
Recent advances in reducible metal oxide catalysts for C1 reactions
Jialu
Li
and
Zhenmeng
Peng
 *
*
Department of Chemical, Biomolecular, and Corrosion Engineering, The University of Akron, Akron, OH 44325, USA. E-mail: zpeng@uakron.edu
First published on 22nd November 2022
Abstract
Chemical conversion of one-carbon (C1) molecules to value-added chemicals and energy fuels has attracted increasing research interest in recent years driven by concerns on the depletion of fossil fuel resources and amplifying environmental issues caused by anthropogenic activities. In this mini-review, we introduce important C1 reactions in CO, CO2, and CH4 conversions via different approaches, including thermally, electrochemically, and photochemically driven processes, and the catalysis mechanism of reducible metal oxide (RMO) materials for use in these reactions. We mainly summarize the latest research advances in RMO catalyst materials and their common functionality in these C1 reactions, discuss the current research status and challenges, and provide a perspective on future research directions and opportunities in this field.
Introduction
One-carbon molecules referred to as C1 chemicals, especially carbon monoxide (CO), carbon dioxide (CO2), and methane (CH4), are primarily sourced from fossil fuels, biomass, and organic wastes, and have importance in many chemical conversions. C1 chemistry has attracted dramatic research interests since concerns on energy shortages and environmental problems have grown. Advanced technologies have been developed and investigated with an aim to transform these inexpensive, abundant C1 feedstocks into value-added chemicals or energy fuels to meet sustainable development. The Fischer–Tropsch (FT) process, water–gas shift reaction (WGSR), and steam methane reforming (SMR) are a few good examples from decades of fundamental studies in the lab to large-scale applications in industry. However, these traditional, thermal reaction routes are typically operated at elevated temperatures and pressures. This is largely due to the inert nature of CH4 and CO2, which makes it hard to activate and break C–H and C–O bonds, and the difficulties of precisely controlling C–C coupling in CO transformation to obtain the target products.1 Catalyst materials that attain good thermal stability are commonly used to adapt the harsh reaction conditions.2,3 Recently, energy-effective strategies have attracted significant attention for C1 chemistry. Utilizing electrocatalysis and photocatalysis that apply electricity and photons as an additional driving force besides thermal energy, researchers have overcome thermodynamic barriers and realized effective C1 conversions under mild reaction conditions.4,5 Many of these studies are still laboratory researched with challenges remaining in reaction activity improvement and selectivity control. Like the crucial role played in thermal C1 conversions, fundamental catalysis study and catalyst material development are again research hotspots to advance these new C1 technologies.Reducible metal oxides (RMOs), such as TiO2, CeO2, MnO2, and V2O5, represent the most important group of C1 catalysts in reducible material categories. In the bigger picture, many oxygen-containing reducible compound catalysts could also be considered the broader family of reducible oxides based on their intrinsic reducibility of active sites, for instance, metal oxide hydroxides and mixed metal complexes (Fig. 1). The RMO active sites would experience a valence oscillation in a catalysis cycle, typically accompanied by losing and regaining lattice oxygen. The term “reducibility” refers to the ability for an active site to get reduced to a lower state or the ability for a local defect vacancy to form during interaction with reacting species (Fig. 1a).6 Unlike RMO catalysts with reversibility of lattice oxygen redox reactions, some other oxides, such as SiO2 and Al2O3, show poor reducibility due to their unfavoured thermodynamics and thus are generally considered as nonreducible oxides.7–9 The interesting redox property creates prosperity in studies on C1 reaction mechanisms, designing conversion routes, developing catalytic structures, and boosting catalysis performance using RMOs.10 RMOs have often been found active in a same reaction via different approaches, i.e. thermally, electrochemically, and photochemically driven processes, implying a common functionality of the RMO active sites regardless of how energy is administered to the reaction system.11 Though there have been a large number of works investigating the unique structures and properties of RMOs with remarkable achievements in recent years,12 the importance of a systematic evaluation of this group of catalyst materials is underestimated. In contrast, there have been many excellent reviews on different catalyst materials, e.g. transition metals and alloys,13,14 metal–organic frameworks (MOFs),15,16 zeolite-based materials,17,18 and single-atom catalysts.19,20 Thus, our review in this article focuses primarily on RMO catalysts in some important C1 reactions that represents a good time to summarize new research progresses and fill the gap in the RMO field. In this mini-review, we introduce important C1 reactions in CO, CO2, and CH4 conversions, including thermal, electrochemical, and photochemical conversions, and summarize the latest understanding of RMO catalysis mechanisms and the catalyst material designs, with the key research challenges and opportunities also discussed.
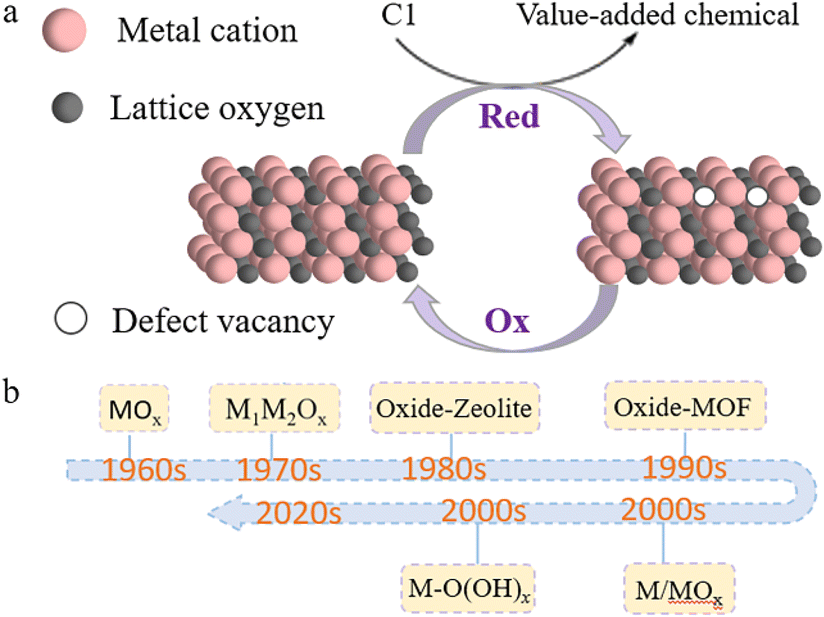 | ||
| Fig. 1 (a) Schematic illustration of RMO catalysts facilitating C1 conversion, and (b) a list of RMO catalyst materials that have been discovered and studied in C1 conversion. | ||
RMO catalysts in CO conversion
The conversion of CO, a colourless, odourless gas that can cause severe health and environmental hazards, has important applications in combustion engineering, gas purification, and fuel cell technologies (Fig. 2). Owing to a weak permanent dipole moment (μ ≈ 0.12 D) and low excitation energy (hν/k ≈ 5.53 K), CO is highly chemically reactive with other reactants, which can lead to a variety of product molecules.21 The research difficulties in CO conversion lie in avoiding overoxidation/overreduction leading to by-product formation.22 | ||
| Fig. 2 Major CO conversion routes. FT: Fischer–Tropsch; WGSR: water–gas shift reaction. | ||
FT synthesis is a classic but complex method for CO hydrogenation to energy fuels and light olefins. An exceptionally selective olefin production can be achieved when RMOs are used, particularly in combination with zeolites, which is attributed to confined C–C coupling by the zeolite acidic sites that remarkably prevent the overhydrogenation on RMO sites. Jiao et al. obtained a C2–C4 hydrocarbons selectivity as high as 94% with 17% CO conversion, compared to the 58% benchmark selectivity, by discovery of an OX–ZEO (oxide–zeolite) catalyst material (Fig. 3a). An important active intermediate, ketene, was identified using highly sensitive synchrotron-based vacuum ultraviolet photoionization mass spectrometry (SVUV-PIMS) (Fig. 3b).23 Zn–Zr–O/SAPO-34, a mixed reducible oxide with zeolite, was reported with high selectivity towards methanol formation, which suggested a bifunctional cooperation mechanism, with ZrO2 activating CO and ZnO accelerating H2 dissociative H2 adsorption.24 Besides zeolites, reducible oxides incorporated with a metal have been often used as cost-effective catalysts in CO conversions. Mejía et al. presented a large improvement in the reaction activity and 80% selectivity for long-chain hydrocarbons (C5+) generation using TiO2 and Nb2O5-supported Co–Ni catalysts. They also found that the oxides got partially reduced to suboxides, which helped modulate the CO adsorption energy and benefitted the following CO dissociation process.12
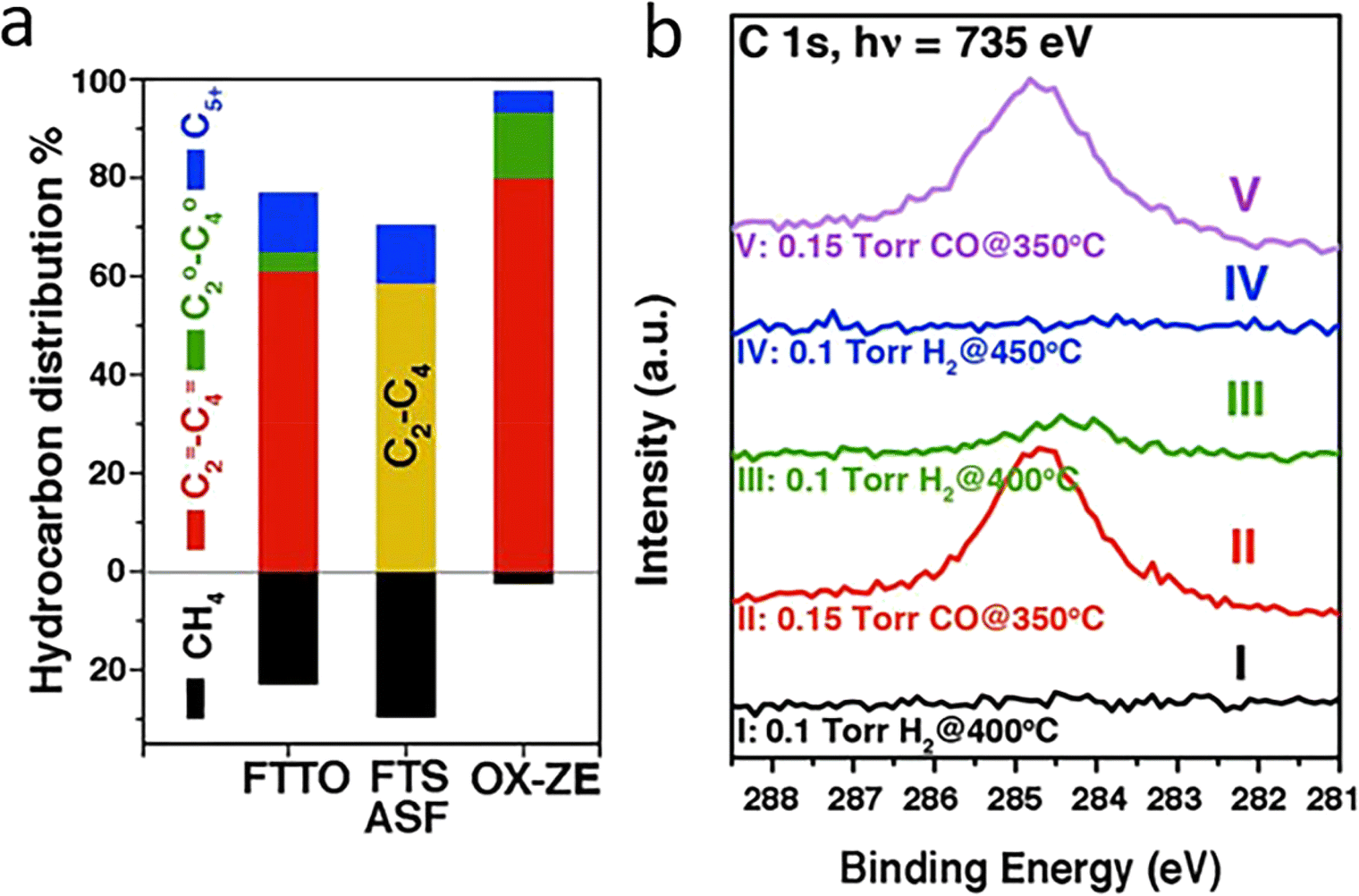 | ||
| Fig. 3 (a) Hydrocarbon distribution in OX–ZEO in comparison to that reported for Fischer–Tropsch to olefins (FTTO) and that in Fischer–Tropsch synthesis (FTS) predicted by the Anderson–Schulz–Flory (ASF) model at a chain growth probability of 0.46, with the yellow bar representing the selectivity of C2–C4 hydrocarbons. (b) In situ study of syngas conversion over ZnCrOx by SVUV-PIMS at hν = 9.72 eV. The insets display the signals of m/z = 42.01 (ketene) and m/z = 42.05 (propene) detected at hν = 9.72 and 11.40 eV, respectively. This figure was adapted from ref. 23 with permission from AAAS, copyright 2016. | ||
Besides the thermal approach that can induce RMO active-site-state oscillation for facilitating CO conversion catalysis, previous studies have discovered many RMOs as effective catalysts in electrochemical and photochemical CO conversion reactions, with similar active-site state oscillation induced by these different energy administration approaches. Many RMOs can serve as photocatalysts or electrocatalysts based on their ability to harvest photons or transfer electronic charges. In electrocatalysis, the electrode potential provides the driving force to fulfil the thermodynamic CO conversion requirement. A high reaction activity and selectivity can be achieved by adjusting the applied potential and tuning the RMO structure, i.e. the oxygen vacancy generation ability.11 In photocatalysis, photon-excited holes/electrons would transfer to RMO active sites and facilitate CO conversion.25 Although the specific CO reaction pathways in the photochemical approach could be different from that in the thermal approach, nevertheless RMOs functions in a similar way, with oxygen vacancy consumption and regeneration being essential to complete the catalysis cycles.26 For instance, Co/Co3O4, Fe/Fe3O4, Ni/NiOx, and Ni2P/TiO2 catalysts were studied in solar-driven CO conversion.27–30 Scanning tunnelling microscopy (STM) characterizations suggested that oxygen vacancies on the RMO surfaces were the primary CO adsorption and activation sites,31 while density functional theory (DFT) calculations suggested that the RMO phase contributed to tuning the electronic properties of metallic components, enhancing C–C coupling towards olefins generation and weakening hydrogenation towards alkanes. These effects collectively resulted in the selective production of light olefins.27–29 A summary of the RMO catalysts and their performance in FT conversion is shown in Table 1.
| Catalysts | Reaction conditions | Product selectivity | ||||||
|---|---|---|---|---|---|---|---|---|
| T (K) | P (MPa) | GHSV (ml gcat−1 h−1) | H2/CO | X CO (%) | C1 | C2–C4 | C5+ | |
| ZnCrOx/MSAPO | 673 | 2.5 | 5143 | 2.5 | 17 | 2 | 94 | 4 (ref. 23) |
| ZrZnOx/SAPO | 673 | 1 | 3600 | 2 | 9.5 | 6 | 92 | 2 (ref. 24) |
| MnOx/SAPO | 400 | 2.5 | 4800 | 2.5 | 7 | 1 | 94 | 5 (ref. 47) |
| Co–Ni/TiO2 | 493 | 0.1 | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
2 | 4.4 | 9 | 15 | 76 (ref. 12) |
| Co–Ni/Nb2O5 | 493 | 0.1 | 20000 |
2 | 3.7 | 9 | 15 | 76 (ref. 12) |
| Fe0–FeOx/ZnO–Al2O3 | 273 (visible light) | 0.18 | 3000 | 3 | 12 | 39 | 50 | 11 (ref. 27) |
| Co–Co3O4/ZnO–Al2O3 | 273 (Xe light) | 0.18 | 6000 | 3 | 15 | 48 | 42 | 10 (ref. 28) |
| Ni/NiOx | 273 (Xe light) | 0.08 | — | 3 | 28 | 41 | 38 | 21 (ref. 29) |
| Ni2P/TiO2 | 478 (Xe light) | 0.18 | 6000 | 3 | 13.3 | 25 | 47 | 28 (ref. 30) |
In WGSR, CO reacts with water and produces H2 and CO2. Being exothermic allows effective CO transformation with high conversion under mild operation conditions, and so this reaction offers an important process for hydrogen production and purification. Reducible metal sulfides and phosphides (MSx, MPx), a subgroup of the broader reducible material family, have been widely exploited for their unique catalytic properties in WGSR.32,33 According to the literature, MSx would generate sulfur vacancies, similar to oxygen vacancies generated in reducible oxides, which can then interact with reacting species and facilitate intermediate and product generation, besides preventing side reactions and sintering.34 Besides WGSR synthesis, MoP can be applied for CO hydrogenation for alcohols, methane, and other hydrocarbons production under room temperature and atmospheric pressure.35
CO oxidation is an important, widely applied reaction in practice for controlling CO emissions by converting the CO to CO2. Many RMOs, like the oxides of Mn, Ce, Cu, Ni, Co, and Ti elements, are good catalyst candidates considering their earth abundance, thus having a low cost, and interesting structure-sensitive catalytic properties.36–39 This reaction was discovered to follow the Mars–van Krevelen mechanism on RMOs, with the catalyst surface being partially reduced by adsorbed CO and then re-oxidized back to its initial state by O2 in a catalysis cycle (i.e. RMOs render a consumption and rejuvenation of oxygen vacancies repeatedly) (Fig. 4).40 Pan et al. investigated the activity property of different RMOs using a combination of experimental measurements and computational simulations. They discovered that the RMO activity can be depicted by two governing parameters: the work function and the work function oscillation of catalyst materials.41 The reducibility of RMO and the corresponding activity property in CO oxidation can be regulated by substitution or incorporation with other cations.42 For example, the study of dopants-modified TiO2 found a boost in activity in CO oxidation, while in situ Fourier-transform infrared spectroscopy (FT-IR) and X-ray photoelectron spectroscopy (XPS) characterizations confirmed that the crystal phase of the oxides has an impact on the catalytic performance.43 Recently, Fe2O3 modified with Pt single atoms was reported with 100% CO conversion at about 70 °C. In this work, monodispersed Pt atoms were found to bring more structure defects in Fe2O3, leading to the generation of more oxygen vacancy sites for catalyzing CO oxidation.44 Some mixed RMOs, like hopcalite (CuMnOx) and modified hopcalite, were also studied and exhibited CO oxidation activity at room temperature.45,46 It was concluded that the lattice oxygen associated with Cu and the mobility of lattice oxygen with Mn enabled the unique properties of hopcalite.
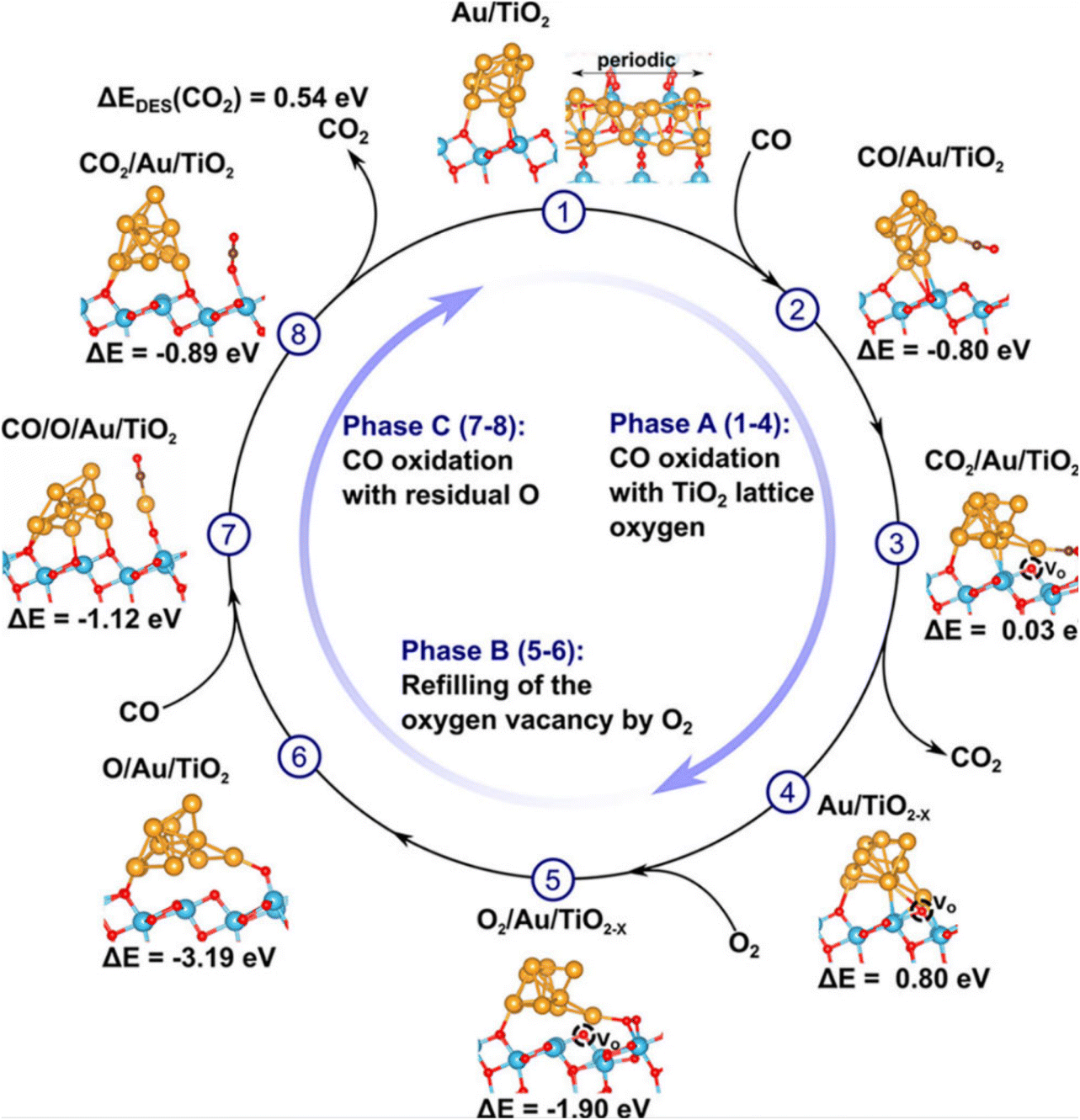 | ||
| Fig. 4 Possible catalytic cycle for CO oxidation on Au10/TiO2 involving the formation of a single oxygen vacancy and one oxygen adatom. In phase A, a titania lattice oxygen is abstracted by CO, forming CO2 and an oxygen vacancy. In phase B, the oxygen vacancy is re-oxidized by O2, leaving an oxygen adatom Oad at the Au/TiO2 interface. In phase C, the oxygen adatom Oad is abstracted by CO, under the formation of the second CO2 molecule. This figure was adapted from ref. 40 with permission from American Chemical Society, copyright 2018. | ||
RMO catalysts in CO2 conversion
CO2 is a natural greenhouse gas and closely connected to human living. Excess CO2 emissions have been generated from anthropogenic activities that are resulting in irreversible environmental problems.48 CO2 conversion offers an imperative strategy to regulate CO2 emissions and has thus attracted significant research interests. The major challenges in CO2 conversion are to overcome the coke effect, particularly for high-temperature reactions, and to activate the thermodynamically uphill reactions.49Thermally driven CO2 hydrogenation, using H2 as the reductant, includes four important reactions: the reverse water–gas shift reaction (RWGS), methanation, FT reaction, and methanol synthesis (Fig. 5).50–52 RMOs embraced by other elements, like perovskite and noble metals, have been found with improved catalytic properties, attributed to a modification in the oxygen mobility by reorganizing the defective oxide structure.6,53–55 Many RMO materials can serve as tandem catalysts in CO2–CO chemistry networks, which help control the reaction pathways in two sequential reactions, avoiding intense energy consumption in one-step conversion,56 and therefore, the catalyst choice and mechanisms for CO2 conversion share some similarities with CO conversion.
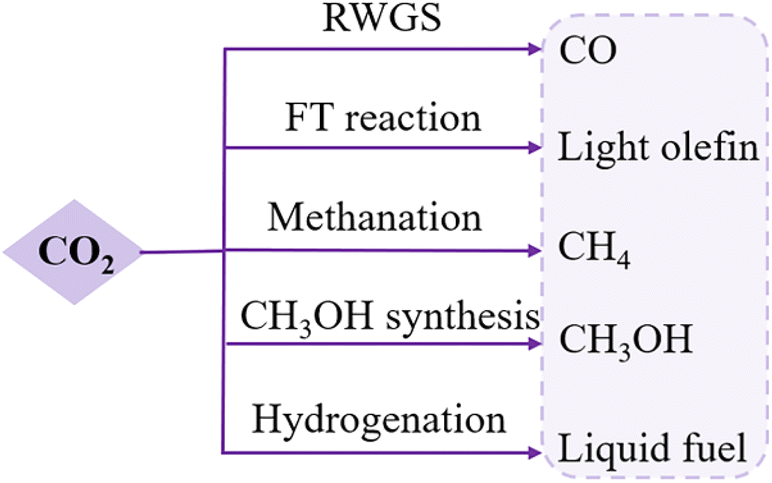 | ||
| Fig. 5 Major CO2 conversion routes. RWGS: reverse water–gas shift; FT: Fischer–Tropsch. | ||
CO2 transformation for methanol synthesis using Cu/ZnO/Al2O3 has attracted substantial attention over decades due to its remarkable properties in a mild temperature range (475 to 575 K).57 Later on, Martin et al. reported an In2O3/ZrO2 catalyst with superior activity and stability and 100% methanol selectivity in the reaction,58 which attracted more investigations into In2O3.59 The introduction of ZrO2 was found to improve the dispersion of In2O3 nanoparticles, leading to more active sties and meanwhile stabilizing them from sintering.58 A following work from the same group unveiled the geometric and interfacial effects of In2O3/ZrO2. It was found that the monoclinic active phase of the ZrO2 support could help the epitaxial alignment of In2O3 to form a monoclinic polymorph that would generate tensile strain and form an improved diverse number of oxygen vacancies.60 Yao et al. performed a study of Cu–In–Zr–O mixed oxide and reported a near 80% methanol selectivity under mild conditions. Metallic Cu sites were found to be responsible for H2 dissociation and active hydrogen formation, which promoted vacancies bonding with intermediates and the hydrogenation kinetics towards methanol formation. In situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) studies suggested the formate–methoxy–methanol formation pathway with this mixed reducible oxide catalyst (Fig. 6).61 The mechanism of the metal promotion effect was further illustrated in the study of Pd/In2O3.62 Frei et al. reported that well-dispersed Pd atoms on In2O3 possessed metal–support interactions that would modify the electronic properties of In2O3. Meanwhile, the stabilized low-nuclearity Pd was found to facilitate H2 splitting by suppressing undesired CO formation. Both XPS O1s and solid-state nuclear magnetic resonance (NMR) spectroscopy indicated the generation of more abundant oxygen vacancies by substituting Pd in In2O3. Co-precipitated catalyst (CP) and dry impregnation catalyst (DI), synthesized with different methods, were demonstrated to have different impacts on the CO2 hydrogenation to methanol performance, which was attributed to a difference in the defect local environment, wherein the CP catalyst exhibited a good activity (0.96 gMeOH h−1 gcat−1) and retained an excellent stability for 500 h.62 More recently, a ternary Pd–In2O3–ZrO2 catalyst with a superior performance (1.3 gMeOH h−1 gcat−1) and improved metal utilization was investigated, with its lower apparent activation energy being responsible for the more facile methanol-generation kinetics. Aberration-corrected scanning electron microscopy (AC-STEM) and energy-dispersive X-ray spectroscopy (EDX) were used to characterize the mixed ZrO2 phase on individual particles and indicated that the pure monoclinic ZrO2 structure was not indispensable for high-performance CO2 conversion. Electron paramagnetic resonance spectroscopy (EPR) has been found to be a powerful technique for helping interpret the catalyst performance in CO2 hydrogenation because EPR is capable of quantifying oxygen vacancies and analysing its dynamics (Fig. 7).63 Besides the cationic component incorporation effect, an anionic P element incorporation into In2O3 was reported to regulate the RMO catalyst activity and selectivity by tuning the catalyst surface-adsorption configuration. In situ DRIFTS spectra indicated that the In–P bond undermined the formation of oxygen vacancies on the In2O3 surface, and thus CO2 was adsorbed on alternative sites and favoured CO generation. In comparison, on pristine In2O3, CO2 took up oxygen vacancies and bonded to the adjacent sites, leading to preferential methanol generation (Fig. 8).64
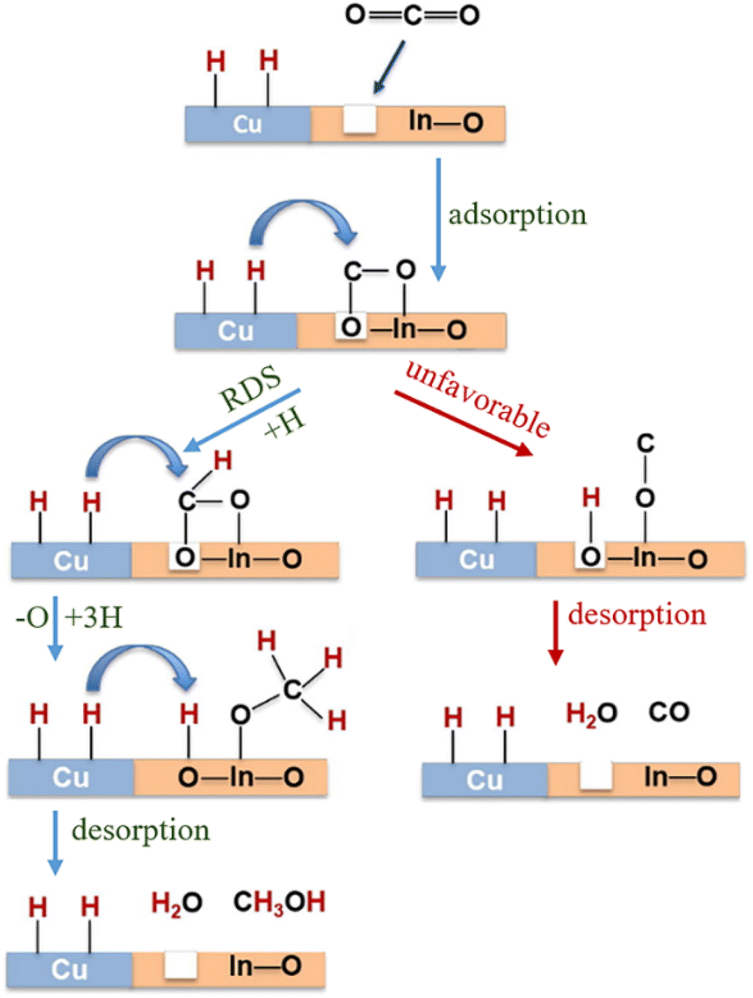 | ||
| Fig. 6 Schematic representation of Cu–In2O3 synergy on Cu0.25–In0.75–Zr0.5–O for methanol synthesis reaction conditions: T = 250 °C, P = 25 bar, GHSV = 18000 ml (gcat−1 h−1). RDS: rate-determining step. This figure was adapted from ref. 61 with permission from Elsevier, copyright 2019. | ||
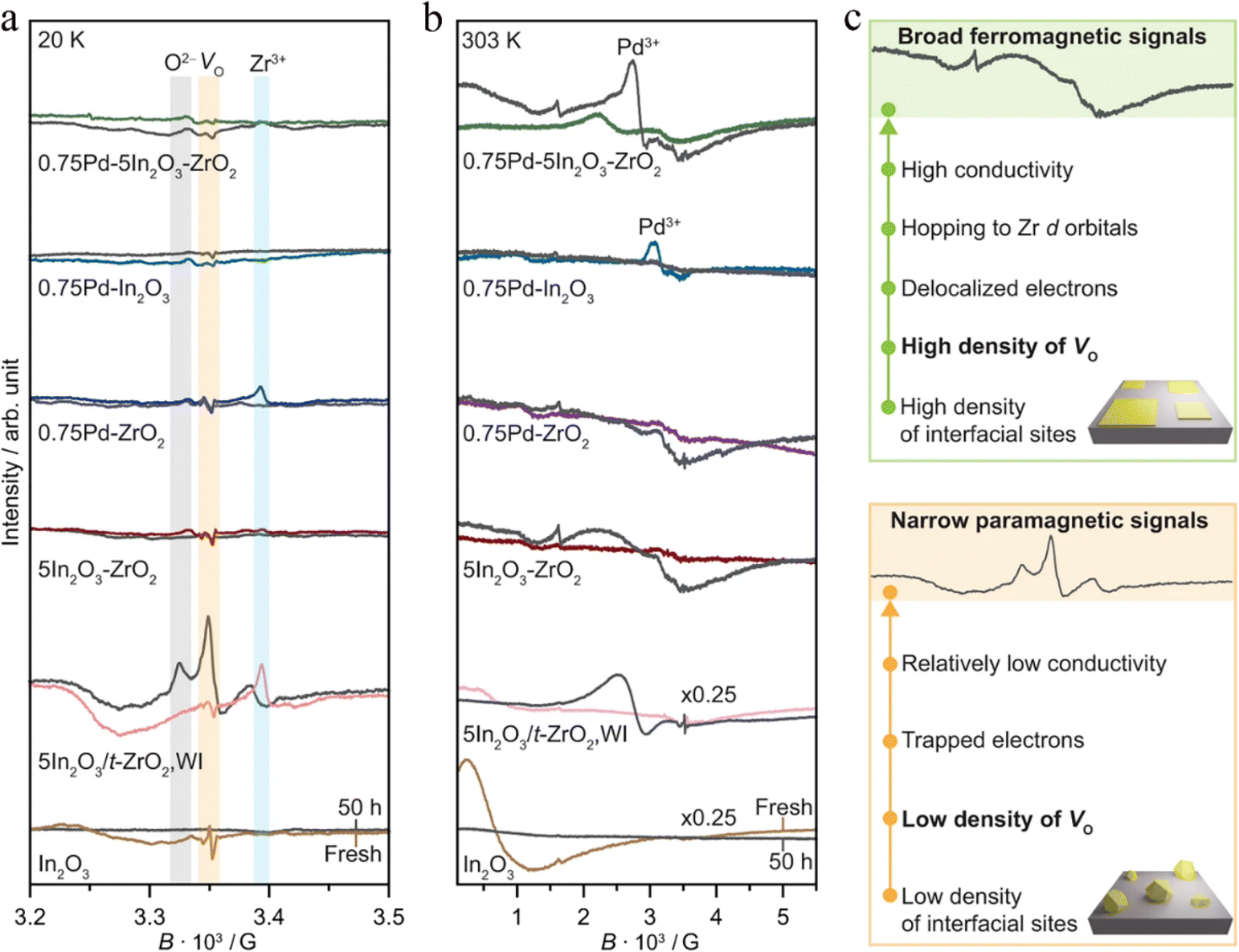 | ||
| Fig. 7
Ex situ EPR spectra of In2O3-based catalysts prepared by FSP and WI in fresh form and after CO2 hydrogenation for 50 h measured at (a) 20 K and (b) 303 K. (c) Scheme describing the nature of the EPR signals generated from catalysts displaying distinct densities of interfacial sites and oxygen vacancies. Activation conditions: T = 553 K, P = 5 MPa, H2/CO2 = 4, and GHSV = 48000 cm3 h−1 gcat−1. This figure was adapted from ref. 63 with permission from Nature, copyright 2022. | ||
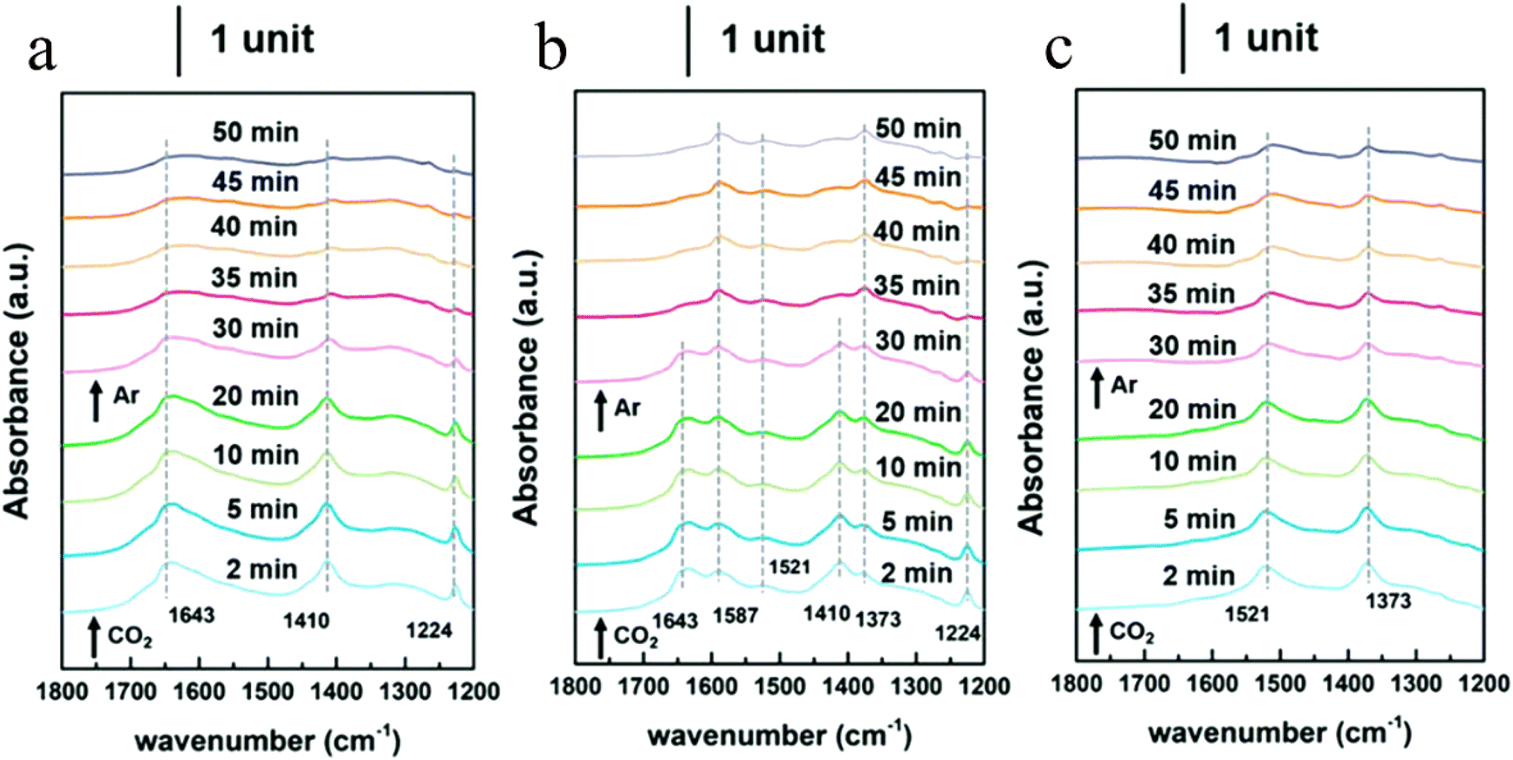 | ||
| Fig. 8 Time-evolved in situ DRIFTS spectra (1800–1200 cm−1) of CO2 adsorption experiments on In2O3 materials. (a) P–In-0, (b) P–In-2, and (c) P–In-3.5 (0, 2, and 3.5 represent the molar ratios between P and In). This figure was adapted from ref. 64 with permission from Royal Society of Chemistry, copyright 2021. | ||
CO2 can also be directly converted to other valuable chemicals, such as ethyl alcohols, dimethyl ether (DME), carboxylic acids, and dimethyl carbonate. For example, in the CO2-to-DME thermal reaction, 65.1% DME selectivity was achieved using a Cu–In–Zr–O/SAPO catalyst. The proximity of the RMO (Cu–In–Zr–O) particles with the zeolite (SAPO) particles made a dramatic difference to the reaction mechanism, with the methoxy–DME shortcut pathway being followed when the two components were in close proximity.65 Photocatalytic and electrocatalytic CO2 reduction have been investigated as alternative methods that can utilize renewable solar/electrical energy to drive the reactions in the presence of RMO catalysts. TiO2, Co3O4, Ga2O3, ZnS, and SnO2 were discovered to have interesting activity properties towards formate formation,66,67 while SnS2, MoS2, and Bi2WO6 were reported as effective catalysts for alcohol generation.68,69
RMO catalysts in CH4 conversion
The selective conversion of methane, the main component of natural gas and an abundant non-renewable resource, to value-added chemicals and liquid fuel has remained a grand challenge in the past century, with difficulties largely caused by the stable characteristic of the CH4 molecule and the need to overcome the 439.3 kJ mol−1 dissociation energy for C–H bond activation and cleavage, as well as the difficulty to control the reaction from overoxidation.70 To date, SMR has been applied as an important industrial process for converting CH4 into syngas, which can be further used as a feedstock for value-added chemicals production. The SMR is endothermic and requires high temperature and pressure conditions (typically over 973 K and 15 bar) for overcoming the thermodynamics and improving the kinetics.71 Direct CH4 conversion to value-added chemicals (Fig. 9) can not only simplify the overall processes but would also be more economically beneficial, theoretically feasible, and has consequently attracted significant, long-time attention. | ||
| Fig. 9 Major CO conversion routes. SMR: steam methane reforming; OCM: oxidative methane conversion; NOCM: non-oxidative methane conversion; EOCM: electrochemical oxidative conversion of methane. | ||
The oxidative conversion of methane (OCM) offers a route that can directly convert CH4 into alkenes and oxygenates by reacting with oxidants like O2 and H2O2. A recent study showed that CH4 could be oxidized to methanol using a Fe-containing zeolite structure, with the Fe sites being identified as active centres that undergo Fe(II)/Fe(IV) redox cycles in catalysis.72 In another study, formic acid production with 90% selectivity was reported by oxidizing CH4 with H2O2 using an FeNx/C-5-700 catalyst, in which the reducible FeNx was found to provide the active sites for hydroxyl radical generation and OCM.73 Although the use of oxidants other than O2 has often been observed to offer better reaction kinetics and selectivity control, it would raise a cost-effectiveness concern for large-scale applications. When O2 is used as the oxidant, the OCM reaction on RMO catalysts follows the typical RMO catalysis mechanism, with the active sites getting reduced by reacting with CH4 and getting re-oxidized by O2 for regeneration in a catalytic cycle. A variety of RMO materials, like IrO2, CeO2, TbOx, TiO2, PdO, La2O3, and mixed oxide MnTiO3, have been explored for their catalytic properties in the OCM reaction.74–76 For example, PdO was found to be effective in CH4 activation following the dissociative adsorption mechanism, in which the stoichiometrically unsaturated Pd sites and surrounding oxygen vacancies were considered to be the active sites.77 Besides, La2O3 was reported with high hydrocarbon production selectivity and CeO2 showed high OCM activity. Meanwhile, the catalytic properties of RMOs can be altered by modifying the catalytic structure. Improvements in both the activity property of La2O3 and the selectivity property of CeO2 in OCM were achieved by doping the two materials.78 More recently, Liu et al. reported OCM to methanol with 95% selectivity on a CeO2/Cu2O/Cu(111) catalyst at low temperature in the presence of water. The introduction of water is found to introduce hydroxyl species that blocked O2 dissociation and favoured the hydrogenation of methoxy groups.79
The non-oxidative conversion of methane (NOCM) provides a potentially more cost-effective, eco-friendly route for CH4 utilization, and circumvents the need to inhibit CO2 generation in OCM, which could otherwise reduce the carbon use efficiency. Earlier in 2015, Gao et al. reported the design of a Mo oxide monomer anchored on Al sites in a zeolite framework as a catalyst for NOCM. It was found that the oxide was first transformed into carbide species by reacting with CH4 under the reaction conditions and then the Mo–C served as active sites that catalyzed C–H bond scissoring and methyl intermediate generation.80 Compared to thermal OCM and NOCM reactions, the electrochemical oxidative conversion of methane utilizes the electrode potential as an additional parameter to drive the reaction, which allows its occurrence under more mild conditions. There have been active studies of EOCM utilizing solid oxide fuel cells (SOFCs) in higher temperature ranges, in which O2 is electrochemically reduced to O2− at the cathode and O2− migrates to the anode and electrochemically reacts with CH4 for generating oxygenates. CeO2 is often used as an anode RMO catalyst owing to its good ionic conductivity and high CH4 oxidation activity.81,82 Surendranath et al. reported the use of a Pd complex in concentrated sulfuric acid electrolytes in EOCM, with the PdII ions serving as the active sites that undergo PdII/PdIII,III2 cycles for catalytic generation of CH3OSO3H and CH3SO3H species (Fig. 10). The interchangeable Pd valence states during the catalytic cycle were evidenced by cyclic voltammetry (CV) and in situ UV-vis spectroelectrochemistry.83 Pt-containing complexes were also found to be effective as catalysts in EOCM with PtII/PtIV reducible sites.84 V2O5 has also attracted interest among RMO catalysts in EOCM for its interesting activity properties in a lower temperature range;85,86 however, the low product selectivity remains an issue. More recently, Li et al. conducted a systematic study of EOCM toward ethanol on an iron-nickel hydroxide (Fe–Ni–OH) nanosheet catalyst under room temperature and atmospheric pressure conditions (Fig. 11). They observed an interesting Fe–Ni–OH composition effect on the catalytic properties, with Fe3Ni7(OH)x showing the best performance and exhibiting excellent activity with 0.26 s−1 turnover frequency (TOFethanol) and 87% ethanol selectivity. The in situ ATR-FTIR experimental results and DFT simulations suggested in situ generated NiIIIOOH were the active sites following the RMO catalysis mechanism, with the Fe incorporation modifying the local configuration of active sites and promoting the EOCM kinetics by lowering the energy barrier.87 A variety of other RMO catalysts, e.g. NiO2,88,89 Co3O4,90 and NiCO2O4,91 have also been discovered with interesting properties in EOCM and were investigated and reported in the literature (Table 2).
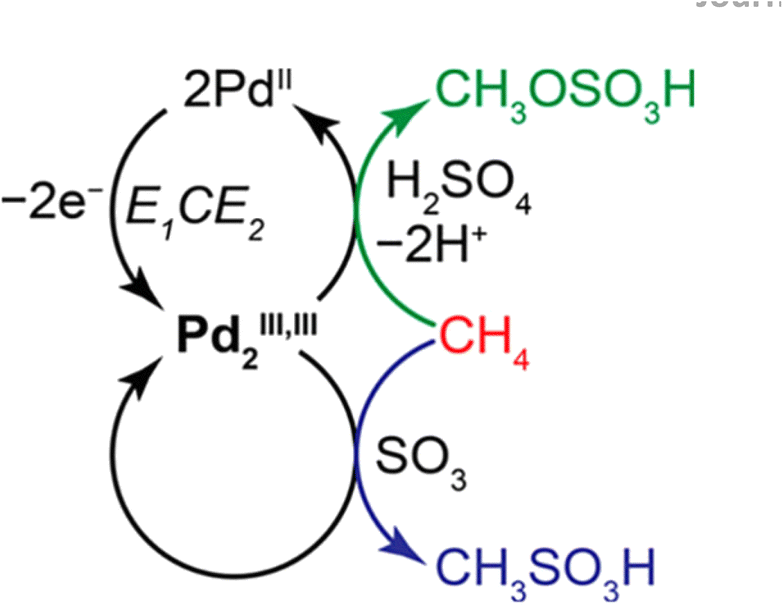 | ||
| Fig. 10 Proposed mechanism for electrochemical methane functionalization by a putative PdIII,III2 intermediate. Green and blue arrows indicate faradaic and non-faradaic reaction pathways, respectively. This figure was adapted from ref. 83 with permission from American Chemical Society, copyright 2017. | ||
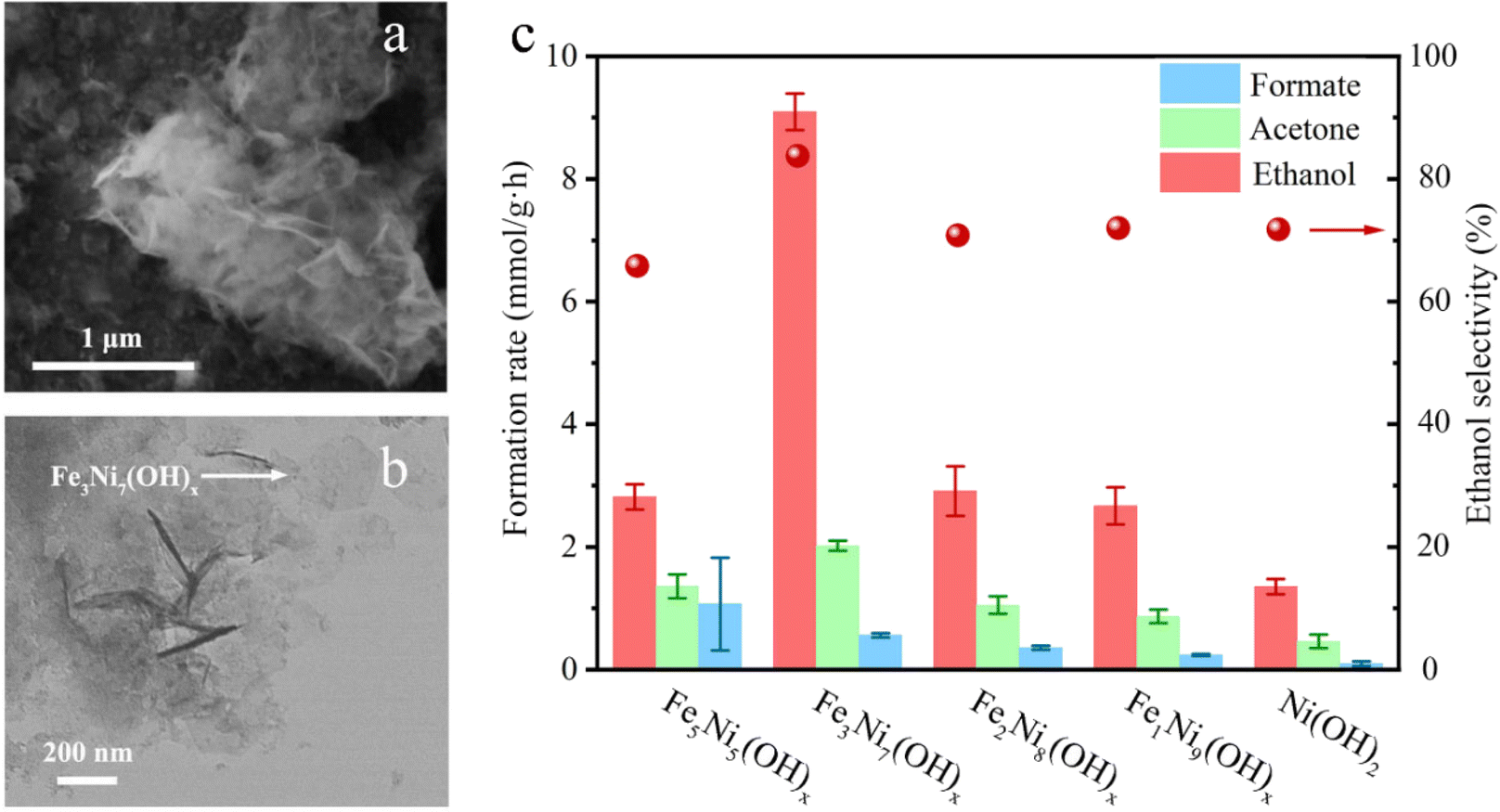 | ||
| Fig. 11 (a) SEM (scanning electron microscopy) images (b) TEM (transmission electron microscopy) images of the synthesized Fe–Ni–OH nanosheets. The arrow points to an individual symmetric hexagonal shape nanosheet. (c) Measured formation rate of different products and ethanol selectivity using different Fe–Ni–OH catalysts. This figure was adapted from ref. 87 with permission from Elsevier, copyright 2022. | ||
| Catalyst | T (K) | Pressure (MPa) | Electrolyte | Potential (V) | Product |
|---|---|---|---|---|---|
| PdII/PdIII,III2 | 375 | 3.5 | 20% SO3/H2SO4 | 2 | Methanol precursors83 |
| Cu/CeO2 | 973 | 0.1 | Yttria-stabilized zirconia (YSZ) | 0.4–1.1 | CO276 |
| PtII/IV | 403 | 3.5 | 10 mM NaCl, 0.5 M H2SO4 | 0.829 (V vs. SHE) | Methanol (80% selectivity)84 |
| V2O5/SnO2 | 373 | 200 | Sn0.9In0.1P2O7 | 0.9 | Methanol (88% selectivity)85 |
| VV/VIV | 273 | 0.1 | 98% H2SO4 | 2.3 (V vs. Hg2SO4/H) | Methyl bisulfate (81% selectivity)86 |
| Fe–Ni–OH | 273 | 0.1 | 0.1 M NaOH | 1.46 (V vs. RHE) | Ethanol (87% selectivity)87 |
| NiO–ZrO2 | 313 | 0.1 | 0.1 M Na2CO3 | 1.8 (V vs. SCE) | Isopropanol, acetate/acetic acid acetone, ethanol, and formate/formic acid88 |
| Rh/ZnO | 273 | 0.1 | 0.1 M KOH | 2.2 (V vs. RHE) | Ethanol (85% selectivity)89 |
| Co3O4/ZrO2 | 273 | 0.1 | 0.5 M Na2CO3 | 2.0 (V vs. Pt) | 2-Propanol and 1-propanol90 |
| ZrO2–NiCo2O4 | 273 | 0.1 | 0.5 M Na2CO3 | 2.0 (V vs. Pt) | Propionic acid, acetic acid and acetone91 |
Outlook and perspective
Sustainable C1 conversion to value-added chemicals and energy fuels offers a promising solution to help alleviate the energy shortages and slow down environmental changes. In this review, we reviewed the important reactions of C1 molecules (CO, CO2, and CH4) and summarized the recent progresses in researching RMO catalyst materials for these reactions. The research progress has significantly advanced C1 chemistry and RMO catalyst development and laid a foundation to guide new catalyst research activities in this field. In the meantime, challenges remain in RMO catalysis research, particularly in the fundamental study of catalysis mechanisms and in the rational discovery of new materials with desired catalytic properties.At the mechanistic study level, previous studies have discovered the general RMO catalysis mechanism in C1 reactions, in which the reducible cations and surrounding vacancies on the RMO surface serve as the active sites. In C1 oxidation, the active sites would react with C1 molecules to generate intermediates/products, accompanied with a reduction in the active sites, i.e. cation valence lowering and vacancy formation. The reduced active sites would then react with the oxidant for regeneration to the initial state, which completes a catalysis cycle. In C1 reduction, the active sites would be reduced by the reductant to a lower state, which would then react with C1 molecules for product generation and meanwhile get oxidized to the initial state. However, it remains challenging to characterize the exact structural configuration and evolution of the working active sites and the interactions with reacting species.
At the RMO catalyst research level, different material parameters, like the composition, crystallinity, and reducibility, have been found to have dramatic effects on the catalytic properties. This implies the existence of an RMO structure–property relationship, which would have significance both in the fundamental interests to understand RMO catalysis properties and in practical applications to guide new catalyst development. However, to date, understanding the exact relationship between RMO material parameters and the catalytic properties remains elusive. RMO catalyst discovery and research still largely rely on experience with trial and error. A lack of explanation of the exact RMO structure–property relationship has been a significant roadblock in new catalyst research and development.
Previous studies have witnessed considerable efforts to explore the RMO structure–property relationship. The research efforts can be traced back to the early twentieth century, with the Brønsted–Evans–Polanyi principle (also referred to as the Bell–Evans–Polanyi or Evans–Polanyi–Semenov principle) finding that the difference in activation energy between two reactions of the same family has a proportional relationship with the difference in their enthalpy of reaction.92 In the following years, researchers continued to study this topic and a series of works were published proposing semiquantitative property–activity relationships to describe RMO catalysis. Most notably, the catalyst activity property was discovered to follow a decent volcano relationship with the enthalpy of transition from a lower to a higher oxidation state for oxides (ΔHF).93 The reaction activation energy was found to show a linear correlation with the oxygen vacancy formation energy of oxides (ΔEvac), also referred to as the surface reducibility. These findings confirmed that the reaction properties are indeed controlled and describable by certain governing parameters of reducible oxides. However, it needs to be noted that these used descriptors are properties of oxide materials, but not at the level of intrinsic oxide material parameters that can be experimentally measured. More recently, RMO material parameters, like the work function94 and occupancy of the eg-symmetry orbital,95 were examined for use as a descriptor for depicting the reaction activity property. Pan et al. developed a theoretical model using the RMO work function (W) and oscillation of the work function (ΔW) for depicting the activity property in CO oxidation as a probe reaction (Fig. 12).41 This model considered the oscillation of active sites between different oxidation states and suggested that the material parameters associated with both states collectively control the activity property. To a certain extent, the used ΔHF and ΔEvac material properties and their correlation with the reaction properties in previous studies already reveal the importance of both active site states during RMO catalysis. The established inverted volcano correlation between (W − ΔW/2) and the activation energy reported by Pan et al. was in decent consistence with the experimental results (Fig. 12a). This theoretical model could possibly be extended to many other RMO catalysis processes. Different C1 reactions, like CO hydrogenation, CO2 hydrogenation, and CH4 oxidation, on RMO catalysts would follow a similar structure–activity property correlation, with the optimal (W − ΔW/2) requirement being different (Fig. 12b). Previous studies have suggested that regardless of how energy is administered to the reaction system, i.e. via thermal, electrochemical, or photochemical approaches, RMO catalysts follow the same function mechanism, with the active site oscillation accompanied by oxygen vacancy consumption and regeneration to complete a catalysis cycle. In this regard, the discovered RMO structure–property relationship in thermal C1 reactions could be possibly extendable to help study electrochemical and photochemical reaction systems.
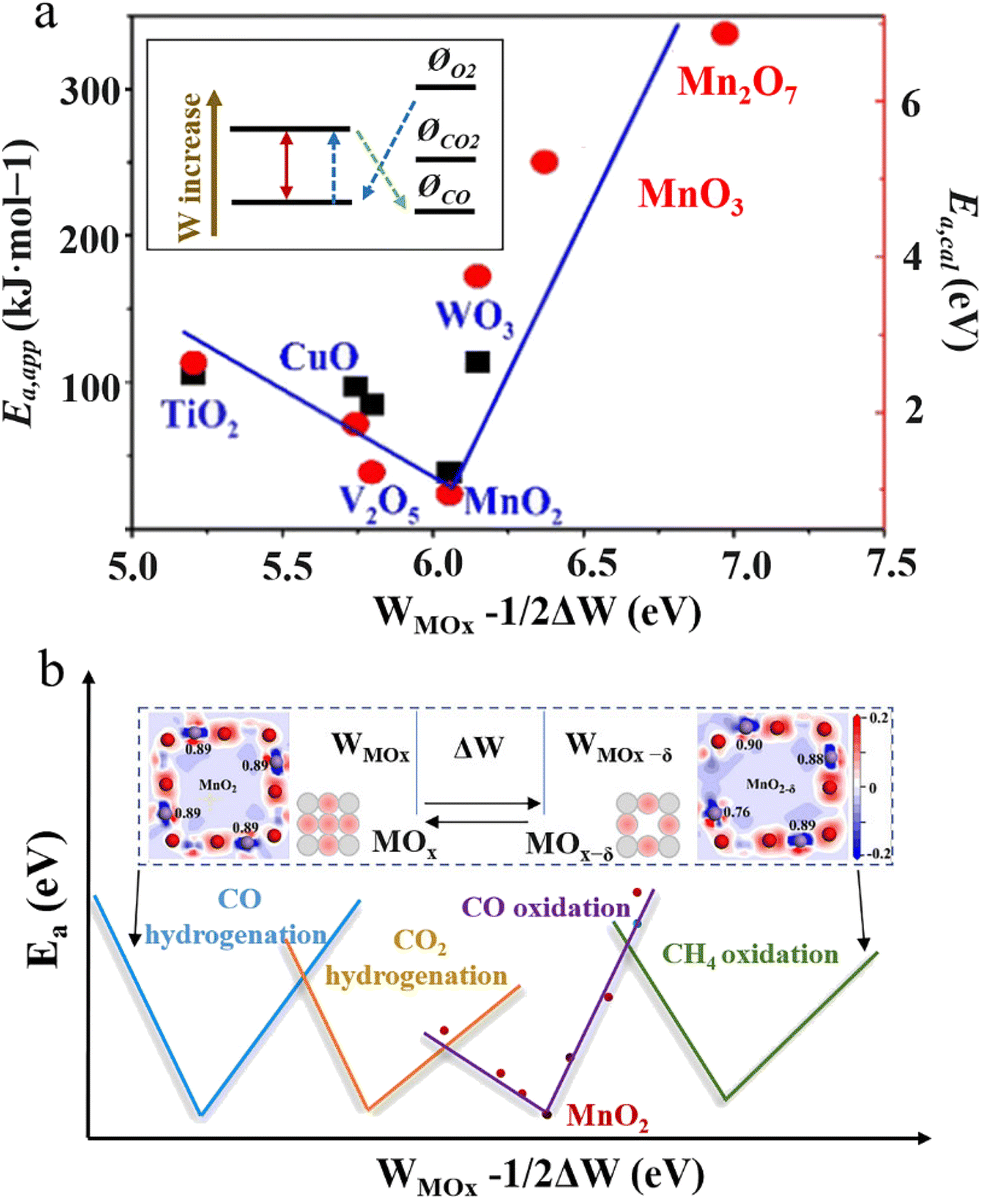 | ||
| Fig. 12 (a) Inverted volcano correlation of (WMOx − ΔW/2) with Ea,app (black square) and Ea,cal (red dot). Inset: relative work function and potential levels of RMOs and reacting species. This figure was adapted from ref. 41 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, copyright 2019. (b) Illustration of (WMOx − 1/2ΔW) as a possible RMO material parameter for depicting the catalytic activity property in different C1 conversion reactions. WMOx represents the work function, ΔW represents the oscillation of the work function, and Ea represents the activation energy. | ||
With the rapid developments in advanced characterization techniques and in computational chemistry,96,97 new approaches have been and will be more extensively utilized to further the RMO catalysis understanding. For example, in situ/operando tools that can spatially and temporally identify and monitor RMO active site,63 atomic-level characterizations that can define the active phase structure,62 and in-depth analyses using electron spin resonance (ESR) spectroscopy, laser-induced fluorescence (LIF), and atomic resonance absorption spectroscopy (ARAS) that can detect active short-life intermediates98 are expected to provide insightful experimental evidence and yield new knowledge of the RMO active site and C1 catalysis mechanisms. These fundamental understandings would also help to identify the governing RMO material parameters and establish exact RMO structure–catalytic property relationships. This would lead to more accurate theoretical models that would not only help to understand RMO catalysis but also provide guidance for new catalyst research and development. Rational RMO catalyst design and high-throughput material screening with a combined theoretical and experimental approach for desired properties will become possible, which would also lead to more efficient C1 chemical conversions to targeted products and eventual applications.
Author contributions
Dr. Zhenmeng Peng provided guidance, conceived, and planned the main context. Jialu Li did literature research and wrote the paper. All authors provided critical feedback and helped shape, finalize the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Science Foundation Support (grant number 1955452) is acknowledged.References
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Homogeneous functionalization of methane, Chem. Rev., 2017, 117, 8521–8573 CrossRef PubMed.
- Y. Song, E. Ozdemir, S. Ramesh, A. Adishev, S. Subramanian, A. Harale, M. Albuali, B. A. Fadhel, A. Jamal and D. Moon, Dry reforming of methane by stable Ni–Mo nanocatalysts on single-crystalline MgO, Science, 2020, 367, 777–781 CrossRef PubMed.
- P. R. Ellis, D. I. Enache, D. W. James, D. S. Jones and G. J. Kelly, A robust and precious metal-free high performance cobalt Fischer–Tropsch catalyst, Nat. Catal., 2019, 2, 623–631 CrossRef.
- J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar and D. Ma, Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide-supported iron species, Nat. Catal., 2018, 1, 889–896 CrossRef.
- Y. Zhou, L. Zhang and W. Wang, Direct functionalization of methane into ethanol over copper modified polymeric carbon nitride via photocatalysis, Nat. Commun., 2019, 10, 506 CrossRef CAS PubMed.
- A. Ruiz Puigdollers, P. Schlexer, S. Tosoni and G. Pacchioni, Increasing oxide reducibility: the role of metal/oxide interfaces in the formation of oxygen vacancies, ACS Catal., 2017, 7, 6493–6513 CrossRef CAS.
- J. Wang, R. You, C. Zhao, W. Zhang, W. Liu, X.-P. Fu, Y. Li, F. Zhou, X. Zheng and Q. Xu, N-coordinated dual-metal single-site catalyst for low-temperature CO oxidation, ACS Catal., 2020, 10, 2754–2761 CrossRef CAS.
- C. H. Wu, C. Liu, D. Su, H. L. Xin, H.-T. Fang, B. Eren, S. Zhang, C. B. Murray and M. B. Salmeron, Bimetallic synergy in cobalt–palladium nanocatalysts for CO oxidation, Nat. Catal., 2019, 2, 78–85 CrossRef CAS.
- Y. Wang, P. Ren, J. Hu, Y. Tu, Z. Gong, Y. Cui, Y. Zheng, M. Chen, W. Zhang and C. Ma, Electron penetration triggering interface activity of Pt-graphene for CO oxidation at room temperature, Nat. Commun., 2021, 12, 5814 CrossRef CAS PubMed.
- A. R. Puigdollers and G. Pacchioni, CO oxidation on Au nanoparticles supported on ZrO2: role of metal/oxide interface and oxide reducibility, ChemCatChem, 2017, 9, 1119–1127 CrossRef CAS.
- S. Xie, W. Ma, X. Wu, H. Zhang, Q. Zhang, Y. Wang and Y. Wang, Photocatalytic and electrocatalytic transformations of C1 molecules involving C–C coupling, Energy Environ. Sci., 2021, 14, 37–89 RSC.
- C. Hernández Mejía, J. E. S. van Der Hoeven, P. E. de Jongh and K. P. De Jong, Cobalt–Nickel Nanoparticles Supported on Reducible Oxides as Fischer–Tropsch Catalysts, ACS Catal., 2020, 10, 7343–7354 CrossRef.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, Active and selective conversion of CO2 to CO on ultrathin Au nanowires, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang and C. Mesters, Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Design and synthesis of an exceptionally stable and highly porous metal-organic framework, Nature, 1999, 402, 276–279 CrossRef CAS.
- J. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu and Y. Pan, Self-adaptive dual-metal-site pairs in metal-organic frameworks for selective CO2 photoreduction to CH4, Nat. Catal., 2021, 4, 719–729 CrossRef CAS.
- C. Wang, W. Fang, Z. Liu, L. Wang, Z. Liao, Y. Yang, H. Li, L. Liu, H. Zhou and X. Qin, Fischer–Tropsch synthesis to olefins boosted by MFI zeolite nanosheets, Nat. Nanotechnol., 2022, 714–720 CrossRef CAS.
- W. Dai, X. Wang, G. Wu, N. Guan, M. Hunger and L. Li, Methanol-to-olefin conversion on silicoaluminophosphate catalysts: Effect of Brønsted acid sites and framework structures, ACS Catal., 2011, 1, 292–299 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts, Science, 2003, 301, 935–938 CrossRef.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Single-atom catalysis of CO oxidation using Pt1/FeOx, Nat. Chem., 2011, 3, 634–641 CrossRef PubMed.
- A. D. Bolatto, M. Wolfire and A. K. Leroy, The CO-to-H2 conversion factor, Annu. Rev. Astron. Astrophys., 2013, 51, 207–268 CrossRef.
- S. Dey and N. S. Mehta, Oxidation of carbon monoxide over various nickel oxide catalysts in different conditions: A review, Chem. Eng. J. Adv., 2020, 1, 100008 CrossRef.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou and M. Li, Selective conversion of syngas to light olefins, Science, 2016, 351, 1065–1068 CrossRef PubMed.
- K. Cheng, B. Gu, X. Liu, J. Kang, Q. Zhang and Y. Wang, Direct and highly selective conversion of synthesis gas into lower olefins: design of a bifunctional catalyst combining methanol synthesis and carbon–carbon coupling, Angew. Chem., 2016, 128, 4803–4806 CrossRef.
- X. Wu, J. Lang, Z. Sun, F. Jin and Y. H. Hu, Photocatalytic conversion of carbon monoxide: from pollutant removal to fuel production, Appl. Catal., B, 2021, 295, 120312 CrossRef CAS.
- D. Glass, R. Quesada-Cabrera, S. Bardey, P. Promdet, R. Sapienza, V. Keller, S. A. Maier, V. Caps, I. P. Parkin and E. Cortés, Probing the Role of Atomic Defects in Photocatalytic Systems through Photoinduced Enhanced Raman Scattering, ACS Energy Lett., 2021, 6, 4273–4281 CrossRef CAS.
- Y. Zhao, Z. Li, M. Li, J. Liu, X. Liu, G. I. N. Waterhouse, Y. Wang, J. Zhao, W. Gao and Z. Zhang, Reductive transformation of layered-double-hydroxide nanosheets to Fe-based heterostructures for efficient visible-light photocatalytic hydrogenation of CO, Adv. Mater., 2018, 30, 1803127 CrossRef PubMed.
- Z. Li, J. Liu, Y. Zhao, G. I. N. Waterhouse, G. Chen, R. Shi, X. Zhang, X. Liu, Y. Wei and X. Wen, Co-based catalysts derived from layered-double-hydroxide nanosheets for the photothermal production of light Olefins, Adv. Mater., 2018, 30, 1800527 CrossRef.
- Y. Zhao, B. Zhao, J. Liu, G. Chen, R. Gao, S. Yao, M. Li, Q. Zhang, L. Gu and J. Xie, Oxide-modified nickel photocatalysts for the production of hydrocarbons in visible light, Angew. Chem., 2016, 128, 4287–4291 CrossRef.
- Z. Li, X. Zhang, J. Liu, R. Shi, G. I. N. Waterhouse, X. Wen and T. Zhang, Titania-Supported Ni2P/Ni Catalysts for Selective Solar-Driven CO Hydrogenation, Adv. Mater., 2021, 33, 2103248 CrossRef CAS.
- R. Mu, A. Dahal, Z.-T. Wang, Z. Dohnálek, G. A. Kimmel, N. G. Petrik and I. Lyubinetsky, Adsorption and photodesorption of CO from charged point defects on TiO2 (110), J. Phys. Chem. Lett., 2017, 8, 4565–4572 CrossRef CAS.
- A. V. Vutolkina, I. G. Baygildin, A. P. Glotov, K. A. Cherednichenko, A. L. Maksimov and E. A. Karakhanov, Dispersed Ni-Mo sulfide catalysts from water-soluble precursors for HDS of BT and DBT via in situ produced H2 under Water gas shift conditions, Appl. Catal., B, 2021, 282, 119616 CrossRef CAS.
- A. V. Vutolkina, A. P. Glotov, A. V. Zanina, D. F. Makhmutov, A. L. Maximov, S. V. Egazar'yants and E. A. Karakhanov, Mesoporous Al-HMS and Al-MCM-41 supported Ni-Mo sulfide catalysts for HYD and HDS via in situ hydrogen generation through a WGSR, Catal. Today, 2019, 329, 156–166 CrossRef CAS.
- A. V. Vutolkina, I. G. Baygildin, A. P. Glotov, K. A. Cherednichenko, A. L. Maksimov and E. A. Karakhanov, Dispersed Ni-Mo sulfide catalysts from water-soluble precursors for HDS of BT and DBT via in situ produced H2 under Water gas shift conditions, Appl. Catal., B, 2021, 282, 119616 CrossRef.
- L. Yao, Y. Pan, X. Shen, D. Wu, A. Bentalib and Z. Peng, Utilizing hydrogen underpotential deposition in CO reduction for highly selective formaldehyde production under ambient conditions, Green Chem., 2020, 22, 5639–5647 RSC.
- S. Dey and N. S. Mehta, Oxidation of carbon monoxide over various nickel oxide catalysts in different conditions: A review, Chem. Eng. J. Adv., 2020, 1, 100008 CrossRef.
- S. Dey, G. C. Dhal, D. Mohan and R. Prasad, Low-temperature complete oxidation of CO over various manganese oxide catalysts, Atmos. Pollut. Res., 2018, 9, 755–763 CrossRef.
- S. Dey and G. C. Dhal, The catalytic activity of cobalt nanoparticles for low-temperature oxidation of carbon monoxide, Mater. Today Chem., 2019, 14, 100198 CrossRef.
- X. Zhang, H. Li, F. Hou, Y. Yang, H. Dong, N. Liu, Y. Wang and L. Cui, Synthesis of highly efficient Mn2O3 catalysts for CO oxidation derived from Mn-MIL-100, Appl. Surf. Sci., 2017, 411, 27–33 CrossRef CAS.
- P. Schlexer, D. Widmann, R. J. Behm and G. Pacchioni, CO oxidation on a Au/TiO2 nanoparticle catalyst via the Au-assisted Mars–van Krevelen mechanism, ACS Catal., 2018, 8, 6513–6525 CrossRef CAS.
- Y. Pan, X. Shen, M. A. Holly, L. Yao, D. Wu, A. Bentalib, J. Yang, J. Zeng and Z. Peng, Oscillation of Work Function during Reducible Metal Oxide Catalysis and Correlation with the Activity Property, ChemCatChem, 2020, 12, 85–89 CrossRef CAS.
- V. M. Shinde and G. Madras, Synthesis of nanosized Ce0. 85M0. 1Ru0. 05O2− δ (M= Si, Fe) solid solution exhibiting high CO oxidation and water gas shift activity, Appl. Catal., B, 2013, 138, 51–61 CrossRef.
- G. Cao, N. A. Deskins and N. Yi, Carbon monoxide oxidation over copper and nitrogen modified titanium dioxide*, Appl. Catal., B, 2021, 285, 119748 CrossRef CAS.
- W. Chen, Y. Ma, F. Li, L. Pan, W. Gao, Q. Xiang, W. Shang, C. Song, P. Tao and H. Zhu, Strong Electronic Interaction of Amorphous Fe2O3 Nanosheets with Single-Atom Pt toward Enhanced Carbon Monoxide Oxidation, Adv. Funct. Mater., 2019, 29, 1904278 CrossRef CAS.
- S. Dey, G. C. Dhal, D. Mohan and R. Prasad, Kinetics of catalytic oxidation of carbon monoxide over CuMnAgOx catalyst, Mater. Discov., 2017, 8, 18–25 CrossRef.
- S. Dey and N. S. Mehta, To optimized various parameters of Hopcalite catalysts in the synthetic processes for low temperature CO oxidation, Applications in Energy and Combustion Science, 2021, 6, 100031 CrossRef.
- Y. Zhu, X. Pan, F. Jiao, J. Li, J. Yang, M. Ding, Y. Han, Z. Liu and X. Bao, Role of manganese oxide in syngas conversion to light olefins, ACS Catal., 2017, 7, 2800–2804 CrossRef CAS.
- V. Masson-Delmotte, P. Zhai, H.-O. Pörtner, D. C. Roberts, J. Skea, P. R. Shukla, A. Pirani, W. Moufouma-Okia, C. Péan and R. Pidcock, Global warming of 1.5° C: Summary for policy makers, IPCC – The Intergovernmental Panel on Climate Change, Geneva, 2018 Search PubMed.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, CO2 reduction: from homogeneous to heterogeneous electrocatalysis, Acc. Chem. Res., 2020, 53, 255–264 CrossRef.
- M. González-Castaño, B. Dorneanu and H. Arellano-García, The reverse water gas shift reaction: a process systems engineering perspective, React. Chem. Eng., 2021, 6, 954–976 RSC.
- W. J. Lee, C. Li, H. Prajitno, J. Yoo, J. Patel, Y. Yang and S. Lim, Recent trend in thermal catalytic low temperature CO2 methanation: A critical review, Catal. Today, 2021, 368, 2–19 CrossRef.
- M. Marchese, G. Buffo, M. Santarelli and A. Lanzini, CO2 from direct air capture as carbon feedstock for Fischer-Tropsch chemicals and fuels: Energy and economic analysis, J. CO2 Util., 2021, 46, 101487 CrossRef.
- H. S. Lim, M. Lee, Y. Kim, D. Kang and J. W. Lee, Low-temperature CO2 hydrogenation to CO on Ni-incorporated LaCoO3 perovskite catalysts, Int. J. Hydrogen Energy, 2021, 46, 15497–15506 CrossRef.
- Y. Ma, Z. Guo, Q. Jiang, K.-H. Wu, H. Gong and Y. Liu, Molybdenum carbide clusters for thermal conversion of CO2 to CO via reverse water-gas shift reaction, J. Energy Chem., 2020, 50, 37–43 CrossRef.
- J. Zhu, P. Wang, X. Zhang, G. Zhang, R. Li, W. Li, T. P. Senftle, W. Liu, J. Wang and Y. Wang, Dynamic structural evolution of iron catalysts involving competitive oxidation and carburization during CO2 hydrogenation, Sci. Adv., 2022, 8, eabm3629 CrossRef PubMed.
- Z. Ma and M. D. Porosoff, Development of tandem catalysts for CO2 hydrogenation to olefins, ACS Catal., 2019, 9, 2639–2656 CrossRef.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr and B.-L. Kniep, The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts, Science, 2012, 336, 893–897 CrossRef.
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Indium oxide as a superior catalyst for methanol synthesis by CO2 hydrogenation, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol, Sci. Adv., 2017, 3, e1701290 CrossRef PubMed.
- M. S. Frei, C. Mondelli, A. Cesarini, F. Krumeich, R. Hauert, J. A. Stewart, D. Curulla Ferré and J. Pérez-Ramírez, Role of zirconia in indium oxide-catalyzed CO2 hydrogenation to methanol, ACS Catal., 2019, 10, 1133–1145 CrossRef.
- L. Yao, X. Shen, Y. Pan and Z. Peng, Synergy between active sites of Cu-In-Zr-O catalyst in CO2 hydrogenation to methanol, J. Catal., 2019, 372, 74–85 CrossRef.
- M. S. Frei, C. Mondelli, R. García-Muelas, K. S. Kley, B. Puértolas, N. López, O. V. Safonova, J. A. Stewart, D. Curulla Ferré and J. Pérez-Ramírez, Atomic-scale engineering of indium oxide promotion by palladium for methanol production via CO2 hydrogenation, Nat. Commun., 2019, 10, 3377 CrossRef.
- T. Pinheiro Araújo, C. Mondelli, M. Agrachev, T. Zou, P. O. Willi, K. M. Engel, R. N. Grass, W. J. Stark, O. V. Safonova and G. Jeschke, Flame-made ternary Pd-In2O3-ZrO2 catalyst with enhanced oxygen vacancy generation for CO2 hydrogenation to methanol, Nat. Commun., 2022, 13, 5610 CrossRef.
- L. Yao, Y. Pan, D. Wu, J. Li, R. Xie and Z. Peng, Approaching full-range selectivity control in CO 2 hydrogenation to methanol and carbon monoxide with catalyst composition regulation, Inorg. Chem. Front., 2021, 8, 2433–2441 RSC.
- L. Yao, X. Shen, Y. Pan and Z. Peng, Unravelling proximity-driven synergetic effect within CIZO–SAPO bifunctional catalyst for CO2 hydrogenation to DME, Energy Fuels, 2020, 34, 8635–8643 CrossRef.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Partially oxidized atomic cobalt layers for carbon dioxide electroreduction to liquid fuel, Nature, 2016, 529, 68–71 CrossRef.
- T. Baran, S. Wojtyła, A. Dibenedetto, M. Aresta and W. Macyk, Zinc sulfide functionalized with ruthenium nanoparticles for photocatalytic reduction of CO2, Appl. Catal., B, 2015, 178, 170–176 CrossRef CAS.
- W. Tu, Y. Li, L. Kuai, Y. Zhou, Q. Xu, H. Li, X. Wang, M. Xiao and Z. Zou, Construction of unique two-dimensional MoS 2–TiO 2 hybrid nanojunctions: MoS 2 as a promising cost-effective cocatalyst toward improved photocatalytic reduction of CO 2 to methanol, Nanoscale, 2017, 9, 9065–9070 RSC.
- J. Wang, S. Lin, N. Tian, T. Ma, Y. Zhang and H. Huang, Nanostructured metal sulfides: classification, modification strategy, and solar-driven CO2 reduction application, Adv. Funct. Mater., 2021, 31, 2008008 CrossRef CAS.
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Direct methane conversion under mild condition by thermo-, electro-, or photocatalysis, Chem, 2019, 5, 2296–2325 CAS.
- E. McFarland, Unconventional chemistry for unconventional natural gas, Science, 2012, 338, 340–342 CrossRef CAS.
- E. Tabor, J. Dedecek, K. Mlekodaj, Z. Sobalik, P. C. Andrikopoulos and S. Sklenak, Dioxygen dissociation over man-made system at room temperature to form the active α-oxygen for methane oxidation, Sci. Adv., 2020, 6, eaaz9776 CrossRef CAS.
- Y. Xing, Z. Yao, W. Li, W. Wu, X. Lu, J. Tian, Z. Li, H. Hu and M. Wu, Fe/Fe3C boosts H2O2 utilization for methane conversion overwhelming O2 generation, Angew. Chem., 2021, 133, 8971–8977 CrossRef.
- P. Wang, G. Zhao, Y. Wang and Y. Lu, MnTiO3-driven low-temperature oxidative coupling of methane over TiO2-doped Mn2O3-Na2WO4/SiO2 catalyst, Sci. Adv., 2017, 3, e1603180 CrossRef.
- Z. Liang, T. Li, M. Kim, A. Asthagiri and J. F. Weaver, Low-temperature activation of methane on the IrO2 (110) surface, Science, 2017, 356, 299–303 CrossRef CAS.
- N. Xu, C. A. Coco, Y. Wang, T. Su, Y. Wang, L. Peng, Y. Zhang, Y. Liu, J. Qiao and X.-D. Zhou, Electro-conversion of methane to alcohols on “capsule-like” binary metal oxide catalysts, Appl. Catal., B, 2021, 282, 119572 CrossRef CAS.
- H. Stotz, L. Maier, A. Boubnov, A. T. Gremminger, J.-D. Grunwaldt and O. Deutschmann, Surface reaction kinetics of methane oxidation over PdO, J. Catal., 2019, 370, 152–175 CrossRef CAS.
- Z.-Q. Wang, D. Wang and X.-Q. Gong, Strategies to improve the activity while maintaining the selectivity of oxidative coupling of methane at La2O3: a density functional theory study, ACS Catal., 2019, 10, 586–594 CrossRef.
- Z. Liu, E. Huang, I. Orozco, W. Liao, R. M. Palomino, N. Rui, T. Duchoň, S. Nemšák, D. C. Grinter and M. Mahapatra, Water-promoted interfacial pathways in methane oxidation to methanol on a CeO2-Cu2O catalyst, Science, 2020, 368, 513–517 CrossRef CAS PubMed.
- J. Gao, Y. Zheng, J.-M. Jehng, Y. Tang, I. E. Wachs and S. G. Podkolzin, Identification of molybdenum oxide nanostructures on zeolites for natural gas conversion, Science, 2015, 348, 686–690 CrossRef CAS.
- S. Park, J. M. Vohs and R. J. Gorte, Direct oxidation of hydrocarbons in a solid-oxide fuel cell, Nature, 2000, 404, 265–267 CrossRef CAS PubMed.
- L. Fan, C. Li, P. V. Aravind, W. Cai, M. Han and N. Brandon, Methane reforming in solid oxide fuel cells: Challenges and strategies, J. Power Sources, 2022, 538, 231573 CrossRef CAS.
- M. E. O'Reilly, R. S. Kim, S. Oh and Y. Surendranath, Catalytic methane monofunctionalization by an electrogenerated high-valent Pd intermediate, ACS Cent. Sci., 2017, 3, 1174–1179 CrossRef.
- R. S. Kim and Y. Surendranath, Electrochemical Reoxidation Enables Continuous Methane-to-Methanol Catalysis with Aqueous Pt Salts, ACS Cent. Sci., 2019, 5, 1179–1186 CrossRef CAS.
- B. Lee and T. Hibino, Efficient and selective formation of methanol from methane in a fuel cell-type reactor, J. Catal., 2011, 279, 233–240 CrossRef CAS.
- J. Deng, S.-C. Lin, J. Fuller, J. A. Iñiguez, D. Xiang, D. Yang, G. Chan, H. M. Chen, A. N. Alexandrova and C. Liu, Ambient methane functionalization initiated by electrochemical oxidation of a vanadium (V)-oxo dimer, Nat. Commun., 2020, 11, 3686 CrossRef CAS.
- J. Li, L. Yao, D. Wu, J. King, S. S. C. Chuang, B. Liu and Z. Peng, Electrocatalytic Methane Oxidation to Ethanol on Iron-Nickel Hydroxide Nanosheets, Appl. Catal., B, 2022, 121657 CrossRef CAS.
- N. Spinner and W. E. Mustain, Electrochemical methane activation and conversion to oxygenates at room temperature, ECS Trans., 2013, 53, 1 CrossRef.
- Y. Song, Y. Zhao, G. Nan, W. Chen, Z. Guo, S. Li, Z. Tang, W. Wei and Y. Sun, Electrocatalytic oxidation of methane to ethanol via NiO/Ni interface, Appl. Catal., B, 2020, 270, 118888 CrossRef CAS.
- M. Ma, B. J. Jin, P. Li, M. S. Jung, J. Il Kim, Y. Cho, S. Kim, J. H. Moon and J. H. Park, Ultrahigh electrocatalytic conversion of methane at room temperature, Adv. Sci., 2017, 4, 1700379 CrossRef PubMed.
- M. Ma, C. Oh, J. Kim, J. H. Moon and J. H. Park, Electrochemical CH4 oxidation into acids and ketones on ZrO2: NiCo2O4 quasi-solid solution nanowire catalyst, Appl. Catal., B, 2019, 259, 118095 CrossRef.
- R. A. Van Santen, M. Neurock and S. G. Shetty, Reactivity theory of transition-metal surfaces: a Brønsted− Evans− Polanyi linear activation energy− free-energy analysis, Chem. Rev., 2009, 110, 2005–2048 CrossRef PubMed.
- S. Trasatti, Electrocatalysis by oxides—attempt at a unifying approach, J. Electroanal. Chem. Interfacial Electrochem., 1980, 111, 125–131 CrossRef CAS.
- G. Kumar, S. L. J. Lau, M. D. Krcha and M. J. Janik, Correlation of methane activation and oxide catalyst reducibility and its implications for oxidative coupling, ACS Catal., 2016, 6, 1812–1821 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles, Science, 2011, 334, 1383–1385 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Towards the computational design of solid catalysts, Nat. Chem., 2009, 1, 37–46 CrossRef.
- J. Li, X. Shen, Y. Pan and Z. Peng, Fingerprinting the Ammonia Synthesis Pathway Using Spatiotemporal Electrostatic Potential Distribution of Intermediates, ACS Omega, 2021, 6, 6292–6296 CrossRef CAS.
- P. Schwach, X. Pan and X. Bao, Direct conversion of methane to value-added chemicals over heterogeneous catalysts: challenges and prospects, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |