-MeO was obtained in 41% yield following the original procedure of Pyrko41 by trimerization of 6-methoxytetralone in the presence of TiCl4 and triethylamine at room temperature.Single-crystal X-ray diffraction measurements reveals that HTX-MeO crystallizes in the centrosymmetric P21/c space group, with two different molecules in the asymmetric unit with same conformations of the methoxy groups (Fig. S1, ESI†). The four aromatic rings are clearly identified by their planar geometry with C–C bonds ranging from 1.374(4) to 1.414(3) whereas the other C–C bond distances in other cycles range from 1.489(4) to 1.539(4). In the crystal, the molecules pack with weak parallel-displaced π-stacking interactions through some external rings and T-shaped C–H⋯π interactions (Fig. 2 and Fig. S2, ESI†).
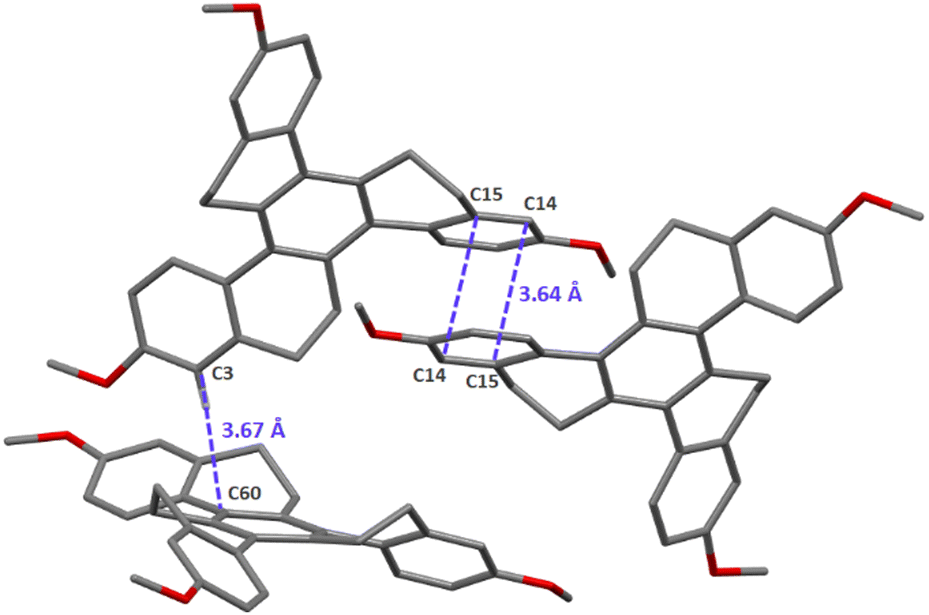 | ||
| Fig. 2 Representation of the intermolecular π stacking interactions. C: grey, O: red. Hydrogen atoms are omitted for clarity, except the one involved in C–H⋯π interaction. | ||
In 2-MeTHF solution at room temperature (RT), HTX-MeO shows a weak absorption band at 345–380 nm (ε = 34–38 cm−1 M−1) (Fig. 3) and a further even weaker band (ε = 7–8 cm−1 M−1) between 385 and 400 nm. According to our previous studies, the band at 345–380 nm is assigned to the S0 → S2 transition, and the absorption at 385–400 nm to the S0 → S1 transition. The absorption spectrum in dilute chloroform solution was measured (Fig. S3, ESI†), and an intense absorption band with a maximum at 283 nm is attributed to π,π* transitions originating mainly from locally excited (LE) states. The steady-state emission spectra of HTX-MeO in dilute 2-MeTHF solution consist of one structured emission peak, with two maxima at about 404 and 422 nm and a shoulder at about 450 nm (Fig. 3). Absorption and emission are slightly dependent on the solvating environment (compare Fig. 3 absorption and emission in 2-MeTHF and toluene). The emission band is oxygen-dependent, and its intensity in 2-MeTHF solution increases by 29% in degassed solution compared to aerated solution. The part suppressed in air is assigned to delayed fluorescence (DF) from triplet–triplet annihilation (TTA), whereas the part observed in air is assigned to PF. At 90 K, a well-structured emission peak attributed to the Ph is observed with two main peaks at 560 and 602 nm and a shoulder close to 655 nm.
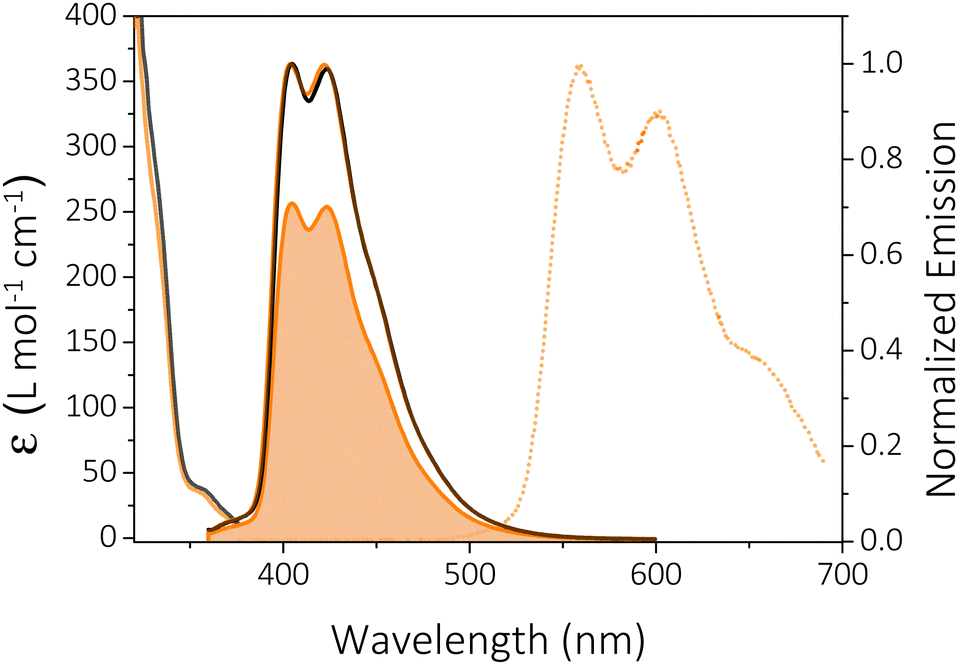 | ||
| Fig. 3 Absorption (10−3 mol L−1) and emission (optic regime conditions) spectra of HTX-MeO in toluene (black line) and 2-MeTHF (light orange line) solutions. The solid lines are the steady-state emission spectra at RT collected in degassed 2-MeTHF solution and the filled area is the spectrum collected in aerated solution. The dashed orange line is the Ph emission collected at 90 K in 2-MeTHF. | ||
The measured PF quantum yields (ΦPF) are 33.2% and 38.7% in toluene and in 2-MeTHF. The DF quantum yield (ΦDF) in 2-MeTHF was determined by the comparison of degassed to aerated emission intensity, showing that the contribution of the oxygen-sensitive delayed emission decreases to 29.5%. TCSPC emission decays from 2-MeTHF solutions collected in the ns range were fitted with tri-exponentials (Fig. S4, ESI†). As previously reported for its non-substituted homolog (HTX), emission occurs from two overlapping states, S2 with τ = 0.94 ns (A1 = 53%) and S1 with τ = 6.65 ns (A2 = 38%).16 The third component with a lifetime of 16.28 ns has a low contribution (9%) and could indicate an aggregate content.
Nanosecond time-resolved delayed emission data for HTX-MeO were obtained using a gated detection technique in degassed 2-MeTHF solution. The decay curves are characterized by a rapid component (in the ns time frame) and a slower one (in the μs to ms time frame) (Fig. 4). At RT from time delay (TD) = 1.1 ns to 41.8 ns (Fig. 4(b), black line), an emission peak at 425 nm (τ = 9.98 ns) with a weak vibronic structure is observed, attributed to decay from the S1 state. The difference between the steady-state and time-resolved spectra is due to the emission of the mixed S1 and S2 states. S2 is a LE state having a radiative rate higher than the S1 CT state. Therefore, S2 largely contributes to the steady-state emission, confirmed by TCSPC measurements, and its emission is not observed in time-resolved measurements, as it decays faster then ns time frame. At TD = 5.9–84.3 μs, the same peak emission (τ = 189 μs) is observed and attributed to DF by TTA. After TD = 84.3 μs, a continuous red-shift until 455 nm is observed (Fig. 4(b), light orange line). This peak at 455 nm is temperature sensitive, and its lifetime increases on cooling (Fig. 4(a)). Therefore, it is attributed to a faster and thermally activated Ph component, as already reported, for its homologs.16 The DF and Ph components disappear in the aerated solution, confirming the triplet state contribution (Fig. 4(c)). Measuring the emission intensity as a function of the laser excitation dose, a value of 1.53 ± 0.05 was obtained, implying a mixture of TTA (with a slope of 2 as required for a bimolecular process) and the competing monomolecular faster Ph decay, which spectrally overlaps with the TTA (Fig. 4(d)). In addition, the larger S1 and T1 energy gap (ΔEST = 0.869 eV), obtained from the on-set of PF and Ph spectra at the high and low temperatures shown in Fig. 3, rules out the thermally activated delayed fluorescence mechanism. The decay curves as a function of temperature showed only a slight evolution of the PF emission (Fig. 3(b), 4(a) and Fig. S5, ESI†). At 90 K, the PF emission at TD = 1.1 ns is slightly blue-shifted, centered at 420 nm. The DF and faster Ph emission component is almost completely gone in the μs to few ms time frame. The DF arising from TTA is frozen out by cooling the sample, and the faster thermally activated Ph is seen as a small component around 490 nm (Fig. 4(b), light orange line). The red-shift observed for the faster Ph can be attributed to the mixed emission from both faster and slower Ph. The dominant slower Ph emission (τ = 27 ms) is observed from TD = 1 ms to 8.4 ms. For HTX-MeO the absorption and steady-state emission of the powder are very similar to the ones in solution, as shown in Fig. S6 (ESI†). The PF from S1 is observed in the time-resolved emission spectra at earlier times with a slightly lower lifetime (τ = 8.95 ns) due to the increase of the PF radiative pathway in solid-state (Table S1, ESI†). Only the slow Ph pathway deactivates the triplet state in the long-lived component (Fig. S7, ESI†). Differences between the Ph emission in the solid-state in Fig. S6 and S7 (ESI†) can be attributed to a loss of vibronic resolution at room temperature. Time-correlated single-photon counting in powder revealed a Ph lifetime of 0.34 s (Fig. S8, ESI†). The ΦPh was not measurable in solution, but in powder, it is 4.7% at RT (Table S1, ESI†), allowing the Ph in the solid to be observed by eye after switching off the excitation.
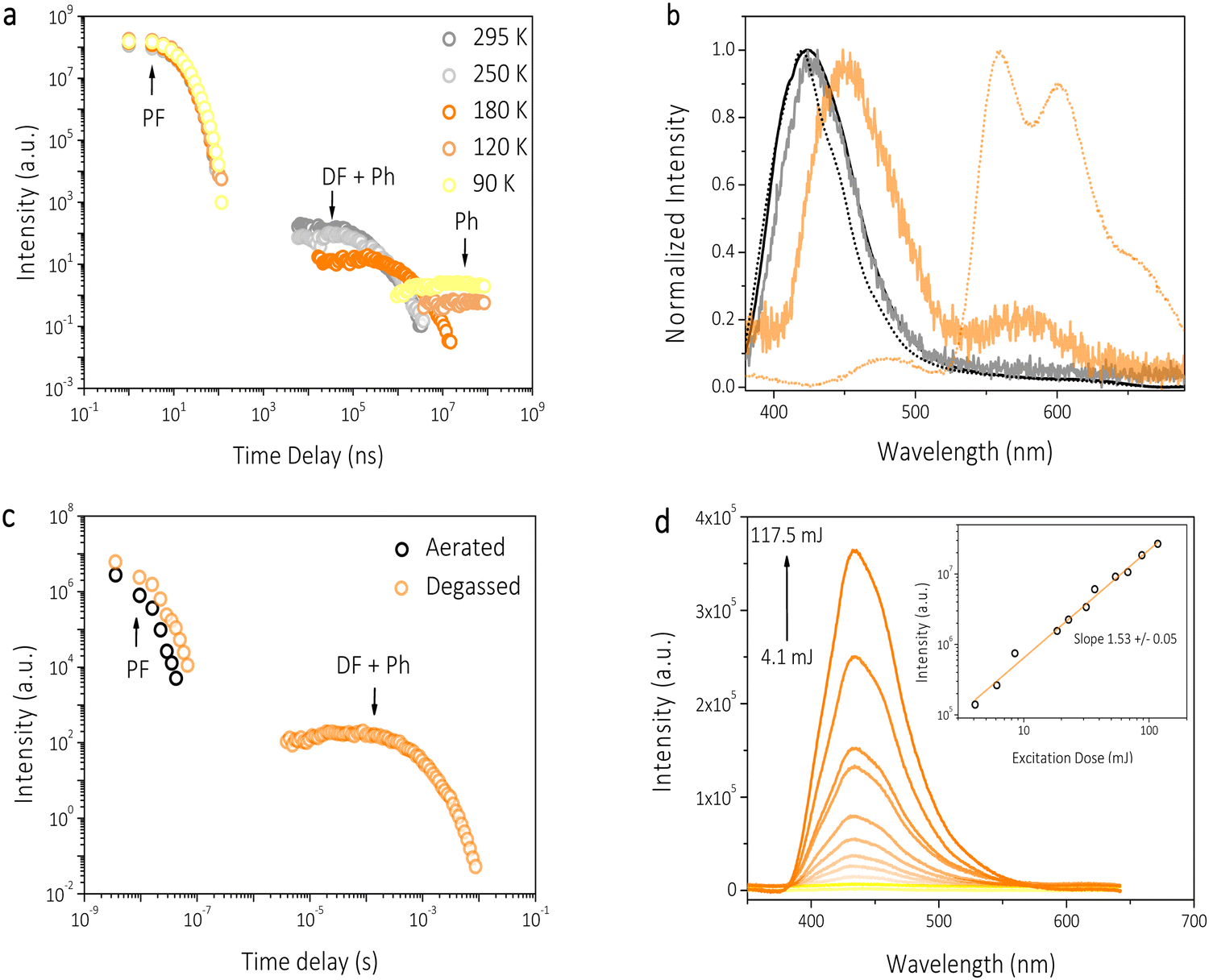 | ||
| Fig. 4 Time-resolved measurements for HTX-MeO. (a) Time-resolved decay in 2-MeTHF as a function of temperature. (b) Normalized spectra were taken at distinct time delays at RT: TD = 1.1–41.8 ns (solid black line), TD = 5.9–84.3 μs (solid grey line) and TD = 969 μs–2.1 ms (solid light orange line); and at 90 K: TD = 1.1–49.1 ns (dotted black line) and TD = 1.1– 8.4 ms (dotted light orange line). (c) Time-resolved decay in degassed and aerated toluene solutions. (d) Emission spectra and intensity as a function of the laser excitation dose with excitation at 355 nm for toluene degassed solution. | ||
Theoretical investigations
By DFT calculations, the ground state geometry was optimized (Fig. S9, ESI†), showing a good agreement with the X-ray crystal structure. The calculated frontier orbitals for all compounds are shown in Fig. 5, where the HOMO and HOMO−1 are almost degenerate π orbitals with contributions on the inner and part of the outer rings. LUMO and LUMO+1 is the corresponding π* orbitals. The HOMO−2 and the LUMO+2, in contrast, are located quite selectively at the outer rings. A significant contribution of the oxygen atom to the HOMO−2, HOMO−1 and HOMO is observed.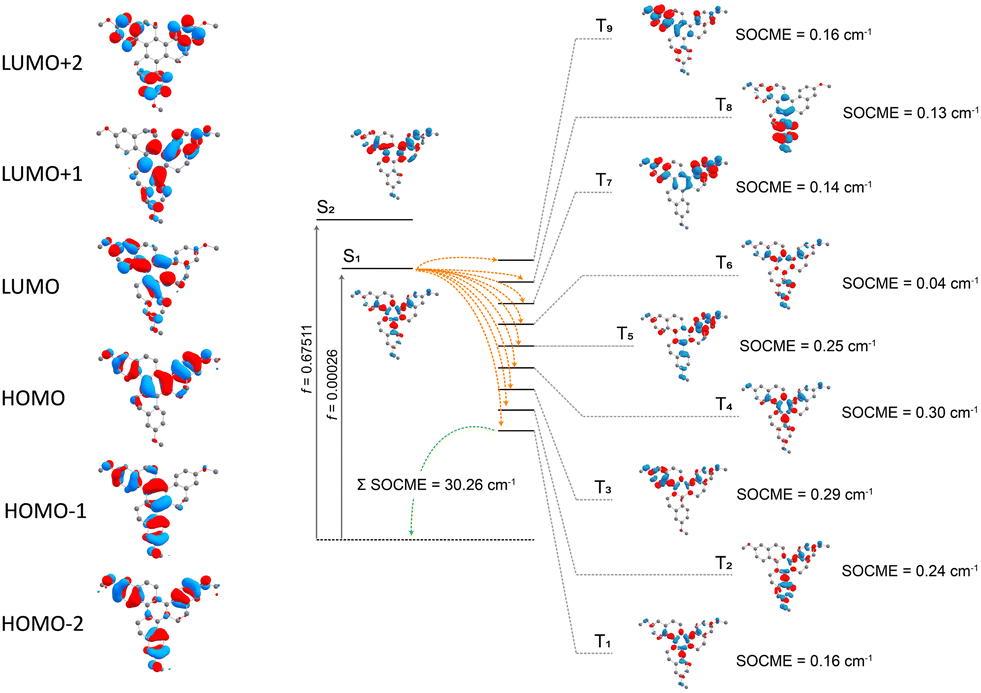 | ||
Fig. 5 Frontier molecular orbitals and SOC-TD-DFT data. (a) Frontier molecular orbitals for HTX-MeO. (b) Jablonski diagram for HTX-MeO and calculated SOCME for S1 → Tn transitions (orange arrows) at the S1 optimized geometry and for the T1 → S0 transition (green arrow) at the optimized T1 geometry. The SOCME were calculated using 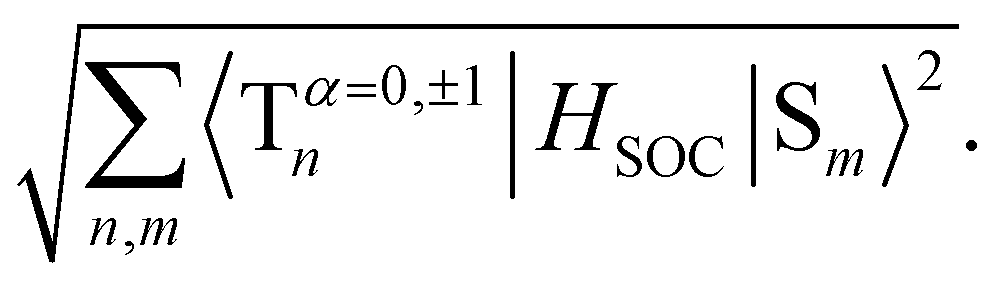 and the ΣSOCME is the sum of all matrix elements between the first ten singlet and triplet states. The vertical excitation arrows indicate the oscillator strength for S1 and S2. and the ΣSOCME is the sum of all matrix elements between the first ten singlet and triplet states. The vertical excitation arrows indicate the oscillator strength for S1 and S2. | ||
The low-lying excited states were modeled using TD-DFT and SOC-TD-DFT approaches (Fig. 5). TD-DFT density difference plots and the oscillator strengths indicate that the S1 arises from π,π* transitions at the central ring with characteristics of a charge-transfer (CT) resonance state, while the upper singlet states S2-S5 are more likely π,π* LE states (Fig. 5 and Table S2, ESI†). HTX-MeO S1 and S2 states have closer energy, and S2 has higher oscillator strength (Table S2, ESI†) in agreement with the dual PF observed experimentally and for its reported homologs.16 Similar to the singlet states, the low-lying triplet states are primarily combinations of the frontier orbitals, with higher contributions from the outer rings and oxygen atoms. For the triplet states, T4 is the first with significantly higher oscillator strength and an orbital configuration similar to S1. Thus the faster Ph emission should arise from this state.16
To evaluate the methoxy group effect on the Ph and the ISC pathways, SOCMEs were calculated for the S1 and T1 geometries. The ISC rate from S1, is proportional to the magnitude of the SOCME between this state and all the triplets below or energetically close, in this case, T1–T9. The sum of the SOCME between these states is 1.71 cm−1, which is relatively low compared to those of metal-containing complexes, but high enough to induce an efficient ISC. The Ph is induced by the SOC between the low-lying singlet and triplet states. 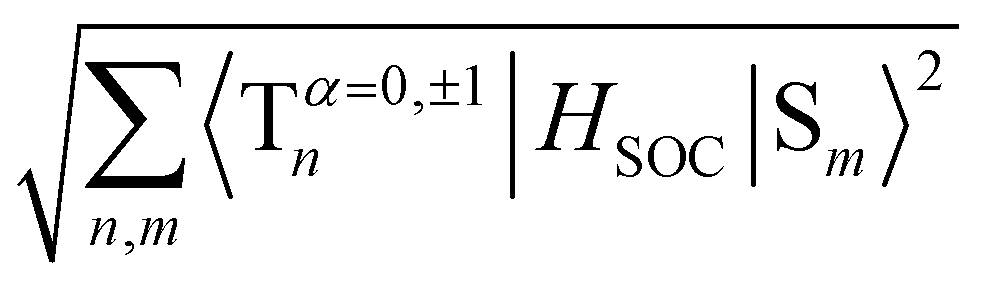 between the first ten singlet and triplet states is 30.26 cm−1. This value of SOCME is mainly attributed to the SOC associated with the transitions inside the inner and other rings in the twisted π-system. This is confirmed by the TD-DFT difference densities, where the contribution of the π-system is much more significant than that of the oxygen atoms.
between the first ten singlet and triplet states is 30.26 cm−1. This value of SOCME is mainly attributed to the SOC associated with the transitions inside the inner and other rings in the twisted π-system. This is confirmed by the TD-DFT difference densities, where the contribution of the π-system is much more significant than that of the oxygen atoms.
Donor–acceptor effect on the twisted π-system
The donor–acceptor effect on this twisted π-system can be discussed by comparing HTX-MeO with its threefold fluorine-substituted (i.e. acceptor-substituted) homolog HTX-F and the unsubstituted homolog HTX.16 In 2-MeTHF diluted solution, HTX-MeO and HTX-F have higher fluorescence and ISC rates than HTX, see Table S1 (ESI†). The kPF is increased due to the higher transition moment dipole when F and O atoms are involved, while kISC is related to the SOC between S1 and the closer triplets and their orbital localization. With the methoxy group, the greater number of triplet states below S1 compared to its non-substituted homolog increases the sum of the SOCME between these states by ≈2. This agrees with the observed increase of the kISC by a factor of 7, as this rate depends on the square of the SOCME.For the isolated molecule of HTX-MeO in solution, a higher kPh is observed compared to its homologs, indicating a larger SOC associated with the low-lying triplets. The Ph is induced by the SOC between the low-lying singlet and triplet states. As with fluorine substitution in HTX-F, relatively little impact on the SOCME is observed with methoxy substitution in HTX-MeO. The HTX-MeO Ph rate increases due to the Herzberg–Teller (HT) vibronic coupling effect, as previously found for HTX and its halogenated homologs.16 The vibronic structure in the emission spectra related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration modes close to 1600 cm−1 confirms that these modes are strongly coupled to the electronic transitions.
C stretching vibration modes close to 1600 cm−1 confirms that these modes are strongly coupled to the electronic transitions.
In the solid state, the fluorescence rate of HTX-MeO is quite similar to the rate in solution, but the Ph lifetime becomes larger than with HTX and HTX-F, and ΦPh increases compared to HTX-F. This significant difference is related to the molecular packing: while HTX and HTX-F form crystals with molecules stacked on top of each other, the presence of the methoxy moiety led to π-stacking interaction with closer distances between them (d(C14-15) = 3.640(3) Å, d(C60-C3) = 3.669(3) Å) (Fig. 2). Such molecular aggregation of HTX-MeO can stabilize the triplet state, increasing its lifetime and allowing the observed RTP.15,30,32 The triplet stabilization through the packing results in a more significant energy difference between the on-set energy for the slower Ph emission in solution at 90 K, ideally from an isolated molecule to solid-state with a π-stacking interaction. The HTX-MeO slower Ph shifts by 0.09 eV, while its homologs HTX and HTX-F shift by 0.01 eV and 0.05 eV, respectively. Furthermore, the reduction observed in the intensity of the 0–1 peak (Fig. S6, ESI†), indicates that the π-stacking packing decreases the HT contribution, probably restricting the CC vibrational modes and reducing the radiative rate from T1. Thus, the donor–acceptor effect observed for this twisted π-system is similar to that described for planar π-systems, and the RTP is due to the molecular packing and aggregation.
Conclusions
In summary, including a donor group in a twisted π-system does not significantly affect the SOCME compared to its non-substituted homolog, but the vibronic coupling enhances its phosphorescence rate. The donor–acceptor effect in this system is similar to molecules with a planar π-system. The packing and the aggregation of HTX-MeO led to a long-lived phosphorescence with a lifetime of 0.34 s and quantum yield of 4.7%, being visible by the eye after switching off the excitation. We conclude that the RTP at the solid-state depends more on the packing than the donor–acceptor substituent effects.Author contributions
CAMS, GF, APM, HB and IHB elaborated the manuscript. LS, FD and HB synthesized the compounds. FD grew the single crystals. PD determined the structure of the single crystals by X-ray analysis. CAMS, GF, and MA measured the photophysical characteristics. GF and BS performed the DFT calculations. BS, HB, APM, and IHB supervised the RTP research. All authors discussed the progress of the research and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to CNPq, FINEP, CAPES (Finance Code 001), FAPESC, INCT-INEO, CAPES-COFECUB (#937/20 and # Ph-C 962/20) and H2020-MSCA-RISE-2017 (OCTA, #778158) for financial support of this work.References
- X. Wang, H. Shi, H. Ma, W. Ye, L. Song, J. Zan, X. Yao, X. Ou, G. Yang, Z. Zhao, M. Singh, C. Lin, H. Wang, W. Jia, Q. Wang, J. Zhi, C. Dong, X. Jiang, Y. Tang, X. Xie, Y. (Michael) Yang, J. Wang, Q. Chen, Y. Wang, H. Yang, G. Zhang, Z. An, X. Liu and W. Huang, Nat. Photonics, 2021, 15, 187–192 CrossRef CAS.
- H.-J. Yu, Q. Zhou, X. Dai, F.-F. Shen, Y.-M. Zhang, X. Xu and Y. Liu, J. Am. Chem. Soc., 2021, 143, 13887–13894 CrossRef CAS PubMed.
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655–660 CrossRef CAS PubMed.
- F. Li, M. Wang, S. Liu and Q. Zhao, Chem. Sci., 2022, 13, 2184–2201 RSC.
- F. Li, C. Qian, J. Lu, Y. Ma, K. Y. Zhang, S. Liu and Q. Zhao, Adv. Opt. Mater., 2022, 10, 2101773 CrossRef CAS.
- Y. Miao, S. Liu, L. Ma, W. Yang, J. Li and J. Lv, Anal. Chem., 2021, 93, 4075–4083 CrossRef CAS PubMed.
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Sci. Adv., 2018, 4, eaas9732 CrossRef PubMed.
- P. She, J. Lu, Y. Qin, F. Li, J. Wei, Y. Ma, W. Wang, S. Liu, W. Huang and Q. Zhao, Cell Rep. Phys. Sci., 2021, 2, 100505 CrossRef CAS.
- A. D. Nidhankar, Goudappagouda, V. C. Wakchaure and S. Santhosh Babu, Chem. Sci., 2021, 12, 4216–4236 RSC.
- Z. Wu, J. Nitsch and T. B. Marder, Adv. Opt. Mater., 2021, 9, 2100411 CrossRef CAS.
- S. Yang, B. Zhou, Q. Huang, S. Wang, H. Zhen, D. Yan, Z. Lin and Q. Ling, ACS Appl. Mater. Interfaces, 2020, 12, 1419–1426 CrossRef CAS PubMed.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- K. Li, Y. Chen, J. Wang and C. Yang, Coord. Chem. Rev., 2021, 433, 213755 CrossRef CAS.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- G. Farias, C. A. M. Salla, M. Aydemir, L. Sturm, P. Dechambenoit, F. Durola, B. de Souza, H. Bock, A. P. Monkman and I. H. Bechtold, Chem. Sci., 2021, 12, 15116–15127 RSC.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- J. Yang, Y. Zhang, X. Wu, W. Dai, D. Chen, J. Shi, B. Tong, Q. Peng, H. Xie, Z. Cai, Y. Dong and X. Zhang, Nat. Commun., 2021, 12, 4883 CrossRef CAS PubMed.
- M. Singh, K. Liu, S. Qu, H. Ma, H. Shi, Z. An and W. Huang, Adv. Opt. Mater., 2021, 9, 2002197 CrossRef CAS.
- W. Wang, Y. Zhang and W. J. Jin, Coord. Chem. Rev., 2020, 404, 213107 CrossRef CAS.
- H. Gao and X. Ma, Aggregate, 2021, 2, e38 Search PubMed.
- D. Wang, H. Wu, J. Gong, Y. Xiong, Q. Wu, Z. Zhao, L. Wang, D. Wang and B. Z. Tang, Mater. Horiz., 2022, 9, 1081–1088 RSC.
- W. Ahmad, J. Wang, H. Li, Q. Ouyang, W. Wu and Q. Chen, Coord. Chem. Rev., 2021, 439, 213944 CrossRef CAS.
- F. Li, S. Guo, Y. Qin, Y. Shi, M. Han, Z. An, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2019, 7, 1900511 CrossRef CAS.
- Y. Hu, Z. Wang, X. Jiang, X. Cai, S.-J. Su, F. Huang and Y. Cao, Chem. Commun., 2018, 54, 7850–7853 RSC.
- C. A. M. Salla, G. Farias, M. Rouzières, P. Dechambenoit, F. Durola, H. Bock, B. de Souza and I. H. Bechtold, Angew. Chem., Int. Ed., 2019, 58, 6982–6986 CrossRef CAS PubMed.
- Z. Wu, J. Nitsch, J. Schuster, A. Friedrich, K. Edkins, M. Loebnitz, F. Dinkelbach, V. Stepanenko, F. Würthner, C. M. Marian, L. Ji and T. B. Marder, Angew. Chem., Int. Ed., 2020, 59, 17137–17144 CrossRef CAS PubMed.
- Q. Liao, Q. Li and Z. Li, ChemPhotoChem, 2021, 5, 694–701 CrossRef CAS.
- H. E. Hackney and D. F. Perepichka, Aggregate, 2022, 3, e123 CrossRef.
- J. S. Ward, R. S. Nobuyasu, M. A. Fox, J. A. Aguilar, D. Hall, A. S. Batsanov, Z. Ren, F. B. Dias and M. R. Bryce, J. Org. Chem., 2019, 84, 3801–3816 CrossRef CAS PubMed.
- J. Chen, N. U. Rahman, Z. Mao, J. Zhao, Z. Yang, S. Liu, Y. Zhang and Z. Chi, J. Mater. Chem. C, 2019, 7, 8250–8254 RSC.
- B. Fang, L. Lai, M. Fan and M. Yin, J. Mater. Chem. C, 2021, 9, 11172–11179 RSC.
- G. M. Sheldrick, SADABS Version 2.03, Bruker Analytical X-Ray Systems Madison, WI, USA, 2000 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- R. Izsák and F. Neese, J. Chem. Phys., 2011, 135, 144105 CrossRef PubMed.
- R. Izsák, F. Neese and W. Klopper, J. Chem. Phys., 2013, 139, 094111 CrossRef PubMed.
- D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156–159 CrossRef CAS.
- F. Neese, J. Chem. Phys., 2005, 122, 034107 CrossRef PubMed.
- A. N. Pyrko, Zh. Org. Khim., 1992, 28, 215–216 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2191273. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cp04658j |
| This journal is © the Owner Societies 2023 |