
DFT computational insight into Pd(0)-catalyzed oxidative cross-coupling of 1,2-allenyl ketones and aryl boronic acids: Pd(II)-carbenoid intermediate versus π-allyl-Pd(II) intermediate†
Jihong
Xu
 ,
Yanhong
Liu
,
Chengbu
Liu
and
Dongju
Zhang
*
,
Yanhong
Liu
,
Chengbu
Liu
and
Dongju
Zhang
*
Key Lab of Colloid and Interface Chemistry, Ministry of Education, Institute of Theoretical Chemistry, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, P. R. China. E-mail: zhangdj@sdu.edu.cn; Fax: +86 0531 88564464; Tel: +86 0531 88365833
First published on 10th November 2022
Abstract
DFT calculations unveil that the Pd(0)-catalyzed oxidative cross-coupling of 1,2-allenyl ketones with aryl boronic acids may either occur via a Pd(II)-carbenoid intermediate or a π-allyl-Pd(II) intermediate, depending on the substituents on 1,2-allenyl ketones. The substituted substrate prefers to undergo the π-allyl-Pd(II) intermediate mechanism over the Pd(II)-carbenoid intermediate one, while the unsubstituted one reverses the preference. The calculations also show a new mechanism for the transformation of Pd(0) to the Pd(II) complex and rationalize the reason for the non-excellent yield for the unsubstituted 1,2-allenyl ketone.
1. Introduction
Furans, one of the most important aromatic compounds, are widely existing in a variety of natural products and synthetic medicines.1–4 Moreover, furan-based functional molecules have potential applications in materials science, especially in the field of furan-based semiconductors for energy conversion and storage.5 The past decades have witnessed great advancements in furan syntheses using various methods,6–10 in which the transition-metal-catalyzed cycloisomerization of 1,2-allenyl ketones has attracted remarkable attention11–15 due to their availability16,17 (Scheme 1a). The efficient syntheses of furan derivatives via the intramolecular nucleophilic cyclization of 1,2-allenyl ketones were initially reported by Marshall13 and Hashmi et al.14 (Scheme 1a with G = H, M = RhI, AgI, or PdII) and subsequently developed by Gevorgyan et al.15 (Scheme 1a with G = alkyl, aryl, halide, silyl, etc., M = AuI, AgI, CuI, etc.) using various transition-metal catalysts. It is believed that the cycloisomerization reactions in these pioneering studies occur via a cyclic metal-carbene intermediate mechanism.18–20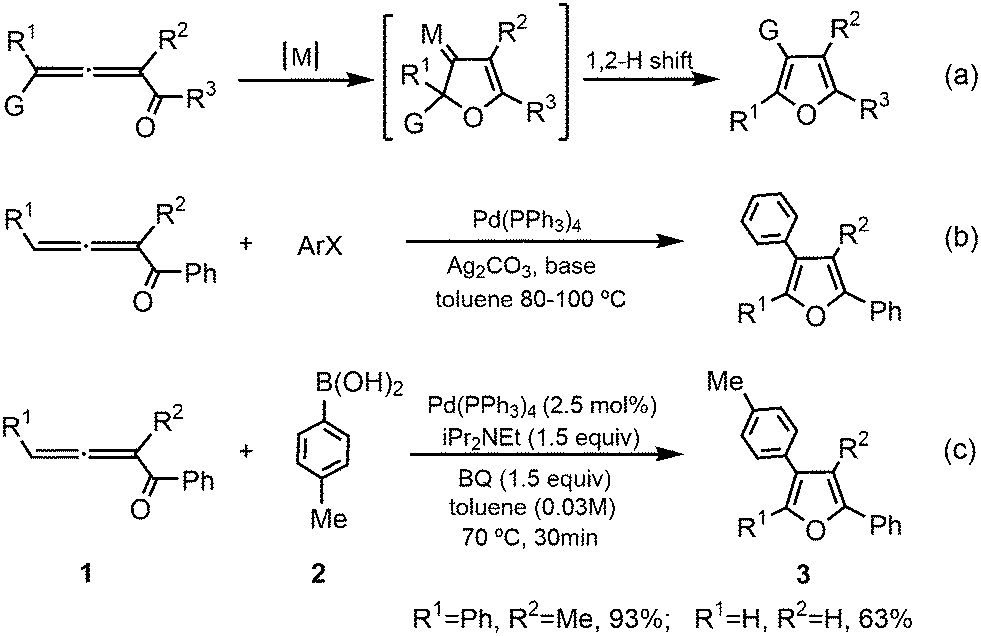 | ||
| Scheme 1 Transition-metal-catalyzed intramolecular cycloisomerization (a) and intermolecular cross-coupling of 1,2-allenyl ketones with aryl halides (b) or aryl boronic acids (c) to synthesize furans. | ||
Another important methodology for the furan synthesis from 1,2-allenyl ketones using catalysis by transition metals, especially palladium(0) species, is either via their intermolecular cross-coupling reactions with electrophiles, such as aryl halides,21 or via their intermolecular oxidative cross-coupling reactions with nucleophiles, for example, aryl boronic acids in the presence of suitable oxidants.22,23 Ma and co-workers24 reported Pd(0)-catalyzed coupling cyclization reactions of 1,2-allenyl ketones with aryl halides to efficiently and conveniently synthesize poly-substituted furans (Scheme 1b). In 2014, Wang et al.25 developed palladium-catalyzed oxidative cross-coupling of 1,2-allenyl ketones with aryl boronic acids (Scheme 1c), which provides a complementary strategy for highly substituted furans, especially tetra-substituted furans.
To account for these intra- and intermolecular cross-coupling reactions, the previous reports have proposed two different mechanistic pathways, which can be referred to as the metal-carbene intermediate mechanism18–20 and the π-allyl-metal intermediate mechanism.26,27 Bao et al.28 have reported an elegant DFT-based computational study on the cross-coupling reaction of 1,2-allenyl ketones with iodobenzene, one of the most commonly used aryl halides. It is found that the viable mechanistic pathway depends on the substituents on either 1,2-allenyl ketones or iodobenzene. Here, we were intrigued by the oxidative cross-coupling reaction of 1,2-allenyl ketones with aryl boronic acids developed by Wang et al.,25 as exemplified by the two reactions in Scheme 1c. According to the experimental results,25 it is not sure whether the reaction proceeds via the metal-carbene intermediate mechanism (pathway I in Scheme 2) or via the π-allyl-metal complex mechanism (pathway II Scheme 2). In other words, exactly how the furan product is formed is uncertain. Moreover, the experimental observations of the different yields using substituted and unsubstituted 1,2-allenyl ketones (excellent vs. good) are still not understood well. These dilemmas prompted us to study the two representative reactions in Scheme 1c by performing DFT calculations. We expect that the calculated results would give a better understanding of the Pd(0)-catalyzed furan synthesis from the oxidative cross-coupling of 1,2-allenyl ketones with aryl boronic acids and provide enlightenment for the design of new catalytic cycles.
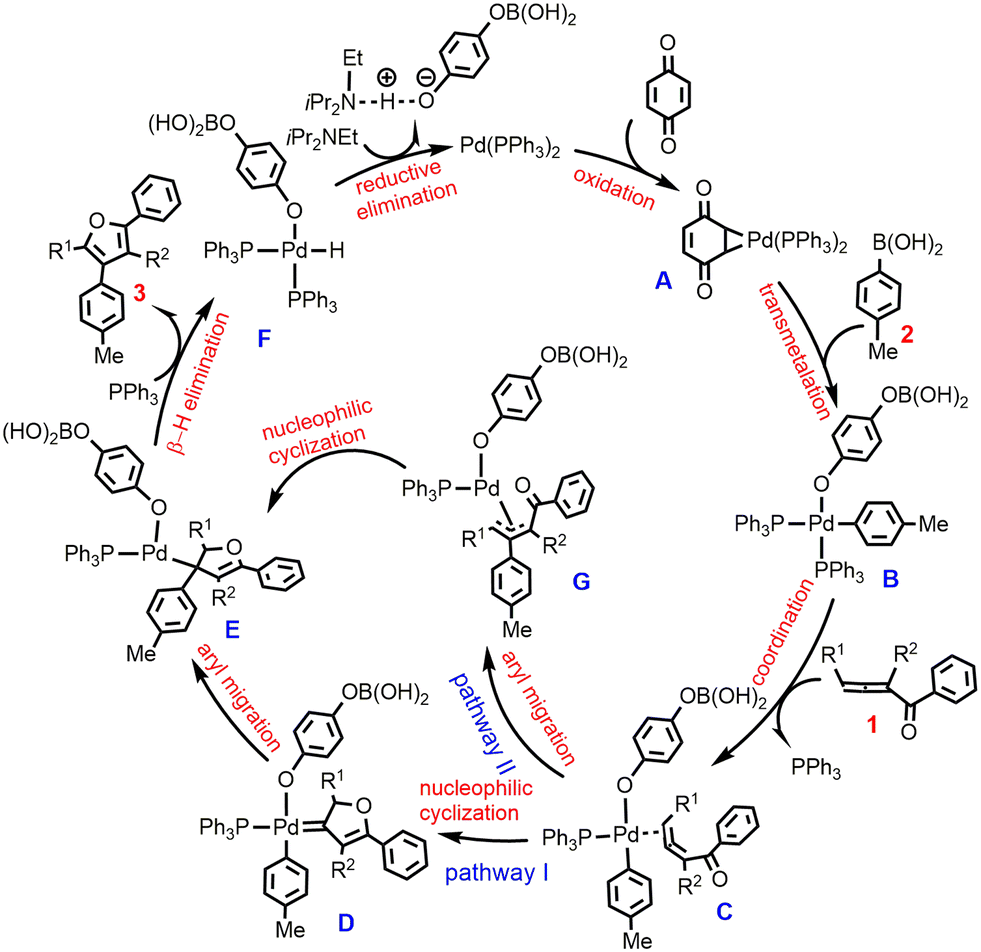 | ||
| Scheme 2 Pd(II)-Carbenoid intermediate mechanism (pathway I) versus π-allyl-Pd(II) intermediate mechanism (pathway II) for Pd(0)-catalyzed oxidative cross-coupling of 1,2-allenyl ketones with aryl boronic acids to form furans. | ||
2. Computational methods
All of the DFT calculations were carried out with the Gaussian 09 program package.29 The geometry optimizations of all stationary points were performed in the gas phase using the M06 functional30 with the LANL2DZ basis set31,32 for the palladium atom and the 6-31G(d) basis set33 for the other atoms. The vibrational frequencies were computed at the same level to verify that the optimized structures correspond to minima (no imaginary frequency) or transition states (one imaginary frequency) and to provide the thermochemical corrections to Gibbs free energies at 298 K. Intrinsic reaction coordinate (IRC)34,35 calculations were used to identify that each transition state connects to its corresponding minima. Solvent effects were considered using the SMD36 continuum solvation model (in toluene) to improve the accuracy of the calculated relative Gibbs energies. Single-point energies were obtained at the M06 levels with the larger SDD basis set37 for palladium and the 6-311++G(d,p) basis set for the other atoms. The Gibbs free energy of each species is obtained by adding the thermal correction obtained in the gas phase to the single-point energy. It should be noted that for reactions in solvent, in particular for situations involving the change of component numbers, thermal corrections based on the ideal gas phase model remarkably overestimate entropy contributions to free energies due to the neglect of the suppressing effect of the solvent on the rotational and transitional freedoms of species.38–40 In the present study, to more accurately evaluate the contribution of entropy in solution, we employed the approach proposed by Martin et al.,41 a correction value of (n–m) × 4.3 kcal mol−1 applies to a transformation from m- to n-components at 298.15 K and 1 atm. The value of 4.3 kcal mol−1 is obtained by considering the actual conditions of a solution reaction medium in the translational entropy. This approximate approach of Martin et al.41 has been proven by Holm and Rybak-Akimova42 and provides reasonable free energies for a process involving multicomponent change.40,43 All the final energies presented are so-corrected Gibbs energies, which were computed by adding the corrections of entropy in solution to the Gibbs free energy at 298.15 K. The 3D structures were prepared using CYLView.44 All the calculated structures and corresponding energies can be found in the ioChem-BD repository45 and can be accessed viahttps://doi.org/10.19061/iochem-bd-6-183.3. Results and discussion
3.1 Formation of the Pd(II)-aryl complex
According to Scheme 2, the reaction starts from the oxidation of the Pd(0) complex by 1,4-benzoquinone (BQ) to form Pd(II) species A, which is then converted into Pd(II)-aryl complex Bvia transmetalation with aryl boronic acid 2. We first preformed calculations for the so-called Pd(II) species A where Pd bonds to two adjacent C atoms of BQ. However, such a Pd(II) species cannot be located, and upon optimization, it always evolves into structure A′, a coordination complex between Pd(0) and BQ, as indicated by calculated structural parameters (Fig. 1). In this case, the formation of Pd(II)-aryl complex B needs to undergo an oxidative transmetalation process46,47 instead of the separated oxidation and transmetalation processes proposed in the experiment (Scheme 2). The calculated results for the formation of Bvia a direct oxidative transmetalation process (i.e. the aryl group directly translates to the Pd center) are shown in Fig. 1, where Pd(PPh3)2 is used as the active species as proposed in previous research.48,49 The formation of the initial coordination complex A′ is calculated to be exergonic by 15.8 kcal mol−1. As indicated in Fig. 1, the present calculations consider two potential oxidative transmetalation pathways, which proceed viaTS1 and TS2, respectively. A′ undergoes a direct oxidative transmetalation with aryl boronic acid 2viaTS1, leading to IM1, which then isomerizes into the Pd(II) species B. This process requires crossing a barrier as high as 64.4 kcal mol−1, suggesting that it is not responsible for the formation of B. Another oxidative transmetalation pathway occurs viaTS2 that is similar to TS1 except for the release of a PPh3 ligand from IM2 upon coordination of aryl boronic acid 2 to give intermediate IM3. The barrier is reduced to 42.9 kcal mol−1, which is still too high to be overcome under the experiment conditions (70 °C).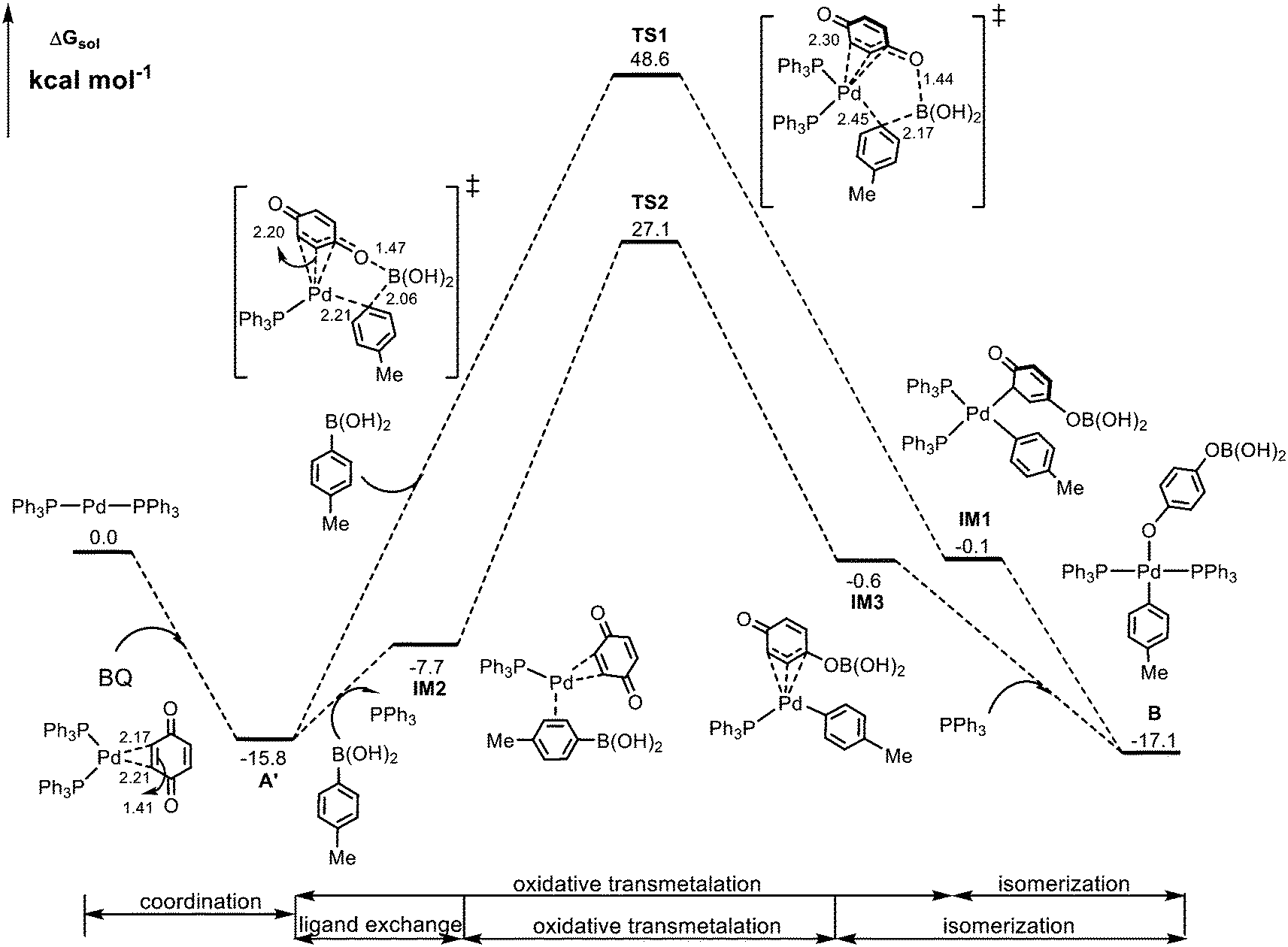 | ||
| Fig. 1 Calculated Gibbs energy profile for the formation of Pd(II)-aryl complex Bvia the direct oxidative transmetalation processes. Selected bond distances are in Å. | ||
In addition, we have also performed calculations for two potential pathways assisted by iPr2NEt. However, as shown in Fig. S1,† these pathways are less favorable than those shown in Fig. 1. Thus, we consider that iPr2NEt does not play a substantial role in the oxidative transmetalation process.
Alternatively, the energetically viable pathway for the formation of Pd(II)-aryl complex B is shown in Fig. 2. In this pathway, from IM2, the initial oxidative transmetalation occurs viaTS3 with the aryl borate translating instead of the aryl group translating to the Pd center discussed above, leading to Pd(II)-borate species IM4 possessing a protonated η3-quinone ligand. Subsequently, a second boronic acid molecule adds to IM4 through O → B coordination interaction to generate IM5, from which the transmetalation of the aryl group from boron to palladium50 takes place viaTS4 to form Pd(II)-aryl species IM6. Then via isomerization of the η3-quinone ligand to p-hydroxyphenoxide and the addition of PPh3, IM6 evolves into a more stable Pd(II)-aryl species IM7, which then undergoes a σ-bond metathesis of the B–O/O–H bonds viaTS5 to give the Pd(II)-aryl complex B. Other energetically less favorable pathways to form Pd(II)-aryl complex B are shown in Fig. S2 and S3.†
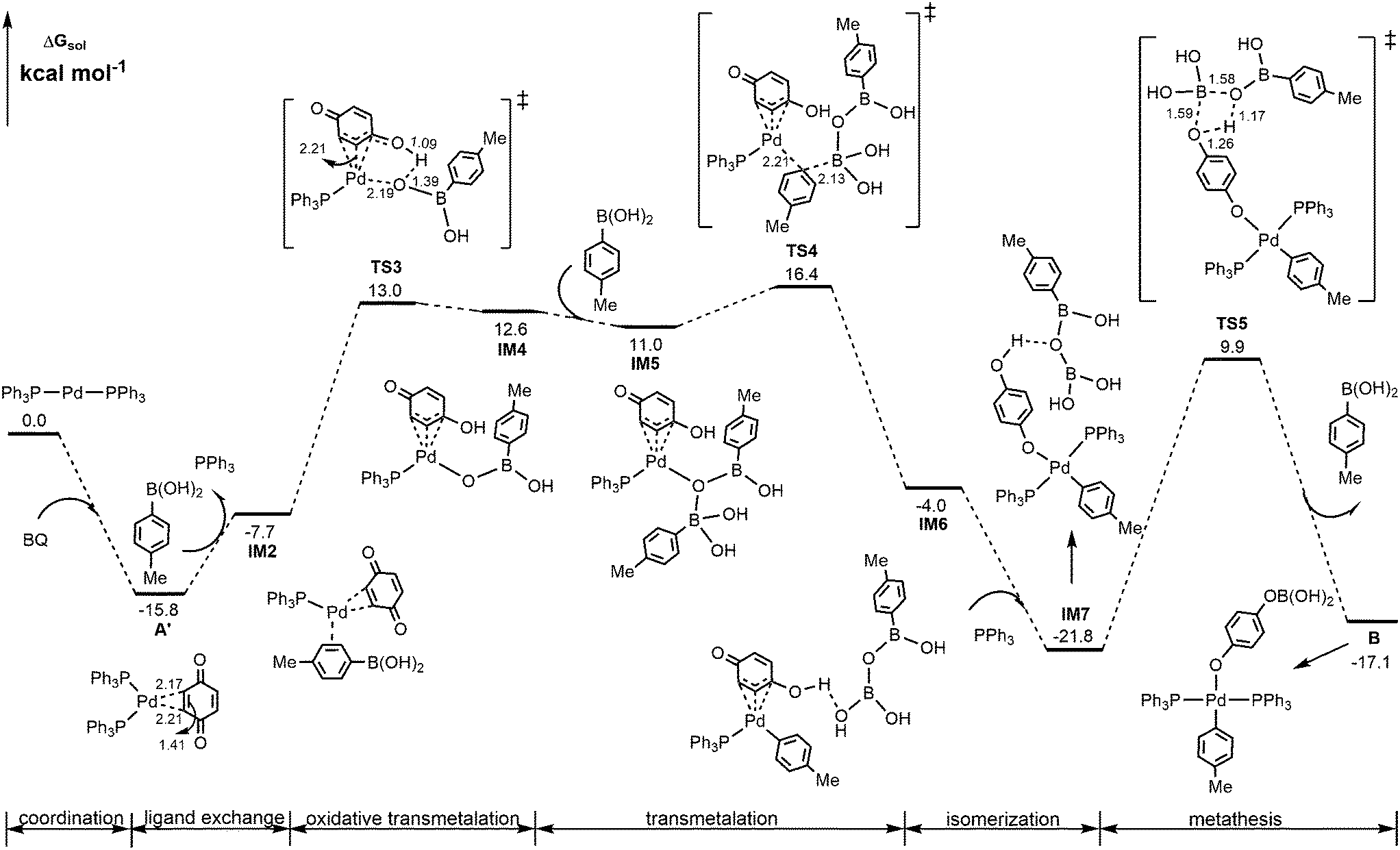 | ||
| Fig. 2 Calculated energetically viable pathway for the formation of Pd(II)-aryl complex B. Selected bond distances are in Å. | ||
From calculated data shown in Fig. 2, it is observed that A′ is the rate-determining intermediate, and TS4 is the rate-determining transition state. Thus, the overall barrier involved in the formation of the Pd(II)-aryl complex B is estimated to be 32.2 kcal mol−1, which is much lower than those shown in Fig. 1, 42.9 and 64.4 kcal mol−1, respectively.
In addition, we also considered the possibility of the 1,2-allenyl ketone for performing the intramolecular nucleophilic cyclization reaction under the catalysis of the reactive Pd(0) species, Pd(PPh3)2 (Fig. S4†). It is found that the barrier of such a process is much higher than that shown in Fig. 2 (39.0 vs. 32.2 kcal mol−1).
3.2 Reaction of substituted 1,2-allenyl ketone 1a
Our attention first focuses on the reaction of the substituted 1,2-allenyl ketone 1a (R1 = Ph, R2 = Me), which led to an excellent furan yield (93%) in the experiment of Wang et al.25 The calculated results are summarized in Fig. 3, where the Pd(II)-carbenoid intermediate pathway (red line) and the π-allyl-Pd(II) intermediate pathway (black line) are compared.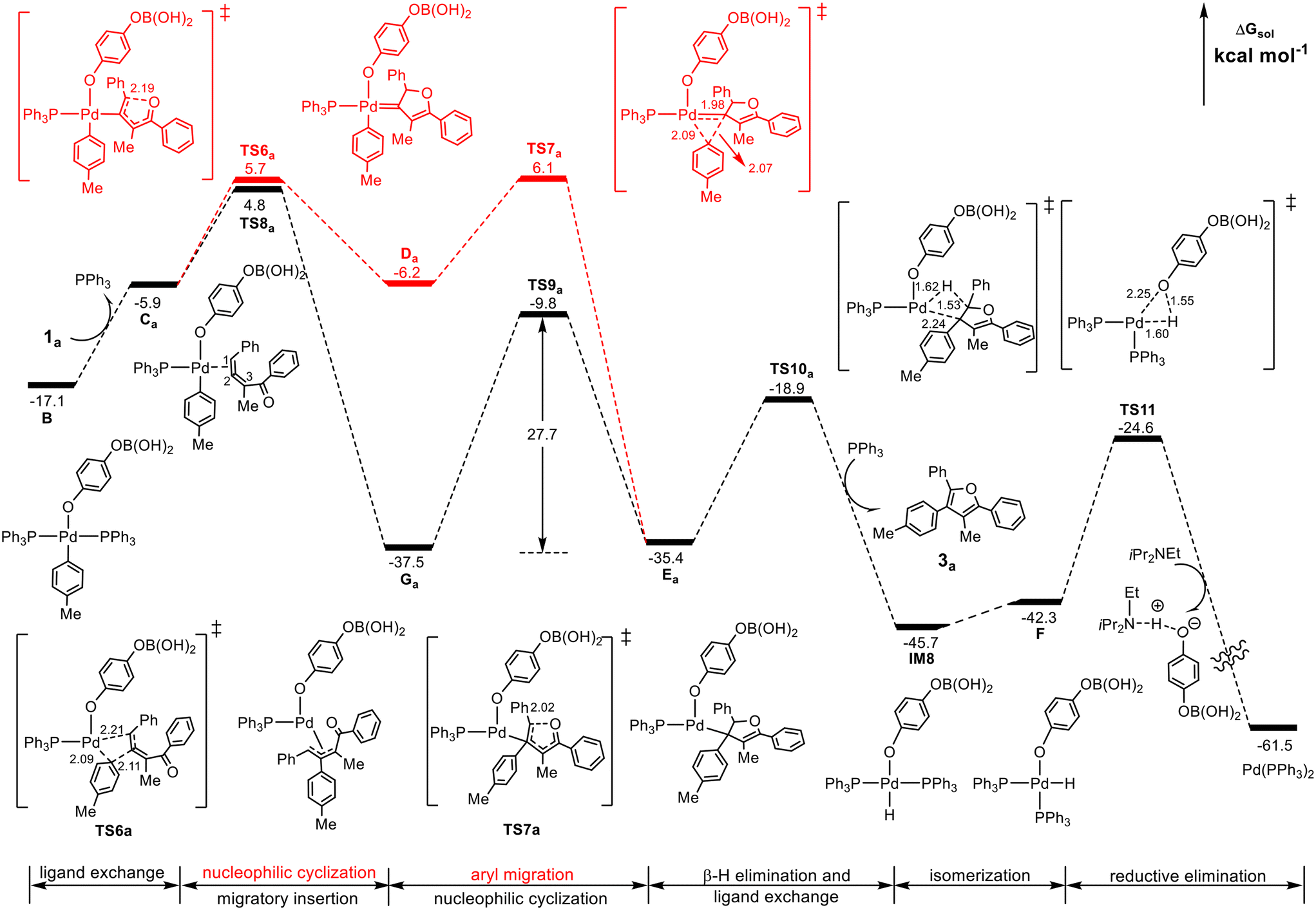 | ||
| Fig. 3 Calculated Gibbs energy profile for the formation of furan 3a from the cross-coupling of substituted 1,2-allenyl ketone 1a with the Pd(II)-aryl complex B either via the Pd(II)-carbenoid intermediate pathway (red line) or via the π-allyl-Pd(II) intermediate pathway (black line). Selected bond distances are in Å. | ||
From the Pd(II)-aryl complex B, the substituted 1,2-allenyl ketone 1a undergoes a ligand exchange with PPh3 to form species Ca, the common precursor for the Pd(II)-carbenoid intermediate pathway and the π-allyl-Pd(II) intermediate pathway. Along the Pd(II)-carbenoid pathway, Ca undergoes intramolecular cyclization viaTS6a by the nucleophilic attack of the carbonyl oxygen atom on the terminal carbon atom of the 1,2-allenyl ketone, leading to the cyclic Pd(II)-carbenoid intermediate Da, from which the aryl group on the Pd center migrates to the carbene carbon atom through TS7a to give species Ea. The relative energies of TS6a and TS7a are 5.7 and 6.1 kcal mol−1, respectively, and thus the predicted barrier for the formation of Ea is 27.9 kcal mol−1, the energy difference between TS7a and IM7 in Fig. 2.
On the other hand, Ca can evolve into a π-allyl-Pd(II) intermediate Gavia the migratory insertion of the aryl group into the internal carbon C2 of the allenyl moiety viaTS8a, which is more stable by 0.9 kcal mol−1 than TS6a (4.8 vs. 5.7 kcal mol−1). Other three migratory insertion transition states of the aryl group into 1,2-allenyl ketone, which are structurally similar to TS8a but energetically less favorable, are shown in Fig. S5.† Subsequently, Ga can be converted to Ea by performing an intramolecular nucleophilic cyclization process viaTS9a with an energy barrier of 27.7 kcal mol−1. From an energy point of view, the barrier of the reaction via the π-allyl-Pd(II) intermediate pathway (27.7 kcal mol−1 from Ga to TS9a) is comparable with that via the Pd(II)-carbenoid intermediate pathway (27.9 kcal mol−1 from IM7 to TS7a). However, it is noted that in the π-allyl-Pd(II) intermediate pathway, the transformation from Ca to Ga is a highly exothermic (irreversible) process, which is in contrast with the almost thermally neutral transformation (reversible) from Ca to Da. In this case, it seems that Ea may be mainly obtained via the π-allyl-Pd(II) intermediate pathway but cannot exclude the Pd(II)-carbenoid pathway.
Once formed, Ea can carry out the β-H elimination viaTS10a to generate the ultimate tetra-substituted furan derivative 3a and IM8 with the addition of the PPh3 ligand. After then, IM8 isomerizes into F, from which the O–H reductive elimination viaTS11 leads to the regeneration of the reactive Pd(PPh3)2 species. It should be noted that during the final reductive elimination process, the appearance of iPr2NEt is substantial, which assists the regeneration of the reactive Pd(PPh3)2 species by forming an acid–base adduct with the generated phenic acid. The calculated barriers of the β-H elimination and the O–H reductive elimination are 18.6 and 21.1 kcal mol−1, respectively.
The above results indicate that for the substituted 1,2-allenyl ketone, the π-allyl-Pd(II) intermediate mechanism with an energy barrier of 27.7 kcal mol−1 may mainly contribute to the formation of the product and the Pd(II)-carbenoid intermediate mechanism also makes partial contributions. This situation is different from the reactions of the substituted 1,2-allenyl ketones with aryl halides reported recently by Bao et al.,28 who showed that the Pd(II)-carbenoid intermediate mechanism is beneficial when the aryl reagent is aryl iodide rather than the present aryl boronic acid.
In addition, from Da, the 1,2-H shift process could directly lead to the tri-substituted side product 2,5-biphenyl-3-methylfuran viaTS15 as shown in Fig. S6;† however, this process requires crossing an energy barrier of 36.1 kcal mol−1 (from IM7 to TS15) and is difficult to occur under the experimental conditions (70 °C), explaining no yield of the side product 2,5-biphenyl-3-methylfuran in the experiment.
3.3 Reaction of unsubstituted 1,2-allenyl ketone 1b
With the clear mechanism in hand, our attention now turns to the reaction of unsubstituted 1,2-allenyl ketone to rationalize the experimental observation that when the R1 and R2 groups of the 1,2-allenyl ketone substrate are replaced with H atoms, denoted as 1,2-allenyl ketone 1b, the furan yield is reduced to 63% from 93%. The calculated results are shown in Fig. 4.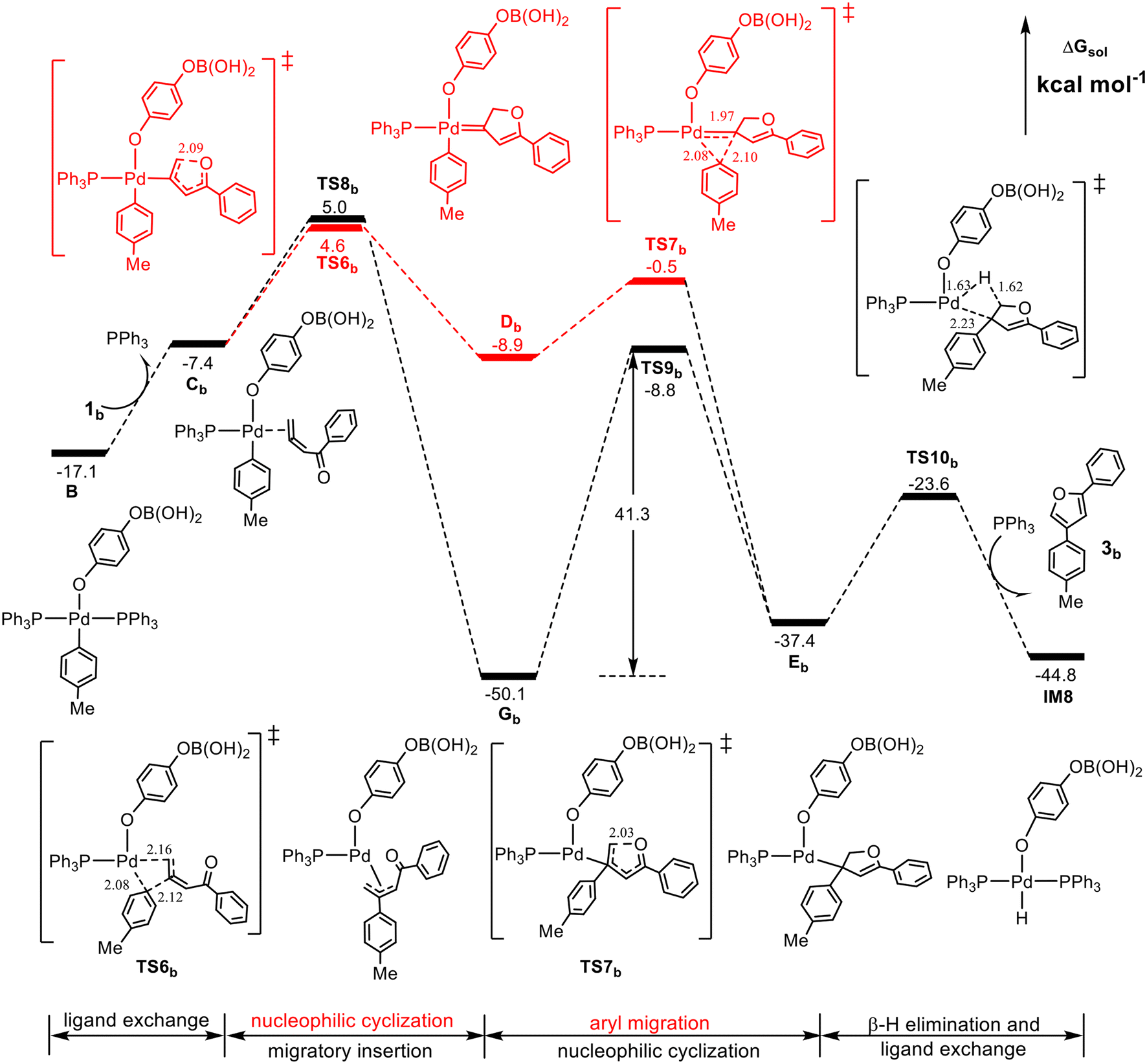 | ||
| Fig. 4 Calculated Gibbs energy profile for the formation of furan 3b from the cross-coupling of unsubstituted 1,2-allenyl ketone 1b with the Pd(II)-aryl complex Bvia the Pd(II)-carbenoid intermediate pathway (red line) or the π-allyl-Pd(II) intermediate pathway (black line). Selected bond distances are in Å. | ||
Intriguingly, in comparison with Fig. 3, there are three essential differences. First, TS6b is now slightly more favorable than TS8b (4.6 vs. 5.0 kcal mol−1), implying that Cb may prefer to form the Pd(II)-carbenoid intermediate Db. Second, TS7b is more stable than TS6b by 5.1 kcal mol−1, and thus once formed, Db would evolve immediately into EbviaTS7b rather than reversibly return to Cb. Third, the barrier from Gb to TS9b is 41.3 kcal mol−1, which is much higher than that in Fig. 3, 27.7 kcal mol−1, and in this case, the further conversion of Gb would be obstructed. These results indicate that for the unsubstituted 1,2-allenyl ketone 1b, the furan product may be only obtained via the Pd(II)-carbenoid intermediate mechanism with a barrier of 26.4 kcal mol−1 (the energy difference between TS6b and IM7), and the remarkable reduction of the furan yield can be attributed to the high stability of the π-allyl-Pd(II) intermediate Gb, whose further conversion involves a too high barrier (41.3 kcal mol−1) to be overcome under the thermal reaction conditions.
3.4 Effects of substituents on 1,2-allenyl ketones
According to the above calculated results, it is very clear that the R1 and R2 groups of the 1,2-allenyl ketone play a substantial role in determining whether the reaction proceeds according to the Pd(II)-carbenoid intermediate mechanism or the π-allyl-Pd(II) intermediate mechanism. When the R1 and R2 groups are H atoms (the unsubstituted 1,2-allenyl ketone), the π-allyl-Pd(II) intermediate mechanism seems impossible due to the high energy barrier (41.3 kcal mol−1) of the nucleophilic cyclization process from Gb to TS9b, and thus the product may be formed via the Pd(II)-carbenoid intermediate mechanism with a barrier of 26.4 kcal mol−1. In contrast, when R1 is a phenyl group and R2 is a methyl group (the substituted 1,2-allenyl ketone), the π-allyl-Pd(II) intermediate mechanism is relatively easier and thermodynamically much favorable, therefore dominantly contributing to the formation of the product with a barrier of 27.7 kcal mol−1 (the energy difference between Ga and TS9a).It should be noted that the calculated rate-determining barriers of the unsubstituted allenyl ketone and the substituted one, 26.4 vs. 27.7 kcal mol−1, seem to be inverse to the corresponding yields observed experimentally, 63% vs. 93%. However, the lower yield of 3b can be attributed to the chemo-divergence in the reaction of the unsubstituted allenyl ketone. As shown in Fig. 4, the pathway viaTS8b competes with that viaTS6b, and from TS8b the reaction delivers the stable intermediate Gb, which cannot be transformed into the desired product due to its high stability. In contrast, for the reaction of the substituted allenyl ketone (Fig. 3), both pathways viaTS6a and TS8a contribute to the product formation, although the pathway viaTS8a is believed to be the decisive pathway. From these calculated thermodynamic and kinetic data, we can qualitatively understand the experimentally observed different product yields from the unsubstituted and substituted allenyl ketones.
The higher barrier from Gb to TS9b than that from Ga to TS9a can be understood by performing the distortion/interaction analysis for these two processes. According to the distortion/interaction model,51 also known as the activation-strain model,52 the activation energy (ΔE‡) is decomposed into the distortion or strain energy (ΔEdist), which is associated with the structural deformation of the reactants/fragments along the reaction coordination, and the interaction energy (ΔEint) between these deformed reactants/fragments. For the present two processes, we used the fragmentation scheme shown in Fig. 5, where by cutting the Pd–C bond each structure is decomposed into two fragments, the palladium catalyst moiety and the allyl substrate moiety. These two fragments are then saturated with hydrogen atoms, whose positions were optimized while all other atoms were kept fixed.53
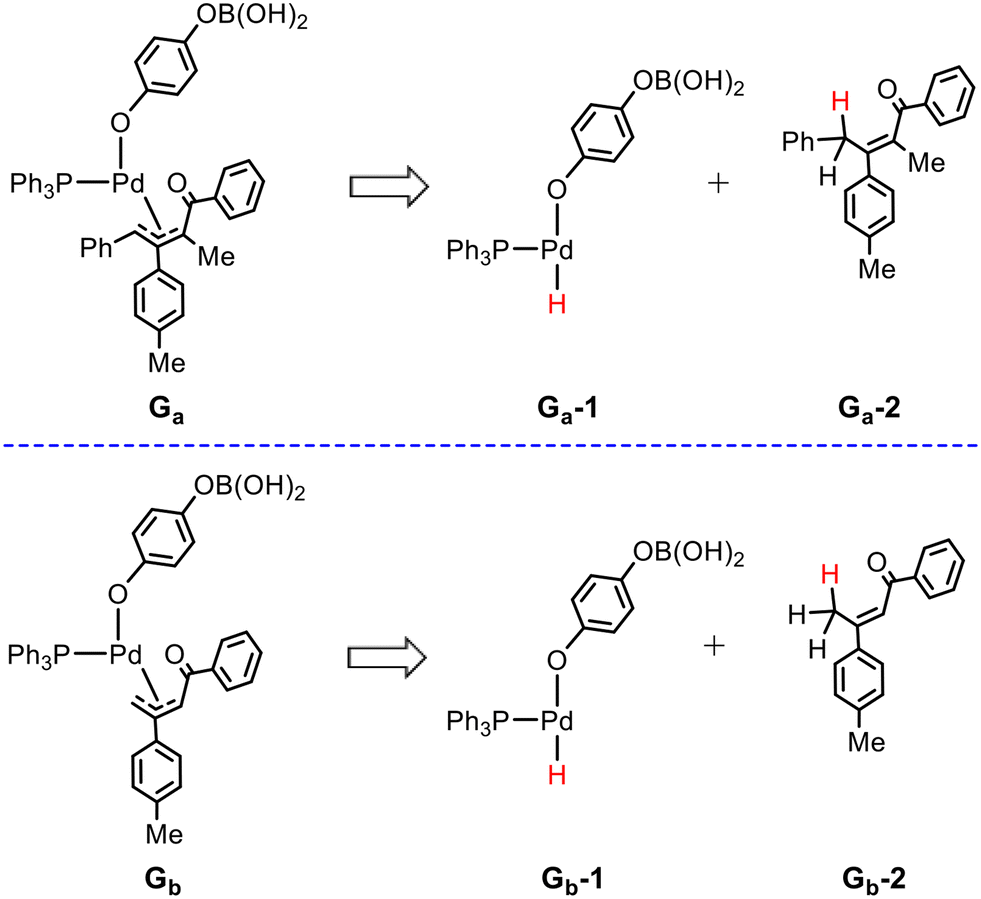 | ||
| Fig. 5 Fragmentation scheme used in distortion/interaction analysis. | ||
Table 1 lists the calculated results of the distortion/interaction analysis for TS9a and TS9b. It is found that the distortion energy of the palladium catalyst fragment in TS9b (28.6 kcal mol−1) is larger than that in TS9a (22.9 kcal mol−1), while the allyl fragment in TS9b (32.7 kcal mol−1) is smaller than that in TS9a (39.9 kcal mol−1). Thus, the total distortion energies are quite close in TS9a (61.8 kcal mol−1) and TS9b (61.3 kcal mol−1). On the other hand, the interaction energy between the two deformed fragments in TS9a (−32.6 kcal mol−1) is remarkably larger in comparison with that in TS9b (−21.8 kcal mol−1), which means the weaker electronic interaction of the two fragments in TS9b and thus does not favor the stabilization of TS9b. Therefore, the weaker interaction between the two fragments contributes to the higher barrier from Gb to TS9b.
| Transition state | ΔE‡ | ΔEdist-1 | ΔEdist-2 | ΔEdist | ΔEint |
|---|---|---|---|---|---|
| TS9 a | 29.2 | 22.9 | 39.9 | 61.8 | −32.6 |
| TS9 b | 39.5 | 28.6 | 32.7 | 61.3 | −21.8 |
4. Conclusion
In conclusion, by performing DFT calculations we have shown the mechanism details of the synthesis of furan derivatives via Pd(0)-catalyzed oxidative cross-coupling of 1,2-allenyl ketones with aryl boronic acids. For the substituted 1,2-allenyl ketone, the reaction proceeds mainly via the π-allyl-Pd(II) intermediate mechanism and partly via the Pd(II)-carbenoid intermediate mechanism. In contrast, for the unsubstituted 1,2-allenyl ketone, the reaction occurs only via the Pd(II)-carbenoid intermediate mechanism. The present calculations also revealed a new mechanism for the formation of the Pd(II)-aryl complex, which involves the key oxidative transmetalation process instead of the prior oxidation of Pd(0) by BQ followed by the transmetalation. The non-excellent yield from the unsubstituted 1,2-allenyl ketone originates from the excessive stability of the π-allyl-Pd(II) intermediate. Expectedly, these mechanistic insights could provide a guide for developing new strategies synthesizing furan derivatives from coupling between 1,2-allenyl ketones and aryl boronic acids.Author contributions
Jihong Xu performed the calculations and wrote the original draft, Yanhong Liu analyzed the results, Chengbu Liu supervised this work, and Dongju Zhang wrote and reviewed this paper.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21833004 and 22273051). We acknowledge the Shandong University Supercomputing Center for providing the computational resources.References
- B. H. Lipshutz, Five-Membered Heteroaromatic Rings as Intermediates in Organic Synthesis, Chem. Rev., 1986, 86, 795–819 CrossRef.
- X. L. Hou, H. Y. Cheung, T. Y. Hon, P. L. Kwan, T. H. Lo, S. Y. Tong and H. N. C. Wong, Regioselective Syntheses of Substituted Furans, Tetrahedron, 1998, 54, 1955–2020 CrossRef.
- R. C. D. Brown, Developments in Furan Syntheses, Angew. Chem., Int. Ed., 2005, 44, 850–852 CrossRef PubMed.
- C. Mitsui, J. Soeda, K. Miwa, H. Tsuji, J. Takeya and E. Nakamura, Naphtho[2,1-b:6,5-b′]difuran: A Versatile Motif Available for Solution-Processed Single-Crystal Organic Field-Effect Transistors with High Hole Mobility, J. Am. Chem. Soc., 2012, 134, 5448–5451 CrossRef PubMed.
- H. Tsuji and E. Nakamura, Design and Functions of Semiconducting Fused Polycyclic Furans for Optoelectronic Applications, Acc. Chem. Res., 2017, 50, 396–406 CrossRef PubMed.
- B. A. Keay, Synthesis of Multi-Substituted Furan Rings: the Role of Silicon, Chem. Soc. Rev., 1999, 28, 209–215 RSC.
- M. Zhang, H.-F. Jiang, H. Neumann, M. Beller and P. H. Dixneuf, Sequential Synthesis of Furans from Alkynes: Successive Ruthenium(II)- and Copper(II)-Catalyzed Processes, Angew. Chem., 2009, 121, 1709–1712 CrossRef.
- C. He, S. Guo, J. Ke, J. Hao, H. Xu, H. Chen and A. Lei, Silver-Mediated Oxidative C−H/C−H Fuctionalization: A Strategy to Construct Polysubstituted Furans, J. Am. Chem. Soc., 2012, 134, 5766–5769 CrossRef CAS PubMed.
- B. Xue, S. Su, Y. Cui, Y. Fei, X. Jia, J. Li and J. Fang, Phosphine-Mediated Sequential Annulations of Allenyl Ketone and Isocyanide: A Bicyclization Strategy to Access a Furan-Fused Eight-Membered Ring and a Spirocycle, Chem. Commun., 2019, 55, 12180–12183 RSC.
- M. He, Y. Yao, C. Ai, Z. Mo, Y. Wu, Q. Zhou, Y. Pan and H. Tang, Electrochemically-Mediated C−H Functionalization of Allenes and 1,3-Dicarbonyl Compounds to Construct Tetrasubstituted Furans, Org. Chem. Front., 2022, 9, 781–787 RSC.
- L. Peng, X. Zhang, M. Ma and J. Wang, Transition-Metal-Catalyzed Rearrangement of Allenyl Sulfides: A Route to Furan Derivatives, Angew. Chem., Int. Ed., 2007, 46, 1905–1908 CrossRef CAS.
- Y. Li, J. P. Brand and J. Waser, Gold-Catalyzed Regioselective Synthesis of 2- and 3-Alkynyl Furans, Angew. Chem., Int. Ed., 2013, 52, 6743–6747 CrossRef CAS PubMed.
- J. A. Marshall and E. D. Robinson, A Mild Method for the Synthesis of Furans. Application to 2,5-Bridged Furano Macrocyclic Compounds, J. Org. Chem., 1990, 55, 3450–3451 CrossRef CAS.
- A. S. K. Hashmi, T. L. Ruppert, T. Knöfel and J. W. Bats, C−C-Bond Formation by the Palladium-Catalyzed Cycloisomerization/Dimerization of Terminal Allenyl Ketones: Selectivity and Mechanistic Aspects, J. Org. Chem., 1997, 62, 7295–7304 CrossRef CAS.
- A. S. Dudnik, A. W. Sromek, M. Rubina, J. T. Kim, A. V. Kel'i and V. Gevorgyan, Metal-Catalyzed 1,2-Shift of Diverse Migrating Groups in Allenyl Systems as a New Paradigm toward Densely Functionalized Heterocycles, J. Am. Chem. Soc., 2008, 130, 1440–1452 CrossRef CAS.
- H. Guo, Y. Fan, Z. Sun, Y. Wu and O. Kwon, Phosphine Organocatalysis, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS.
- J. Son, T. W. Reidl, K. H. Kim, D. J. Wink and L. L. Anderson, Generation and Rearrangement of N,O-Dialkenylhydroxylamines for the Synthesis of 2-Aminotetrahydrofurans, Angew. Chem., Int. Ed., 2018, 57, 6597–6600 CrossRef PubMed.
- Y. Xia, A. S. Dudnik, V. Gevorgyan and Y. Li, Mechanistic Insights into the Gold-Catalyzed Cycloisomerization of Bromoallenyl Ketones: Ligand-Controlled Regioselectivity, J. Am. Chem. Soc., 2008, 130, 6940–6941 CrossRef.
- A. S. Dudnik, Y. Xia, Y. Li and V. Gevorgyan, Computation Guided Development of Au-Catalyzed Cycloisomerizations Proceeding via 1,2-Si or 1,2-H Migrations: Regiodivergent Synthesis of Silylfurans, J. Am. Chem. Soc., 2010, 132, 7645–7655 CrossRef PubMed.
- M. Rajzmann, J. Wang and S. Humbel, Metal-Catalyzed Rearrangement of Allenylsulfides to Furan: A Theoretical Mechanistic Approach, Mol. Catal., 2017, 443, 148–154 CrossRef.
- S. Ma and J. Zhang, Pd0-Catalyzed Cyclization Reaction of Aryl or Alk-1-Enyl Halides with 1,2-Dienyl ketones: A General and Efficient Synthesis of Polysubstituted Furans, Chem. Commun., 2000, 117–118 RSC.
- C. Peng, Y. Wang and J. Wang, Palladium-Catalyzed Cross-Coupling of α-Diazocarbonyl Compounds with Arylboronic Acids, J. Am. Chem. Soc., 2008, 130, 1566–1567 CrossRef CAS PubMed.
- X. Zeng, G. Cheng, J. Shen and X. Cui, Palladium-Catalyzed Oxidative Cross-Coupling of N-Tosylhydrazones with Indoles: Synthesis of N-Vinylindoles, Org. Lett., 2013, 15, 3022–3025 CrossRef CAS PubMed.
- S. Ma, J. Zhang and L. Lu, Pd0-Catalyzed Coupling Cyclization Reaction of Aryl or 1-alkenyl Halides with 1,2-Allenyl Ketones: Scope and Mechanism. An Efficient Assembly of 2,3,4-, 2,3,5-Tri- and 2,3,4,5-Tetrasubstituted Furans, Chem. – Eur. J., 2003, 9, 2447–2456 CrossRef CAS.
- Y. Xia, Y. Xia, R. Ge, Z. Liu, Q. Xiao, Y. Zhang and J. Wang, Oxidative Cross-Coupling of Allenyl Ketones and Organoboronic Acids: Expeditious Synthesis of Highly Substituted Furans, Angew. Chem., Int. Ed., 2014, 53, 3917–3921 CrossRef CAS PubMed.
- S. Ma and L. Li, Palladium-Catalyzed Cyclization Reaction of Allylic Bromides with 1,2-Dienyl Ketones. An Efficient Synthesis of 3-Allylic Polysubstituted Furans, Org. Lett., 2000, 2, 941–944 CrossRef CAS.
- S. Ma and Z. Yu, Oxidative Cyclization–Dimerization Reaction of 2,3-Allenoic Acids and 1,2-Allenyl Ketones: An Efficient Synthesis of 4-(3′-Furanyl)butenolide Derivatives, Angew. Chem., Int. Ed., 2002, 41, 1775–1778 CrossRef CAS.
- K. Lv, P. Dai and X. Bao, Mechanistic Understanding of the Pd(0)-Catalyzed Coupling Cyclization of 1,2-Allenyl Ketones with Aryl Halides: A Computational Study, ACS Catal., 2020, 10, 13202–13212 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision B.01), Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- P. J. Hay and W. R. Wadt, Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Hollwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, A Set of f-Polarization Functions for Pseudo Potential Basis Sets of the Transition Metals Sc-Cu, Y-Ag and La-Au, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- S. Huzinaga, Basis Sets for Molecular Calculations, Comput. Phys. Rep., 1985, 2, 281–339 CrossRef.
- K. Fukui, A Formulation of the Reaction Coordinate, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef.
- K. Fukui, The Path of Chemical Reactions−The IRC Approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Energy-Adjustedab Initio Pseudopotentials for the Second and Third Row Transition Elements, Theor. Chem. Acc., 1990, 77, 123–144 Search PubMed.
- A. A. C. Braga, G. Ujaque and F. Maseras, A DFT Study of the Full Catalytic Cycle of the Suzuki−Miyaura Cross-Coupling on a Model System, Organometallics, 2006, 25, 3647–3658 CrossRef.
- T. Tuttle, D. Wang, W. Thiel, J. Köhler, M. Hofmann and J. Weis, Mechanism of Olefin Hydrosilylation Catalyzed by RuCl2(CO)2(PPh3)2: A DFT Study, Organometallics, 2006, 25, 4504–4513 CrossRef CAS.
- S. Qu, Y. Dang, C. Song, M. Wen, K. Huang and Z. Wang, Catalytic Mechanisms of Direct Pyrrole Synthesis via Dehydrogenative Coupling Mediated by PNP-Ir or PNN-Ru Pincer Complexes: Crucial Role of Proton-Transfer Shuttles in the PNP-Ir System, J. Am. Chem. Soc., 2014, 136, 4974–4991 CrossRef CAS PubMed.
- R. L. Martin, P. J. Hay and L. R. Pratt, Hydrolysis of Ferric Ion in Water and Conformational Equilibrium, J. Phys. Chem. A, 1998, 102, 3565–3573 CrossRef CAS.
- D. Huang, O. V. Makhlynets, L. L. Tan, S. C. Lee, E. V. Rybak-Akimova and R. H. Holm, Fast Carbon Dioxide Fixation by 2,6-Pyridinedicarboxamidato-Nickel(II)-Hydroxide Complexes: Influence of Changes in Reactive Site Environment on Reaction Rates, Inorg. Chem., 2011, 50, 10070–10081 CrossRef CAS PubMed.
- J. Yu, S. Zhang and X. Hong, Mechanisms and Origins of Chemo- and Regioselectivities of Ru(II)-Catalyzed Decarboxylative C−H Alkenylation of Aryl Carboxylic Acids with Alkynes: A Computational Study, J. Am. Chem. Soc., 2017, 139, 7224–7243 CrossRef.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, (https://www.cylview.org) Search PubMed.
- M. Álvarez-Moreno, C. de Graaf, N. López, F. Maseras, J. M. Poblet and C. Bo, Managing the Computational Chemistry Big Data Problem: The ioChem-BD Platform, J. Chem. Inf. Model., 2015, 55, 95–103 CrossRef.
- Y. Yamamoto, The First General and Selective Palladium(II)-Catalyzed Alkoxycarbonylation of Arylboronates: Interplay among Benzoquinone-Ligated Palladium(0) Complex, Organoboron, and Alcohol Solvent, Adv. Synth. Catal., 2010, 352, 478–492 CrossRef.
- A. Vasseur, J. Muzart and J. L. Bras, Ubiquitous Benzoquinones, Multitalented Compounds for Palladium-Catalyzed Oxidative Reactions, Eur. J. Org. Chem., 2015, 19, 4053–4069 CrossRef.
- C. Amatore and F. Pfluger, Mechanism of Oxidative Addition of Palladium(0) with Aromatic Iodides in Toluene, Monitored at Ultramicroelectrodes, Organometallics, 1990, 9, 2276–2282 CrossRef.
- K. C. Lam, T. B. Marder and Z. Lin, DFT Studies on the Effect of the Nature of the Aryl Halide Y-C6H4-X on the Mechanism of Its Oxidative Addition to Pd0L versus Pd0L2, Organometallics, 2007, 26, 758–760 CrossRef CAS.
- T. Zhou, P. Xie, C. Ji, X. Hong and M. Szostak, Decarbonylative Suzuki-Miyaura Cross-Coupling of Aroyl Chlorides, Org. Lett., 2020, 22, 6434–6440 CrossRef CAS PubMed.
- I. Fernández, F. P. Cossío and F. M. Bickelhaupt, Aromaticity and Activation Strain Analysis of [3 + 2] Cycloaddition Reactions between Group 14 Heteroallenes and Triple Bonds, J. Org. Chem., 2011, 76, 2310–2314 CrossRef.
- I. Fernández and F. M. Bickelhaupt, The Activation Strain Model and Molecular Orbital Theory: Understanding and Designing Chemical Reactions, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC.
- T. J. Seguin and S. E. Wheeler, Competing Noncovalent Interactions Control the Stereoselectivity of Chiral Phosphoric Acid Catalyzed Ring Openings of 3-Substituted Oxetanes, ACS Catal., 2016, 6, 7222–7228 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional computational results and Cartesian coordinates of all optimized structures located in this work (PDF). See DOI: https://doi.org/10.1039/d2cy01506d |
| This journal is © The Royal Society of Chemistry 2023 |