
Stabilization of the Li metal anode through constructing a LiZn alloy/polymer hybrid protective layer towards uniform Li deposition†
Zihao
Wang
ab,
Zhicui
Song
ab,
Yuchi
Liu
ab,
Jianxiong
Xing
ab,
Chaohui
Wei
a,
Wei
Zou
c and
Jingze
Li
 *ab
*ab
aYangtze Delta Region Institute (Huzhou), University of Electronic Science and Technology of China, Huzhou 313001, P. R. China. E-mail: lijingze@uestc.edu.cn
bSchool of Materials and Energy, University of Electronic Science and Technology of China, Chengdu 611731, P. R. China
cResearch and Development Center, Tianqi Lithium Co., Ltd., Chengdu 610093, P. R. China
First published on 21st November 2022
Abstract
Constructing an artificial solid electrolyte interphase (SEI) layer is an effective strategy for solving uncontrolled Li dendrite growth resulting from an unstable and heterogeneous Li/electrolyte interface. Herein, we develop a hybrid layer of a LiZn alloy and a polyethylene oxide (PEO) polymer to protect the Li metal anode for achieving a Li dendrite-free Li metal anode surface. The LiZn alloy is advantageous for fast Li+ transport, and is uniformly dispersed in the PEO matrix to regulate electronic and Li+ ion flux distributions homogeneously. Furthermore, the flexible PEO network can alleviate the volume change during cycling. The synergistic effect enables Li deposition underneath the hybrid film. Hence, the hybrid protection film results in significantly improved cycling stability with respect to the pristine Li metal anode. A symmetric Li/Li cell with a composite protective layer can be cycled for over 1000 h at a current density of 1 mA cm−2 with a fixed capacity of 1 mA h cm−2, and a full cell with a high areal capacity of the LiFePO4 (2.45 mA h cm−2) cathode exhibits an outstanding cycling performance.
Introduction
Lithium metal batteries (LMBs) have emerged as promising candidates for next-generation energy storage devices due to their high theoretical specific capacity (3860 mA h g−1) and the lowest electrochemical potential (−3.04 V vs. S.H.E) of the metallic Li anode.1–3 However, the highly reactive Li metal spontaneously reacts with the electrolyte to form an inhomogeneous and fragile solid electrolyte interface (SEI), which leads to non-uniform Li nucleation and the formation of Li dendrites. In addition, the infinite volume change of the Li metal anode and the continuous growth of Li dendrites during cycling can lead to fast capacity fading of LMBs.4–8 Therefore, the practical application of rechargeable LMBs has been severely hindered.Many strategies have been developed to solve side reactions between metallic Li and the electrolyte and suppress the Li dendrite growth.9–13 Constructing an artificial SEI layer at the Li anode/electrolyte interface has been certified as an effective and facile method for achieving stable Li metal anodes.14–21 Inorganic materials are attractive because of their high Li+ conductivity and mechanical strength.22,23 Many inorganic materials have been used as artificial protective layers (such as Al2O3,24,25 Li3PO4,26,27 LiF,28–31 Li3N,32,33etc.). Kozen et al.24 directly deposited an Al2O3 protective layer on the Li metal by atomic layer deposition (ALD) to alleviate side reactions between Li and the electrolyte and successfully suppressed the shuttle effect in Li–S batteries. Yuan et al.31 proposed to form a LiF layer with good chemical stability to restrain Li dendrite growth by using ammonium hydrogen difluoride (NH4HF2) as the fluorine source, in which the low Li+ diffusion barrier of LiF enabled the bottom-up Li deposition. Nevertheless, the weak contact between the inorganic layer and the Li metal inevitably increases the interfacial impedance. Additionally, the intrinsically brittle nature of inorganic matter makes it a real challenge to adapt to the volume change of the Li anode, resulting in fragmentation during cycling.34–36 Therefore, it is very difficult to achieve protective effects while using inorganic films solely.
Recently, researchers have incorporated inorganic fillers into the polymer matrix to construct a composite layer for protecting the Li metal anode ideally.37–41 The high flexibility of the polymer enables the conformal coverage of the Li metal anode and accommodates volume changes during cycling.42–44 Alternatively, inorganic fillers enhance the mechanical properties to block the growth of Li dendrites. This strategy is succeeded in preparing a robust protective layer with high modulus, flexibility and favorable Li+ transport ability.45 Among all the polymers, polyethylene oxide (PEO) has been widely used as the primary material of solid polymer electrolytes because of its good interfacial stability toward the Li metal.46–48 PEO has quick segmental motion, which effectively promotes the transport of Li+ in the polymeric matrix.49 Yang et al.50 reported a hybrid film which consists of Li7La3Zr1.75Nb0.25O12 (LLZNO) and a PEO-based polymer electrolyte. Because of the flexibility of the PEO matrix, the hybrid film can easily accommodate the considerable volume change during Li stripping/plating. Meanwhile, the ceramic LLZNO particles significantly improve the mechanical properties and suppress the growth of Li dendrites because of the superior Li+ conductivity, enabling Li deposition under the hybrid film. Similarly, Fu et al.51 combined LiF and PEO as a nanocomposite protective layer. The nanoscale LiF particles with a high interfacial area provide Li+ transport pathways throughout the composite and exhibit a low barrier of surface diffusion for Li+, which is favorable for the diffusion of Li+ at the LiF/PEO interface. As a consequence, the performance of the Li metal anode was improved to some extent. Unfortunately, these inorganic fillers suffer from high surface energy and poor compatibility with polymer matrices and are prone to agglomeration in the polymer, resulting in an uneven Li+ flux distribution.34,52 Furthermore, most of these hybrid films are pure ionic conductors, in which poor charge transport and limited rate performance lead to the cracking of the protective layer after limited cycles.53,54
Li alloy films have been widely recognized as one of the promising candidates for protecting the Li metal anode, attributed to their unique transportation capability of mixed ions/electrons, which is different from traditional inorganic materials.55–59 Li alloys can effectively control the uniform Li deposition due to their excellent Li+ diffusion ability and lithiophilic properties. Moreover, the Li alloy artificial layer can be in tight contact with the Li metal, which is ascribed to the alloying reaction between the plated metal and Li. Deng et al.60 sputtered a Zn layer on the surface of the Li metal to form a LiZn alloy film. Benefiting from a high Li+ diffusion coefficient (4.7 × 10−8 cm2 s−1) of the LiZn alloy, the alloy-protected Li anode exhibits better kinetic properties, enabling a longer cycle lifespan at a larger current density. Furthermore, the PEO-based polymer solid-state battery can be cycled for more than 900 h, indicating that the Li alloy film has good compatibility and an interfacial contact with the PEO matrix. Compared with the physical vapor deposition method, the chemical method is more convenient for preparing an advanced artificial SEI layer. Recently, Hu et al.61 constructed an artificial SEI layer by combing the Li3Sb alloy with a LiF insulator through a simple liquid-phase reaction. The Li3Sb phase can facilitate the rapid diffusion of Li+ ions so as to eliminate Li+ diffusion barriers within the interphase layer while LiF with an electronically insulating character can prevent electron tunneling across the artificial SEI layer, achieving the uniform Li deposition at the SEI/Li metal interface. It should be noted that the chemically prepared Li alloy layer usually exhibits a 3D array structure. Its high electrical conductivity effectively reduces the local current density and inhibits the growth of Li dendrites.
Inspired by the above studies, herein, an artificial hybrid film is constructed using a composite of LiZn–LiCl–PEO on the Li metal surface (Li@LZP). The mixed solution of ZnCl2 and PEO is directly coated on the Li metal surface through a doctor-blade, forming an alloy–inorganic polymer hybrid layer. Because of the favorable compatibility of the LiZn alloy with PEO, the LiZn particle array is uniformly dispersed in the PEO matrix to produce uniform electronic and Li+ flux distributions, achieving a dendrite-free Li deposition morphology. Moreover, LiCl can serve as a Lewis acid to decrease PEO crystallization, which is of benefit for the Li+ transfer. On the other hand, inorganic LiCl, as the electronic blocking component, can improve the mechanical properties of the composite films and effectively inhibit the Li dendrite generation. The excellent flexibility of the PEO matrix alleviates the volume change during cycling. Therefore, the Li@LZP anode exhibits outstanding cyclic stability for more than 1000 h under conditions of 1 mA cm−2 and 1 mA h cm−2, and the LFP-based full cell shows excellent performance with a high areal capacity of 2.45 mA h cm−2.
Experimental
Preparation of the Li@LZP anode
Bare Li foil was first polished to remove surface contaminants in an argon-filled glovebox with less than 1 ppm of moisture and oxygen before use. For the fabrication of a hybrid film, 0.01 M ZnCl2 and 0.1 g of PEO were dissolved in dimethyl sulfoxide (DMSO). The homogeneous solution was obtained under magnetic stirring overnight. Then, 50 μL of the PEO–ZnCl2 suspension was cast onto the Li metal using a doctor-blade and dried in the glovebox overnight. Finally, the Li alloy–polymer interphase layer on the Li electrode was obtained. The pristine LiZn/LiCl or PEO layer was obtained under the same conditions.Materials characterization
X-ray diffraction (XRD) patterns were carried out using Cu Kα (λ = 0.154056 nm) radiation at a scanning rate of 10° min−1 over a 2θ range from 10 to 90°. The morphology and elemental mapping images were obtained using a scanning electron microscope (FE-SEM, Hitachi, S3400N) at 20 kV.Electrochemical measurements
Coin cells (CR-2032) were used to evaluate the electrochemical performance using a CT2001A cell test instrument (LAND Electronic Co. Ltd) at a constant temperature (25 °C). The symmetric cells were assembled in the Ar-filled glove box, and the electrolyte was 1.0 M Li hexafluorophosphate (LiPF6) in ethylene carbonate (EC)/diethyl carbonate (DEC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) with 5 vol% fluoroethylene carbonate (FEC). The amount of electrolyte in each cell was fixed at 100 μL. The separator is Celgard 2325. Electrochemical impedance spectroscopy (EIS) measurements were carried out using a CHI660E electrochemical workstation in the frequency range of 1 MHz to 100 mHz. And, the rest time of the cells before performing the EIS measurement is 6 h. The LFP cathode for the full cell test was paired with a Li or Li@LZP anode. The LFP cathode was obtained by coating the mixture of active material powders, super P and polyvinylidene fluoride (8:1:1, in N-methyl pyrrolidone), onto the carbon-coated Al foil. The areal capacity of LFP was about 2.45 mA h cm−2 and the mass loading was 17.1 mg cm−2. The mixture was dried at 110 °C under vacuum for 12 h. The electrolyte used was the same as that in the symmetric cells. The full cells were cycled between 2.2 and 3.85 V.
:1, v/v) with 5 vol% fluoroethylene carbonate (FEC). The amount of electrolyte in each cell was fixed at 100 μL. The separator is Celgard 2325. Electrochemical impedance spectroscopy (EIS) measurements were carried out using a CHI660E electrochemical workstation in the frequency range of 1 MHz to 100 mHz. And, the rest time of the cells before performing the EIS measurement is 6 h. The LFP cathode for the full cell test was paired with a Li or Li@LZP anode. The LFP cathode was obtained by coating the mixture of active material powders, super P and polyvinylidene fluoride (8:1:1, in N-methyl pyrrolidone), onto the carbon-coated Al foil. The areal capacity of LFP was about 2.45 mA h cm−2 and the mass loading was 17.1 mg cm−2. The mixture was dried at 110 °C under vacuum for 12 h. The electrolyte used was the same as that in the symmetric cells. The full cells were cycled between 2.2 and 3.85 V.
Results and discussion
The hybrid protective layer of the LiZn alloy/LiCl with PEO was formed by a spontaneous reaction between Li and the mixed solution of ZnCl2/PEO on the Li metal (Fig. 1(a)). As shown in Fig. 1(b), the nano-sized particles appear on the surface of the Li@LZP electrode, which are uniformly distributed on the Li metal illustrating the grey color according to the digital picture shown in the inset. EDS mapping demonstrates that these nano-particles are a composite of the LiZn alloy and LiCl compound, which is homogeneously dispersed in the PEO polymer film (Fig. 1(d), (e) and Fig. S1, ESI†). Thus, a robust coating layer is fabricated on the Li metal, reflecting that the LiZn alloy has good compatibility with the PEO matrix. The relevant reaction equations are as follows:| ZnCl2 + Li → Zn + LiCl | (1) |
| Li + Zn → LiZn | (2) |
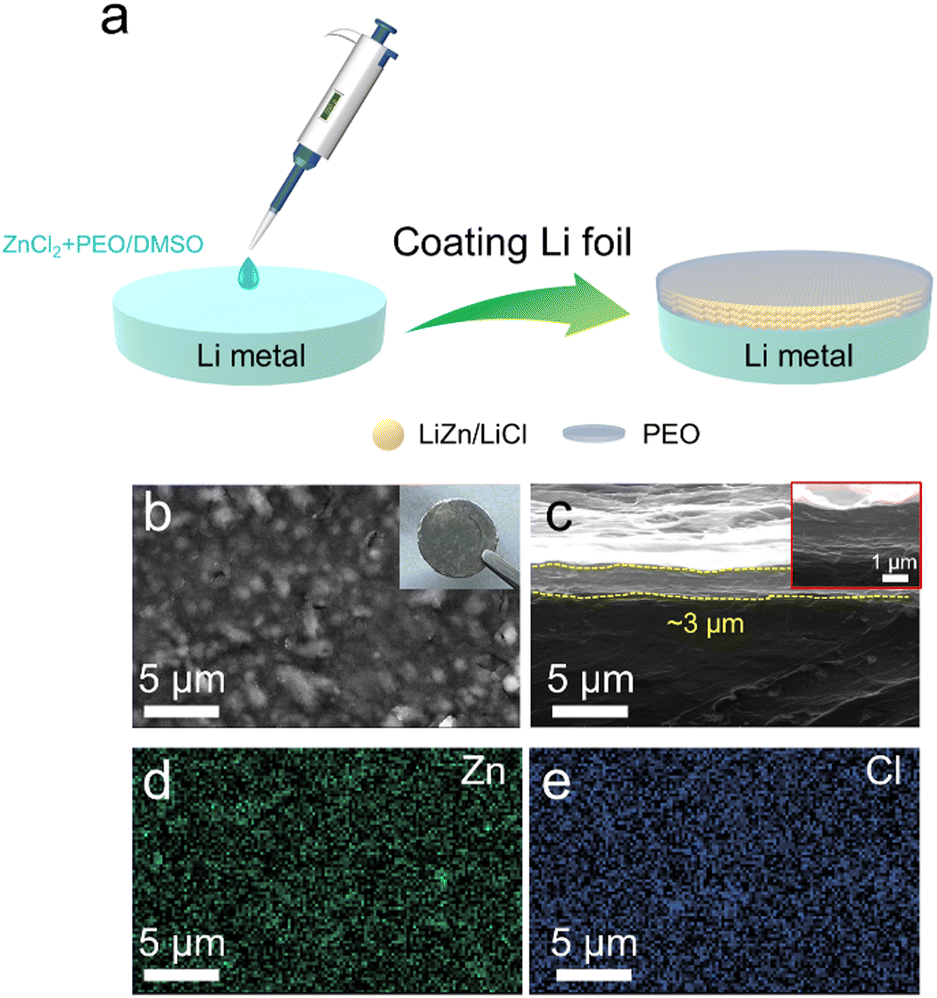 | ||
| Fig. 1 (a) Schematic diagram of the formation process of a hybrid film on Li foil. (b) Top-view and (c) side-view SEM images of the Li@LZP anode, and the inset is the corresponding high-magnification SEM image. The corresponding (d) Zn and (e) Cl signals on the Li@LZP surface. | ||
The corresponding side-view SEM image (Fig. 1(c)) illustrates that the hybrid film coated on the Li foil is about 3 μm in thickness and keeps a compact contact with bulk Li. This thin protective layer is beneficial to accelerate Li+ transport, which does not greatly reduce the battery's energy density. In order to highlight the advantages of the hybrid layer, we coated a LiZn/LiCl layer on the Li metal (named Li@LiZn) by using the same concentration of ZnCl2/DMSO solution. As shown in Fig. S2 (ESI†), the surface of Li@LiZn exhibits a rough surface with spherical particles of micro-sized scale. Moreover, some agglomeration and cracks can be observed in the Li@LiZn surface. Compared with Li@LZP, the size of the LiZn alloy particles is reduced and the composite layer surface is more uniform after the LiZn alloy is dispersed into the PEO polymer matrix, which benefits the uniform distribution of electronic and Li+ flux. The characteristic peak of LiZn appears in both the XRD patterns of Li@LZP and Li@LiZn, indicating that the alloying reaction successfully occurs and a protective layer with the LiZn alloy is formed on the Li surface (Fig. S3, ESI†). Furthermore, there is no PEO diffraction peak in Li@LZP, indicating that the crystallinity of the PEO phase is reduced and the polymer matrix becomes amorphous, which can aid for Li+ migration quickly. In addition, the FWHM of the peak of LiZn in the Li@LZP anode is 0.011 rad and its counterpart in Li@LiZn is 0.008 rad. Therefore, the average grain size of LiZn in Li@LZP (14.9 nm) and Li@LiZn (20.5 nm) can be calculated according to the Scherrer formula, which proves that the grain size of the LiZn alloy is reduced by the introduced PEO matrix. That is to say, a smaller grain size of the LiZn alloy is synthesized in the PEO polymer matrix, which is in line with the above-mentioned SEM observation. Hence, the hybrid protective membrane with nanoparticles of the LiZn alloy can ensure more effectively homogenized Li+ flux and induce uniform Li deposition.
The role of the Li@LZP anode in suppressing the Li dendrite growth was further verified by SEM measurements. The reduced Li was continuously plated on the bare Li or Li@LZP at a current density of 1 mA cm−2. As shown in Fig. 2(b), Li dendrites and cracked Li appear on the surface of the Li electrode after the deposition of 3 mA h cm−2 of Li, which is attributed to the uneven electric field distribution and uncontrolled Li dendrite growth on the pristine Li electrode.62 As shown in the corresponding cross-sectional SEM images (Fig. 2(c) and (d)), the appearance of large Li dendrite clusters is clearly evidenced with around ∼30 μm in height. Herein, these loose Li dendrites certainly promote the risk of battery short circuit. In contrast, when 3 mA h cm−2 Li is deposited on the Li@LZP electrode, the hybrid layer on the surface of the Li anode remains intact, where the LiZn alloy particles are uniformly dispersed in the PEO matrix. Furthermore, the reduced Li is deposited underneath the hybrid layer instead of on the top surface of the protection film without any signal of Li dendrites (Fig. 2(e)). The compact and dense morphology is illustrated in the corresponding side-view SEM images (Fig. 2(f)). Moreover, the corresponding cross-sectional EDS mapping images as illustrated in Fig. 2(f) reflects that both Zn and Cl elements are concentrated in the top surface area within the hybrid layer, which further evidences that the protective layer can maintain geometric integrity and the Li deposition occurs under the hybrid film (Fig. S5, ESI†). Notably, the plated Li layer that forms under the hybrid film is flat with a thickness of approximately 15 μm, which is equal to the theoretical thickness of the reduced Li. The spatial location of the deposited Li under the composite layer can effectively eliminate the possibility for the generation of Li dendrites. These results demonstrate that the LiZn alloy can act as a favorable Li+ conductor with fast Li+ transport channels and it is well-dispersed in the PEO matrix ensuring the uniformity of the Li+ flux. Moreover, both PEO and LiCl are electronic insulators, eliminating the occurrence of Li+ reduction inside the protective layer. The synergistic effect of the three components can regulate uniform Li deposition underneath the protective layer. In addition, the excellent flexibility of PEO has a good contact with the deposited Li metal and alleviates the volume change, preventing structural damage for the hybrid layer. Furthermore, when 3 mA h cm−2 Li was stripped in Li@LZP (Fig. S4, ESI†), the film maintained a uniform and dense morphology without cracks and thickness change, indicating the superiority of the hybrid protective layer for structural stability.
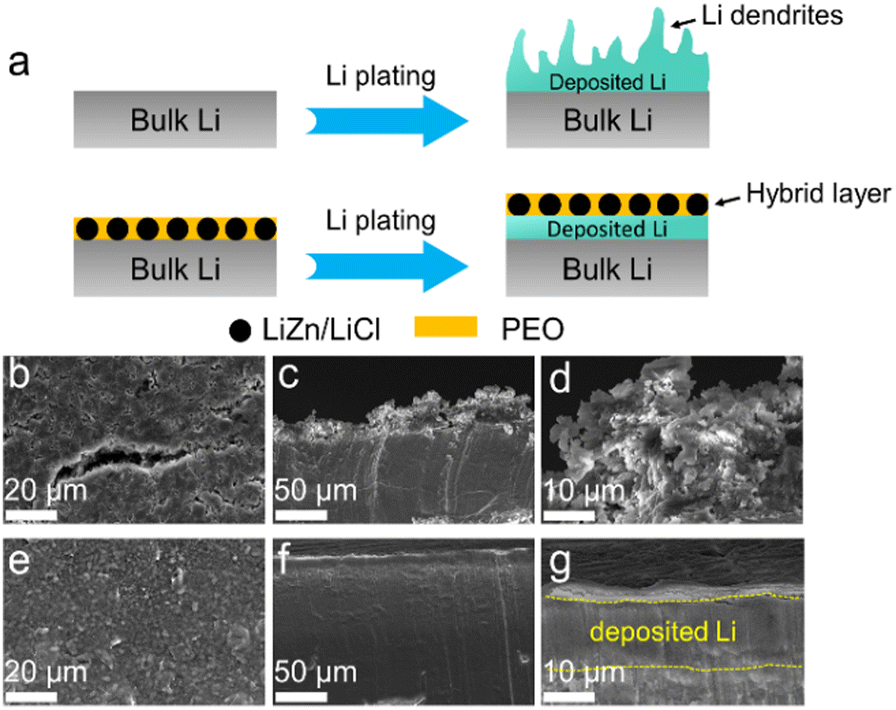 | ||
| Fig. 2 (a) Schematic illustration of Li deposition on bare Li and the Li@LZP anode. (b) Top-view and (c) and (d) side-view SEM images of Li deposition on bare Li and (e)–(g) Li@LZP with a plating capacity of 3 mA h cm−2 at a current density of 1 mA cm−2. | ||
In order to confirm the advantages of the Li@LZP anode, the electrochemical performance of the Li–Li symmetric cells was investigated in a carbonate-based electrolyte, which was assembled by two identical Li@LZP electrodes. The Li/Li, Li@LiZn/Li@LiZn, and Li@PEO/Li@PEO symmetric cells were also assembled for comparison. The time-voltage profile of the symmetric cells reflects the cycling stability of the electrodes. As shown in Fig. 3(a), the symmetric cell with the Li@LZP anode exhibits steady and low Li plating/stripping overpotentials of ∼35 mV at a current density of 1 mA cm−2 with a fixed Li deposition capacity of 1 mA h cm−2, which is significantly lower than that of Li (150 mV), verifying fast Li+ migration kinetics and superior interface properties. Moreover, the cell with the Li@LZP anode can be cycled stably for 1050 h. By comparison, the cell with bare Li displayed larger overpotentials of 500 mV after 200 h, indicating that a large amount of “dead Li” is accumulated on the Li foil, leading to unstable Li plating/stripping. For the cell with the Li@PEO anode, the cycle lifetime is extended to a certain extent. However, the poor mechanical properties of the soft polymer can hardly inhibit the growth of Li dendrites during cycling, and the polarization increases sharply around 300 h. In contrast, when a composite layer of LiZn/LiCl is employed to protect the Li metal, the cell shows a stable voltage profile over 400 h due to the superior Li transport ability and lithiophilicity of the Li alloy, which reduces the overpotential and suppresses the formation of Li dendrites. However, the intrinsic brittleness of the large alloy particles and the weak bonding force between the LiZn alloy and LiCl compound can endure the volume change during repeated Li stripping/plating, resulting in irreversible damage for the Li alloy layer, thereby the polarization voltage of the Li@LiZn anode shows a sudden increase and the cell fails after 450 h. The same tendency is repeated for cycling at a current density of 3 mA cm−2 (Fig. 3(b)). The Li@LZP anode exhibits superior cycling stability with a voltage hysteresis of 120 mV for 240 h, whereas the pristine Li metal anode suffers from short circuit even after 80 h. Moreover, Li@PEO and Li@LiZn batteries fail at 100 h and 130 h, respectively. Moreover, the hybrid layers with different thicknesses were also fabricated as shown in Fig. S6 (ESI†). Then the corresponding Li symmetric cell measurements were further conducted to evaluate the performance. Apparently, the hybrid layer with a thickness of 3 μm achieves the longest cycling lifespan (Fig. S7, ESI†). The optimal thickness of the hybrid layer is 3 μm due to the limited Li+ transport in the thicker layer and the poor suppression ability of the thinner layer against Li dendrite growth. Therefore, the artificial hybrid layer with a moderate thickness (3 μm) modified Li anode exhibits superior cycling stability. While the current density and areal capacity are increased to 3 mA cm−2 and 3 mA h cm−2 (Fig. S8, ESI†), respectively, the Li@LZP anode still can cycle up to 190 h, exhibiting a longer cycle life than others and indicating that the hybrid layer can suppress the Li dendrites growth even under the larger area capacity and higher current density. Additionally, the cycling performance of the Li@LZP electrode in the symmetric cell is more excellent with respect to the recent reports in carbonate-based electrolytes (Table S1, ESI†). Apparently, the growth of Li dendrites become severe at a higher current density, and a single protection layer of the polymer or Li alloy coating is difficult to achieve the desired performance.
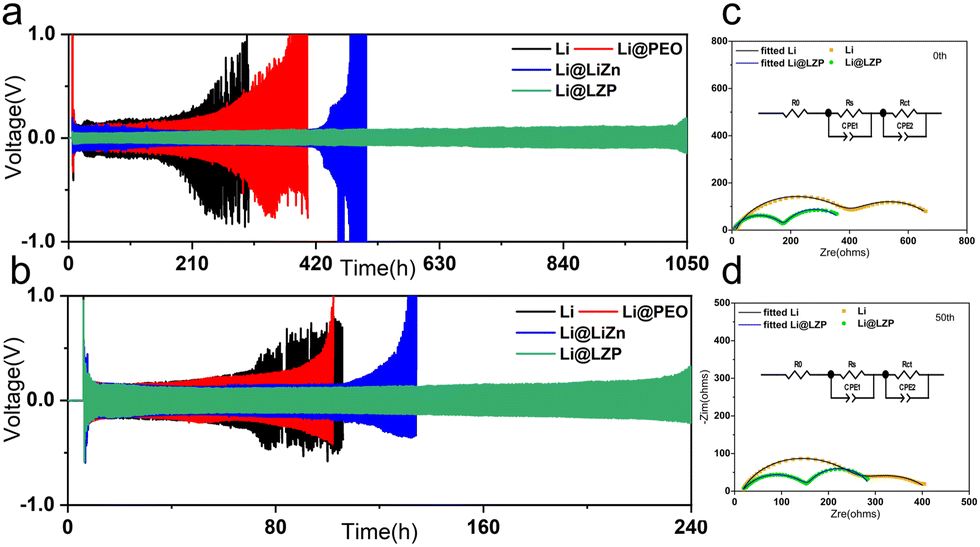 | ||
| Fig. 3 Cycling performance of the symmetric cells with Li, Li@PEO, Li@LiZn and the Li@LZP anode at current densities of (a) 1 mA cm−2 and (b) 3 mA cm−2 for 1 mA h cm−2. The fitted line and the equivalent circuit of Li@LZP and Li symmetric cells (c) before and (d) after 50 cycles with 1 mA h cm−2 at 1 mA cm−2. | ||
To illustrate the effect of the LiZn/PEO hybrid layer and the kinetics of Li+ transport at the electrode/electrolyte interface, EIS tests were examined. In the EIS plot, the high frequency semicircle and mid-frequency semicircle are associated with SEI resistance (Rs) and charge transfer resistance (Rct), respectively. The values of the Rs and Rct of Li or Li@LZP-based symmetrical cells can be obtained by fitting the EIS curves to the equivalent circuit (Fig. 3(b) and (c)). The Li@LZP based cell exhibits low Rs (174.7 Ω) and Rct (210.8 Ω) values, while both the Rs and Rct of the bare Li based cell (387.1 Ω and 309.7 Ω) are larger than those of the Li@LZP based cell, revealing that the hybrid layer can facilitate a stable electrode/electrolyte interface with weakened parasitic reactions. After 50 cycles under conditions of 1 mA h cm−2 and 1 mA cm−2, the Rs (143 Ω) and Rct (131.4 Ω) of the Li@LZP based cell are much lower than those of the bare Li based cell (Rs 236.1 Ω and Rct 164.7 Ω), reflecting that the hybrid protection film can facilitate Li plating/stripping in a compact way and the electrode/electrolyte interface is quite stable with respect to that of the bare Li electrode.
The top-view and cross-sectional morphologies of the anodes are declared by SEM imaging. The surface of the Li metal anode shows a typical loose and rough morphology with a large number of Li dendrites after 50 cycles at a current density of 1 mA cm−2 with a fixed areal capacity of 1 mA h cm−2, which is direct evidence of the poor Li dendrite inhibition ability regarding the intrinsic SEI layer (Fig. 4(a)). The corresponding side-view SEM images exhibit Li dendrites and “dead Li” above the bulk Li with a huge thickness change (Fig. 4(b) and (c)). The surface of the bare Li presents a circulating loose Li layer nearly 20 μm thick, further deteriorating the batteries’ performance. In addition, mossy Li is deposited on the top surface of the Li@LiZn anode with a porous and loose morphology (Fig. S10a, ESI†). It can also be observed from the sectional-view SEM images that the Li–Zn alloy layer is destroyed by deposited Li (Fig. S10b and c, ESI†). In contrast, the introduction of the LiZn/PEO hybrid film on the Li metal anode surface results in a relatively uniform and dense Li surface without any signal of Li dendrites. Notably, the alloy particles were still clearly visible, which were uniformly anchored in the PEO matrix (Fig. 4(d)). The surface of the Li@LZP electrode maintains a similar morphology compared to that of the electrode before cycling, indicating that the Li stripping/plating behavior is physically limited under the protective layer, which effectively avoids the short circuit caused by Li dendrite growth. As shown in the corresponding side-view SEM images, the protective film maintains good geometric integrity with a stable thickness and structure, which indicates the capability of LiZn/PEO for restricting the Li dendrite growth and volume change, realizing a dense and compact pattern of the deposited Li under the protective layer. In addition, the XRD profile shows that the compositions of the Li@LZP anode after 50 cycles are LiZn and Li, reflecting that the complete alloying reaction occurred favoring the structural stability of LiZn alloy particles distributed in the PEO matrix (Fig. S9, ESI†).
 | ||
| Fig. 4 The surface and cross-section morphology of (a)–(c) pristine Li and (d) and (e) Li@LZP after 50 cycles at 1 mA cm−2 and 1 mA h cm−2. | ||
To evaluate the protective effect of the composite layer under practical conditions, the LFP cathode was chosen to match with the Li@LZP anode for assembling the full cell. The areal capacity of the LFP cathode used was about 2.45 mA h cm−2. When charged and discharged at a rate of 1C (equivalent to a current density of 2.45 mA cm−2), the Li@LZP/LFP full cell can provide a long-term stable cycling of up to 200 cycles with a capacity retention rate of 82.2% (Fig. 5(a)). In contrast, the capacity of the Li/LFP battery decreased sharply within 100 cycles with a retention rate of 27.7%, reflecting an extremely unstable electrode/electrolyte interface caused by the random deposition of Li. Fig. 5(b) shows the rate performances of Li/LFP and Li@LZP/LFP full cells. Both cells deliver almost the similar specific discharge capacity at the lowest current rate of 0.1C, implying that the anode itself and the interfacial condition between the anode and electrolyte are not the determined factor for the electrochemical behavior. With gradually increasing the rate from 0.1 to 2C, the discharge specific capacity shows a decreasing trend. However, the Li@LZP/LFP cell capacities are always higher than those of the Li/LFP cell, with the values of 148, 132 and 71 mA h g−1 at 0.5C, 1C, and 2C, respectively. Notably, the Li@LZP/LFP full cell has good reversibility when the charge/discharge rate is recovered from 2C to 1C, providing a specific capacity of 122 mA h g−1. Compared with the unprotected Li anode, the performance of Li@LZP is improved significantly due to the inhibition of electrolyte consumption and suppression of Li dendrite formation, indicating its great potential in high-energy Li batteries.
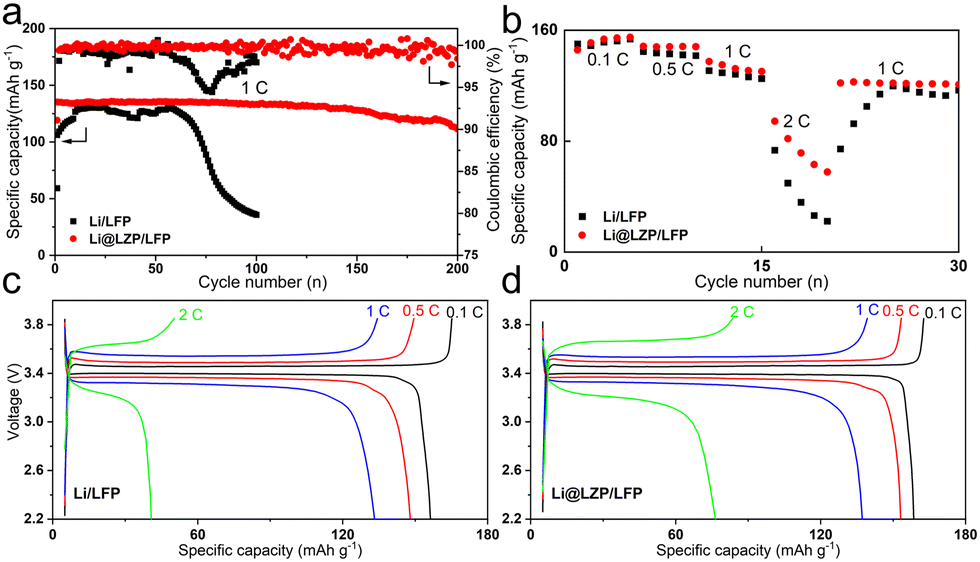 | ||
| Fig. 5 (a) Cycling performance of Li/LFP and Li@LZP/LFP full cells at a rate of 1C. (b) Rate performance of LFP-based full cells with the Li and Li@LZP anode. (c) and (d) The corresponding characteristic charge/discharge curves at various rates. | ||
Conclusions
In summary, a hybrid thin film consisting of a LiZn alloy mixed conductor, LiCl insulating particles, and a PEO soft polymer matrix was constructed on a Li metal anode as an effective protection layer by a simple coating method. The LiZn alloy is a mixed electronic/ionic conductor, featuring both high electronic conductivity and fast Li+ transport capability. LiCl is an inorganic insulator with high mechanical strength preventing the reduction of Li+ in the artificial SEI layer and inhibiting the penetration of Li dendrites. The ion-conductive PEO network with good flexibility ensures a good interfacial contact between the hybrid film and metallic Li and relieves the volume change during cycling. Consequently, the synergistic effect among the three components enables the hybrid film as a superior artificial SEI layer. The LZP composite membrane can ensure Li+ migration capability and regulate Li deposition in a dense and compact manner. The plated Li is accumulated under the hybrid protection film, which can efficiently prevent the fresh Li from being exposed to the electrolyte, significantly improving the reversibility of electrochemical plating/stripping. Furthermore, the PEO matrix can act as an adhesive binder, which plays an important role in maintaining the dimensional integrity of the hybrid buffer layer and the anode. Therefore, the modified cell in the symmetric configuration can be cycled beyond 1000 h under conditions of 1 mA cm−2 and 1 mA h cm−2, and LFP-based full cells demonstrate an excellent cycling performance and rate performance even with a high areal capacity of 2.45 mA h cm−2. This work provides a feasible pathway for the protection of Li metal anodes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the National Natural Science Foundation of China (No. 21673033 and 52172184), and the Suining Science and Technology Program (2019ZDCGZH003).Notes and references
- Z. A. Ghazi, Z. Sun, C. Sun, F. Qi, B. An, F. Li and H. M. Cheng, Small, 2019, 15, e1900687 CrossRef.
- X. He, D. Bresser, S. Passerini, F. Baakes, U. Krewer, J. Lopez, C. T. Mallia, Y. Shao-Horn, I. Cekic-Laskovic, S. Wiemers-Meyer, F. A. Soto, V. Ponce, J. M. Seminario, P. B. Balbuena, H. Jia, W. Xu, Y. Xu, C. Wang, B. Horstmann, R. Amine, C.-C. Su, J. Shi, K. Amine, M. Winter, A. Latz and R. Kostecki, Nat. Rev. Mater., 2021, 6, 1036–1052 CrossRef CAS.
- H. Huo, J. Gao, N. Zhao, D. Zhang, N. G. Holmes, X. Li, Y. Sun, J. Fu, R. Li, X. Guo and X. Sun, Nat. Commun., 2021, 12, 176 CrossRef CAS.
- B. Horstmann, J. Shi, R. Amine, M. Werres, X. He, H. Jia, F. Hausen, I. Cekic-Laskovic, S. Wiemers-Meyer, J. Lopez, D. Galvez-Aranda, F. Baakes, D. Bresser, C.-C. Su, Y. Xu, W. Xu, P. Jakes, R.-A. Eichel, E. Figgemeier, U. Krewer, J. M. Seminario, P. B. Balbuena, C. Wang, S. Passerini, Y. Shao-Horn, M. Winter, K. Amine, R. Kostecki and A. Latz, Energy Environ. Sci., 2021, 14, 5289–5314 RSC.
- D. Lin, Y. Liu, A. Pei and Y. Cui, Nano Res., 2017, 10, 4003–4026 CrossRef CAS.
- S. Park, H. J. Jin and Y. S. Yun, Adv. Mater., 2020, 32, e2002193 CrossRef PubMed.
- K. Sun and Z. Peng, InfoMat, 2021, 3, 1083–1109 CrossRef CAS.
- F. Ren, Z. Li, L. Huai, Z. Ren, M. Wang, J. Liu, H. Lin, Z. Peng and D. Wang, Carbon, 2021, 171, 894–906 CrossRef CAS.
- W. Jia, Y. Liu, Z. Wang, F. Qing, J. Li, Y. Wang, R. Xiao, A. Zhou, G. Li, X. Yu, Y.-S. Hu, H. Li, Z. Wang, X. Huang and L. Chen, Sci. Bull., 2020, 65, 1907–1915 CrossRef CAS.
- X. Wang, K. Luo, L. Xiong, T. Xiong, Z. Li, J. Sun, H. He, C. Ouyang and Z. Peng, Energy Environ. Mater., 2022, 1–11 Search PubMed.
- Z. Wang, J. Xue, Y. Liu, J. Xing, A. Zhou, J. Li, W. Zou, F. Zhou and H. Li, Sci. China Mater., 2021, 65, 69–77 CrossRef.
- D. Wang, A. Zhou, Z. Yao, X. Xia and Y. Zhang, Materials, 2021, 14, 6131 CrossRef CAS PubMed.
- H. Huo, Y. Chen, R. Li, N. Zhao, J. Luo, J. G. Pereira da Silva, R. Mücke, P. Kaghazchi, X. Guo and X. Sun, Energy Environ. Sci., 2020, 13, 127–134 RSC.
- X. Zhang, Y. Yang and Z. Zhou, Chem. Soc. Rev., 2020, 49, 3040–3071 RSC.
- K. Li, Y. Wang, W. Jia, S. Qu, Z. Yao, R. Cui, W. Zou, F. Zhou and J. Li, ACS Appl. Mater. Interfaces, 2020, 12, 2285–2292 CrossRef CAS.
- Q. Jin, X. Zhang, H. Gao, L. Li and Z. Zhang, J. Mater. Chem. A, 2020, 8, 8979–8988 RSC.
- D. Kang, S. Sardar, R. Zhang, H. Noam, J. Chen, L. Ma, W. Liang, C. Shi and J. P. Lemmon, Energy Storage Mater., 2020, 27, 69–77 CrossRef.
- S. Qu, W. Jia, Y. Wang, C. Li, Z. Yao, K. Li, Y. Liu, W. Zou, F. Zhou, Z. Wang and J. Li, Electrochim. Acta, 2019, 317, 120–127 CrossRef CAS.
- J. F. Ding, R. Xu, X. X. Ma, Y. Xiao, Y. X. Yao, C. Yan and J. Q. Huang, Angew. Chem., Int. Ed., 2022, 61, e202115602 CAS.
- X. Shen, X.-Q. Zhang, F. Ding, J.-Q. Huang, R. Xu, X. Chen, C. Yan, F.-Y. Su, C.-M. Chen, X. Liu and Q. Zhang, Energy Mater. Adv., 2021, 2021, 1–15 CrossRef.
- Y. Xiao, R. Xu, L. Xu, J.-F. Ding and J.-Q. Huang, Energy Mater., 2022, 1, 100013 CrossRef.
- D. Han, X. Wang, Y. N. Zhou, J. Zhang, Z. Liu, Z. Xiao, J. Zhou, Z. Wang, J. Zheng, Z. Jia, B. Tian, J. Xie, Z. Liu and W. Tang, Adv. Energy Mater., 2022, 12, 2201190 CrossRef CAS.
- Y. Lin, Z. Wen, J. Liu, D. Wu, P. Zhang and J. Zhao, J. Energy Chem., 2021, 55, 129–135 CrossRef CAS.
- A. C. Kozen, C. F. Lin, A. J. Pearse, M. A. Schroeder, X. Han, L. Hu, S. B. Lee, G. W. Rubloff and M. Noked, ACS Nano, 2015, 9, 5884–5892 CrossRef CAS PubMed.
- L. Chen, J. G. Connell, A. Nie, Z. Huang, K. R. Zavadil, K. C. Klavetter, Y. Yuan, S. Sharifi-Asl, R. Shahbazian-Yassar, J. A. Libera, A. U. Mane and J. W. Elam, J. Mater. Chem. A, 2017, 5, 12297–12309 RSC.
- L. Wang, Q. Wang, W. Jia, S. Chen, P. Gao and J. Li, J. Power Sources, 2017, 342, 175–182 CrossRef CAS.
- X. Wang, J. Zhuang, M. Liu, C. Wang, Y. Zhong, H. Wang, X. Cheng, S. Liu, G. Cao and W. Li, J. Mater. Chem. A, 2019, 7, 19104–19111 RSC.
- S. Choudhury and L. A. Archer, Adv. Electron. Mater., 2016, 2, 1500246 CrossRef.
- D. Lin, Y. Liu, W. Chen, G. Zhou, K. Liu, B. Dunn and Y. Cui, Nano Lett., 2017, 17, 3731–3737 CrossRef CAS PubMed.
- G. Wang, X. Xiong, D. Xie, X. Fu, Z. Lin, C. Yang, K. Zhang and M. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 4962–4968 CrossRef CAS.
- Y. Yuan, F. Wu, Y. Bai, Y. Li, G. Chen, Z. Wang and C. Wu, Energy Storage Mater., 2019, 16, 411–418 CrossRef.
- Y. Liu, D. Lin, P. Y. Yuen, K. Liu, J. Xie, R. H. Dauskardt and Y. Cui, Adv. Mater., 2017, 29, 1605531 CrossRef.
- K. Chen, R. Pathak, A. Gurung, E. A. Adhamash, B. Bahrami, Q. He, H. Qiao, A. L. Smirnova, J. J. Wu, Q. Qiao and Y. Zhou, Energy Storage Mater., 2019, 18, 389–396 CrossRef.
- S. Li, J. Huang, Y. Cui, S. Liu, Z. Chen, W. Huang, C. Li, R. Liu, R. Fu and D. Wu, Nat. Nanotechnol., 2022, 17, 613–621 CrossRef CAS PubMed.
- R. Xu, Y. Xiao, R. Zhang, X. B. Cheng, C. Z. Zhao, X. Q. Zhang, C. Yan, Q. Zhang and J. Q. Huang, Adv. Mater., 2019, 31, e1808392 CrossRef.
- X. Song, H. Wang, H. Wu, J. Zou, F. Zhao, H. Wu and X. Zeng, Appl. Surf. Sci., 2021, 565, 150589 CrossRef CAS.
- Y. Cai, Q. Jin, K. Zhao, K. Shen, L. Wu and X. Zhang, J. Alloys Compd., 2022, 900, 163444 CrossRef CAS.
- S. Li, Y. Huang, W. Ren, X. Li, M. Wang and H. Cao, Chem. Eng. J., 2021, 422, 129911 CrossRef CAS.
- K. Liao, Z. Li, S. Gu, Y. Li, L. Kong, N. Qin, H. Huang, S. Wu, J. Chen, Q. Gan, K. Zhang and Z. Lu, ACS Appl. Mater. Interfaces, 2021, 13, 45416–45425 CrossRef CAS.
- R. Xu, X.-Q. Zhang, X.-B. Cheng, H.-J. Peng, C.-Z. Zhao, C. Yan and J.-Q. Huang, Adv. Funct. Mater., 2018, 28, 1705838 CrossRef.
- Z. Jiang, L. Jin, Z. Han, W. Hu, Z. Zeng, Y. Sun and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 11374–11378 CrossRef CAS.
- Y. Liang, Y. Xiao, C. Yan, R. Xu, J.-F. Ding, J. Liang, H.-J. Peng, H. Yuan and J.-Q. Huang, J. Energy Chem., 2020, 48, 203–207 CrossRef.
- Y. Feng, C. Zhang, X. Jiao, Z. Zhou and J. Song, Energy Storage Mater., 2020, 25, 172–179 CrossRef.
- N. W. Li, Y. Shi, Y. X. Yin, X. X. Zeng, J. Y. Li, C. J. Li, L. J. Wan, R. Wen and Y. G. Guo, Angew. Chem., Int. Ed., 2018, 57, 1505–1509 CrossRef CAS.
- W. Cao, J. Lu, K. Zhou, G. Sun, J. Zheng, Z. Geng and H. Li, Nano Energy, 2022, 95, 106983 CrossRef CAS.
- A. A. Assegie, J. H. Cheng, L. M. Kuo, W. N. Su and B. J. Hwang, Nanoscale, 2018, 10, 6125–6138 RSC.
- A. Storelli, S. Rousselot, N. Alzate-Carvajal, V. Pelé and M. Dollé, J. Electrochem. Soc., 2021, 168, 040505 CAS.
- K. N. Wood, E. Kazyak, A. F. Chadwick, K. H. Chen, J. G. Zhang, K. Thornton and N. P. Dasgupta, ACS Cent. Sci., 2016, 2, 790–801 CrossRef CAS.
- S. Cho, S. Kim, W. Kim, S. Kim and S. Ahn, Polymers, 2018, 10, 1364 CrossRef PubMed.
- C. Yang, B. Liu, F. Jiang, Y. Zhang, H. Xie, E. Hitz and L. Hu, Nano Res., 2017, 10, 4256–4265 CrossRef CAS.
- C. Fu and C. Battaglia, ACS Appl. Mater. Interfaces, 2020, 12, 41620–41626 CrossRef CAS PubMed.
- Y. Zhang, W. Lv, Z. Huang, G. Zhou, Y. Deng, J. Zhang, C. Zhang, B. Hao, Q. Qi, Y.-B. He, F. Kang and Q.-H. Yang, Sci. Bull., 2019, 64, 910–917 CrossRef CAS.
- C. Ma, C. Liu, Y. Zhang, X. Zhang, Z. Zhao, T. Song, B. Wu and D. Mu, Chem. Eng. J., 2022, 434, 134637 CrossRef CAS.
- Y. Lin, Z. Wen, C. Yang, P. Zhang and J. Zhao, Electrochem. Commun., 2019, 108, 106565 CrossRef CAS.
- S. Qian, C. Xing, M. Zheng, Z. Su, H. Chen, Z. Wu, C. Lai and S. Zhang, Adv. Energy Mater., 2022, 12, 2103480 CrossRef CAS.
- G. Wang, M. Zhu, Y. Zhang, C. Song, X. Zhu, Z. Huang, Y. Zhang, F. Yu, G. Xu, M. Wu, H. K. Liu, S. X. Dou and C. Wu, InfoMat, 2022, 4, e12293 CAS.
- Y. Zhang, G. Wang, L. Tang, J. Wu, B. Guo, M. Zhu, C. Wu, S. X. Dou and M. Wu, J. Mater. Chem. A, 2019, 7, 25369–25376 RSC.
- B. Sun, J. Lang, K. Liu, N. Hussain, M. Fang and H. Wu, Chem. Commun., 2019, 55, 1592–1595 RSC.
- F. Ren, Z. Li, Y. Zhu, P. Huguet, S. Deabate, D. Wang and Z. Peng, Nano Energy, 2020, 73, 104746 CrossRef CAS.
- J. Deng, Y. Wang, S. Qu, Y. Liu, W. Zou, F. Zhou, A. Zhou and J. Li, Batteries Supercaps, 2020, 4, 140–145 CrossRef.
- A. Hu, W. Chen, X. Du, Y. Hu, T. Lei, H. Wang, L. Xue, Y. Li, H. Sun, Y. Yan, J. Long, C. Shu, J. Zhu, B. Li, X. Wang and J. Xiong, Energy Environ. Sci., 2021, 14, 4115–4124 RSC.
- Z. Wang, T. Chen, Y. Liu, J. Xing, A. Zhou, J. Li, W. Zou and F. Zhou, Chem. Eng. J., 2022, 430, 132970 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04787j |
| This journal is © the Owner Societies 2023 |