
Catalytic upgrading of ethanol to higher alcohols over nickel-modified Cu–La2O3/Al2O3 catalysts†
Jinfeng
He
 a,
Xiuzhen
Li
b,
Jianyao
Kou
a,
Tingjie
Tao
a,
Xinyue
Shen
a,
Dahao
Jiang
*a,
Lili
Lin
a and
Xiaonian
Li
a
a,
Xiuzhen
Li
b,
Jianyao
Kou
a,
Tingjie
Tao
a,
Xinyue
Shen
a,
Dahao
Jiang
*a,
Lili
Lin
a and
Xiaonian
Li
a
aInstitute of Industrial Catalysis, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: dhjiang@zjut.edu.cn
bFujian Runhua Chemical Co., Ltd., Nanping 354003, China
First published on 7th November 2022
Abstract
A series of nickel and nickel free modified Cu–La2O3/Al2O3 catalysts were prepared using a simple wetness impregnation method and applied for the catalytic upgrading of ethanol to higher alcohols. 56.7% ethanol conversion, 76.1% total selectivity to higher alcohols and 43.1% total yield thereof were achieved over the optimal 0.65%NiO–Cu–La2O3/Al2O3 catalyst. The addition of an appropriate amount of NiO effectively promoted ethanol upgrading to higher alcohols by increasing the amount of surface Cu active species and optimizing the acid–base properties of Cu-based catalysts, thus obviously enhancing the dehydrogenation/hydrogenation capacity, as well as the activity of aldol condensation of the catalysts. This study will provide inspiration or direction for the design of more efficient Cu-based catalysts for the catalytic upgrading of ethanol.
Introduction
Bioethanol has attracted extensive attention as an alternative to traditional fossil fuels in recent years, particularly because of concerns over energy security and climate change. However, some limitations are still attached to the usage of ethanol as a fuel additive owing to the low energy density, high water solubility and corrosive behavior of ethanol.1 In contrast, n-butanol as a biofuel has obvious advantages over ethanol and is considered an ideal biofuel or fuel additive.2,3 In the last decades, it has been proved that n-butanol could be obtained by catalytic upgrading of ethanol via the Guerbet pathway (Scheme S1†), which generally proceeds via four successive steps: 1) ethanol dehydrogenation, 2) aldol condensation of acetaldehyde, 3) dehydration of 3-hydroxybutyraldehyde, and 4) hydrogenation of crotonaldehyde.4 Other higher alcohols such as 2-ethyl-1-butanol, 1-hexanol, 2-ethyl-1-hexanol, and 1-octanol could also be produced simultaneously during ethanol upgrading through a similar mechanism. Different kinds of catalysts, including metal oxides or mixed metal oxides,5–8 basic zeolites,9 and hydroxyapatite (HAP),10–13 have been investigated in the last decades, but their catalytic performance was unsatisfactory even under harsh reaction conditions. Incorporation of transition metals into porous acid–base materials has been proven to significantly increase the yields of higher alcohols, mainly because the dehydrogenation/hydrogenation steps, which were accomplished previously with acid–base centers, are greatly improved.14,15 Among the catalysts containing transition metals, both Cu-based and Ni-based catalysts are promising systems for ethanol upgrading since Cu and Ni are not only well-known active components for ethanol dehydrogenation but also have excellent hydrogenation capability, which can further boost the tandem catalytic process. Our group had demonstrated that Cu–CeO2/AC catalysts could exhibit an ethanol conversion of 46.2%, a selectivity to n-butanol of 41.3% and a total selectivity to C4–C8 alcohols of 65% at 523 K and 2 MPa,16,17 while ordered mesoporous Cu–La–Al composite metal oxides exhibited an ethanol conversion of 52.2%, a selectivity to n-butanol of 51.9% and a total selectivity to higher alcohols of 72.2%.18 Yang et al. firstly investigated the one-step synthesis of n-butanol from ethanol over nickel supported gamma alumina catalysts, which exhibited an ethanol conversion of 19.1% and a selectivity to n-butanol of 64.3% at 473 K.19 Ghaziaskar and Xu also investigated the performance of Ni/γ-Al2O3 with different nickel contents of 8, 17and 27 wt% and achieved 35% ethanol conversion and 83% C4+ alcohol selectivity at 250 °C and 176 bar.20 Unfortunately, due to the strong C–C cleavage ability of metal Ni, there were always CH4, CO, CO2 and other cracking products detected in the tail gas, which led to a lower yield of liquid products.21 On the other hand, the Cu based catalyst had higher ethanol dehydrogenation activity and zero cleavage capability of the C–C bond under normal reaction conditions but with relatively low hydrogenation activity.22 In the current study, the addition of nickel oxide was found to be an effective method to further improve the catalytic performance of Cu-based catalysts for ethanol upgrading to higher alcohols. Furthermore, a nickel promoter at low loadings, mainly in the form of highly dispersed nickel oxide, showed an extremely low capability of cracking the C–C bond of ethanol, acetaldehyde, or other reaction intermediates, thereof avoiding the relatively large loss of liquid products. The action mechanism of a nickel oxide promoter was subsequently investigated by a series of characterization techniques such as N2-adsorption, XRD, HRTEM, H2-TPR, XPS, NH3-TPD and CO2-TPD. This study will provide inspiration or direction for the design of more efficient Cu-based catalysts for the catalytic upgrading of ethanol.Experimental
Catalyst preparation
All of chemicals used in this experiment were of analytical grade and used without further purification. A commercial alumina support was purchased from Shandong Aluminum Industry Co., Ltd.The xNiO–Cu–La2O3/Al2O3 catalysts, where x (0.32–2.55%) represented the mass fraction of NiO in the catalysts, were prepared by a simple wetness impregnation method. The Cu loadings of all the catalysts were fixed at 6 wt%, while the Cu/La molar ratios were 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5. In a typical procedure, stoichiometric nitrate precursors were dissolved in 10 mL ethanol absolute, and a 2 g Al2O3 support was added into the above solution. After impregnating at room temperature overnight, the resultant composite was dried in an electric thermostatic drying oven at 383 K for 4 h and subsequently calcined at 723 K for 3 h under an air atmosphere using a muffle furnace. Cu–La2O3/Al2O3, NiO/Al2O3 and NiO–La2O3/Al2O3 catalysts were prepared using a similar method as reference.
:1.5. In a typical procedure, stoichiometric nitrate precursors were dissolved in 10 mL ethanol absolute, and a 2 g Al2O3 support was added into the above solution. After impregnating at room temperature overnight, the resultant composite was dried in an electric thermostatic drying oven at 383 K for 4 h and subsequently calcined at 723 K for 3 h under an air atmosphere using a muffle furnace. Cu–La2O3/Al2O3, NiO/Al2O3 and NiO–La2O3/Al2O3 catalysts were prepared using a similar method as reference.
Catalysts characterization
The BET surface areas and the parameters of the pore structure of catalysts were determined by N2 adsorption using a Quantachrome ASIQC0 FV100·2 apparatus with liquid-N2 at the temperature of 77 K. The samples were outgassed at 473 K for 4 h prior to analysis.X-ray diffraction (XRD) data were collected on an X'Pert PRO X-ray diffractometer between 2θ = 10° and 80° at 2° min−1 employing a Cu-Kα radiation source (λ = 1.5406 Å). The average crystal diameters of the nanoparticles were calculated using the Scherrer formula.
High resolution transmission electron microscopy (HRTEM) was performed using a Talos S-FEG operated at 300 kV from Philips-FEI Company. Before microscopy examination, the samples were dispersed onto a copper grid coated with a thin holey carbon film.
H2 temperature-programmed reduction (H2-TPR) and H2 temperature-programmed desorption (H2-TPD) were performed on a Micromeritics Autochem II 2920 chemisorber equipped with a thermal conductivity detector (TCD) and an OmniStar GSD320 mass spectrometer (MS). For H2-TPR, the sample (100 mg) was pretreated in a quartz U-tube reactor with a He gas stream (30 mL min−1) at 473 K for 1 h. After the temperature decreased to 323 K, a 10%H2–Ar flow (50 mL min−1) was introduced, and the sample was heated from room temperature to 1273 K (10 K min−1), while desorbed species were monitored using the TCD and MS in real time. For H2-TPD, the catalyst was first pre-reduced at 523 K (10 K min−1) in a flow of 10%H2–Ar (50 mL min−1) for 1 h, purged with Ar (40 mL min−1) at the same temperature for 1 h, and then cooled down to 323 K in an Ar atmosphere. The H2 uptake of the reduced catalyst was achieved by introducing 10%H2–Ar till H2 adsorption saturation. After the H2 adsorption, the catalyst was purged again with Ar for 30 min at 323 K; then the temperature was linearly increased from 323 K to 1073 K at a rate of 10 K min−1, while the H2-TPD profiles were recorded with the TCD and MS in real time.
X-ray photoelectron spectroscopy (XPS) spectra were obtained with an ESCALab220i-XL electron spectrometer from VG Scientific using 300 W AlKα radiation and deconvoluted using XPS PEAK41 software. The catalyst after the reaction was transferred into the XPS chamber under Ar protection. The binding energies (BE) were referenced to the graphitic C 1s peak at 284.6 eV, providing accuracy within ±0.2 eV.
CO2 temperature-programmed desorption (CO2-TPD) and NH3 temperature-programmed desorption (NH3-TPD) were also performed on a Micromeritics Autochem II 2920 chemisorber equipped with a TCD and MS. For CO2-TPD experiments, the sample (100 mg) was pretreated in a quartz U-tube reactor with a He gas stream (30 mL min−1) at 473 K for 1 h. When the temperature decreased to 323 K, the CO2 saturation uptake of the pretreated catalysts was achieved by passing pure CO2 for 0.5 h at 30 ml min−1. After the CO2 adsorption, the catalyst was purged with Ar flow (40 mL min−1) for 30 min at 323 K. Then the temperature was linearly increased from 323 K to 1073 K (10 K min−1) in the Ar flow while the desorbed species were monitored using the TCD and MS in real time. NH3-TPD experiments were carried out by a similar procedure to that of CO2-TPD experiments except for using 10% NH3/Ar gas mixture as the adsorbate.
Catalysts evaluation
The catalytic tests were performed in a fixed-bed reactor. Typically, 1 g of the catalyst was placed in a tubular reactor. The reaction was carried out under the following conditions: 523 K, 3 MPa, LHSV = 2 ml gcat−1 h−1 and N2/ethanol = 250:1 (v/v). The liquid products were analyzed by a gas chromatograph (GC) with a flame ionization detector (FID) and an HP-5 column (30 m, 0.25 mm inner diameter). 1-pentanol was used as the internal standard for the quantification of liquid products. The outlet gases from the condenser were analyzed using the GC with a TCD and an HP-PLOT/Q column (30 m, 0.32 mm inner diameter).
The ethanol conversion, selectivity and yield of products were calculated as follows:
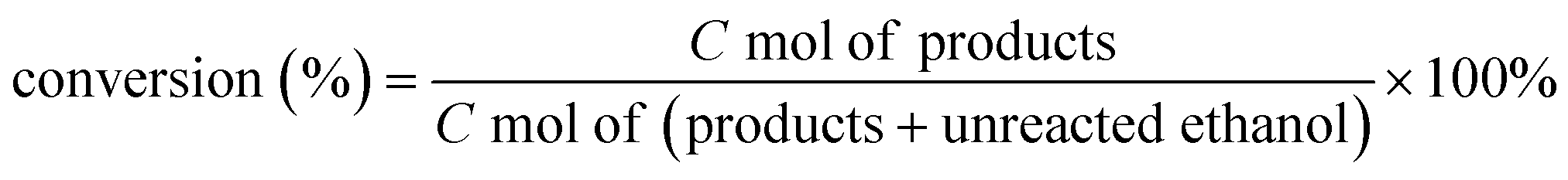
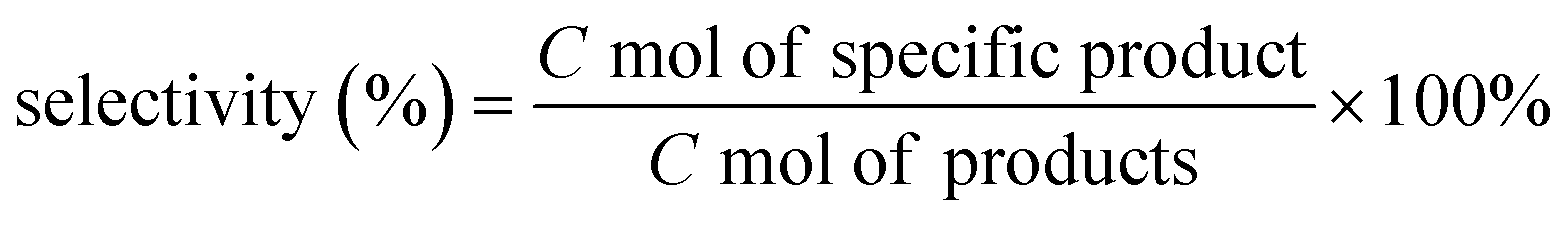
| Product yield (%) = Ethanol conversion × Product selectivity |
Results and discussion
XRD analysis of the NiO–Cu–La2O3/Al2O3 catalysts
The XRD patterns of this series of catalysts after the reaction are displayed in Fig. 1. The diffraction peaks at 2θ = 43.3°, 50.4° and 74.1° are characteristic of the Cu crystal phase, while the others are attributed to the γ-alumina support. It can be found that the nickel-modified Cu–La2O3/Al2O3 catalysts except for the 2.55%NiO–La2O3/Al2O3 catalyst exhibited relatively weak metal Cu diffraction peaks in comparison with the Cu–La2O3/Al2O3 catalyst, suggesting that the addition of an appropriate amount of nickel improved the dispersion of copper; moreover, the intensity of metal Cu diffraction peaks reached the lowest at 0.65% of the NiO content, indicating the highest Cu dispersion for 0.65%NiO–Cu–La2O3/Al2O3. As confirmed by the results of the FTIR characterization of the Al2O3 support and the CuO/Al2O3, NiO/Al2O3 and CuO–NiO/Al2O3 catalysts with different CuO and/or NiO loadings (Fig. S1–S3 and Tables S1–S3†), both CuO and NiO preferentially bonded with the coordinatively unsaturated Al3+,23,24 and then –OH on the surface of the Al2O3 support.25 Thus, the initial enhancement in Cu dispersion with the increase of NiO loading might be attributed to the separation effect of the NiO promoter on CuO species on the surface of catalysts, while the subsequent decrease in Cu dispersion with the excessive loading of NiO should be ascribed to the overweight of competitive deposition of CuO and NiO over the separation effect of the NiO promoter on CuO species. Additionally, the characteristic diffraction peaks of metal Ni, NiO, La2O3 or other crystal phases of nickel and lanthanum species were not observed in the XRD patterns, indicating that the nickel and lanthanum species over these catalysts were highly dispersed or existed in amorphous forms.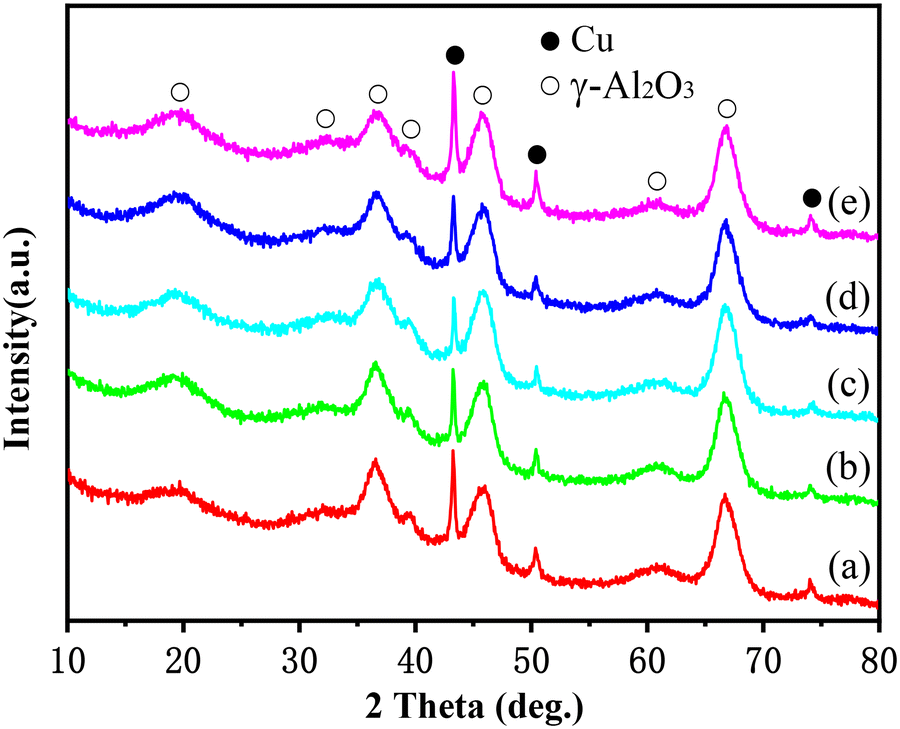 | ||
| Fig. 1 XRD patterns of catalysts after reaction: (a) Cu–La2O3/Al2O3, (b) 0.32%NiO–Cu–La2O3/Al2O3, (c) 0.65%NiO–Cu–La2O3/Al2O3, (d) 1.27%NiO–Cu–La2O3/Al2O3, and (e) 2.55%NiO–Cu–La2O3/Al2O3. | ||
TEM analysis of the NiO–Cu–La2O3/Al2O3 catalysts
As shown in Fig. 2A–D, there are no distinguishable metal Ni, NiO or La2O3 or other nanoparticles of nickel and lanthanum species except Cu nanoparticles found in typical HRTEM images of Cu–La2O3/Al2O3 and 0.65%NiO–Cu–La2O3/Al2O3 catalysts, suggesting that the nickel and lanthanum species were highly dispersed or existed in amorphous forms, which is consistent with the XRD results. The HRTEM and the corresponding EDX mapping images (Fig. 2E) further suggest that a small amount of copper existed in the form of Cu nanoparticles, while most of the Cu species were ultra-highly dispersed. In particular, the EDX mapping photographs show that nickel and lanthanum species were not only highly dispersed but also contacted intimately with Cu species or partially covered Cu nanoparticles which may play an important role in preventing the sintering of the ultra-highly dispersed Cu species or Cu nanoparticles. Additionally, the average size of the Cu nanoparticles over the 0.65%NiO–Cu–La2O3/Al2O3 catalyst was smaller than that over the Cu–La2O3/Al2O3 catalyst (25 vs. 42 nm), further confirming that the addition of nickel oxide improved the dispersion of Cu species effectively.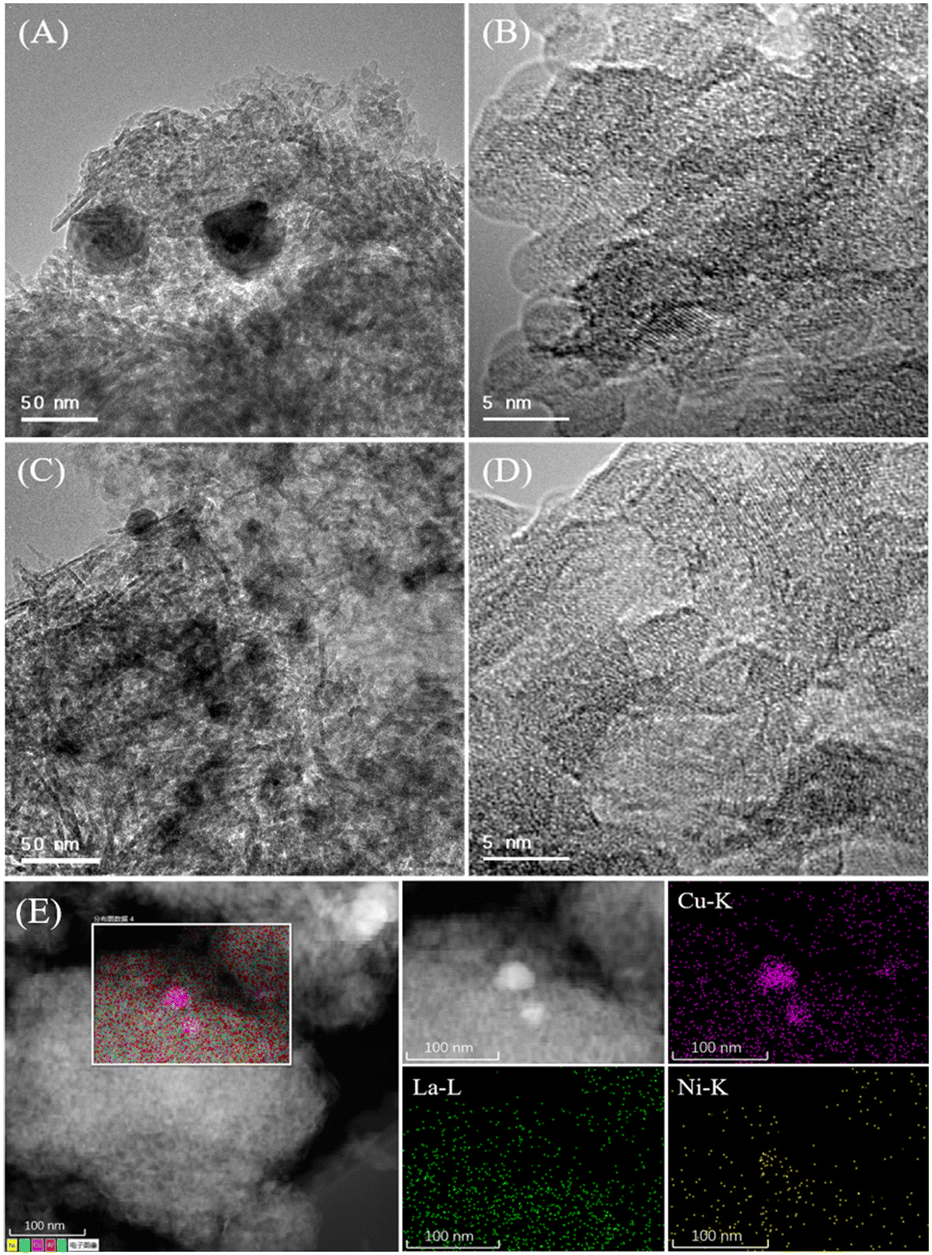 | ||
| Fig. 2 (A) and (B) HRTEM images of the Cu–La2O3/Al2O3 catalyst after reaction, (C) and (D) HRTEM images, and (E) HRTEM and the corresponding EDX mapping images of the 0.65%NiO–Cu–La2O3/Al2O3 catalyst after reaction. | ||
H2-TPR analysis of the NiO–Cu–La2O3/Al2O3 catalysts
The H2-TPR profiles of this series of catalysts are shown in Fig. 3. It can be seen that each sample showed two H2 consumption peaks. The peaks at low temperature were attributed to the reduction of CuO species on the surface of these catalysts,26–28 while the H2 consumption peaks in the range of 800 K to 1000 K probably corresponded to the reduction of Cu–Al mixed oxides such as CuAl2O4 or CuAlO2.29,30 Area of the peak at low temperature first increased with the NiO content from 0 to 0.65% and then decreased with the further increase of the NiO content to 2.55%. The maximum area of the H2 consumption peak was observed for the 0.65%NiO–Cu–La2O3/Al2O3 catalyst, indicating the maximum amount of reduced CuO species over the surface of this catalyst. Moreover, the reduction peaks at both low and high temperatures shifted to a higher temperature with the increase of the NiO content, implying the existence of a strong interaction between Cu and Ni species, which will improve the stability of Cu species. Additionally, it can be found from the H2-TPR profile of the 2.55%NiO/Al2O3 catalyst (Fig. S4†) that the H2 consumption peak of NiO species on the surface of NiO/Al2O3 catalyst ranged from 750 K to 1000 K, with the maximum at 851 K, while the NiO reduction peak shifted to high temperature with the addition of lanthanum and almost overlapped with the H2 consumption peak of Cu–Al spinel on the Cu–La2O3/Al2O3 and NiO–Cu–La2O3/Al2O3 catalysts. Furthermore, the intensity of the high temperature peak in the TPR spectra of the NiO–Cu–La2O3/Al2O3 catalysts slightly increased with the increase of NiO loading (Fig. 3). These results indicated that the reduction peaks of NiO or Ni–Al spinel species on the NiO–Cu–La2O3/Al2O3 catalysts were probably included in the high temperature peaks and overlapped with the H2 consumption peaks of the Cu–Al spinels.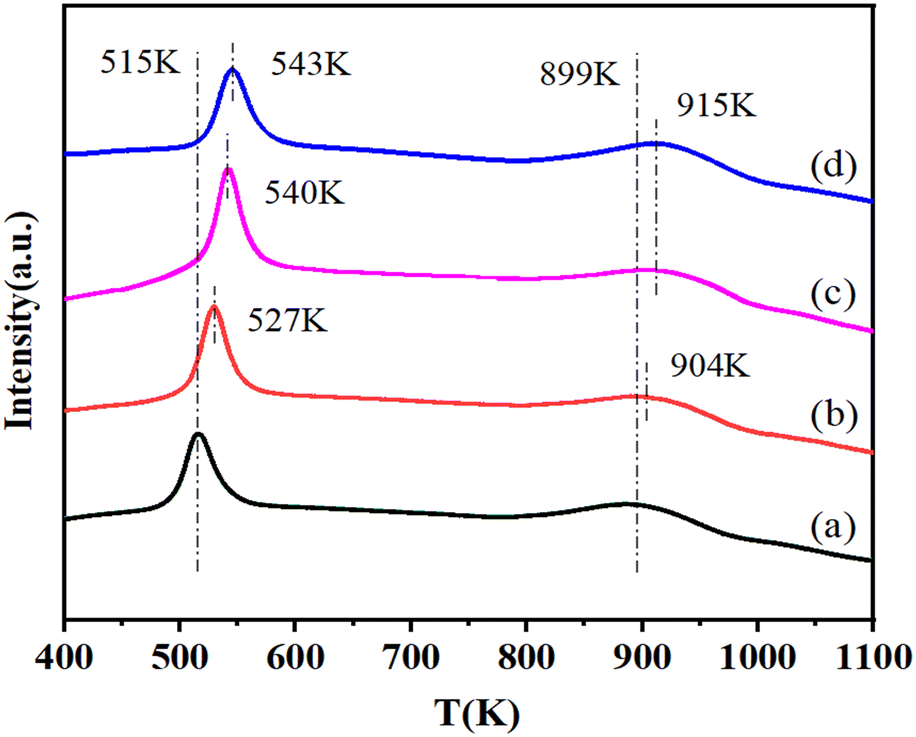 | ||
| Fig. 3 H2-TPR profiles of calcined catalysts: (a) Cu–La2O3/Al2O3, (b) 0.32%NiO–Cu–La2O3/Al2O3, (c) 0.65%NiO–Cu–La2O3/Al2O3, and (d) 2.55%NiO–Cu–La2O3/Al2O3. | ||
XPS analysis of the NiO–Cu–La2O3/Al2O3 catalysts
In order to further identify the chemical states of Ni and Cu species, the Cu–La2O3/Al2O3 and 0.65%NiO–Cu–La2O3/Al2O3 catalysts after reaction was characterized by XPS. As shown in Fig. 4A, the peaks at 855.5 and 873.8 eV along with the satellite peaks at 862.6 and 879.1 eV are respectively characteristic of Ni2+ 2p3/2 and 2p1/2 in the form of NiO, while there were no peaks characteristic of metallic Ni (at ca. 853.0 eV) found in the Ni 2p XPS spectrum.31–33 These results indicate that the nickel species on the surface of the catalyst mainly existed in the form of NiO. Additionally, the peaks at 852.0 and 856.7 eV were ascribed to La3+ 3d3/2, while the ones at 838.5 and 835.6 eV were ascribed to La3+ 3d5/2 (Fig. 4A and S5 and Table S4†).34 Furthermore, the binding energies of La 3d5/2 (835.6 eV) were substantially higher than the values measured for bulk La2O3 (833.2 eV) standards, indicating a high dispersion of La2O3 on the surface of the catalysts,35 which was consistent with the former XRD and HRTEM results. As shown in Fig. 4B, the Cu–La2O3/Al2O3 and 0.65%NiO–Cu–La2O3/Al2O3 catalysts exhibited a similar Cu 2p XPS spectrum in which two peaks at ca. 932.1 and 952.3 eV corresponded to Cu+ or Cu0,36–38 while the peaks located at ca. 933.4 and 953.4 eV with a satellite peak at 940–945 eV confirmed the existence of Cu2+.39,40 The Cu LMM AES spectrum of the catalysts (Fig. 4C) further confirmed that the low-valence Cu species comprised two kinds of Cu species, i.e., Cu+ and Cu0. The ratios of Cu2+, Cu+ and Cu0 were calculated to be 1.2:1.2:1 and 2.6:5.6:1 for the Cu–La2O3/Al2O3 and 0.65%NiO–Cu–La2O3/Al2O3 catalysts, respectively, while the ratio of the total amount of Cu species detected in the two catalysts was 0.96:1, indicating that the addition of NiO suppressed the progressive reduction of Cu2+ → Cu+ → Cu0 and increased significantly the relative amount of the Cu+ species. This phenomenon might be explained as follows: 1) the existence of a strong interaction between Cu and Ni species as confirmed by the XRD results; 2) the creation of more vacancies by adding NiO on the surface of the catalyst, which was beneficial to the stabilization of Cu+ active species.41 Ramasamy et al. concluded that the presence of the atomically dispersed Cu1+ species over the low copper loaded MgAl mixed oxide catalysts for conversion of ethanol to higher alcohols is a key factor in achieving a high dehydrogenation rate while minimizing side product formation.42 In our recent study, octahedrally coordinated Cu2+ was demonstrated to be highly active for dehydrogenation/hydrogenation during ethanol upgrading.18 Furthermore, Cu0 has been generally considered the active center of ethanol dehydrogenation and crotonaldehyde hydrogenation.22 Thus, all of Cu2+, Cu+ and Cu0 should be active sites for the dehydrogenation and hydrogenation steps during the Guerbet coupling of ethanol over the NiO–Cu–La2O3/Al2O3 catalyst. Moreover, the increase of the total amount of Cu2+, Cu+ and Cu0 on the surface of the 0.65%NiO–Cu–La2O3/Al2O3 catalyst compared with the Cu–La2O3/Al2O3 catalyst implied that the addition of an appropriate amount of NiO increased the amount of Cu active sites, which was consistent with the former XRD and TEM results.
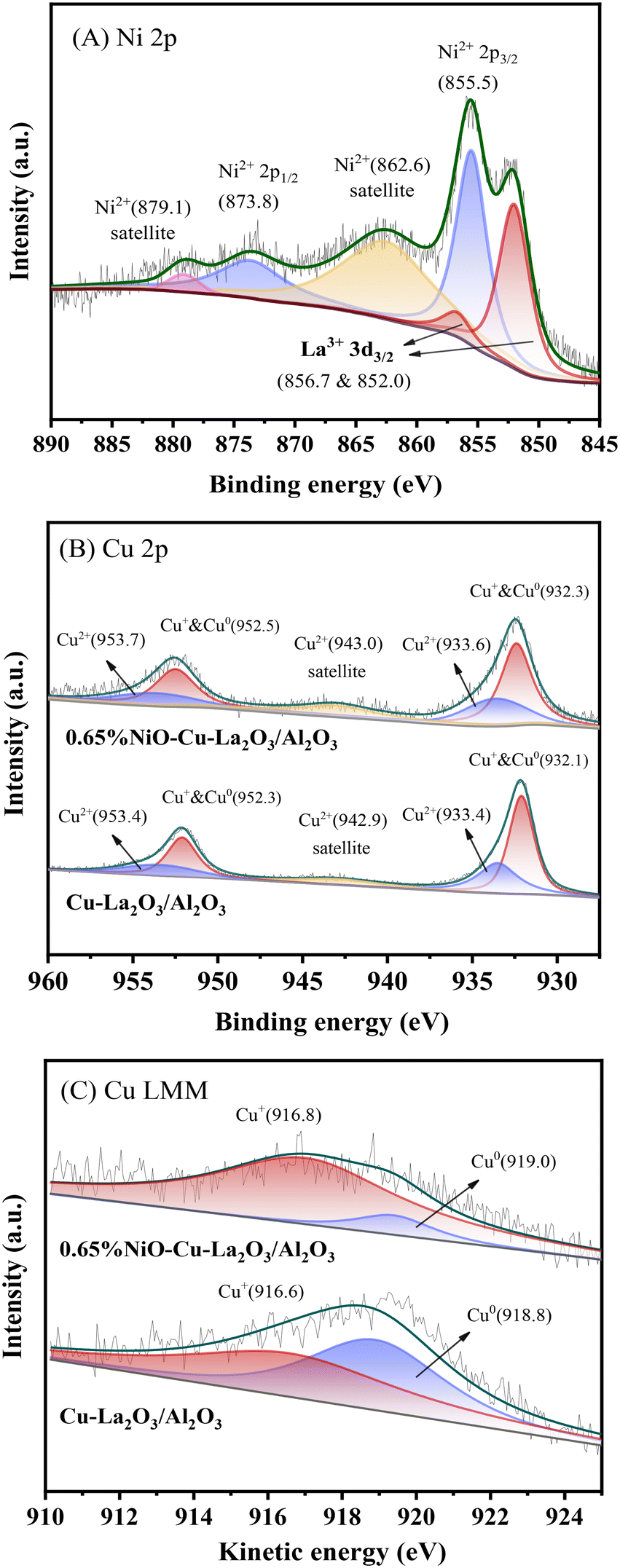 | ||
| Fig. 4 (A) Ni 2p XPS spectrum of the 0.65%NiO–Cu–La2O3/Al2O3 catalyst after the reaction. (B) Cu 2p XPS spectra and (C) Cu LMM AES spectra of the Cu–La2O3/Al2O3 and 0.65%NiO–Cu–La2O3/Al2O3 catalysts after the reaction. | ||
H2-TPD analysis of the NiO–Cu–La2O3/Al2O3 catalysts
The H2-TPD profiles of this series of catalysts are presented in Fig. 5. Two H2 desorption peaks at ca. 364 K and 728 K were observed for all samples. The peak at low temperature was attributed to desorption of H2 adsorbed on the Cu active sites, while the peak at high temperature could be attributed to the desorption of hydrogen spilled over lanthanum oxide or an Al2O3 support.43,44 The amount of H2 desorbed from Cu active sites increased as follows: Cu–La2O3/Al2O3 (0.058 mmol g−1) < 0.32%NiO–Cu–La2O3/Al2O3 (0.104 mmol g−1) < 2.55%NiO–Cu–La2O3/Al2O3 (0.233 mmol g−1) < 0.65%NiO–Cu–La2O3/Al2O3 (0.330 mmol g−1), implying the trend of the capability for hydrogen adsorption and activation. As confirmed by XRD, H2-TPR and XPS results, 0.65%NiO–Cu–La2O3/Al2O3 catalyst had the highest Cu dispersion and the largest amount of Cu active species, which were responsible for the highest capability for hydrogen activation.45,46 Though the amounts of the spilled hydrogen on the NiO-containing catalysts were less than those on the Cu–La2O3/Al2O3 catalyst, the spilled hydrogen had lower desorption temperature, probably indicating that the addition of NiO increased the activity of the spilled hydrogen.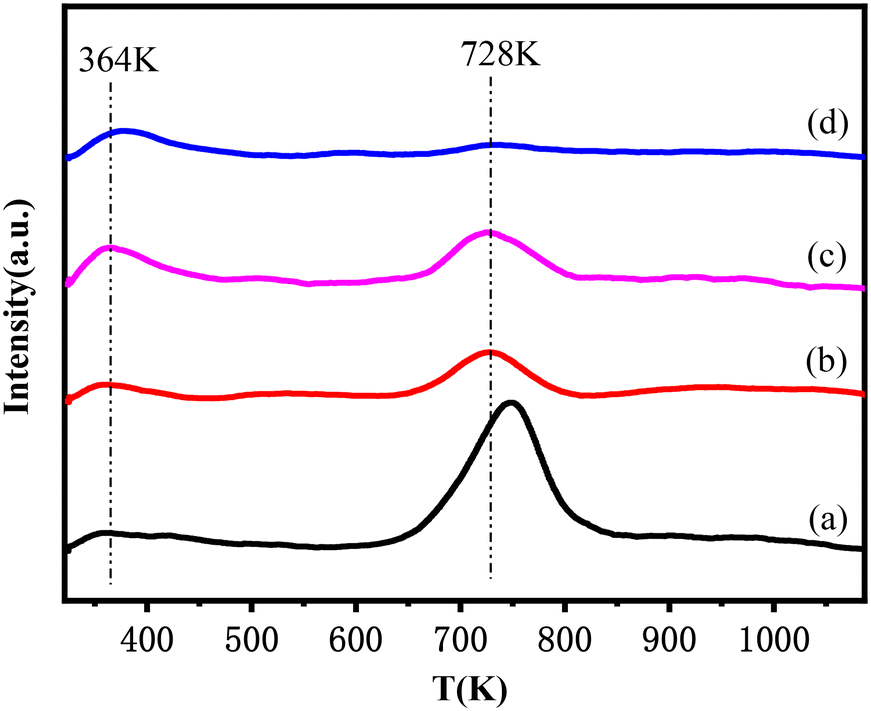 | ||
| Fig. 5 H2-TPD profiles of the reduced catalysts: (a) Cu–La2O3/Al2O3, (b) 0.32%NiO–Cu–La2O3/Al2O3, (c) 0.65%NiO–Cu–La2O3/Al2O3, and (d) 2.55%NiO–Cu–La2O3/Al2O3. | ||
NH3-TPD and CO2-TPD analysis of the NiO–Cu–La2O3/Al2O3 catalysts
The NH3-TPD and CO2-TPD profiles of this series of catalysts are presented in Fig. 6. The Cu–La2O3/Al2O3 catalyst exhibited three NH3 desorption peaks centered at 377 K, 538 K and 630 K (Fig. 6A), which were assigned to the weak, medium and strong acidic sites, respectively.47 These acidic sites were related to Al3+ with different coordination structural patterns and aluminum hydroxyl groups on the surface of the Al2O3 support.48–50 According to Fig. 6A and Table S5,† the strong acidic sites disappeared, while the total amount of weak and medium acidic sites slightly decreased with the addition of NiO, which is understandable given the weak alkalinity of nickel oxide.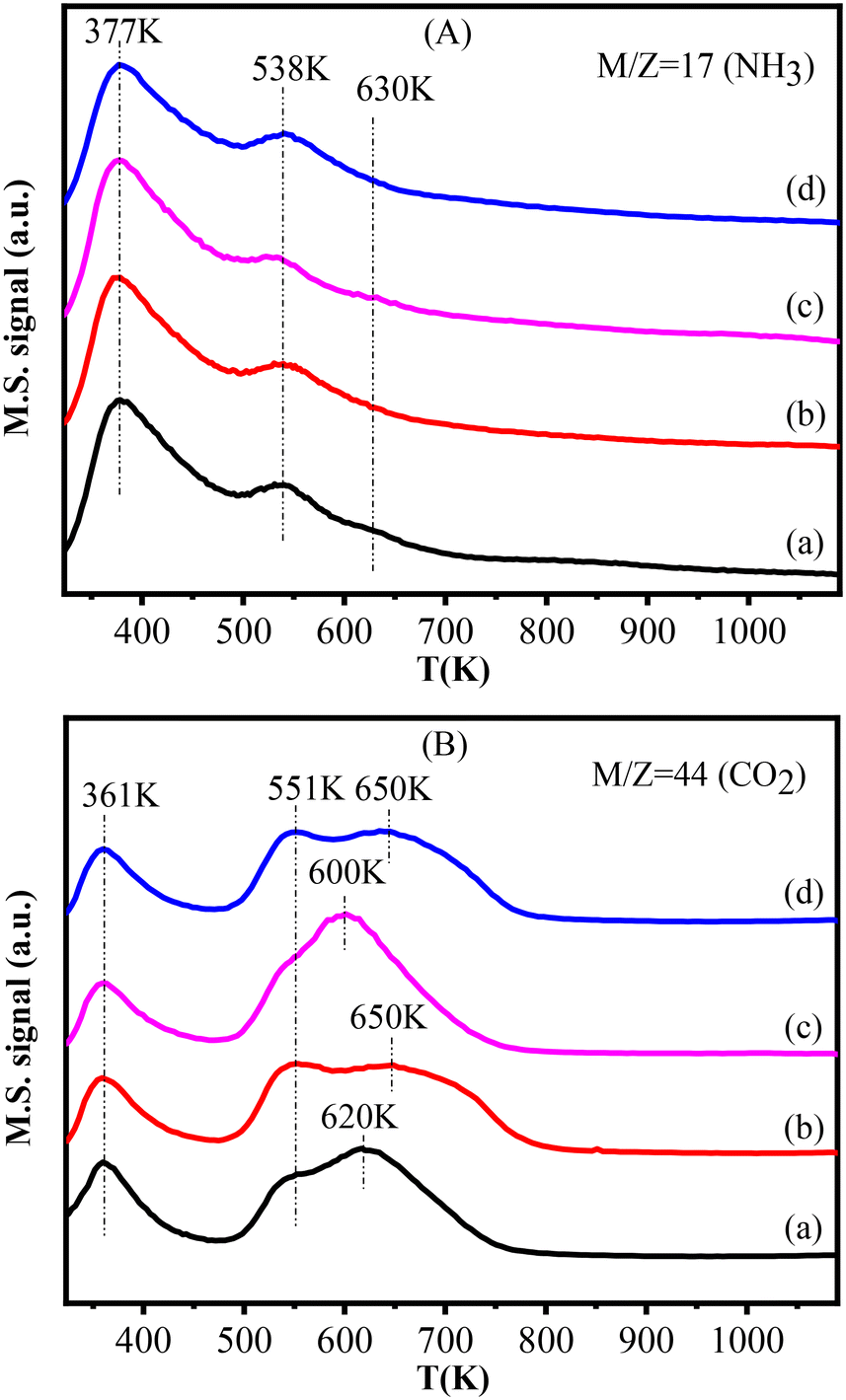 | ||
| Fig. 6 NH3-TPD (A) and CO2-TPD (B) profiles of the catalysts: (a) Cu–La2O3/Al2O3, (b) 0.32%NiO–Cu–La2O3/Al2O3, (c) 0.65%NiO–Cu–La2O3/Al2O3, and (d) 2.55%NiO–Cu–La2O3/Al2O3. | ||
As shown in Fig. 6B, there were three CO2 desorption peaks at 361 K, ∼550 K and 600–650 K observed for each catalyst. The first CO2 desorption peak corresponding to weak basic sites was associated to the surface basic hydroxyl groups (–OH), while the subsequent two peaks corresponding respectively to medium strength and strong basic sites might be related to metal–oxygen pairs (Al–O and La–O) and low coordination surface O2−.51–55 It can be seen from Fig. 6B and Table S5† that the amount of the weak basic sites almost did not change, while that of the medium and strong basic sites slightly increased after introducing nickel oxide. In particular, for the 0.65%NiO–Cu–La2O3/Al2O3 catalyst (Fig. 6B(c)), the number of strong bases was the largest, and the distribution was most concentrated, probably implying the highest and most uniform dispersion of La species.54,55 Thus, the addition of an appropriate amount of NiO optimized the acid–base distribution and promoted the balance of acid–base sites, which is essential for aldol condensation of acetaldehyde, thus improving the efficiency of catalysts for ethanol upgrading to higher alcohols.
Catalytic performance of the NiO–Cu–La2O3/Al2O3 catalyst
The catalytic performances of this series of catalysts for upgrading of ethanol to higher alcohols are presented in Table 1. As shown in Table 1, Cu–La2O3/Al2O3 exhibited 50.9% ethanol conversion, 67.9% total selectivity to higher alcohols (ST), and 34.6% total yield thereof (YT). The ethanol conversion and YT first increased and then decreased with the increase of NiO. The optimal 0.65wt%NiO–Cu–La2O3/Al2O3 catalyst exhibited an ethanol conversion of 56.7%, a ST of 76.1%, and YT of up to 43.1%. What's more remarkable was that the 0.65%NiO–Cu–La2O3/Al2O3 catalyst exhibited excellent stability: ethanol conversion, ST and YT remained almost the same during 200 h on-stream evaluation (Fig. S6†). Additionally, the liquid yield of the Cu–La2O3/Al2O3 catalyst was 99.3%, while those of the Cu–La2O3/Al2O3 catalysts modified with 0.32%, 0.65%, and 1.27% NiO were 98.3%, 98.0% and 97.8%, respectively. The cracking rates of liquid products calibrated by decreasing the liquid loss during the collection of samples were below 1.5%, indicating that the highly dispersed NiO species on the NiO–Cu–La2O3/Al2O3 catalysts with NiO loadings of 0.32%, 0.65%, and 1.27% had extremely low activity of cracking the C–C bond. However, the liquid yield of the 2.55%NiO–Cu–La2O3/Al2O3 catalyst was 95.8%, corresponding to the cracking rate of 3.5%, which might be caused by the reduction of a small amount of nickel oxide to metallic nickel during ethanol upgrading.19–21 Since NiO itself showed no catalytic activity for catalytic upgrading of ethanol to higher alcohols (Table S6†), the introduction of an appropriate amount of NiO actually increased the amount of Cu active sites and therefore enhanced the ethanol dehydrogenation activity of the Cu-based catalyst, which can be reflected by the increase of ethanol conversion and YT with the addition of NiO. The increase of the amount of Cu active sites also enhanced the hydrogen adsorption and activation ability of the Cu-based catalyst, as directly confirmed by the H2-TPD results. Furthermore, the nickel species in the form of NiO might be favorable for the adsorption and activation of esters and aldehydes by interacting with their oxygen atoms on carbonyl groups56,57 and promote their reduction to alcohols by the hydrogen from adjacent Cu active sites. Besides ethanol dehydrogenation and hydrogenation of crotonaldehyde, acetaldehyde condensation is the other key elementary step in the ethanol to n-butanol process, which is highly dependent on the acidic and basic properties of catalysts. Moreover, besides solo acid or base sites, acid–base pairs demonstrated elevated activity in condensation reactions.18,58 As confirmed by the results of NH3-TPD and CO2-TPD, the addition of NiO reduced slightly the number of acidic sites on the surface of the Cu-based catalyst while increased the amount of basic sites, especially strong basic sites, forming abundant acid–base pairs, which promoted the acetaldehyde condensation reactions and then the ethanol upgrading to higher alcohols.| Catalyst | Conv. (%) | Sel. (%) | S T | Y T | |||||
|---|---|---|---|---|---|---|---|---|---|
| Acetaldehyde | Ethyl acetate | Butyraldehyde | Butanol | C6–C8 alcoholsb | Othersc | ||||
|
a Conversion and selectivity are obtained at steady-state; reaction conditions: catalyst, 1.0 g; 523 K, 3 MPa (N2), LHSV = 2 ml gcat−1 h−1, N2/ethanol (v/v) = 250:1.
b C6–C8 alcohols include 2-ethylbutanol, 1-hexanol, 2-ethylhexanol and 1-octanol.
c Other products include diethyl ether, 1,1-diethoxyethane, butyl acetate, etc.
d
S
T represents the total selectivity to higher alcohols.
e
Y
T represents the total yield to higher alcohols.
|
|||||||||
| Cu–La2O3/Al2O3 | 50.9 | 1.7 | 6.5 | 4.2 | 48.6 | 19.3 | 19.7 | 67.9 | 34.6 |
| 0.32%NiO–Cu–La2O3/Al2O3 | 55.3 | 1.8 | 3.1 | 3.8 | 48.5 | 26.1 | 16.7 | 74.6 | 41.3 |
| 0.65%NiO–Cu–La2O3/Al2O3 | 56.7 | 1.9 | 3.1 | 3.5 | 49.7 | 26.4 | 15.4 | 76.1 | 43.1 |
| 1.27%NiO–Cu–La2O3/Al2O3 | 55.9 | 1.5 | 2.7 | 3.3 | 49.8 | 27.1 | 15.6 | 76.9 | 43.0 |
| 2.55%NiO–Cu–La2O3/Al2O3 | 51.3 | 1.6 | 2.1 | 3.1 | 50.5 | 28.9 | 13.8 | 79.4 | 40.7 |
In summary, the catalytic performance of the NiO–Cu–La2O3/Al2O3 catalysts were obviously improved by the addition of nickel oxide, and the 0.65%NiO–Cu–La2O3/Al2O3 catalyst exhibited excellent catalytic activity and the highest total yield of higher alcohols in continuous catalytic upgrading of ethanol. The addition of an appropriate NiO amount effectively not only improved the dehydrogenation/hydrogenation ability of the catalyst by improving the dispersion of Cu species, but also optimized the acid–base properties of the catalysts, therefore promoting the aldol condensation of acetaldehyde.
Conclusions
A series of NiO–Cu–La2O3/Al2O3 catalysts with varying NiO contents (0–2.55 wt%) were prepared using a simple wetness impregnation method and applied for the catalytic upgrading of ethanol to higher alcohols. The catalytic performance of the Cu–La2O3/Al2O3 catalyst was obviously influenced by addition of NiO, and the optimal 0.65%NiO–Cu–La2O3/Al2O3 catalyst exhibited 56.7% ethanol conversion, 76.1% total selectivity to higher alcohols and up to 43.1% total yield thereof. The characterization results proved that addition of an appropriate amount of NiO not only increased the amount of surface Cu active species, thus improving the dehydrogenation/hydrogenation capability of the catalyst, but also adjusted the acid–base properties of the catalysts, thus facilitating aldol condensation of acetaldehyde. This study demonstrated that the introduction of metal oxide promoters with specific adjustment functions can be a feasible approach to achieve more efficient Cu-based catalysts for catalytic upgrading of ethanol to higher alcohols.Author contributions
Jinfeng He: investigation, resources, analysis, data curation, and writing – original draft; Xiuzhen Li: investigation and data curation; Jianyao Kou: investigation, analysis, and resources; Tingjie Tao: investigation and resources; Xinyue Shen: investigation and data curation; Dahao Jiang: writing – review & editing, project administration, supervision, and funding acquisition; Lili Lin: resources and analysis; Xiaonian Li: supervision and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the Natural Science Foundation of Zhejiang Province (LY19B030007).References
- P. Durre, Biotechnol. J., 2007, 2, 1525–1534 CrossRef PubMed.
- B. Kolesinska, J. Fraczyk, M. Binczarski, M. Modelska, J. Berlowska, P. Dziugan, H. Antolak, Z. J. Kaminski, I. A. Witonska and D. Kregiel, Materials, 2019, 12, 350–371 CrossRef PubMed.
- M. Uyttebroek, W. Van Hecke and K. Vanbroekhoven, Catal. Today, 2015, 239, 7–10 CrossRef.
- T. J. Jordison, C. T. Lira and D. J. Miller, Ind. Eng. Chem. Res., 2015, 54, 10991–11000 CrossRef.
- A. S. Ndou, N. Plint and N. J. Coville, Appl. Catal., A, 2003, 251, 337–345 CrossRef.
- T. W. Birky, J. T. Kozlowski and R. J. Davis, J. Catal., 2013, 298, 130–137 CrossRef.
- D. L. Carvalho, R. R. Avillez, M. T. Rodrigues, L. E. P. Borges and L. G. Appel, Appl. Catal., A, 2012, 415, 96–100 CrossRef.
- I. C. Marcu, N. Tanchoux, F. Fajula and D. Tichit, Catal. Lett., 2012, 143, 23–30 CrossRef.
- C. Yang and Z. Y. Meng, J. Catal., 1993, 142, 37–44 CrossRef CAS.
- T. Tsuchida, S. Sakuma, T. Takeguchi and W. Ueda, Ind. Eng. Chem. Res., 2006, 45, 8634–8642 CrossRef CAS.
- T. Tsuchida, J. Kubo, T. Yoshioka, S. Sakuma, T. Takeguchi and W. Ueda, J. Catal., 2008, 259, 183–189 CrossRef CAS.
- S. Ogo, A. Onda and K. Yanagisawa, Appl. Catal., A, 2011, 402, 188–195 CrossRef CAS.
- S. Hanspal, Z. D. Young, H. Shou and R. J. Davis, ACS Catal., 2015, 5, 1737–1746 CrossRef CAS.
- I. Nezam, J. Zak and D. J. Miller, Ind. Eng. Chem. Res., 2020, 59, 13906–13915 CrossRef CAS.
- F. Y. Fan, Q. Zhang, X. Wang, Y. H. Ni, Y. Q. Wu and Z. B. Zhu, Fuel, 2016, 186, 11–19 CrossRef CAS.
- D. H. Jiang, X. Y. Wu, J. Mao, J. Ni and X. N. Li, Chem. Commun., 2016, 52, 13749–13752 RSC.
- X. Y. Wu, G. Q. Fang, Z. Liang, W. H. Leng, K. Y. Xu, D. H. Jiang, J. Ni and X. N. Li, Catal. Commun., 2017, 100, 15–18 CrossRef.
- H. J. Zhao, X. Y. Shen, J. F. He, J. Y. Kou, T. J. Tao, Y. Can, H. Huang, D. H. Jiang, L. L. Lin and X. N. Li, Appl. Surf. Sci., 2022, 599, 153851–153862 CrossRef.
- K. W. Yang, X. Z. Jiang and W. C. Zhang, Chin. Chem. Lett., 2004, 15, 1497 Search PubMed.
- H. S. Ghaziaskar and C. Xu, RSC Adv., 2013, 3, 4271–4280 RSC.
- T. Riittonen, E. Toukoniitty, D. K. Madnani, A. R. Leino, K. Kordas, M. Szabo, A. Sapi, K. Arve, J. Warnå and J. P. Mikkola, Catalysts, 2012, 2, 68–84 CrossRef.
- X. Y. Wu, G. Q. Fang, Y. Q. Tong, D. H. Jiang, Z. Liang, W. H. Leng, L. Liu, P. X. Tu, H. J. Wang, J. Ni and X. N. Li, ChemSusChem, 2018, 11, 71–85 CrossRef CAS.
- E. Borello, A. Cimino, G. Ghiotti, M. Lo Jacono, M. Schiavello and A. Zecchina, Discuss. Faraday Soc., 1971, 52, 149–160 RSC.
- C. Zhao, Y. Z. Yu, A. Jentys and J. A. Lercher, Appl. Catal., B, 2013, 132, 282–292 CrossRef.
- Y. M. Zhang, Y. Zu, D. D. He, J. Liang, L. H. Zhu, Y. Mei and Y. M. Luo, Appl. Catal., B, 2022, 315, 121539 CrossRef CAS.
- G. Águila, F. Gracia, J. Cortés and P. Araya, Appl. Catal., B, 2008, 77, 325–338 CrossRef.
- F. E. López-Suárez, A. Bueno-López and M. J. Illán-Gómez, Appl. Catal., B, 2008, 84, 651–658 CrossRef.
- I. Gandarias, J. Requies, P. L. Arias, U. Armbruster and A. Martin, J. Catal., 2012, 290, 79–89 CrossRef CAS.
- S. Sato, M. Iijima, T. Nakayama, T. Sodesawa and F. Nozaki, J. Catal., 1997, 169, 447–454 CrossRef CAS.
- S. K. Lee and W. H. Tuan, Int. J. Appl. Ceram. Technol., 2013, 10, 780–789 CrossRef.
- L. Fu, Z. X. Liu, X. Li, Y. F. Li, H. Y. Yang and Y. J. Liu, Fuel, 2022, 322, 124027–124041 CrossRef.
- X. Y. Lin, H. Zhu, M. Huang, C. S. Wan, D. L. Li and L. L. Jiang, Fuel Process. Technol., 2022, 233, 10721–10733 CrossRef.
- Y. Duan, R. Wang, Q. H. Liu, X. Y. Qin and Z. H. Li, Front. Chem., 2022, 10, 857199–857208 CrossRef PubMed.
- G. Garbarino, C. Y. Wang, T. Cavattoni, E. Finocchio, P. Riani, M. Flytzani-Stephanopoulos and G. Busca, Appl. Catal., B, 2019, 248, 286–297 CrossRef.
- L. P. Haack, J. E. deVries, K. Otto and M. S. Chattha, Appl. Catal., A, 1992, 82, 199–214 CrossRef.
- I. Platzman, R. Brener, H. Haick and R. Tannenbaum, J. Phys. Chem. C, 2008, 112, 1101–1108 CrossRef.
- Y. T. Wang, W. Zhou, R. R. Jia, Y. F. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef PubMed.
- S. D. Jones, L. M. Neal and H. E. Hagelin-Weaver, Appl. Catal., B, 2008, 84, 631–642 CrossRef.
- H. W. Zhang, H. R. Tan, S. Jaenicke and G. K. Chuah, J. Catal., 2020, 389, 19–28 CrossRef.
- S. Q. Xu, H. X. Zhu, W. R. Cao, Z. B. Wen, J. N. Wang, C. P. François-Xavier and T. Wintgens, Appl. Catal., B, 2018, 234, 223–233 CrossRef.
- Y. J. Liu, S. J. Qing, X. N. Hou, F. J. Qin, X. Wang, Z. X. Gao and H. W. Xiang, ChemCatChem, 2018, 10, 5698–5706 CrossRef.
- M. F. Guo, M. J. Gray, H. Job, C. Alvarez-Vasco, S. Subramaniam, X. Zhang, L. Kovarik, V. Murugesan, S. Phillipsa and K. K. Ramasamy, Green Chem., 2021, 23, 8030–8039 RSC.
- S. Ewald, S. Standl and O. Hinrichsen, Appl. Catal., A, 2018, 549, 93–101 CrossRef.
- J. Zelin, S. A. Regenhardt, C. I. Meyer, H. A. Duarte, V. Sebastian and A. J. Marchi, Chem. Eng. Sci., 2019, 206, 315–326 CrossRef.
- M. Dan, M. Mihet, Z. Tasnadi-Asztalos, A. Imre-Lucaci, G. Katona and M. D. Lazar, Fuel, 2015, 147, 260–268 CrossRef.
- Z. H. Ren, M. N. Younis, H. Zhao, C. S. Li, X. G. Yang, E. Q. Wang and G. Y. Wang, Chin. J. Chem. Eng., 2020, 28, 1612–1622 CrossRef.
- O. V. Larina, K. V. Valihura and T. Čendak, React. Kinet., Mech. Catal., 2020, 132, 359–378 CrossRef.
- P. J. Chupas, K. W. Chapman and G. J. Halder, J. Am. Chem. Soc., 2011, 133, 8522–8524 CrossRef.
- Z. C. Wang, Y. J. Jiang, A. Baiker and J. Huang, Acc. Chem. Res., 2020, 53, 2648–2658 CrossRef PubMed.
- H. Q. Ma, Y. N. Guan, W. Y. Chen, Z. J. Sui, G. Qian, D. Chen, X. G. Zhou and X. Z. Duan, Catal. Today, 2021, 365, 310–317 CrossRef CAS.
- M. Y. Byun, J. S. Kim, D. W. Park and M. S. Lee, Korean J. Chem. Eng., 2018, 35, 1083–1088 CrossRef CAS.
- R. Daroughegi, F. Meshkani and M. Rezaei, Chem. Eng. Sci., 2021, 230, 116194–116206 CrossRef CAS.
- P. F. Cao, H. T. Zhao, S. Adegbite, B. L. Yang, E. Lester and T. Wu, Fuel, 2021, 298, 120599–120607 CrossRef CAS.
- J. Liu, C. Liu, G. Zhou, S. T. Shen and L. Rong, Green Chem., 2012, 14, 2499–2505 RSC.
- J. Ni, L. W. Chen, J. Y. Lin, M. K. Schreyer, Z. Wang and S. Kawi, Int. J. Hydrogen Energy, 2013, 38, 13631–13642 CrossRef CAS.
- J. Pritchard, A. Ciftci, M. W. G. M. Verhoeven, E. J. M. Hensen and E. A. Pidko, Catal. Today, 2017, 279, 10–18 CrossRef CAS.
- C. Thunyaratchatanon, A. Luengnaruemitchai, N. Chollacoop, S. Y. Chen and Y. Yoshimura, Fuel Process. Technol., 2018, 179, 422–435 CrossRef CAS.
- J. Sun, R. A. L. Baylon, C. Liu, D. Mei, K. J. Martin, P. Venkitasubramanian and Y. Wang, J. Am. Chem. Soc., 2016, 138, 507–517 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy01554d |
| This journal is © The Royal Society of Chemistry 2023 |