
Atomically dispersed s-block metal calcium site modified mesoporous g-C3N4 for boosting photocatalytic N2 reduction†
Guoliang
Pan‡
a,
Wensheng
Zhang‡
b,
Tianren
Liu
a,
Qingmei
Tan
a,
Binzhe
Wei
a,
Kaihang
Ye
a,
Yingxin
Yang
a,
Dongxue
Han
 *ac,
Zhenbang
Liu
*d and
Li
Niu
a
*ac,
Zhenbang
Liu
*d and
Li
Niu
a
aSchool of Chemistry and Chemical Engineering Guangzhou Key Laboratory of Sensing Materials & Devices, Center for Advanced Analytical Science, Guangzhou University, Guangzhou 510006, P. R. China. E-mail: dxhan@gzhu.edu.cn
bSchool of Civil Engineering c/o Center for Advanced Analytical Science, Guangzhou University, Guangzhou 510006, P. R. China
cGuangdong Provincial Key Laboratory of Psychoactive Substances Monitoring and Safety, Anti-Drug Tethnology Center of Guangdong Province, Guangzhou 510230, P. R. China
dSchool of Computer Science and Cyber Engineering, Guangzhou University, Guangzhou 510006, P. R. China. E-mail: cczbliu@gzhu.edu.cn
First published on 14th November 2022
Abstract
Alkaline-earth metal elements in the s-block of the periodic table have rarely been studied as active sites for nitrogen (N2) photofixation. Herein, we report a single-atom calcium (Ca)-modified mesoporous g-C3N4 (Ca/m-g-C3N4) for promoting the photocatalytic N2 reduction reaction (pNRR) under ambient conditions. Moreover, the atomically dispersed Ca as active sites of Ca/m-g-C3N4 could achieve high adsorption of N2 molecules, which could be confirmed by nitrogen temperature-programmed desorption test (N2-TPD). In this regard, Ca single atoms can not only serve as the active centers of N2 but also optimize the energy band structure of m-g-C3N4, facilitating the photocatalytic synthesis of ammonia (NH3). Therefore, the optimal 0.5 Ca/m-g-C3N4 demonstrates a remarkable NH3 generation amount of 42.23 μg gcat.−1 h−1, which is 2.1 times that of pure g-C3N4 (20.41 μg gcat.−1 h−1). Furthermore, the present 0.5 Ca/m-g-C3N4 demonstrates good stability, and the NH3 production is still remarkably unvaried after four cycling tests. More interestingly, it was also found that other alkaline earth metal elements, including magnesium (Mg), strontium (Sr), and barium (Ba), can also activate N2 molecules. Accordingly, the photocatalytic NH3 synthesis yield of Ba/m-g-C3N4 is 29.52 μg gcat.−1 h−1, slightly more than that of Mg/m-g-C3N4 (20.52 μg gcat.−1 h−1) and Sr/m-g-C3N4 (20.88 μg gcat.−1 h−1). We hope that this work could provide a novel insight for developing other high-performance N2-photofixation systems based on low-cost s-block alkaline-earth metal materials in the future.
Introduction
Photocatalytic synthesis of ammonia (NH3) offers a promising prospect for the conversion of nitrogen (N2) to NH3 through solar energy with low energy consumption and zero carbon emissions. Since the triple bond of N2 molecule is extremely stable and the binding energy is as high as 941 kJ mol−1, the effective activation of N2 molecule is a great bottleneck.1,2 Inspired by the biomimetic mechanism of the Fe–Mo–S cofactor of nitrogenase, many transition metals have been reported to form a strong bond with N2 (transition metals–N2, TM–N2), which can effectively weaken the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond. Many reports have confirmed that there is a strong electron back-donation into the antibonding orbitals of the TM, between the d orbital electron of the TM and the p orbitals of the N2 molecule, where the N2 molecule could accept electrons from the metal sites and thus induce polarization for the adsorption and activation of N2.3–5 For example, Skulason et al.6,7 demonstrated that scandium (Sc), yttrium (Y), titanium (Ti), and zirconium (Zr) show strong coordination bonding of N2, which is conducive for the capture and activation of N2 molecules. Unfortunately, the main-group elements do not have the structural characteristics of the d orbitals of the transition metal valence layer; thus, it has long been believed that the activation of N2 molecules using the main-group elements is not feasible.8
N bond. Many reports have confirmed that there is a strong electron back-donation into the antibonding orbitals of the TM, between the d orbital electron of the TM and the p orbitals of the N2 molecule, where the N2 molecule could accept electrons from the metal sites and thus induce polarization for the adsorption and activation of N2.3–5 For example, Skulason et al.6,7 demonstrated that scandium (Sc), yttrium (Y), titanium (Ti), and zirconium (Zr) show strong coordination bonding of N2, which is conducive for the capture and activation of N2 molecules. Unfortunately, the main-group elements do not have the structural characteristics of the d orbitals of the transition metal valence layer; thus, it has long been believed that the activation of N2 molecules using the main-group elements is not feasible.8
Recently, some interesting work has proved that the p-block element boron (B) can also activate N2 molecules and further enable the complete cyclic process of NH3 formation.9,10 For example, the Holger Braunschweig's group introduced a series of remarkable achievements using main-group element compounds in the field of nitrogen fixation. They also showed that the p-block elements can not only activate N2 molecules in the form of transition metals but also achieve a coupling reaction of nitrogen dimer reduction that is not yet achievable through TM-based catalysts.3,9 However, the main-group element materials that can adsorb and activate N2 molecules are limited to some p-block elements, but for s-block elements, the related reports are few or only limited to lithium and its nitrides, such as Li3N.11 For a long time, other s-block elements including alkali metals and alkaline-earth metals, except Li, were not considered to be effective for adsorbing and activating N2 molecules. Until 2021, Sjoerd Harder and co-workers used stable monovalent calcium complexes to activate N2 molecules.10 They confirmed that the calcium (Ca) sites of the alkaline-earth metal elements can use its p orbitals to participate in the adsorption and activation of N2 molecules, just like the d orbital electron action of TM. The discovery of Ca active sites has become a new breakthrough in the field of N2 fixation. Since then, Chen et al.12 confirmed that the unique p-orbital electronic structure of s-block metals, such as the Ca active centers, could enhance the adsorption of intermediates and accelerate electron transfer during the ORR reaction process. Afterward, Liu and co-workers13 reported the efficient electrocatalytic reduction of CO2 by loading s-block magnesium (Mg) single atoms on g-C3N4. Concomitantly, Qiao's group14 evaluated the NRR activity of the main-group elements through the functional mechanism selectively promoting NRR and inhibiting HER, which remarkably accelerated the development of main-group elements for N2 fixation reaction. A valuable study for developing s-block element-based NRR system with satisfactory properties, therefore, seems imperative.
Herein, the Ca single-atom-modified mesoporous g-C3N4 nanosheets (denoted as Ca/m-g-C3N4) was synthesized through calcination and continuous stirring approach. Undoubtedly, the single mesoporous g-C3N4 (m-g-C3N4) generally restricts the surface active sites, which limits its N2 photofixation activity. In this regard, the modification strategy using the s-block Ca single-atom endowed Ca active sites to achieve the effective enhancement of the pNRR performance of m-g-C3N4. The Ca single atoms were decorated on the surface of m-g-C3N4 as active sites to realize high adsorption and activation of the N2 molecules. Furthermore, the N2-TPD data further proved that the Ca single-atom sites can significantly promote the N2 chemisorption of g-C3N4 and improve its pNRR activity. Simultaneously, from the energy band structure, the VB potential of Ca/m-g-C3N4 clearly has a greater potential to produce hydrogen protons, promoting the process of electron-coupled hydrogenation during pNRR. Using the advantage of the Ca single atoms as the active centers together with the sufficient proton source derived from the valence band oxidation process, N2 could be effectively reduced to NH3 over Ca/m-g-C3N4 under ambient conditions.
Experimental section
Preparation of mesoporous g-C3N4 nanosheets
Mesoporous g-C3N4 nanosheets (m-g-C3N4) were prepared according to previous reports with minor modifications.15 In general, 1.0 g melamine was mixed with 5.0 g cyanuric acid and then ground for 30 min with the addition of 2.0 mL ethanol. The mixture was then placed in a tube furnace and subjected to a two-stage heating process under N2 atmosphere, starting with calcination at 400 °C for 10 min at a heating rate of 5.0 °C min−1, followed by calcination at the same rate to 550 °C for 4.0 h. Finally, the light-yellow product was ground into a powder to produce mesoporous g-C3N4 nanosheets (denoted as m-g-C3N4).Preparation of Ca single-atom-modified m-g-C3N4 nanosheets
The Ca single-atom-modified mesoporous g-C3N4 nanosheets was synthesized via calcination and continuous stirring method (for detailed steps, see Fig. S1†). In brief, 100 mg m-g-C3N4 was dispersed in 60 mL water. Subsequently, different masses of calcium chloride dihydrate (the added amounts of Ca were controlled as 30 mg, 50 mg, 100 mg, and 200 mg) were added to the above dispersion. The mixture was stirred for 24 h and then collected by centrifugation and washing several times. Next, the collected materials were dried at 60 °C in vacuum for 4 h. Finally, the powder was calcined at 300 °C for 1 h in an air atmosphere with a heating rate of 2.0 °C min−1. g-C3N4 loaded with different mass ratios of Ca was denoted as 0.3 Ca/m-g-C3N4, 0.5 Ca/m-g-C3N4, 1.0 Ca/m-g-C3N4, and 2.0 Ca/m-g-C3N4, respectively.Preparation of Mg, Sr, or Ba single-atom-modified g-C3N4 nanosheets
Mg, Sr, and Ba-modified m-g-C3N4 (denoted as Mg/m-g-C3N4, Sr/m-g-C3N4, and Ba/m-g-C3N4, respectively) were prepared with a similar method as the synthesis of Ca/m-g-C3N4, except that CaCl2·2H2O was replaced with MgCl2·6H2O, SrCl2·6H2O, and BaCl2·2H2O.Apparatus
The crystal phase of the as-prepared catalysts was evaluated using a PANalytical X'Pert powder X-ray diffractometer (XRD, Netherlands). Scanning electron microscopy (SEM, JEOL JSM-7001F) and transmission electron microscopy (TEM, JEOL 2100F) were used to analyze the porous morphology, and the accompanying energy dispersive X-ray spectroscopy (EDS) was used to analyze the elemental composition of the samples. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM, Titan Cubed Themis G230) was used to analyze the surface single-metal atoms. Further, the surface element properties were investigated using a Thermo Fisher Scientific K-alpha X-ray photoelectron spectrometer (XPS, Thermo ESCALAB250Xi), and all binding energies were referenced to the C 1s peak at 284.8 eV. The N2 temperature-programmed desorption assessments were performed (Micromeritics Auto Chem II) by implementing a TCD as the detector. Transient and steady photoluminescence for the as-fabricated samples were achieved under 800 nm excitation on a FLS1000 fluorescence lifetime spectrophotometer (Edinburgh Instruments, UK). The specific surface area (BET) was recorded using an ASAP2460 instrument (Micromeritics).Results and discussion
Characterization
The structures and morphologies were investigated by the means of TEM and HAADF-STEM. As shown in Fig. 1a, the m-g-C3N4 sample exhibits a bubbly-fold nanosheet structure with rich pores. Notably, no obvious single atoms were observed in Fig. 1b. Furthermore, Fig. 1c exhibits that the whole structure of m-g-C3N4 did not change after Ca single-atom modification, indicating that the structure of the catalyst is relatively stable. As presented by the HAADF-STEM image for 0.5 Ca/m-g-C3N4, many bright spots (i.e., Ca single atoms) could be seen in Fig. 1d, suggesting that monatomic Ca is successfully decorated on the surface of m-g-C3N4 nanosheets. Besides, the corresponding EDS mapping characterization was used to analyze the distribution of Ca, N, and C elements in 0.5 Ca/g-C3N4, and it was clearly observed that the Ca single atoms were uniformly distributed (Fig. 1e and f). Thus, the special porous sheet structure of the present 0.5 Ca/g-C3N4 sample can facilitate the rapid transfer of reactants and products during photocatalytic reactions.16–18 | ||
| Fig. 1 TEM (a) and HAADF-STEM (b) images of m-g-C3N4. TEM (c), HAADF-STEM (d), and corresponding EDS-mapping images (e and f) of 0.5 Ca/m-g-C3N4. | ||
The XRD patterns of the as-prepared samples are displayed in Fig. 2a. In Fig. 2a, two characteristic diffraction peaks at ∼13.1° and ∼27.4° appeared in all m-g-C3N4-based samples, which correspond to the (100) crystal surface of the tri-s-triazine structural unit of g-C3N4 and the (002) crystal surface with aromatic stacking, respectively (Fig. 2a).19 It is worth noting that after doping Ca single atoms, the diffraction peak became sharper and stronger, indicating that Ca single atoms could improve the crystallinity of m-g-C3N4. Crucially, the enhancement of crystallinity accelerates the movement of photogenerated electrons on the nanosheets, further improving the photocatalytic activity of g-C3N4.20 No diffraction peak of calcium oxide (CaO) was observed in all the samples, indicating that Ca does not exist as an oxide. Moreover, with the increase in the Ca content, the typical (002) diffraction peak of m-g-C3N4 was significantly shifted to a lower value, which is caused by the successful modification of Ca single atoms into the lattice of m-g-C3N4. We used FTIR spectroscopy to further analyze the surface groups of these catalysts, as shown in Fig. 2b. The prominent absorption at about 808 cm−1 is due to out-of-plane bending vibration of tri-s-triazine. The strong bands at 1100–1700 cm−1 were assigned to the typical stretching vibration of the heptazine ring. The broad weak band between 3000 and 3500 cm−1 could be ascribed the presence of NH and/or NH2 groups.19 Compared to the single m-g-C3N4, the NH2 peak of Ca/m-g-C3N4 was slightly enhanced, indicating that there are more C vacancies on Ca/m-g-C3N4.21 The above data confirm the existence of a bonding force between calcium monoatomic Ca and m-g-C3N4, showing that the single-atom structure of 0.5 Ca/m-g-C3N4 has good stability.
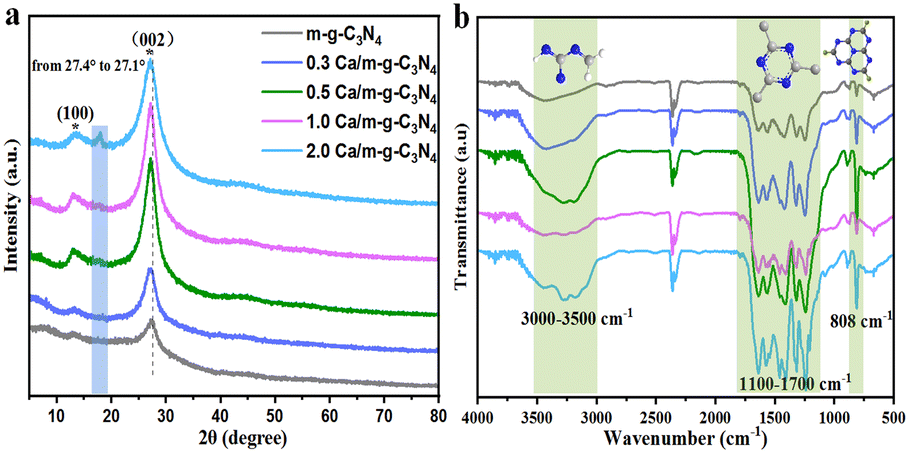 | ||
| Fig. 2 The XRD patterns (a) and FTIR spectra (b) of the as-prepared m-g-C3N4, 0.3 Ca/m-g-C3N4, 0.5 Ca/m-g-C3N4, 1.0 Ca/m-g-C3N4, and 2.0 Ca/m-g-C3N4 samples. | ||
The high-resolution spectra of XPS for C 1s, O 1s, N 1s, and Ca 2p were recorded particularly for studying the chemical state and surface composition of m-g-C3N4 and 0.5 Ca/m-g-C3N4 catalysts. In the high-resolution spectrum of C 1s (Fig. 3a), the peak at 288.3 eV originates from the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N structure, and the peak at 284.8 eV is due to graphitic carbon species (Csp2–Csp2 bonds). Compared to m-g-C3N4, the graphitic carbon peak intensity of 0.5 Ca/C3N4 was significantly lower, which helps to optimize the charge distribution of the aromatic π-conjugated system to promote charge separation and reduce the centers of charge recombination in the conjugated system.19 This indicates that the modified Ca atoms can effectively promote the separation and migration of photogenerated carriers. Notably, relative to m-g-C3N4, the peak assigned to the C–NHx species in 0.5 Ca/m-g-C3N4 showed a slight blue shift, indicating a decrease in the charge density at the edges of tri-s-triazine, which is caused by the exposed N defects in tri-s-triazine.17 Relative to m-g-C3N4, a characteristic peak at ∼533.5 eV of 0.5 Ca/m-g-C3N4 was attributed to the water (H2O) adsorbed on the surface in the high-resolution O 1s XPS spectrum (Fig. 3b). This data indicates that the 0.5 Ca/m-g-C3N4 sample may possess better hydrophilicity and more easily adsorb H2O molecules, which will be conducive to the consumption of photogenerated holes. In the N 1s spectra, the existence of the C–NC structure is demonstrated by the binding energy at ∼398.8 eV (Fig. 3c). Also, the peak at 399.9 eV is the signal of N connecting three C atoms (N–(C)3), whereas the contribution at ∼401.2 eV may be caused by the amino group.18 Notably, the spectrum of N 1 s shows that the ratio of peak areas between CN–C and N–C3 decreases from 5.53 to 4.45 after the Ca atoms are loaded on m-g-C3N4, indicating that the Ca sites may be coordinated with CN–C in 0.5 Ca/m-g-C3N4.14 Simultaneously, the typical 2p orbital signal of Ca element was detected (Fig. 3d), indicating that the Ca single atoms are present in a bivalent state, firmly confirming that Ca single atoms of the 0.5 Ca/m-g-C3N4 material exist in coordination.11,22 Meanwhile, the peak of N–(C)3 displayed a slight red shift, which was caused by Ca–N coordination. Presumably, the partial valence electron of the Ca atoms shifts to the tri-s-triazine unit structure in the g-C3N4 framework, resulting in an increase in the electron density of N in N–(C)3.23 Thus, the XPS characterization results prove that Ca single atoms mainly coordinated to the cavity edge N formed by the tri-s-triazine ring.24
C–N structure, and the peak at 284.8 eV is due to graphitic carbon species (Csp2–Csp2 bonds). Compared to m-g-C3N4, the graphitic carbon peak intensity of 0.5 Ca/C3N4 was significantly lower, which helps to optimize the charge distribution of the aromatic π-conjugated system to promote charge separation and reduce the centers of charge recombination in the conjugated system.19 This indicates that the modified Ca atoms can effectively promote the separation and migration of photogenerated carriers. Notably, relative to m-g-C3N4, the peak assigned to the C–NHx species in 0.5 Ca/m-g-C3N4 showed a slight blue shift, indicating a decrease in the charge density at the edges of tri-s-triazine, which is caused by the exposed N defects in tri-s-triazine.17 Relative to m-g-C3N4, a characteristic peak at ∼533.5 eV of 0.5 Ca/m-g-C3N4 was attributed to the water (H2O) adsorbed on the surface in the high-resolution O 1s XPS spectrum (Fig. 3b). This data indicates that the 0.5 Ca/m-g-C3N4 sample may possess better hydrophilicity and more easily adsorb H2O molecules, which will be conducive to the consumption of photogenerated holes. In the N 1s spectra, the existence of the C–NC structure is demonstrated by the binding energy at ∼398.8 eV (Fig. 3c). Also, the peak at 399.9 eV is the signal of N connecting three C atoms (N–(C)3), whereas the contribution at ∼401.2 eV may be caused by the amino group.18 Notably, the spectrum of N 1 s shows that the ratio of peak areas between CN–C and N–C3 decreases from 5.53 to 4.45 after the Ca atoms are loaded on m-g-C3N4, indicating that the Ca sites may be coordinated with CN–C in 0.5 Ca/m-g-C3N4.14 Simultaneously, the typical 2p orbital signal of Ca element was detected (Fig. 3d), indicating that the Ca single atoms are present in a bivalent state, firmly confirming that Ca single atoms of the 0.5 Ca/m-g-C3N4 material exist in coordination.11,22 Meanwhile, the peak of N–(C)3 displayed a slight red shift, which was caused by Ca–N coordination. Presumably, the partial valence electron of the Ca atoms shifts to the tri-s-triazine unit structure in the g-C3N4 framework, resulting in an increase in the electron density of N in N–(C)3.23 Thus, the XPS characterization results prove that Ca single atoms mainly coordinated to the cavity edge N formed by the tri-s-triazine ring.24
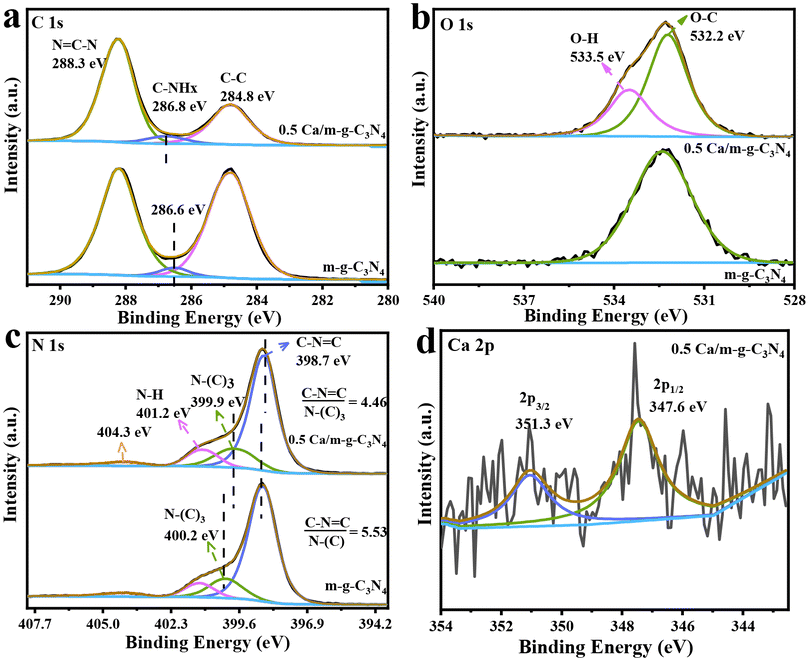 | ||
| Fig. 3 High-resolution XPS spectra of C 1s (a), O 1s (b), N 1s (c), and Ca 2p (d) of the as-synthesized photocatalysts. | ||
The specific surface area (SBET) was investigated by N2 adsorption–desorption isotherm analysis (Fig. S2†). The SBET values of m-g-C3N4 and 0.5 Ca/m-g-C3N4 are 71.1 m2 g−1 and 40.9 m2 g−1, respectively. The two samples belong to type IV isotherms, indicating that our samples are mesoporous materials and have comparable average pore sizes. Although the introduction of Ca single atoms decreased the specific surface area and pore volume, the activity of pNRR was significantly enhanced, demonstrating that the SBET is not the main factor. In addition, to further understand the pNRR activity of 0.5 Ca/m-g-C3N4, the essence of photocatalytic active sites was revealed by the N2-TPD test (Fig. 4). The N2 desorption peak at ∼150 °C corresponds to the physisorption peak of N2 molecules and ∼420 °C corresponds to the chemisorption peak of N2 molecules. As shown in Fig. 4, the sample of 0.5 Ca/m-g-C3N4 demonstrates an obvious chemisorption of N2 molecules, corresponding to the desorption peak at ∼420 °C, while m-g-C3N4 lacks this effective chemisorption of N2. The N2-TPD data confirms that the 0.5 Ca/m-g-C3N4 catalyst could provide a large amount of chemical adsorption sites for capturing N2 from water. Since N2 adsorption is the first step for NH3 synthesis, the remarkable N2 absorption on 0.5 Ca/m-g-C3N4 will be beneficial for the whole pNRR procedure.
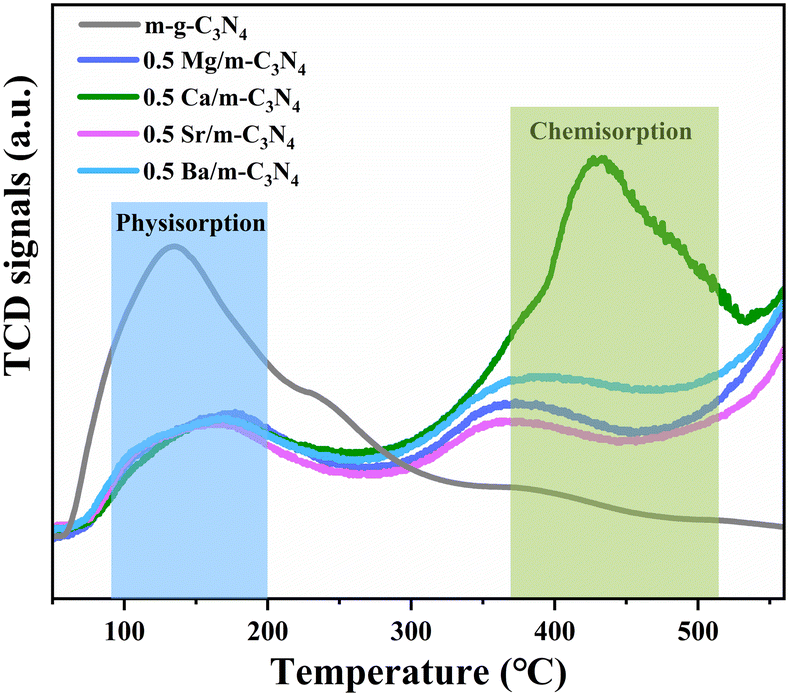 | ||
| Fig. 4 N2-TPD spectra of the as-prepared various photocatalysts. | ||
Photocatalytic activities
The photocatalytic N2 fixation performance of the as-prepared samples was determined by employing water as the solvent and proton source under full spectrum by the spectrophotometric measurement of the generated NH3 with indophenol indicator (Fig. S3†).25 The 0.5 Ca/m-g-C3N4 sample showed an excellent NH3 production rate of 42.23 μg gcat.−1 h−1 under full-spectrum light irradiation (Fig. 5a and b), which is about 2.1 times higher than that of m-g-C3N4 (20.41 μg gcat.−1 h−1). Meanwhile, under 5 h light irradiation, the synthetic NH3 yield in per unit time exhibits a “volcanic” relationship with Ca content, as shown in Fig. 5b, the optimal 0.5 Ca/m-g-C3N4 exhibiting excellent pNRR performance. Thus, the optimized catalyst of the 0.5 Ca/m-g-C3N4 sample was conducted for other related characterizations and experiments. Accordingly, no N2H4 was detected during the process (Fig. S4† and 5c), demonstrating an excellent selectivity for NH3 production.26–29 After four cycles, the activity of Ca/m-g-C3N4 for reducing N2 to NH3 decreased slightly, indicating remarkable catalytic stability (Fig. 5d). Owing to the successful modification of the surface single-atom Ca sites, the 0.5 Ca/m-g-C3N4 catalyst demonstrates excellent pNRR activity.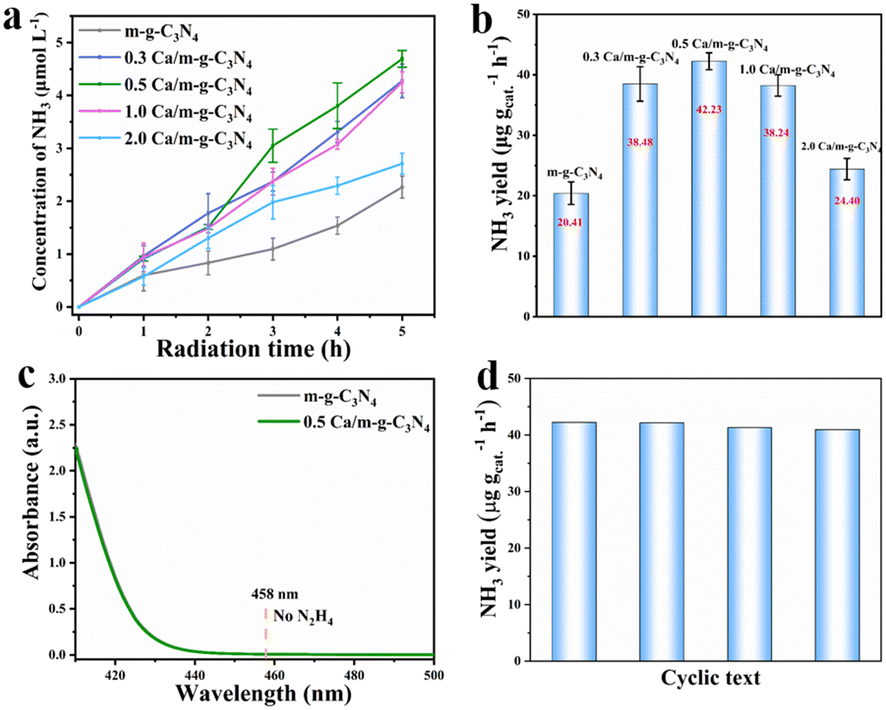 | ||
| Fig. 5 (a) Quantitation measurement of the generated NH3 by full spectra illumination with an indophenol indicator over different systems in N2. (b) Comparison of the photocatalytic NH3 production rate within 5.0 h of the corresponding catalysts. (c) UV-vis absorption spectra of the reaction solution by different catalysts. No N2H4 was detected at 458 nm. (d) Photocatalytic cycling tests for 0.5 Ca/m-g-C3N4. | ||
Next, several rigorous contrast experiments were conducted for investigating the NRR activity of 0.5 Ca/m-g-C3N4 different conditions (Fig. S5†). When the NRR process was carried out under Ar atmosphere (without N2), under dark or without catalyst, and only trace NH3 could be detected, confirming that the produced NH3 over 0.5 Ca/m-g-C3N4 indeed originates from the photocatalytic reaction. As the quencher for the photo-induced hole, CH3OH is critical for sufficient charge separation and efficient NH3 production. Adding CH3OH (20 vol%) as the solvent, NH3 formation demonstrates a significant enhancement.
Moreover, to further confirm whether photocatalytic N2 fixation on the 0.5 Ca/m-g-C3N4 was authentic, photocatalytic N2 fixation under 15N isotope-labeled N2 was conducted (with purity not less than 99%). The produced NH4+ could react with phenol and hypochlorite for the formation of 15N-labeled indophenol,30,31 which could be assayed accurately by liquid chromatography-mass spectrometry (LC-MS). Fig. S6a† displays a potent mass spectroscopy signal of 14N-labeled indophenol anion at about 198 m/z in LC-MS research when using 14N2 as the feeding gas. Notably, 15N-labeled indophenol negative anion displays a remarkable enhanced mass spectrum signal at about 199 m/z in LC-MS analysis (Fig. S6b†). The signal gives a higher intensity relative to the 14 N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 N natural abundance ratio after 30 min illumination. These data identify that the generate NH4+ ions detected in this work originated from N2 photofixation.
:15 N natural abundance ratio after 30 min illumination. These data identify that the generate NH4+ ions detected in this work originated from N2 photofixation.
Possible mechanisms
With the intention of exploring the kinetic behaviors of the carriers in-depth, steady-state photoluminescence (PL) measurements and time-resolved photoluminescence decay measurements were carried out. In comparison to the potent emission peak of m-g-C3N4, the 0.5 Ca/m-g-C3N4 composites demonstrated a rather weak PL peak, illustrating that the modification of Ca single atoms can effectively reduce the recombination velocity of electrons and holes. (Fig. 6a). Relative to the time-resolved photoluminescence decay measurements of g-C3N4 (Fig. 6b), 0.5 Ca/m-g-C3N4 displayed longer average decay times of 9.6 ns, indicating the better condition for carrier separation in 0.5 Ca/m-g-C3N4. A series of electrochemical characterizations were carried out to evaluate the photocatalytic performance of the as-synthesized samples.32,33 As shown in Fig. 6c, the electrochemical impedance spectroscopy (EIS) of 0.5 Ca/m-g-C3N4 exhibits a smaller semicircle than that of the pure m-g-C3N4, indicating that 0.5 Ca/m-g-C3N4 presents a lower charge transference resistance and a greater charge transfer rate. In contrast to m-g-C3N4, the transient photocurrent response of 0.5 Ca/m-g-C3N4 under full-spectrum light irradiation was significantly stable over 11 on–off cycles, indicating a stable photocatalytic process (Fig. 6d). As shown in Fig. 6d, the catalysts demonstrate a rapid light current response to periodic light on–off. Among these, 0.5 Ca/m-g-C3N4 displays a remarkably higher photocurrent intensity relative to m-g-C3N4, providing that 0.5 Ca/m-g-C3N4 takes possession of more beneficial carrier separation. This provides firm evidence that the modification of Ca single-atom sites can enhance the activity of g-C3N4. Hence, the modification of Ca single-atom sites presents greatly promoted carrier separation efficiency and effective N2 adsorption sites, which are beneficial for pNRR.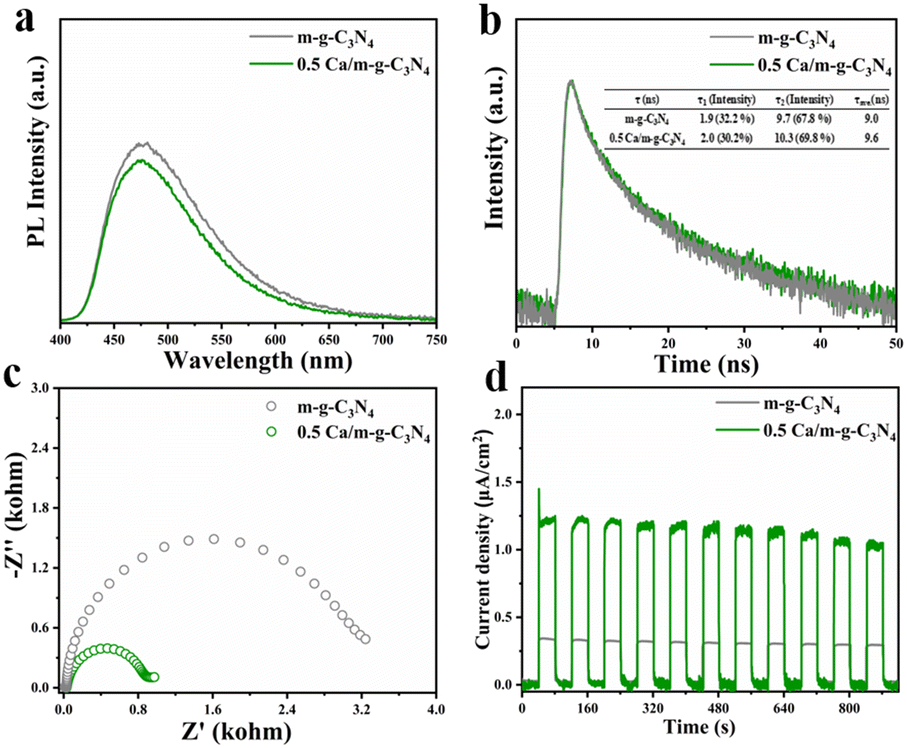 | ||
| Fig. 6 (a) The steady-state photoluminescence spectra (PL) and (b) time-resolved transient photoluminescence decay spectra of m-g-C3N4 and 0.5 Ca/m-g-C3N4, respectively. (c) Electrochemical impedance spectroscopy response for m-g-C3N4 and 0.5 Ca/mg-C3N4 samples in 0.1 M Na2SO4 aqueous solution. (d) The transient photocurrent spectra of the as-prepared samples. | ||
Intriguingly, the pNRR performance of other alkaline-earth metal elements (e.g., Mg, Sr, and Ba) was also probed. The NH3 concentration over Mg/m-g-C3N4 and Sr/m-g-C3N4 samples increased with increasing irradiation time, indicating that the modified Mg and Sr can also improve the pNRR activity of m-g-C3N4 to a certain extent (Fig. S7a†). In contrast, the loading of Ca or Ba atoms can significantly promote the NRR performance of m-g-C3N4, which may be attributed to the more effective adsorption and activation ability of Ca and Ba atoms to N2 molecules. At the same time, no N2H4 was detected during the pNRR process (Fig. S7b†). We use UV-vis diffuse reflectance spectra (DRS) to estimate the light absorption performance and calculate the band gap of the samples. Compared with m-g-C3N4, the 0.5 Ca/m-g-C3N4 sample possesses a wider light absorption, demonstrating that its solar energy utilization is further improved (Fig. 6a). Moreover, the ability of 0.5 Ca/m-g-C3N4 to absorb light is also improved in the UV region, which could imply enhancements in the photocatalytic activity. The bandgap of 0.5 Ca/m-g-C3N4 is 2.62 eV, which could be obtained from the Tauc diagram transformed from UV-vis DRS (Fig. S8a†). It increases a little by 0.09 eV compared with m-g-C3N4 (2.53 eV). The flat-band potential of the materials can be measured by the Mott-Schottky test to further investigate the valence band (VB) and conduction band (CB) position (Fig. S8c and d†). Conversion between the measured potential (vs. Ag/AgCl) and NHE is achieved through the following equation.34
| ENHE = EAg/AgCl + 0.1976(25 °C) |
Therefore, the CB potential of m-g-C3N4 and 0.5 Ca/m-g-C3N4 is −1.35 V (vs. NHE) and −1.32 V (vs. NHE), respectively. Based on Eg = EVB − ECB, the valence band (VB) potential can be estimated as 1.18 V (vs. NHE, m-g-C3N4) and 1.30 V (vs. NHE, 0.5 Ca/m-g-C3N4), respectively. Apparently, the VB potential deviation of both was 0.12 eV, which is consistent with the results of the XPS valence band spectrum (Fig. S8b†).19 Notably, although the photogenerated electrons of m-g-C3N4 have a stronger N2 reduction potential than 0.5 Ca/m-g-C3N4, the opposite results are obtained by yield experiments. This is owing to the fact that the valence band of 0.5 Ca/m-g-C3N4 is +1.30 V, which the photogenerated holes could oxidize H2O to produce more H protons (H2O − 4e− − O2 + H+, +1.23 V vs. NHE), whereas the VB potential of m-g-C3N4 (+1.18 V vs. NHE) was not. It is inferred that the chelated Ca single atoms not only serve as the active sites for N2 molecules but also optimize the bandgap energy structure of m-g-C3N4. The VB potential is positively shifted to produce more H+, thus significantly facilitating the coupled proton-electron process of pNRR compared to g-C3N4.
From the above analysis, a continuous hydrogenation pathway of N2 molecules over the 0.5 Ca/m-g-C3N4 catalyst was proposed (Fig. 7). Because no N2H4 was detected during the pNRR process (Fig. 5c), it can be assumed that the N2 molecules reduction pathway is mainly the associative distal pathway. During the pNRR process, the adsorption of N2 molecules is accompanied by chemical reactions. In the associative distal pathway,35–38 the N2 molecules adsorb on the Ca single-atom sites in an end-on configuration, which causes the NN bond to be weakened. Simultaneously, the N atom farthest from the catalytic surface in the adsorbed N2 molecule prefers to conjugate H+ and couple the photogenerated electrons, facilitating the process of hydrogenation of the N2 molecules to form an NH3 molecule (*NN, *NNH, *N–NH2, *N–NH3, NH3). Then, one NH3 is released and the other N atom of the adsorbed state proceeds to the hydrogenation reduction process. Finally, another NH3 molecule was formed by the same hydrogenation process.
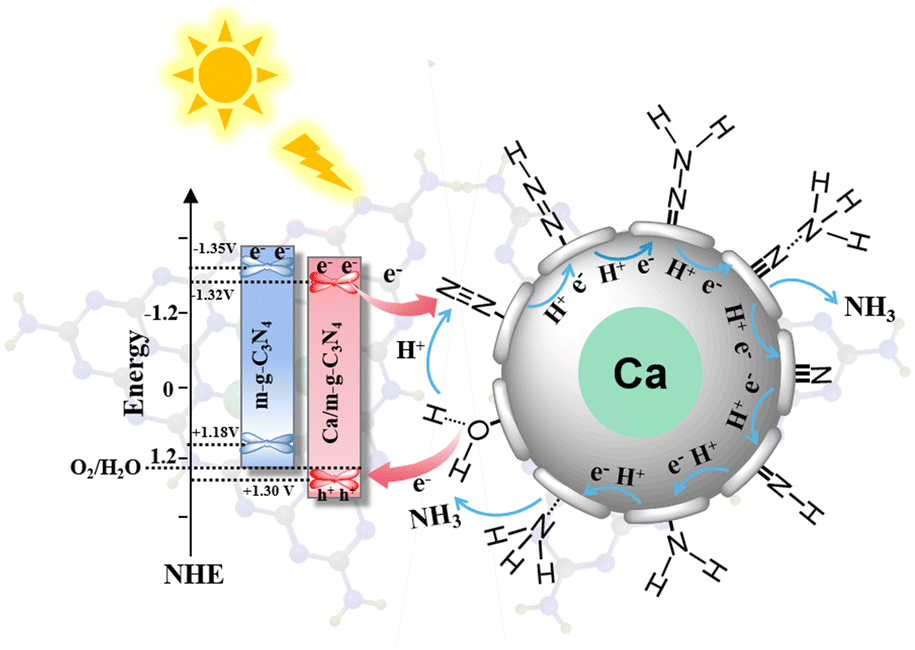 | ||
| Fig. 7 The energy band structure and the proposed pNRR path of the continuous hydrogenation process on the Ca active sites of 0.5 Ca/m-g-C3N4. | ||
Conclusion
In summary, the photocatalytic activity of m-g-C3N4 driven by sunlight for N2 fixation was significantly boosted after the modification of Ca single atoms. The reinforced activity is due to the high-efficiency separation of photogenerated charges and the improvement of surface Ca sites; 0.5 Ca/m-g-C3N4 demonstrates an excellent pNRR activity. Accordingly, it is confirmed that the Ca single atom can not only act as an active site to realize high adsorption and activation of N2 molecules but also improve the band gap structure of m-g-C3N4 to induce more hydrogen protons and promote the hydrogenation process during the pNRR procedure. Therefore, 0.5 Ca/m-g-C3N4 displays an excellent NH3 generation amount of 42.23 μg gcat.−1 h−1, which is 2.1 times than that of m-g-C3N4 (20.41 μg gcat.−1 h−1). Besides, we also found that other alkaline-earth metals (e.g., Mg, Sr, and Ba.) can also promote the pNRR perfomance of m-g-C3N4. It is expected that our study will provide insights into the future development of the photocatalytic synthesis of ammonia by s-block metal materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was sponsored by the National Natural Science Foundation of China (22172040, 21974031), the Department of Science and Techniques of Guangdong Province (2021A1515010180, 2019B010933001), Guangzhou Municipal Science and Technology Bureau (202102010449), and the Department of Guangdong Provincial Public Security (GZQC20-PZ11-FD084). We also thank Shiyanjia Lab platform (https://www.shiyanjia.com/) for providing the XPS and N2-TPD experimental testing and analysis services.References
- Y. Xin, S. M. Wang, H. B. Yuan, T. T. Hou, W. K. Zhu, Y. X. Liu, Y. Yao, W. H. Zhang, S. Q. Liang and L. B. Wang, Chem, 2021, 7, 2118–2136 Search PubMed.
- J. H. Yang, H. Y. Bai, Y. Z. Guo, H. Zhang, R. B. Jiang, B. C. Yang, J. F. Wang and J. C. Yu, Angew. Chem., Int. Ed., 2021, 60, 927–936 CrossRef PubMed.
- M. A. Legare, G. Belanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–899 CrossRef PubMed.
- H. Tanaka, Y. Nishibayashi and K. Yoshizawa, Acc. Chem. Res., 2016, 49, 987–995 CrossRef PubMed.
- W. S. Zhang, D. F. Han, M. J. Dai, Y. Y. Fan, G. L. Pan, W. Q. Liang, Q. T. Zheng, D. D. Qin, D. X. Han, Y. He and L. Niu, ACS Sustainable Chem. Eng., 2021, 9, 15331–15343 CrossRef.
- Y. Abghoui, A. L. Garden, V. F. Hlynsson, S. Bjorgvinsdottir, H. Olafsdottir and E. Skulason, Phys. Chem. Chem. Phys., 2015, 17, 4909–4918 RSC.
- E. Skulason, T. Bligaard, S. Gudmundsdottir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jonsson and J. K. Norskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- M. A. Legare, G. Belanger-Chabot, M. Rang, R. D. Dewhurst, I. Krummenacher, R. Bertermann and H. Braunschweig, Nat. Chem., 2020, 12, 1076–1080 CrossRef CAS.
- M. A. Legare, M. Rang, G. Belanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 CrossRef CAS.
- B. Rosch, T. X. Gentner, J. Langer, C. Farber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Science, 2021, 371, 1125–1128 CrossRef CAS PubMed.
- J. G. Kay, Science, 1964, 144, 703 CrossRef CAS.
- Z. Y. Lin, H. Huang, L. Cheng, W. Hu, P. P. Xu, Y. Yang, J. M. Li, F. Y. Gao, K. Yang, S. Liu, P. Jiang, W. S. Yan, S. Chen, C. L. Wang, H. G. Tong, M. X. Huang, W. Zheng, H. Wang and Q. W. Chen, Adv. Mater., 2021, 33, 7103–7113 Search PubMed.
- Q. Y. Wang, K. Liu, J. W. Fu, C. Cai, H. J. W. Li, Y. Long, S. Y. Chen, B. Liu, H. M. Li, W. Z. Li, X. Q. Qiu, N. Zhang, J. H. Hu, H. Pan and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 25241–25245 CrossRef CAS.
- L. Q. Li, C. Tang, H. Y. Jin, K. Davey and S. Z. Qiao, Chem, 2021, 7, 3232–3255 CAS.
- H. Y. Niu, W. J. Zhao, H. Z. Lv, Y. L. Yang and Y. Q. Cai, Chem. Eng. J., 2021, 411, 400–411 CrossRef.
- X. S. Wang, C. Zhou, R. Shi, Q. Q. Liu, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Nano Res., 2019, 12, 2385–2389 CrossRef.
- S. Y. Gao, X. Y. Wang, C. J. Song, S. J. Zhou, F. Yang and Y. Kong, Appl. Catal., B, 2021, 295, 3873–3883 Search PubMed.
- W. J. Luo, X. J. Chen, Z. Wei, D. Liu, W. Q. Yao and Y. F. Zhu, Appl. Catal., B, 2019, 255, 117761 CrossRef.
- X. J. Chen, R. Shi, Q. Chen, Z. J. Zhang, W. J. Jiang, Y. F. Zhu and T. R. Zhang, Nano Energy, 2019, 59, 644–650 CrossRef.
- Y. Zhang, L. L. Wu, X. Y. Zhao, Y. N. Zhao, H. Q. Tan, X. Zhao, Y. Y. Ma, Z. Zhao, S. Y. Song, Y. H. Wang and Y. G. Li, Adv. Energy Mater., 2018, 8, 1–9 Search PubMed.
- S. H. Cao, B. Fan, Y. C. Feng, H. Chen, F. Jiang and X. Wang, Chem. Eng. J., 2018, 353, 147–156 CrossRef CAS.
- H. J. Chen, Z. Chen, G. X. Zhao, Z. B. Zhang, C. Xu, Y. H. Liu, J. Chen, L. Zhuang, T. Haya and X. K. Wang, J. Hazard. Mater., 2018, 347, 67–77 CrossRef CAS.
- S. Z. Hu, X. Chen, Q. Li, F. Y. Li, Z. P. Fan, H. Wang, Y. J. Wang, B. H. Zheng and G. Wu, Appl. Catal., B, 2017, 201, 58–69 CrossRef CAS.
- Y. T. Xiao, G. H. Tian, W. Li, Y. Xie, B. J. Jiang, C. G. Tian, D. Y. Zhao and H. G. Fu, J. Am. Chem. Soc., 2019, 141, 2508–2515 CrossRef CAS PubMed.
- S. Z. Andersen, V. Colic, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, F. S. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Norskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- Z. F. Zhao, H. J. Ren, D. Yang, Y. Han, J. F. Shi, K. An, Y. Chen, Y. H. Shi, W. J. Wang, J. D. Tan, X. Xin, Y. Zhang and Z. Y. Jiang, ACS Catal., 2021, 11, 9986–9995 CrossRef CAS.
- G. W. Watt and J. D. Chrisp, Anal. Chem., 1952, 24, 2006–2008 CrossRef CAS.
- J. X. Zhao, X. J. Liu, X. Ren, X. Sun, D. X. Tian, Q. Wei and D. Wu, Appl. Catal., B, 2021, 284, 1–8 CrossRef.
- W. B. Qiu, X. Y. Xie, J. D. Qiu, W. H. Fang, R. P. Liang, X. Ren, Q. X. Ji, G. W. Cui, A. M. Asiri, G. L. Cui, B. Tang and X. P. Sun, Nat. Commun., 2018, 9, 3485–3493 CrossRef.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef.
- G. H. Dong, W. K. Ho and C. Y. Wang, J. Mater. Chem. A, 2015, 3, 23435–23441 RSC.
- K. Chen, X. Zhao, X. J. Zhang, W. S. Zhang, Z. F. Wu, H. Y. Wang, D. X. Han and L. Niu, Catal. Sci. Technol., 2021, 11, 2713–2717 RSC.
- X. J. Zhang, D. F. Han, M. J. Dai, K. Chen, Z. Y. Han, Y. Y. Fan, Y. He, D. X. Han and L. Niu, Catal. Sci. Technol., 2021, 11, 6248–6256 RSC.
- X. Yang, X. J. Lian, S. J. Liu, J. Tian, C. P. Jiang and G. Wang, Int. J. Electrochem. Sci., 2013, 8, 3721 CAS.
- S. Zhang, Y. X. Zhao, R. Shi, G. I. N. Waterhouse and T. R. Zhang, EnergyChem, 2019, 1, 1–40 Search PubMed.
- T. T. Hou, Y. Xiao, P. X. Cui, Y. N. Huang, X. P. Tan, X. S. Zheng, Y. Zou, C. X. Liu, W. K. Zhu, S. Q. Liang and L. B. Wang, Adv. Energy Mater., 2019, 9, 2319–2327 Search PubMed.
- C. X. Guo, J. R. Ran, A. Vasileff and S. Z. Qiao, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- Y. N. Bo, H. Y. Wang, Y. X. Lin, T. Yang, R. Ye, Y. Li, C. Y. Hu, P. Y. Du, Y. G. Hu, Z. Liu, R. Long, C. Gao, B. J. Ye, L. Song, X. J. Wu and Y. J. Xiong, Angew. Chem., 2021, 60, 16085–16092 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy01507b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |