
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective hydrogenation of highly concentrated acetylene streams over mechanochemically synthesized PdAg supported catalysts†
Klara Sophia
Kley‡
 ,
Jacopo
De Bellis‡
and
Ferdi
Schüth
*
,
Jacopo
De Bellis‡
and
Ferdi
Schüth
*
Department of Heterogeneous Catalysis, Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470, Mülheim an der Ruhr, Germany. E-mail: schueth@kofo.mpg.de
First published on 24th November 2022
Abstract
The selective hydrogenation of acetylene to ethylene in equimolar acetylene–ethylene streams is challenging due to the extreme exothermicity of the reaction. However, it could be necessary to convert the product stream of plasma-induced methane pyrolysis processes, where large amounts of acetylene are typically formed, to pure ethylene. This would allow a new production pathway for ethylene, but new catalyst materials need to be designed for this specific purpose. To this end, several Pd–Ag bimetallic catalysts supported on α-Al2O3 were obtained by ball milling and conventional wet impregnation, and their performance was compared. In general, higher amounts of Ag in the alloy were beneficial to attain high selectivity levels in the reaction, irrespective of the preparation method used. However, if the synthesis method little affected the selectivity, it greatly affected the stability of the catalysts under reaction conditions. While catalysts prepared via wet impregnation were typically stable for 5–10 h depending on the conditions, ball-milled catalysts exhibited no signs of deactivation for as long as tested (up to 15 h). The best performing formulation of the catalyst was reproduced with a mixed synthesis approach to understand possible underlying effects. This work shows that catalysts prepared by ball milling are the most suitable choice for such a challenging reaction.
Introduction
Ethylene is a primary building block in the chemical industry as it is mainly involved in the mass production of polyethylene, the worldwide most used polymer.1 However, ethylene also has multiple other uses. For instance, it is the starting material for short-chain linear oligomers, used directly as polyethylene comonomers or as plasticizers, lubricants, and detergents after suitable functionalization. In addition, it could be converted into ethylene oxide, a convenient starting material to manufacture surfactants and detergents. Likewise, when converted to vinyl chloride (via halogenation and hydrohalogenation) or styrene (via alkylation), ethylene serves as the primary precursor of other valuable polymeric materials, such as polyvinylchloride and polystyrene. Thus, the worldwide production volume of ethylene was as high as 214 Mt in 2021,2 a value expected to grow even further due to the increasing demand and use of polymeric materials and other products.3The most convenient way to produce ethylene on a large scale is via the steam cracking of naphtha. In several countries, especially in the US, light alkanes have become an increasingly important feedstock due to the low cost of shale gas. Other production processes include methanol to olefins (MTO) and Fischer–Tropsch (FT) synthesis.4 In addition, pilot productions have started or are projected to synthesize renewable ethylene from ethanol.
Another possible production path, connecting methane directly to ethylene, involves the conversion of biogas, natural gas, or biomass in an electric plasma arc. In such a process, the hydrocarbon feed is cracked into a mixture of acetylene, ethylene, and hydrogen, leaving carbon black as a solid product. This process is known since 1940, i.e., as the Hüls process, and has been long used to produce acetylene, although from coal and not natural gas.5–8 However, acetylene must be selectively hydrogenated to obtain high-grade ethylene, directly in the plasma reactor,9,10 or in a dedicated catalytic reactor.11,12 Recently, Delikonstantis and co-workers reported on the success in converting methane to ethylene through the cracking of methane in a non-thermal plasma reactor and subsequent selective hydrogenation of the gas feed over a commercial supported Pd catalyst.13,14 The process appears to be a more suitable production pathway than thermally-driven and other processes, according to the latest life-cycle assessments.15 However, since the hydrogenation conditions vary severely from those typically applied for the selective hydrogenation of acetylene in industry, i.e., elevated levels of acetylene and high acetylene to ethylene ratios in the gas feed, a catalyst specifically designed for this purpose would be beneficial to improve the overall efficiency of the process.
Typically, the semi-hydrogenation of acetylene is required to purify ethylene before further utilization. Since acetylene is a potent poison for the downstream polymerization catalysts, the levels obtained at the exit of a steam cracker need to be reduced significantly, typically from values as high as 2% to less than 1 ppm, as current specifications would require.16 To this end, several catalytic systems have been investigated, especially Pd-based catalysts have received the most attention. Supported forms of Pd are generally highly active but not very selective; hence ethylene is typically fully hydrogenated to ethane unless promoters are used.17 Among other strategies, the selectivity can be enormously improved by exploiting the so-called “ensemble effect”.18 Briefly, it was postulated that mixing of Pd with a second metal (e.g., Cu, Ag, Au) could help reduce the concentration of large Pd ensembles primarily responsible for the complete hydrogenation to ethane and thus improve selectivity.19,20 At the same time, catalyst stability would also be enhanced because the formation of oligomers (green oils) and other carbonaceous deposits is similarly moderated.21 Similar effects were observed with newly designed Pd-based catalysts promoted by Zn or Ga.22,23 However, improved selectivity comes with the compromise of having fewer active and stable palladium sites. Due to the significant economic implications and potential for further progress, developing more selective and longer stable hydrogenation catalysts still represents a very active area of research.18 Present trends include single-atom catalysts and noble-metal-free formulations.24–26
The scope of the present research work is to investigate the selective hydrogenation of acetylene under strongly different conditions than previously described and implemented in practice. High partial pressures and equivalent amounts of acetylene and ethylene will be explored, conditions that will significantly challenge catalyst stability and heat management. As a starting point, the behavior of classical supported PdAg catalysts, either prepared via conventional wet impregnation (and drying) or a novel mechanochemical approach, will be investigated. Briefly, when the coarse powders of metal and oxide support are energetically ball-milled together, supported metal nanoparticles are formed.27,28 In a follow-up study, it was also found that combining two metal sources results in alloyed nanoparticles under similar conditions.29 The method proved to be somewhat flexible concerning the choice of metals and oxide support.30 In fact, compatibly with the scope of the study, it was used to prepare a variety of supported PdAg catalysts with different relative content of Pd and Ag. The effect of composition on catalytic behavior was studied in detail. In addition, the role of the support (e.g., low- and high-surface-area α-Al2O3) and the possible impact of alternative metal precursors (e.g., metal chlorides and/or nitrates) used during the synthesis was considered. Thus, the study could highlight which material features contribute the most to the overall performance and durability of the catalysts under the harsh reaction conditions explored.
Experimental
Materials
Boehmite (γ-AlOOH) was obtained from Sasol (DISPAL 11 N7-80, Lot No. S4486J) as nanopowder (71 m2 g−1, measured) and used as received. Low-surface-area (LSA) α-Al2O3 was obtained via the thermal decomposition of γ-AlOOH at 1200 °C (10 h, 2 °C min−1). High-surface-area (HSA) α-Al2O3 was obtained from the mechanochemically-induced dehydration of γ-AlOOH, i.e., upon ball milling, according to a variation of the method by Amrute et al. (vide infra).31 Palladium powder (Alfa Aesar, −200 mesh, 99.95%, metal basis), palladium(II) chloride powder (Sigma Aldrich, ≥99.9%), and palladium(II) nitrate dihydrate crystalline powder (Sigma Aldrich, ∼40% Pd basis) were used as received. Silver powder (Alfa Aesar, −120 +325 mesh, atomized, 99%, metals basis), silver(I) chloride powder (Sigma Aldrich, 99.999%, trace metal basis), and silver(I) nitrate crystalline powder (Sigma Aldrich, 99.9999%, trace metal basis) were used as received.Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y) / HSA-α-Al2O3 and 800 mg of HSA-α-Al2O3 (from 6 h of milling) were milled at 450 rpm for 1 h (4 cycles of 15 min milling followed by a 5 min break) with three big grinding balls (15 mm Ø). The same protocol was used to synthesize a material containing 1 wt% of Ag.
:y) / HSA-α-Al2O3 and 800 mg of HSA-α-Al2O3 (from 6 h of milling) were milled at 450 rpm for 1 h (4 cycles of 15 min milling followed by a 5 min break) with three big grinding balls (15 mm Ø). The same protocol was used to synthesize a material containing 1 wt% of Ag.
Characterization methods
Catalyst testing and recovery
The catalytic tests were carried out in a pressurized continuous-flow fixed-bed reactor. Handling acetylene under pressure requires extreme caution. Hence, several safety measures were implemented upon the operation of the setup. More detailed information can be found in the dissertation of I.-T. Trotus.33 All gases (e.g., hydrogen, acetylene, ethylene, and methane) were obtained from Air Liquide. Except for acetylene, all other gases were used without further purification. Acetylene was purified from possible acetone impurities (acetylene is dissolved in liquid acetone in the storage cylinders) by selective adsorption over alumina and zeolite A extrudates. Next, acetylene was compressed from 1.5 bar to 20 bar with a two-stage compressor. All gas streams were controlled with pressure reducers, mass flow controllers, and magnetic valves. Additional check valves and safety valves were implemented. The reactor tube (8 mm i.d.) was loaded with 0.1 g of catalyst diluted in 3.0 g silicon carbide (Alfa Aesar, 46 grit). The catalyst bed was supported on a metal mesh and held in place with a quartz wool plug. The reactor was heated to a set temperature, and the temperature inside the catalytic bed was monitored with a thermocouple. The pressure upstream of the reactor (including the reactor itself) was maintained around a fixed value through a temperature-conditioned back pressure regulator (BPR). Methane was added downstream from the BPR as an internal standard for gas chromatographic (GC) analysis. All setup parts downstream from the reactor, including lines and connections, were conditioned and maintained at around 150 °C to prevent condensation of possible low boiling side products. The gas mixture leaving the reactor was analyzed via online gas chromatography, i.e., Agilent 7890B. In detail, the GC device was equipped with three columns (e.g., RT-alumina BOND/Na2SO4, Rxi-5Sil MS, and RT-Msieve 5A from RESTEK) and three detectors (e.g., two flame ionization detectors, FID, and one thermal conductivity detector, TCD) to resolve and correctly discriminate between short-chain hydrocarbons, long-chain hydrocarbons (>C4), and inert gases, respectively. A complete schematic of the setup is provided in Fig. S4.† Typically, the setup was operated under an overall pressure of about 10 bar. The gas flows were customarily set around 100 mL min−1 for hydrogen, 20 mL min−1 for ethylene and acetylene, and 10 mL min−1 for methane. Thus, the catalysts were tested at a total weight hourly space velocity (WHSV) of 90000 cm3 h−1 g−1cat. For the catalytic testing, the reactor was heated to 50, 100, or 150 °C.
The conversion (X) was calculated by relating the amount of acetylene leaving the reactor (nC2H2,out) to the amount of acetylene injected (nC2H2,in), as in eqn (a). Ethylene selectivity (SC2H4) was found by relating the excess of ethylene found in the stream leaving the reactor (nC2H4,out − nC2H4,in) and the total amount of acetylene converted (nC2H2,out − nC2H2,in), as in eqn (b). Ethane selectivity (SC2H6) is represented by the fraction of ethane (nC2H6,out) originating from the conversion of acetylene (nC2H2,out − nC2H2,in), as in eqn (c).
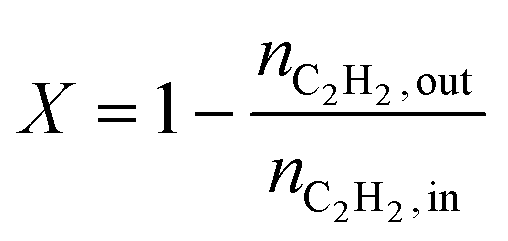 | (a) |
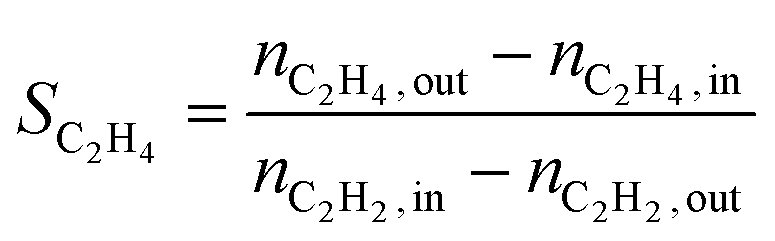 | (b) |
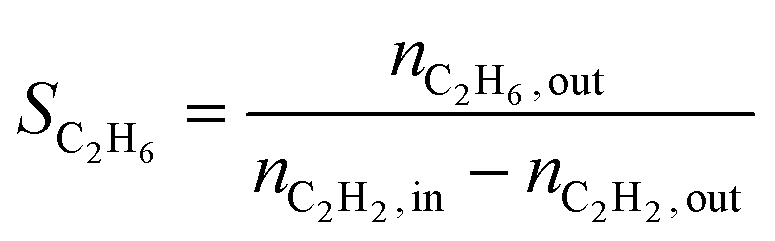 | (c) |
Results and discussion
Direct dry synthesis of the supported PdAg catalysts
Concerning the mechanochemical synthesis, each material was prepared in two steps. First, a concentrated batch, i.e., including about 5 wt% of Pd and Ag, was obtained upon ball milling by combining the coarse metal powders and the α-Al2O3 support precursor (boehmite, γ-AlOOH) in one pot. Reaction conditions were optimized to observe the comminution and alloying of the metal components, while high-surface-area (HSA) α-Al2O3 was formed via the mechanochemically-induced decomposition of γ-AlOOH.34–36 Particularly, the powders were milled for 6 h in yttria-stabilized zirconia milling jars with a few big balls of the same material at a milling frequency of about 500 rpm. It has already been reported that under suitable conditions, the dehydration of boehmite leads to the formation of α-Al2O3 with remarkably high specific surface area upon ball milling.31 Accordingly, the process resulted in the formation of Pd–Ag alloy nanoparticles on HSA-α-Al2O3. However, the target material, i.e., including 1 wt% of Pd and Ag, was obtained by diluting the concentrated batch with freshly prepared HSA-α-Al2O3 again upon ball milling.For the dilution, similar conditions were applied: zirconia-based milling equipment and a bulky grinding medium were again used, but a lower milling frequency was selected (450 rpm instead of 500 rpm) to boost specific surface area values by breaking large aggregates and at the same time limit abrasion of the zirconia jar and balls. Interestingly, the average particle size distribution is maintained upon dilution, and the metal nanoparticles are similarly homogeneously dispersed over the oxide support. This was especially demonstrated in the case of PdAg (1:1) supported nanoparticles (Fig. S1†). The synthesis of concentrated batches and subsequent dilution was applied to prepare materials with different Ag-to-Pd molar ratios (e.g., 1, 5, and 9) and one containing only Ag. A thermal treatment was applied to all the materials (vide infra) before catalyst testing. The following notation will be used to identify the materials: PdAg (1:1), PdAg (1:5), PdAg (1:9), and Ag / HSA-α-Al2O3 _BM. All the characterization results are summarized in Table 1.
:y, as specified) catalysts supported on LSA/HSA-α-Al2O3 prepared either by wet impregnation (WI) or ball milling (BM)
| Catalyst ID | Phase composition (XRD) | Pd + Ag load. (EDX, wt%) | Bulk Ag/Pd (EDX) | Surface Ag/Pd (XPS) | Surf. Area (BET, m2 g−1) | d-NP ± σ (TEM, nm) |
|---|---|---|---|---|---|---|
| Ag LSA-α_WI | Ag, α-Al2O3 | 1.5 | No Pd | Only Ag | 8 | 9 ± 11 |
| Ag HSA-α_BM | Ag, γ/α-Al2O3, (t-ZrO2, m-ZrO2) | 1.5 | No Pd | Only Ag | 81 | 8 ± 3 |
| PdAg (1:9) LSA-α _WI |
PdAg, α-Al2O3 | 1.0 | 18 | Low Pd | 9 | 11 ± 6 |
| PdAg (1:9) HSA-α _BM |
PdAg, γ/α-Al2O3, (t-ZrO2, m-ZrO2) | 1.3 | 10 | Only Ag | 73 | 6 ± 3 |
| PdAg (1:5) LSA-α_WI |
PdAg, α-Al2O3 | 1.6 | 3.4 | Low Pd | 6 | 11 ± 6 |
| PdAg (1:5) HSA-α _BM |
PdAg, γ/α-Al2O3, (t-ZrO2, m-ZrO2) | 1.4 | 5.1 | Only Ag | 78 | 6 ± 4 |
| PdAg (1:1) LSA-α_WI |
PdAg, α-Al2O3 | 1.5 | 1.5 | 2 | 8 | 12 ± 12 |
| PdAg (1:1) HSA-α_BM |
PdAg, γ/α-Al2O3, (t-ZrO2, m-ZrO2) | 1.5 | 0.9 | Only Ag | 69 | 9 ± 4 |
Synthesis of the PdAg catalysts via impregnation and drying
As one of the most commonly applied methods to prepare supported metal catalysts, wet impregnation (and drying) was chosen as an alternative synthesis protocol.37 In this way, a homologous series of supported catalysts was synthesized by impregnating the α-Al2O3 support, i.e., low-surface-area (LSA) α-Al2O3 (<10 m2 g−1), with an aqueous solution of the Pd and/or Ag nitrate salts, followed by a suitable drying procedure. The resulting materials were then reduced in a pure hydrogen atmosphere (150 °C, 3 h) and finally annealed under argon at a higher temperature (600 °C, 10 h). Reduction and annealing temperatures were optimized to ensure that Pd and Ag would alloy upon the thermal treatment. As already mentioned, the thermal treatment was also applied to the ball-milled materials, even though no reduction and annealing would be required, to have all the materials exposed to the same conditions prior to the catalysis. As before, three materials with different Ag-to-Pd molar ratios (e.g., 1, 5, and 9) and one containing only Ag were obtained. The following notation will be used to address the materials obtained via impregnation and drying: PdAg (1:1), PdAg (1:5), PdAg (1:9), and Ag / LSA- α-Al2O3 _WI. All the characterization results are reported in Table 1.
Characterization of wet-impregnated and ball-milled materials
From the characterization of the samples, some distinctive features were noted. The most obvious differences concern the α-Al2O3 support. Wet impregnation was carried out on α-Al2O3 with a relatively low specific surface area, i.e., <10 m2 g−1, a feature preserved after the functionalization with metal nanoparticles (Table 1). In contrast, the materials obtained by ball milling are characterized by a much higher specific surface area, i.e., <110 m2 g−1, which is also reflected in a reduction in the average size of the alumina particles by about one order of magnitude. From Table 1, it is also evident that the specific surface area of all metal-functionalized nanopowders obtained by ball milling is systematically lower than the one found for the bare support (upper limit previously reported), probably due to particle agglomeration (Fig. S3e–S3h†). This phenomenon is known to occur upon extended milling and could be limited but not avoided entirely.38 A similar drop in the specific surface area was observed for the bare support when milled for an equivalent amount of extra time, i.e., from 110 m2 g−1 after 6 h of milling to 94 m2 g−1 after 7 h.As shown in Fig. S2a,† the XRD patterns of the materials obtained via wet impregnation are dominated by the sharp diffraction lines of the highly crystalline α-Al2O3. In contrast, in the case of the ball-milled materials, broad, low-intensity diffraction lines characterize the α-Al2O3 phase (Fig. S2c†). Therefore, a twofold magnification was necessary to identify all relevant features of the samples compared to the former case. The presence of small amounts of γ-Al2O3 in ball-milled samples, resulting from the crystallization of partly amorphous or simply unprocessed material, should be noted. Finally, ZrO2 was also detected due to some abrasion of the milling equipment (Fig. S2c†). However, according to energy-dispersive X-ray (EDX) elemental analysis, the samples contain only a minimal amount of ZrO2, i.e., from 0.7 to 1.2 wt%.
The typical reflexes of the metallic components are detected in the XRD patterns of all the materials. Most importantly, the position of the reflections is consistent with the existence of alloy phases. A clear shift in the position of the reflex maxima towards lower 2θ-angles with increasing content of Ag was observed, both in the case of wet-impregnated (Fig. S2b†) and ball-milled materials (Fig. S2d†), in agreement with expectations according to Vegard's law.39 However, unfortunately, the overlapping of dominant reflections, such as those from α-Al2O3 and γ-Al2O3, prevented a more accurate determination of the average composition of the metal particles from the fitting of corresponding diffraction line profiles. Still, EDX bulk elemental analysis reported Pd and Ag levels in the samples compatible with target values (Table 1), which, combined with the detection of only one metallic crystalline phase from XRD, would support the formation of supported metal particles with the desired compositions. However, it is important to note that a somewhat more significant deviation from expected compositions was found for the samples obtained via wet impregnation, possibly, due to the problematic manipulation of Pd(II)-nitrate as a source of small amounts of Pd due to its highly hygroscopic character. The broad shape of the diffraction lines for the metallic species supports the existence of nanosized crystalline domains. This notion was corroborated by the direct observation of metal nanoparticles in the electron microscope (Fig. S3†). EDX line scans were also performed for selected systems to confirm alloying of Pd and Ag (Fig. S18†). No surface segregation was noted irrespective of the synthesis method used.
The most noticeable differences between impregnated and ball-milled materials were observed when corresponding metal particle size distributions were compared (Table 1 and Fig. S3†). In fact, for each given composition, the impregnated materials present larger average supported metal nanoparticles than the mechanochemically synthesized counterparts. In addition, the size distribution is broader in the former case. Thus, average particle sizes from 9 to 12 nm and standard deviations as high as 12 nm were estimated for impregnated materials (Fig. S3a–S3d†). In contrast, average particle sizes in the 6 to 9 nm range and standard deviations from 3 to 4 nm were observed in the case of ball-milled materials (Fig. S3e–S3h†). This is probably due to the different properties of the α-Al2O3 support for the two synthetic pathways. The higher specific surface area of the support is generally beneficial to improve the metal dispersion, in line with the observations described above. Further, it is also possible that the surface properties of the HSA-α-Al2O3 support may be involved.40–42 It has already been demonstrated that different alumina phases enable a different level of dispersion of metal species, depending on their surface acidity.43
Another peculiar feature of ball-milled materials compared to impregnated counterparts is the Pd and Ag surface distribution. For impregnated materials, the surface Ag-to-Pd molar ratio, determined via X-ray photoelectron spectroscopy (XPS), was compatible with the bulk values estimated via EDX elemental analysis (Table 1). In contrast, in the case of ball-milled materials, Pd could not be detected. This was somewhat unexpected, considering that both impregnated and ball-milled materials were subjected to the same thermal treatment and thus exposed to the same conditions before the analysis was carried out so that no preferential segregation would be expected. On the other hand, primary differences in the samples, possibly intrinsic to the method used for the synthesis, could account for the differences observed in the surface distribution of Pd and Ag when not moderated by the thermal treatment. However, the experimental evidence is clear, and further investigation would be necessary to understand the phenomenon in detail.
Catalytic performance of impregnated and ball-milled materials
All the materials were tested for the selective hydrogenation of acetylene. The reaction was performed in a continuous flow fixed bed reactor at equal partial pressures of ethylene and acetylene, as detailed in the Experimental section, under a total pressure of 10 bar. Reaction products were analyzed by online gas chromatography. The setup was built with several control measures to safely work with highly concentrated and pressurized acetylene streams. The complete schematic of the setup is represented in Fig. S4.† Irrespective of the safety measures applied, working at the setup required an operator to be present at all times. Thus, the catalytic reaction could be carried out only for a limited time, typically up to 8 h, or until the catalyst had become inactive, as indicated by the absence of hydrogenation products. However, some catalysts were active for longer than 8 h. In these cases, no sign of deactivation was observed for as long as they were tested.For each material investigated, the overall performance, as activity and selectivity in the reaction, was evaluated at 50 °C, 100 °C, and 150 °C. However, the extreme exothermicity of the reaction complicated the fine-tuning of the temperature inside the catalytic bed, especially during the initial stages of the reaction when the catalyst is first brought in contact with the reactive gas mixture. This resulted in severe overheating with temperature spikes of 150 °C above the set temperature in the first minutes on stream. The complete data log, including conversion and selectivity levels, the temperature of the reactor and catalytic bed versus time on stream, are provided as supporting material (from Fig. S5–S10†).
In Fig. 1a, the selectivity of the reaction towards the formation of ethane, ethylene, and C4 hydrocarbons after 30 min on stream is reported for different reaction temperatures and each catalytic system. In Fig. 1b and c, the average selectivity is similarly presented after 2–3 h and 5–8 h. Every time, only catalysts showing a conversion above 95% during the entire specified time on stream are considered. Accordingly, the results for Pd-free materials are not reported as no activity was observed up to 150 °C, irrespective of the preparation method used. This proves that the presence of Pd is necessary for any activity and that Ag alone (or the oxide support) is insufficient to catalyze the reaction to an appreciable extent under the reaction conditions studied.
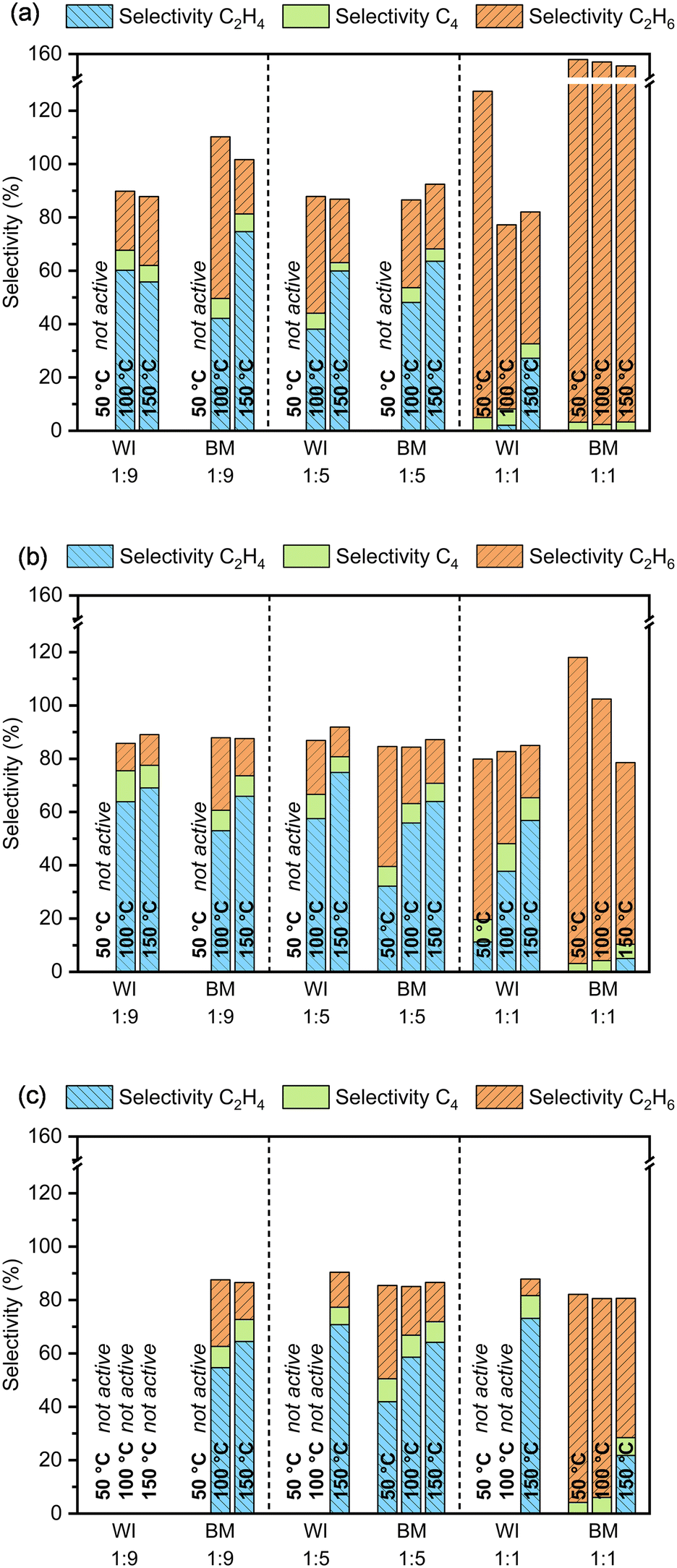 | ||
| Fig. 1 Selectivity towards ethylene (C2H4), ethane (C2H6), and C4 hydrocarbons after 30 min (a) and average selectivity after 2–3 h (b) and 5–8 h (c) for 1 wt% PdAg (x:y = 1:1; 1:5; 1:9) catalysts supported on α-Al2O3 catalysts prepared by wet impregnation (WI, LSA) and ball milling (BM, HSA). Selectivity levels are shown only for the materials exhibiting acetylene conversion above 95% for the entire period specified at 50, 100, and 150 °C (reactor temperature). | ||
As shown in Fig. 1, the materials with the highest Pd content, i.e., Ag/Pd molar ratio around unity, are not suitable for the selective hydrogenation of acetylene, independently of the preparation method used, as ethane is mainly obtained. These catalysts were so active that acetylene and ethylene were both hydrogenated to ethane, resulting in apparent ethane selectivity above 100% (NB the selectivity was referred to the conversion of acetylene; more information in the Experimental section). However, for the same composition, the materials prepared by wet impregnation were moderately more selective towards ethylene than ball-milled counterparts, probably due to the slightly higher Ag content (Table 1). Interestingly, the selectivity seems to improve over time on stream, especially for the catalysts designed with a Pd/Ag molar ratio of 1, probably due to the strong initial temperature excursion. This feature becomes more evident when the selectivity patterns after 30 min (Fig. 1a) and after several hours on stream are compared (Fig. 1b and c). While the selectivity improves for catalysts with high Pd content, the conversion remains at 100%, meaning that ethylene hydrogenation is moderated and consequently less ethane is produced. However, it is also possible for this behavior to depend on the gradual accumulation of changes in the catalyst material under reaction conditions, including those leading to its deactivation. Such changes for some time hardly affect the conversion, when the conversion of the target substrate is essentially complete, and thus the reaction is substrate limited.
On the other hand, the catalysts with increased Ag content, i.e., Ag/Pd molar ratio around 5 and 9, show good selectivity towards ethylene, with the latter being slightly more performant after some time on stream. This is in line with previously reported results on different Ag/Pd ratios for hydrogenation of trace amounts of acetylene, showing that an increase in Ag content improves selectivity.44
A trend towards improved selectivity was also noticed with increasing reaction temperature, consistently found for all tested catalysts. Hence, the best ethylene selectivity was observed when the reaction was performed at 150 °C. In contrast, when the reaction was carried out at 50 °C, the materials with a high Ag content were only slightly, if at all, active in the reaction. Only the material with an Ag/Pd ratio around 5, obtained by ball milling, grew more active within the first 30 min on stream, from only 20% to 100% conversion, after a sufficiently high temperature was attained in the catalytic bed (Fig. S8a†). Interestingly, it was also observed that spent catalysts, i.e., inactive at low temperature, do not recover catalytic performance when exposed to higher temperatures under reaction conditions, even though they are known to be active at such temperatures from directly testing them at higher temperatures. This is because the contact with the reactants at a relatively low temperature alters the catalyst irreversibly, possibly due to the deposition of green oils and/or other carbonaceous deposits on the metal nanoparticles. Not surprisingly, all catalysts proved to be longer stable at increasing reaction temperatures where the desorption/deconstruction of possible precursors of such polymeric species is likely favored.
The synthesis method did not show any distinctive influence on ethylene selectivity. Overall, the results indicate that the higher the Ag content, the smaller the difference in selectivity when comparing wet-impregnated and ball-milled materials. Only for the catalysts with the highest Pd content, wet impregnation seemed beneficial to obtain higher selectivity levels, but small differences in the composition of catalyst materials could also account for the observed behavior. Yet, a low Ag/Pd ratio of 1:1 is not advantageous to enhance ethylene selectivity in the first place. Interestingly, the selectivity towards C4 hydrocarbons was somewhat independent of the catalyst system or reaction conditions and was initially estimated to be between 3–5% and around 8% after a few hours on stream for some catalysts. Notably, for catalysts with an Ag/Pd molar ratio around 5 and 9, the selectivity towards C4 hydrocarbons was slightly higher at 50 °C than at 100 and 150 °C, suggesting that the formation of long-chain hydrocarbons is more likely at low temperatures.
The selectivity profile tends to change with time on stream; the selectivity towards ethylene formation at 150 °C typically increases, as shown in Fig. 2. All catalysts were initially rather unselective, showing ethylene selectivities below 50%. For all the materials containing more Ag than Pd, stable ethylene selectivity levels above 60% were attained under reaction conditions within the first 30 min. In contrast, in the case of the catalysts containing about the same amount of Pd and Ag, no conversion to ethylene was observed for short reaction times: these catalysts are too active so that acetylene and ethylene are both hydrogenated to ethane. However, the selectivity of these catalysts slowly improves over time. This evidence might be related to the gradual formation of subsurface PdCx species, as it has already been reported in the literature.45 Briefly, if the accumulation of C species in the subsurface regions is observed, the formation of interstitial hydrides is likely hindered. As a result, fewer activated hydrogen species are locally available, and over-hydrogenation is less probable.
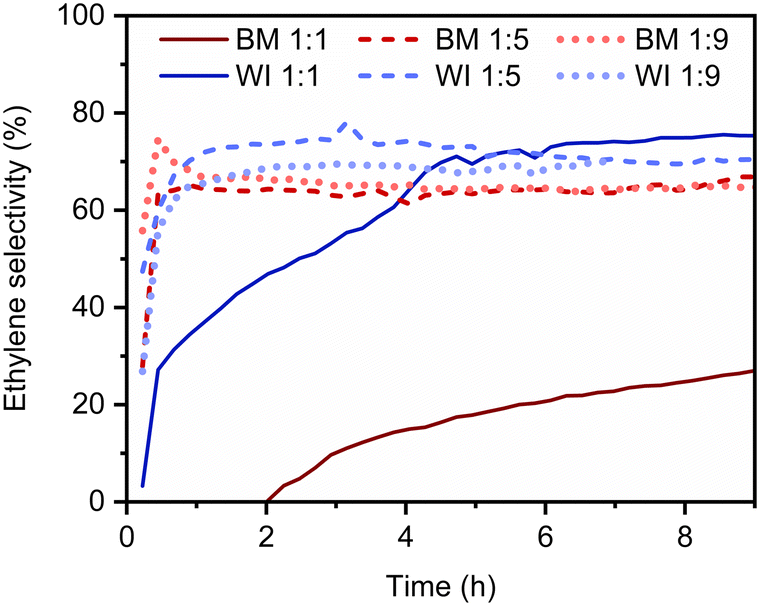 | ||
| Fig. 2 Selectivity towards ethylene formation vs. time on stream for 1 wt% PdAg (x:y = 1:1; 1:5; 1:9) catalysts supported on α-Al2O3 prepared by wet impregnation (WI, LSA) and ball milling (BM, HSA) at 150 °C (reactor temperature). | ||
The most striking difference in catalytic performance when synthesis methods are compared is the long-term stability. This can be seen in Fig. 3, where the activity time, defined as the time during which conversion levels remain above 95%, is presented. Indeed, while most catalysts prepared via wet impregnation turned inactive within 5–9 h, ball-milled counterparts remained active for as long as tested, sometimes up to 15 h without deactivation. Interestingly, the drop in conversion when the catalyst loses its activity is relatively steep for wet-impregnated materials and declines from 100% conversion to below 10% within 15–30 min, in contrast with ball-milled material, if any deactivation was observed at all (Fig. S9 vs. S10†). These results indicate that catalysts prepared by ball milling are more suitable under the harsh reaction conditions determined by high acetylene partial pressures. Possibly, the larger specific surface area, along with the smaller average metal particle sizes and narrow size distributions, account for the superior stability of the materials prepared by ball milling. However, it is also possible that surface phenomena might be involved. To gain further insight into the deactivation mechanism of the catalysts, some of the spent catalysts were characterized, as will be discussed later.
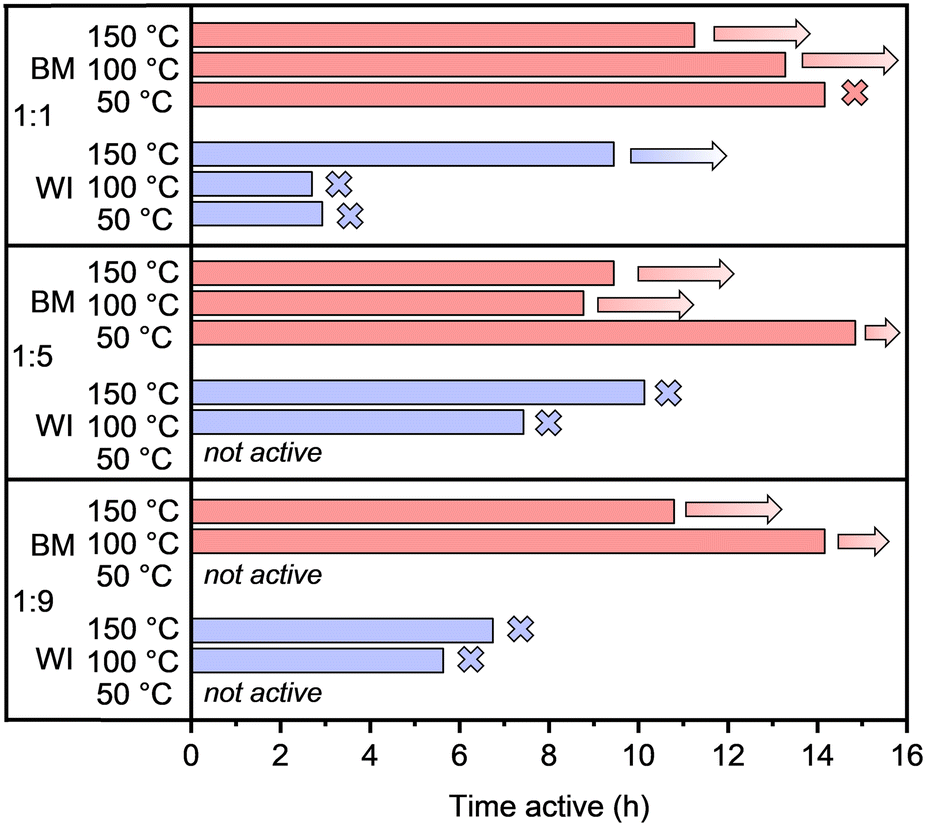 | ||
| Fig. 3 Activity time (above 95% conversion of acetylene) of PdAg (x:y = 1:1; 1:5; 1:9) catalysts supported on α-Al2O3 prepared by wet impregnation (WI, LSA) and ball milling (BM, HSA) at 50, 100, and 150 °C (reactor temperature). Crosses mark catalysts for which deactivation was observed; arrows are assigned to catalysts for which no sign of deactivation (conversion drop below 95%) was observed before the reaction was interrupted. | ||
Impact of metal precursors and support on material features
To study whether the choice of metal precursors and the nature of the α-Al2O3 support could impact catalyst performance, wet impregnation was also carried out on HSA-α-Al2O3 as support. The mechanochemical synthesis was also adapted to use metal chlorides and/or nitrates, corresponding to the wet impregnation synthesis, as the metal precursors and LSA- or HSA-α-Al2O3 as the support (more information in the Experimental section). It is important to note that only the most performant formulation of the catalyst, i.e., Ag/Pd molar ratio of around 9, was prepared using the mixed synthesis approach. The palladium(II) nitrate salt was not used as the solid precursor for Pd in the mechanochemical synthesis due to its hygroscopic character.As shown in Table 2, the composition estimated via EDX bulk elemental analysis agreed reasonably well with nominal values. However, low Pd levels resulted in high uncertainty on the Ag/Pd ratios. Still, the formation of alloyed species could be confirmed via XRD (Fig. S11b and S12b†). Likewise, it was found that low and high surface area materials share a similar phase composition (Fig. S11a and S12a†). Independently, N2-physisorption showed that specific surface area values were generally retained (Table 2). The main differences were again observed when metal nanoparticle size distributions were compared, with the trends described above still being valid. Low surface area materials (Fig. S13a and S13b†) present larger metal particles on average compared to their high surface area counterparts (Fig. S13c–S13e†), irrespective of the synthesis procedure applied. No systematic trend was noted when the specific combination of metal precursors was considered within the same group of materials. XPS analysis found only a negligible amount of Pd (below the quantification limit) in all cases.
:9) catalysts supported on LSA/HSA-α-Al2O3 prepared either by wet impregnation or ball milling with metals (m), chlorides (c), and nitrates (n) as metal precursors for Pd and Ag, respectively
| Catalyst ID | Phase composition (XRD) | Pd + Ag load. (EDX, wt%) | Bulk Ag/Pd (EDX) | Surface Ag/Pd (XPS) | Surf. area (BET, m2 g−1) | d-NP ± σ (TEM, nm) |
|---|---|---|---|---|---|---|
| PdAg (1:9) LSA-α _WI (n,n) |
PdAg, α-Al2O3 | 1.0 | 18 | Low Pd | 9 | 11 ± 6 |
| PdAg (1:9) LSA-α _BM (c,c) |
PdAg, α-Al2O3, (t-ZrO2, m-ZrO2) | 1.3 | 5 | Low Pd | 10 | 10 ± 4 |
| PdAg (1:9) LSA-α _BM (c,n) |
PdAg, α-Al2O3, (t-ZrO2, m-ZrO2) | 1.4 | 7 | Low Pd | 7 | 8 ± 3 |
| PdAg (1:9) HSA-α _WI (n,n) |
PdAg, γ/α-Al2O3, (t-ZrO2, m-ZrO2) | 1.3 | 8 | Low Pd | 161 | 7 ± 5 |
| PdAg (1:9) HSA-α _BM (c,c) |
PdAg, γ/α-Al2O3, (t-ZrO2, m-ZrO2) | 1.7 | 8 | Low Pd | 107 | 5 ± 3 |
| PdAg (1:9) HSA-α _BM (c,n) |
PdAg, γ/α-Al2O3, (t-ZrO2, m-ZrO2) | 1.4 | 9 | Low Pd | 102 | 5 ± 2 |
| PdAg (1:9) HSA-α _BM (m,m) |
PdAg, γ/α-Al2O3, (t-ZrO2, m-ZrO2) | 1.3 | 10 | Low Pd | 73 | 6 ± 3 |
The same procedures were applied to investigate the catalytic performance of the latter materials. However, the reaction was only performed at 150 °C under the best possible conditions (Fig. S16 and S17†). In Fig. 4, the selectivity after 30 min is compared with the average selectivity over 5–8 h on stream (when active). Furthermore, the results from the testing of the materials with the same nominal composition, obtained via the impregnation of LSA-α-Al2O3 and the original mechanochemical synthesis, are also compared. Finally, the time active on stream is presented in Fig. 5 for the same set of materials.
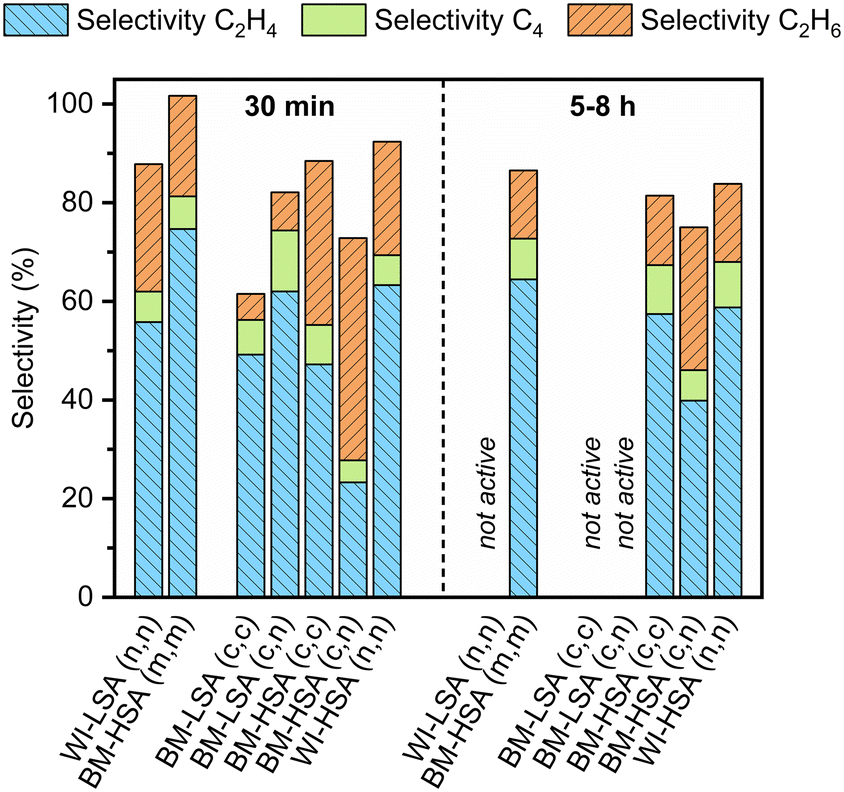 | ||
| Fig. 4 Selectivity towards ethylene (C2H4), ethane (C2H6), and C4 hydrocarbons formation after 30 min and average selectivity after 5–8 h for 1 wt% PdAg (1:9) catalysts supported on LSA/HSA-α-Al2O3 prepared either by wet impregnation (WI) or ball milling (BM) with metals (m), chlorides (c) and nitrates (n) as metal precursors for Pd and Ag, respectively. Selectivity levels are shown only for the materials exhibiting acetylene conversion above 95% for the entire period specified at 150 °C (reactor temperature). | ||
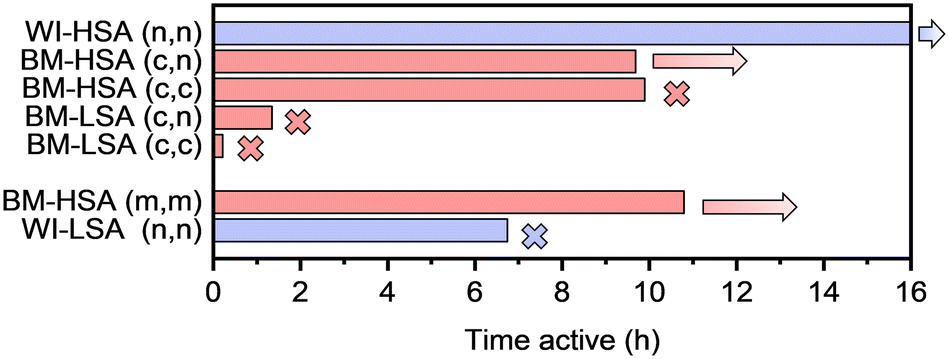 | ||
| Fig. 5 Activity time (above 95% conversion for acetylene) of 1 wt% PdAg (1:9) catalysts supported on LSA/HSA-α-Al2O3 prepared either by wet impregnation (WI) or ball milling (BM) with metals (m), chlorides (c), and nitrates (n) as metal precursors for Pd and Ag, respectively. In all cases, the temperature of the reactor was set at 150 °C. Crosses mark catalysts for which deactivation was observed; arrows are assigned to catalysts for which no sign of deactivation (conversion drop below 95%) was observed before the reaction was interrupted. | ||
The catalyst stability correlates with the specific surface area of the material. For example, all LSA catalysts obtained via ball milling lose their activity within 2 h on stream. In contrast, the HSA catalysts were stable the entire time they were tested. Notably, deactivation of mechanochemically synthesized LSA catalysts occurred much earlier than for their impregnated counterpart. It was also observed that ball-milled HSA catalysts prepared using nitrates and chlorides as metal precursors were less selective towards ethylene formation than the standard ball-milled material, especially during the first stages of the reaction. Possibly, wet impregnation is a more suitable method to deposit inorganic metal salts on the alumina support than ball milling without a good optimization of milling parameters. Hence, the choice of the metal precursor seems important when targeting selective catalysts, according to the preparation method used.
Characterization of the catalysts post-reaction
A few selected systems were characterized after the catalytic reaction to investigate possible deactivation pathways. In particular, PdAg (1:1) / LSA-α-Al2O3_WI after testing at 100 °C and PdAg (1:1) / HSA-α-Al2O3_BM after testing at 50 and 100 °C were studied in detail. As shown in Fig. S9b,† the impregnated catalyst tested at 100 °C deactivated rather quickly (after about 2.5 h) under reaction conditions and was completely inactive after about 4 h, when the reaction was interrupted. On the other hand, the ball-milled counterpart tested at 50 °C was much longer active on stream (above 8 h, Fig. S10a†) but died after about 14 h. The same material, tested at 100 °C, was recovered before deactivation, after about 13 h on stream. The Experimental section provides more information on the recovery of the materials from the catalyst bed.
According to XRD, the phase composition of the materials was preserved after the reaction. However, a significant amount of SiC, used to dilute the catalyst bed, was found in the recovered powders (Fig. S16a†), hampering further analysis. For the supported metal particles, the diffraction lines were centered around a position compatible in a reasonable approximation with the initial Ag/Pd ratio, even though a slight shift towards higher Ag content was observed in all cases, which would point at the surface segregation of Pd to some extent (Fig. S16b†). Nonetheless, the XPS analysis of the samples did not report any surface segregation.
On the other hand, the metal particle size distributions were essentially unchanged within the error margin (Fig. S17†). Hence, catalyst deactivation could not be attributed to the sintering of metal particles. Deactivation might have instead been caused by surface coverage of Pd sites with oligomers, including green oils and other carbonaceous deposits. If this was the case, deactivation should proceed faster for the materials obtained via wet impregnation than ball-milled counterparts, as the surface concentration of Pd is higher in the former case (fresh catalyst). Unfortunately, the formation of carbonaceous deposits could neither be determined from the analysis of leached samples, probably due to their low amount.
Summary of main results and discussion
Considering the results of the present study, a few crucial correlations between material features and performance in the catalytic reaction can be outlined.Most importantly, the specific surface area of the oxide support appears to have an enormous impact on the metals' overall dispersion. The catalytic performance was also affected, especially the stability of the catalysts. As previously reported, on LSA-α-Al2O3, larger metal nanoparticles were systematically obtained, in contrast with the cases where HSA-α-Al2O3 was the support, irrespective of the synthetic approach. Thus, for all the materials prepared via wet impregnation, particles with an average size from 9 to 12 nm were observed with LSA-α-Al2O3 as the support. Similarly, average particle sizes between 8 and 10 nm were determined for the materials obtained via ball milling with metal chlorides and nitrates as the metal precursors and LSA-α-Al2O3 as the support. On the other hand, all systems supported on HSA-α-Al2O3 were characterized by metal nanoparticles with an average size lower than 9 nm and the most frequent size between 5 and 7 nm. In general, this evidence would corroborate the notion that using high-surface-area supports is beneficial to attain high levels of metal dispersion. In our case, higher dispersion, or smaller supported metal nanoparticles, was obtained over the high-surface-area support, where a relatively large interparticle distance would hinder sintering upon heating at high temperature. Along these lines, all the materials based on high-surface-area supports were more active with stable activity for a longer time on stream. In contrast, for most of the catalysts supported on LSA-α-Al2O3, deactivation occurred before 10 h on stream.
However, the particle size distribution for spent catalysts was unchanged. This means that the thermal treatment, following wet impregnation or the mechanochemical synthesis, resulted in robust solid catalysts, namely materials stable towards deactivation via sintering under reaction conditions. This also implies that sintering is not the reason for the deactivation of the catalysts, and thus other phenomena must be involved. The higher surface concentration of Pd species observed for wet-impregnated materials possibly accounts for their lower stability on stream. As previously discussed, the formation of green oils and other carbonaceous deposits, common poisons for the catalytic reaction, would be favored on Pd sites, and faster deactivation should be observed. The formation of such deposits would also explain why the catalyst stability appears to be enhanced when the reaction is performed at higher temperatures because the desorption of organic fragments is likely promoted under these conditions. Nonetheless, post-characterization of the materials did not evidence the formation of such species, probably due to their low amount, which could nevertheless be sufficient to block reactive sites.
Apart from stability and overall activity, no particular correlation was noted between support surface area and catalyst selectivity towards ethylene formation. Indeed, the composition of the alloy is the determining parameter, with a higher Ag content being beneficial to achieve high selectivity levels, independently of the preparation method used, as has already been reported before for selective hydrogenation of low acetylene concentrations in ethylene streams. Yet, no significant difference was observed between PdAg (1:5) and PdAg (1:9) supported catalysts. However, considering that Pd is the only active metal, the ethylene formation per active site potentially available is much higher for the PdAg (1:9) catalysts, and thus they appear to be more interesting from a cost perspective. Interestingly, relatively high reaction temperatures favored higher ethylene selectivities in all cases, as full-hydrogenation to ethane was observed to a lesser extent. On the other hand, for the catalysts with a low Pd content, higher reaction temperatures were essential to observe any activity at all. However, in general, higher temperatures improved stability on stream for all the systems investigated. In contrast, reaction temperatures as low as 50 °C seemed to favor catalyst deactivation.
The previous findings are quite interesting, especially because low reaction temperatures are typically used for the selective hydrogenation of acetylene under conventional conditions.44,46 This is done to limit ethylene runaway and consequent formation of a high volume of ethane, when acetylene levels are low and a large excess of ethylene is present in the gas stream.47 However, especially in tail-end type reactors, it would actually be beneficial to operate the reaction at higher temperatures to improve the stability of the catalysts, because the low hydrogen partial pressures result in rapid catalyst deactivation.19 Besides, when acetylene concentration is significantly increased, as in our case, near-complete equilibrium conversion is more easily attained at higher operating temperatures. Improved selectivity at higher operating temperatures has been observed before under similar conditions.12 The temperature during acetylene hydrogenation is one of the most important factors. However, due to the complex interplay of gas feed composition and reaction temperature on the catalysis, the most suitable temperature range results from a compromise between ethylene selectivity and catalyst stability under reaction conditions.
Overall, the work indicates that similar trends in the catalytic behavior of promoted Pd-based catalysts, particularly PdAg supported catalysts, apply in the selective hydrogenation of acetylene with ethylene present, irrespective of the different reaction conditions operated during our study.47Per se, this is an important piece of information, as little is known so far on the crucial features of catalysts, which are most suitable, when the reaction is carried out with high concentrations of acetylene and ethylene at the same time, as in this study.
Incidentally, as mentioned before, ball-milled materials were recognized as systematically longer stable than conventionally synthesized counterparts. This could simply be due to the higher initial activity of ball-milled catalysts (smaller metal particles), so that a sufficient number of active sites for full acetylene conversion is available for a longer time. It has also been observed that only a negligible amount of Pd is present on the surface of ball-milled materials (below the quantification limit), even for Pd-rich formulations, thus offering a more stable catalyst configuration.20,48 This could represent an intrinsic feature of the methodology. We hope to corroborate this hypothesis by expanding the scope of materials in a follow-up investigation.
At any rate, since no substantial difference was noted between ball-milled and impregnated materials concerning conversion and selectivity levels, ball milling represents a valid alternative to solution-based methods, which are normally applied by default for catalyst synthesis, especially for the synthesis of supported metal catalysts. Indeed, ball milling has many intrinsic advantages: firstly, it is a very effective means to the nanostructuring of materials for catalytic applications,32 including supported catalysts, being more straightforward and faster to operate. Secondly, ball milling is characterized by a relatively low environmental impact, due to the limited use of solvents and hazardous substances.49 Finally, the materials obtained via ball milling are often more performant than their conventionally synthesized counterparts obtained, for example, by relying on solution-based methods,50 as it partly occurred in our case with respect to stability.
Conclusions
Bimetallic Pd–Ag catalysts supported on α-Al2O3 were tested in the selective hydrogenation of acetylene in the presence of equimolar amounts of ethylene at different temperatures. High concentrations of acetylene and ethylene were used to meet the requirements of a hypothetical plasma-assisted methane-to-ethylene conversion plant where acetylene would mostly form from methane pyrolysis. Due to the high exothermicity of the hydrogenation step, a robust catalyst needs to be designed, which would allow a new pathway for ethylene production based on methane as feedstock. The catalysts were obtained either by conventional wet impregnation or ball milling. Different catalyst formulations were explored, including low and high surface area supports, various Pd-to-Ag molar ratios, and alternative metal precursors.PdAg (1:9) was recognized as the most performant composition for the reaction, independently of the preparation methods used, as high selectivity towards ethylene formation was measured, and the highest conversion per active site potentially available was noted. Reaction temperatures as high as 150 °C were beneficial for activity and catalyst stability on stream. The synthesis method little affected the selectivity but significantly affected catalyst stability. The catalysts prepared via wet impregnation were typically stable only up to 5–10 h, depending on the conditions, in contrast to ball-milled counterparts, showing no signs of deactivation as long as they were tested (up to 15 h). The characterization of the catalysts post-reaction helps to better understand possible underlying effects. Since the PdAg particle size distribution was unchanged after the catalytic reaction, deactivation was attributed to other phenomena, such as surface poisoning via the formation of green oils or other carbonaceous deposits. Therefore, catalysts prepared by ball milling appear the most suitable choice for this challenging reaction; moreover, the method carries the intrinsic advantages of ball milling compared to traditional solution-based methods. More catalyst formulations will be explored in the future, e.g., supported PdAu, PdZn, and PdGa catalysts, all conveniently obtained by ball milling under suitable conditions. Finally, the results showed that the selective acetylene hydrogenation of highly concentrated acetylene streams is feasible upon catalyst optimization, and some important features were highlighted.
Author contributions
Conceptualization (KSK, JDB, FS). Data curation, formal analysis, investigation (KSK, JDB). Methodology and project administration (KSK, JDB, FS). Data interpretation (KSK, JDB, FS). Validation and visualization (KSK, JDB). Supervision (FS). Writing – original draft (KSK, JDB). Writing – review & editing (KSK, JDB, FS).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Eko Budiyanto, Alexander Hopf, Adrian Schlüter, Silvia Palm, and Norbert Pfänder (MPI-CEC) for the support with the electron microscopy analysis, as well as to Sebastian Leitnig, Jan Ternieden, and Claudia Weidenthaler for the XRD and XPS analysis of the samples. We are also thankful to Nguyen Khang Tran for supporting the preparation of the samples before characterization and catalysis testing. Finally, the assistance of the fine mechanic workshop led by Wolfgang Kersten and the technical laboratories led by Niels Thyssen for the support with the design, construction, and maintenance of the setup. Open Access funding provided by the Max Planck Society.Notes and references
- H. Zimmermann and R. Walzl, Ullmann's Encyclopedia of Industrial Chemistry, Ethylene, (accessed August 2022), DOI:10.1002/14356007.a10_045.pub3.
- Statista, Production capacity of ethylene worldwide from 2018 to 2021, https://www.statista.com/statistics/1067372/global-ethylene-production-capacity/, (accessed August 2022).
- Deloitte, The Future of Petrochemicals: Growth Surrounded by Uncertainty, https://www2.deloitte.com/content/dam/Deloitte/us/Documents/energy-resources/us-the-future-of-petrochemicals.pdf, (accessed August 2022).
- I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, Engineering, 2017, 3, 171–178 CrossRef.
- P. Baumann, Am. Ethnol., 1948, 10, 257–259 Search PubMed.
- H. Gladisch, Chem. Ing. Tech., 1969, 41, 204–208 CrossRef.
- H. Brachold, C. Peuckert and H. Regner, Chem. Ing. Tech., 1993, 65, 293–297 CrossRef.
- P. Pässler, W. Hefner, K. Buckl, H. Meinass, A. Meiswinkel, H.-J. Wernicke, G. Ebersberg, R. Müller, J. Bässler, H. Behringer and D. Mayer, Ullmann's Encyclopedia of Industrial Chemistry, Acetylene, (accessed August 2022), DOI:10.1002/14356007.a01_097.pub4.
- C. L. Gordon, L. L. Lobban and R. G. Mallinson, Catal. Today, 2003, 84, 51–57 CrossRef.
- S. Kado, Y. Sekine, K. Urasaki, K. Okazaki and T. Nozaki, Stud. Surf. Sci. Catal., 2004, 147, 577–582 CrossRef CAS.
- K. Wang, X. Li and A. Zhu, Plasma Sci. Technol., 2011, 13, 77–81 CrossRef CAS.
- B. Wang and H. M. Guan, Catal. Lett., 2016, 146, 2193–2199 CrossRef CAS.
- E. Delikonstantis, M. Scapinello and G. D. Stefanidis, Fuel Process. Technol., 2018, 176, 33–42 CrossRef CAS.
- E. Delikonstantis, M. Scapinello and G. D. Stefanidis, Processes, 2019, 7, 68 CrossRef CAS.
- E. Delikonstantis, E. Igos, M. Augustinus, E. Benetto and G. D. Stefanidis, Sustainable Energy Fuels, 2020, 4, 1351–1362 RSC.
- C. Urmès, J.-M. Schweitzer, A. Cabiac and Y. Schuurman, Catalysts, 2019, 9, 180 CrossRef.
- G. C. Bond, D. A. Dowden and N. Mackenzie, Trans. Faraday Soc., 1958, 54, 1537–1546 RSC.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- A. Borodziński and G. C. Bond, Catal. Rev.: Sci. Eng., 2006, 48, 91–144 CrossRef.
- A. Borodziński and G. C. Bond, Catal. Rev.: Sci. Eng., 2008, 50, 379–469 CrossRef.
- A. Pachulski, R. Schödel and P. Claus, Appl. Catal., A, 2011, 400, 14–24 CrossRef.
- M. Armbrüster, K. Kovnir, M. Behrens, D. Teschner, Y. Grin and R. Schlögl, J. Am. Chem. Soc., 2010, 132, 14745–14747 CrossRef.
- H. Zhou, X. Yang, L. Li, X. Liu, Y. Huang, X. Pan, A. Wang, J. Li and T. Zhang, ACS Catal., 2016, 6, 1054–1061 CrossRef.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Science, 2008, 320, 1320–1322 CrossRef.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef PubMed.
- F. Huang, Y. Deng, Y. Chen, X. Cai, M. Peng, Z. Jia, P. Ren, D. Xiao, X. Wen, N. Wang, H. Liu and D. Ma, J. Am. Chem. Soc., 2018, 140, 13142–13146 CrossRef CAS PubMed.
- M. Danielis, S. Colussi, C. de Leitenburg, L. Soler, J. Llorca and A. Trovarelli, Angew. Chem., Int. Ed., 2018, 57, 10212–10216 CrossRef CAS.
- H. Schreyer, R. Eckert, S. Immohr, J. De Bellis, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2019, 58, 11262–11265 CrossRef CAS.
- J. De Bellis, M. Felderhoff and F. Schüth, Chem. Mater., 2021, 33, 2037–2045 CrossRef CAS.
- J. De Bellis, H. Petersen, J. Ternieden, N. Pfänder, C. Weidenthaler and F. Schüth, Angew. Chem., Int. Ed., 2022, 61, e202208016 CrossRef CAS.
- A. P. Amrute, Z. Łodziana, H. Schreyer, C. Weidenthaler and F. Schüth, Science, 2019, 366, 485–489 CrossRef CAS.
- A. P. Amrute, J. De Bellis, M. Felderhoff and F. Schüth, Chem. – Eur. J., 2021, 27, 6819–6847 CrossRef PubMed.
- I.-T. Trotus, PhD Thesis, Ruhr-Universität Bochum, 2016 Search PubMed.
- A. Tonejc, C. Kosanović, M. Stubićar, A. M. Tonejc, B. Subotić and I.
![[S with combining umlaut]](https://www.rsc.org/images/entities/char_0053_0308.gif) mit, J. Alloys Compd., 1994, 204, L1–L3 CrossRef.
mit, J. Alloys Compd., 1994, 204, L1–L3 CrossRef. - A. Tonejc, M. Stubicar, A. M. Tonejc, K. Kosanović, B. Subotić and I. Smit, J. Mater. Sci. Lett., 1994, 13, 519–520 CrossRef.
- A. Tonejc, A. M. Tonejc, D. Bagović and C. Kosanović, Mater. Sci. Eng., A, 1994, 181–182, 1227–1231 CrossRef.
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef PubMed.
- P. Balaz, M. Achimovicova, M. Balaz, P. Billik, Z. Cherkezova-Zheleva, J. M. Criado, F. Delogu, E. Dutkova, E. Gaffet, F. J. Gotor, R. Kumar, I. Mitov, T. Rojac, M. Senna, A. Streletskii and K. Wieczorek-Ciurowa, Chem. Soc. Rev., 2013, 42, 7571–7637 RSC.
- I. Karakaya and W. T. Thompson, Bull. Alloy Phase Diagrams, 1988, 9, 237–243 CrossRef.
- C.-F. Mao and M. A. Vannice, Appl. Catal., A, 1994, 111, 151–173 CrossRef CAS.
- C.-F. Mao and M. A. Vannice, Appl. Catal., A, 1995, 122, 41–59 CrossRef CAS.
- C.-F. Mao and A. M. Vannice, Appl. Catal., A, 1995, 122, 61–76 CrossRef.
- S. Komeili, M. T. Ravanchi and A. Taeb, Appl. Catal., A, 2015, 502, 287–296 CrossRef.
- M. Kuhn, M. Lucas and P. Claus, Ind. Eng. Chem. Res., 2015, 54, 6683–6691 CrossRef.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Hävecker, A. Knop-Gericke, D. S. Jackson and R. Schlögl, Science, 2008, 320, 86–89 CrossRef.
- H. Zhang, J. Cao, B. Wu, W. Dai, Z. Chen and M. Ma, RSC Adv., 2016, 6, 57174–57182 RSC.
- T. D. Shittu and O. B. Ayodele, Front. Chem. Sci. Eng., 2022, 16, 1031–1059 CrossRef CAS.
- G. X. Pei, X. Y. Liu, A. Wang, A. F. Lee, M. A. Isaacs, L. Li, X. Pan, X. Yang, X. Wang, Z. Tai, K. Wilson and T. Zhang, ACS Catal., 2015, 5, 3717–3725 CrossRef CAS.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS.
- K. Ralphs, C. Hardacre and S. L. James, Chem. Soc. Rev., 2013, 42, 7701–7718 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Complete characterization data of the samples via XRD and electron microscopy; scheme of the experimental setup used for the catalysis testing; time profile of the catalytic behaviour of all the samples at different temperatures of the reactor. See DOI: https://doi.org/10.1039/d2cy01424f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |