
Dynamic environment at the Zr6 oxo cluster surface is key for the catalytic formation of amide bonds†
Yujie
Zhang
a,
Ismail Y.
Kokculer
 ab,
Francisco
de Azambuja
*a and
Tatjana N.
Parac-Vogt
*a
ab,
Francisco
de Azambuja
*a and
Tatjana N.
Parac-Vogt
*a
aDepartment of Chemistry, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: francisco.deazambuja@kuleuven.be; tatjana.vogt@kuleuven.be
bDepartment of Chemistry, Bilkent University, 06800 Ankara, Turkey
First published on 28th October 2022
Abstract
Zirconium compounds are an attractive alternative to costly, low abundant metals for the development of inexpensive, readily available, and robust catalysts. The air and moisture stable Zr oxo clusters such as the Zr6O8 species, are of particular interest as they are key building blocks of several Zr-based metal–organic framework (Zr-MOF) catalysts. However, broader use of these cluster-based materials as catalysts is still hampered by the modest understanding of their fundamental reactivity. To bridge this gap, we report on the activity of a soluble Zr6O8 cluster, [Zr6(OH)4O4(OMc)12] (OMc = methacrylate) (Zr6), as a discrete molecular catalyst for the atom-economic formation of amide bonds. This reaction demands two completely different substrates interacting with the Zr6 catalyst, a key step rarely addressed in MOF catalyzed reactions. Remarkably, Zr6 catalyzes the formation of amide bonds directly from non-activated carboxylic acid and amine substrates in ethanol, without requiring anhydrous conditions or water scavenging to achieve good yields. As shown by a series of kinetic, and mechanistic experiments, this promising reactivity arises from a dynamic environment at the cluster surface, where the essential coordination of both substrates requires an excess of amine to enhance the reaction output. Strikingly, Zr6 catalyst tolerates a range of substrates, including (hetero)aromatic, aliphatic, and α-branched acids, even though their nature directly impacts the reaction efficiency. Further, insights for the future design of catalysts based on Zr oxo cluster are discussed through a detailed comparison of Zr6 reactivity with a related Zr12 cluster, and Zr-MOF catalysts. Considering the advantages of zirconium, and the relevance of discrete Zr oxo clusters as building blocks of several MOF materials of varied utility, the molecular level understanding disclosed here contributes at large to the development of novel catalytic entities, and sustainable approaches to synthetic chemistry.
Introduction
Group IV metal oxo clusters, usually synthesized by mixing a metal alkoxide with carboxylic acids, have been extensively investigated in the development of new inorganic–organic hybrid materials; however, their catalytic ability as discrete species remains underexplored.1,2 Polymer materials' thermal stability, and mechanical properties like strength, hardness, and brittleness, have been enhanced by the addition of diverse group IV metal oxo clusters.3–5 Hexanuclear metal oxo clusters of the type [Zr6O4(OH)4(OOCR)12] (OOCR = carboxylate) have also been used as building blocks of Zr-based metal–organic frameworks (Zr-MOFs) (Fig. 1).6–9 Notably, several Zr-MOFs feature catalytic active metal sites in their Zr6O8 cluster nodes, either intrinsically or due to missing linker defects.8,10–12 In our work, we have leveraged these active sites to develop novel classes of metal oxo cluster based catalysts for the cleavage,9,13–19 or formation20 of amide bonds with a unique tolerance to the water formed during the reaction.21–23 Recently, we have also showcased that zirconium oxo clusters (ZrOC) can be used as discrete molecular catalysts. A dodecanuclear ZrOC composed of two Zr6O8 units, [Zr6(OH)4O4(OAcr)12]2 (OAcr = acrylate) (Zr12), catalyzed the direct formation of amide bonds, remarkably without requiring water scavenging or anhydrous conditions.24 Considering the difficulties faced on elucidating molecular aspects of the reactivity in our previous work with Zr-MOF catalysts,25 this pioneer work on Zr12 prompted us to explore more of the fundamental reactivity of Zr6O8 clusters without common constrains arising from the heterogeneous nature of MOFs (e.g., diffusion of substrates and products), leveraging the possibility of investigating them as discrete molecular species for the catalytic direct intermolecular formation of amide bonds.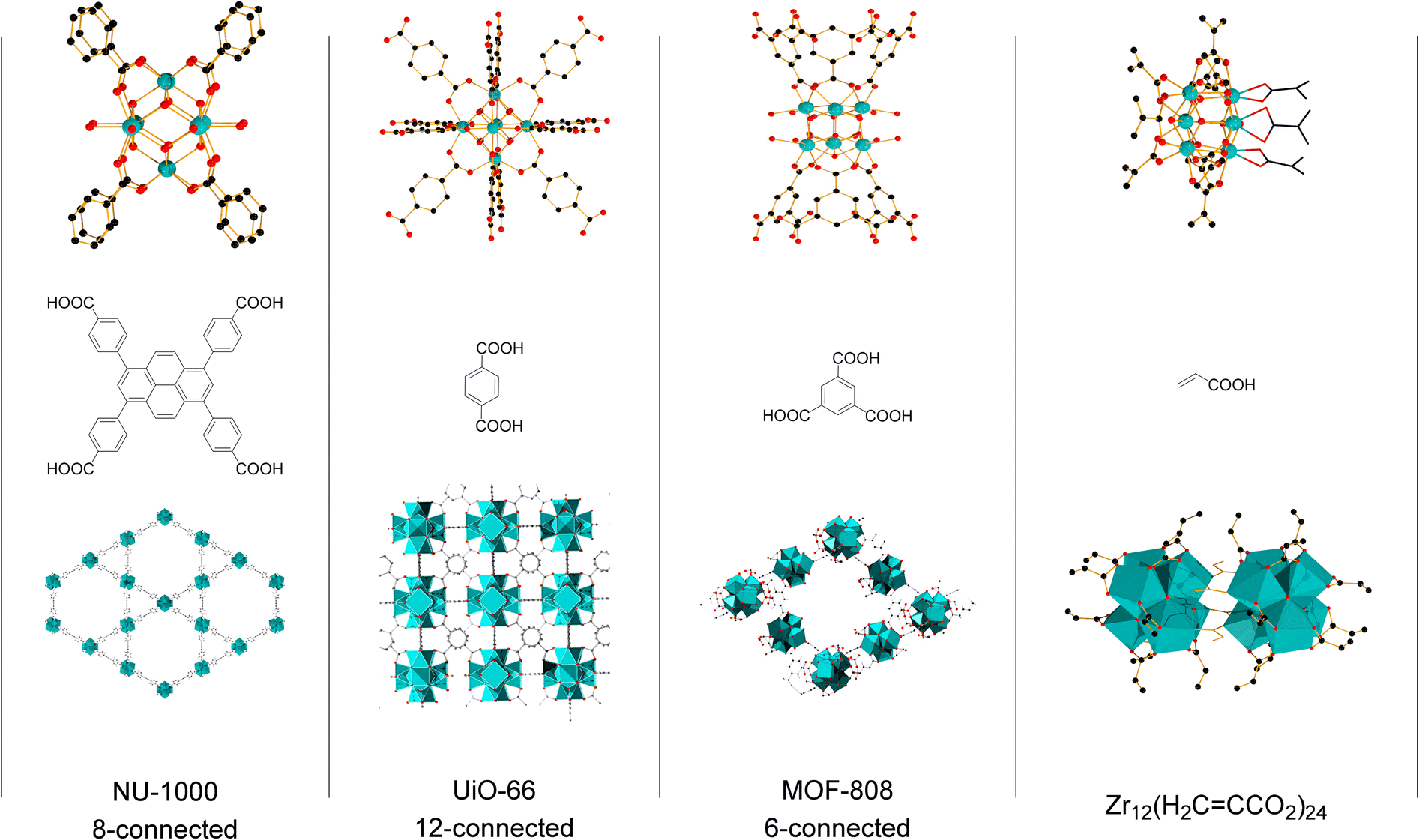 | ||
| Fig. 1 Zr6O8 clusters are key components of highly useful Zr-based metal–organic frameworks (Zr-MOFs) such as NU-1000, UiO-66, and MOF-808, and well-defined Zr-based nanoclusters such as Zr12(acrylate)24. Color code: Zr (teal), O (red), C (black). Hydrogen atoms omitted for clarity. | ||
Accordingly, the newly developed reactivity of [Zr6(OH)4O4(OMc)12] (OMc = methacrylate) (Zr6) cluster towards amide bond formation is presented here. Amide bonds are pivotal structural motifs in biomolecules, drugs, and synthetic materials.26 However, their synthesis frequently generates large amounts of waste, which also complicates product purification.27–31 The ideal amide synthesis directly from non-activated carboxylic acids and free amines, which generates water as the sole by-product, requires catalysis to efficiently circumvent its high energy barrier.32 However, most catalysts are sensitive to the water generated in the reaction, making water scavenging strategies needed not only to drive the equilibrium towards the products, but also to prevent catalyst deactivation.33,34 Thus, air and moisture stable ZrOCs like the Zr6 studied here could offer valuable insights for the future design of more robust, and efficient amidation catalysts. More importantly, Zr6 homogeneous, and discrete nature allows for detailed mechanistic studies into its structure and reactivity, providing an entry to insights typically difficult, if not impossible, to obtain with invaluable heterogeneous catalysts based on Zr6 clusters such as Zr-MOFs. For instance, the challenging amide bond formation reaction requires two substrates of completely different nature to interact with the ZrOC based catalyst, an uncommon but highly desired feature in MOF catalyzed reactions whose focus is mostly on the conversion of a single substrate.12,35–37 In line with this expectation, reactivity, and NMR data collected in this work revealed a dynamic environment at the Zr6 cluster surface, with an excess of amine enhancing the reaction output, while the nature of substrates directly impacts the reaction efficiency. Moreover, a comparative analysis of these findings with our previous reports using a Zr12 cluster, and Zr-MOFs also suggests that developing ways to control ligand equilibrium at the cluster surface should boost the reactivity of sustainable catalysts relying on the Zr6O8 unit such as Zr-MOFs.20,24
Results and discussion
[Zr6(OH)4O4(OMc)12] (OMc = methacrylate) (Zr6) was chosen as a representative Zr6O8 cluster because of its straightforward synthesis,38 and the Zr6O8 core stability upon exchange of its surface ligands (Fig. 2).39 This cluster can be seen as a Zr6O8 octahedral core coordinated by twelve carboxylate ligands. The synthesis of Zr6 was done by mixing Zr(OPr)4 in 70% w propanol with a 4-fold excess of methacrylic acid in a Schlenk flask under N2 atmosphere. After several days, crystals precipitated from the solution were collected through filtration, and washed with CH2Cl2, and toluene to remove the excess of methacrylic acid.38,39 Analysis by 1H nuclear magnetic resonance (NMR), and infrared (IR) spectroscopies were consistent with previous reports (Fig. S1–S4†).39,40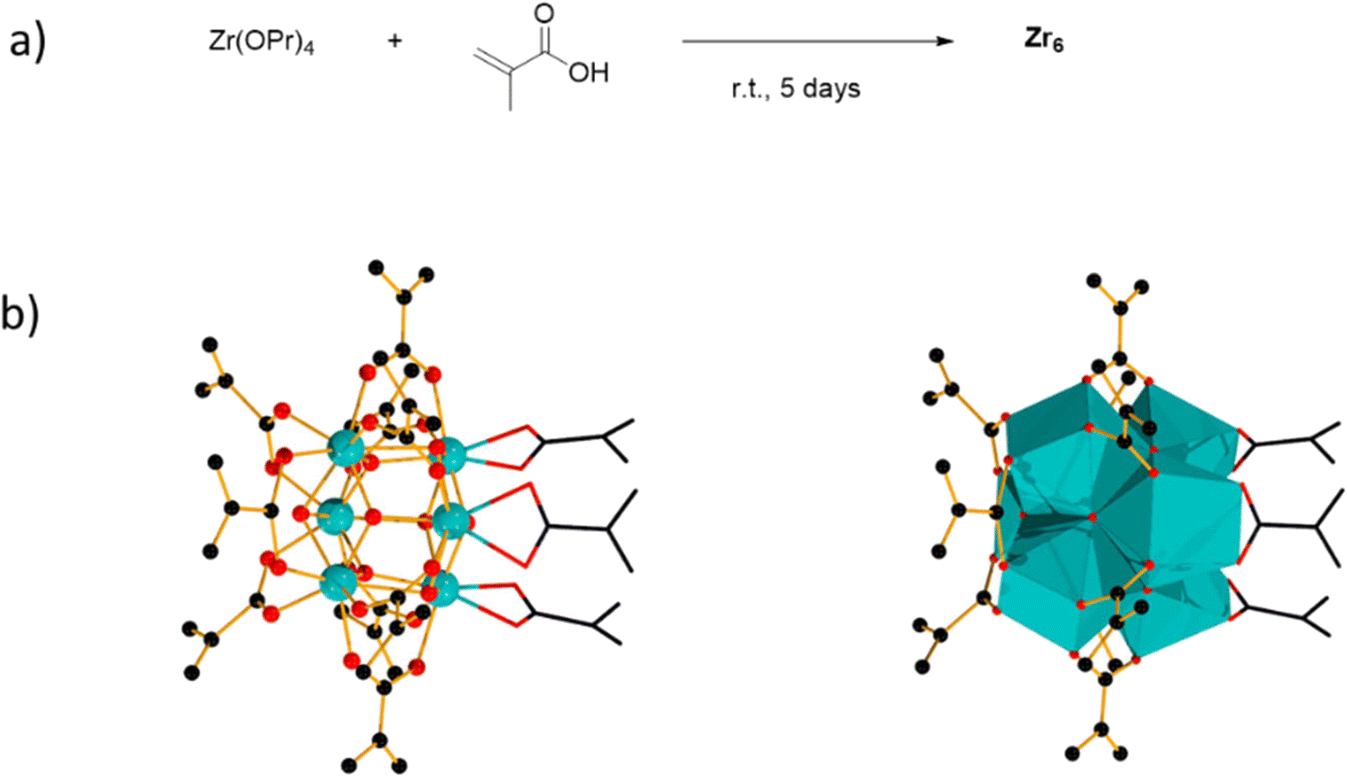 | ||
| Fig. 2 a) Synthesis of [Zr6(OH)4O4(OMc)12]2 (OMc = methacrylate) (Zr6). b) Ball and stick representation of Zr6 on the left, and the polyhedron representation of Zr6 on the right. Three chelating carboxylate ligands are represented by two-color wires. Color code: Zr (teal), O (red), C (black). Hydrogen atoms omitted for clarity. | ||
To investigate the Zr6 reactivity, we opted for an intermolecular amide bond formation using a model reaction between phenylacetic acid (1), and benzylamine (2) (Table 1).20,24 Initially, we studied the influence of solvent, and global reaction concentration on the yield of product 3 (Table 1). Firstly, we probed different solvents at 70 °C. Good to excellent yields were observed in dimethyl sulfoxide (DMSO), 1,4-dioxane, acetonitrile, toluene, diethyl ether, and tetrahydrofuran (Table 1, entries 1–6). Interestingly, a promising yield was also observed in ethanol, a biomass-derived green solvent rarely used in amidations (Table 1, entry 7).34 In addition, we investigated the effect of reaction concentration on the reaction yield (expressed as the concentration of the limiting reagent phenylacetic acid). In general, a concentration increase results in a yield increase, with the lowest amount of solvent ([1] = 3.3 M) providing the highest yield (Table 1, entry 7).21,24 These results are in line with our previous observations using metal oxo cluster based catalysts.20,21,24 Moreover, they underline ZrOCs' unique ability to provide good to excellent amide yields in alcoholic solvents, in sharp contrast with other group IV metal based catalysts.41–47 As most of the waste generated in a chemical reaction relates to the solvent used, the development of reactions using minimal amounts of benign solvents (i.e., higher reaction concentration) is highly desired from a sustainability point of view.48 Therefore, we continued our study using 3.3 M ethanolic solution as a standard condition.
|
|
|||
|---|---|---|---|
| Entry | Solvent | Conc. (mol L−1) | Yield of 3 (%) |
| a Zr6 = [Zr6(OH)4O4(OMc)12]; OMc = methacrylate. | |||
| 1 | DMSO | 3.33 | 58 |
| 2 | 1,4-Dioxane | 3.33 | 91 |
| 3 | MeCN | 3.33 | 71 |
| 4 | Toluene | 3.33 | 80 |
| 5 | Et2O | 3.33 | 89 |
| 6 | THF | 3.33 | 84 |
| 7 | EtOH | 3.33 | 65 |
| 8 | EtOH | 1.67 | 54 |
| 9 | EtOH | 0.83 | 51 |
| 10 | EtOH | 0.42 | 47 |
| 11 | EtOH | 0.21 | 23 |
The effect of the stoichiometry between acid 1 and amine 2 was also evaluated (Fig. 3). Up until 5 equivalents of 2, the yield of 3 increases, which is in line with the positive effect of an excess of amine previously reported.49 When 6 equivalents of amine were used, the yield of 3 decreased. In this case, the high concentration of amine in the solution could favor its coordination to the cluster, inducing the formation of an off-cycle species featuring no sites available for the coordination of a carboxylic acid. This subsequently affects the ability of the carboxylic acid to react with amine, and afford product. Similarly, an excess of acid 1 had a deleterious effect over the yield of 3, which is in line with the negative influence of the acid substrate on the yield of 3 reported previously.21,24,49 An excess of 1 probably has a similar effect as the excess of amine, leading to the formation of an ‘acid-rich’ off-cycle inactive species. Together, these data suggest that there is an optimal ratio that allows both substrates to interact with the cluster, which enhances the formation of product 3 (Scheme 1).
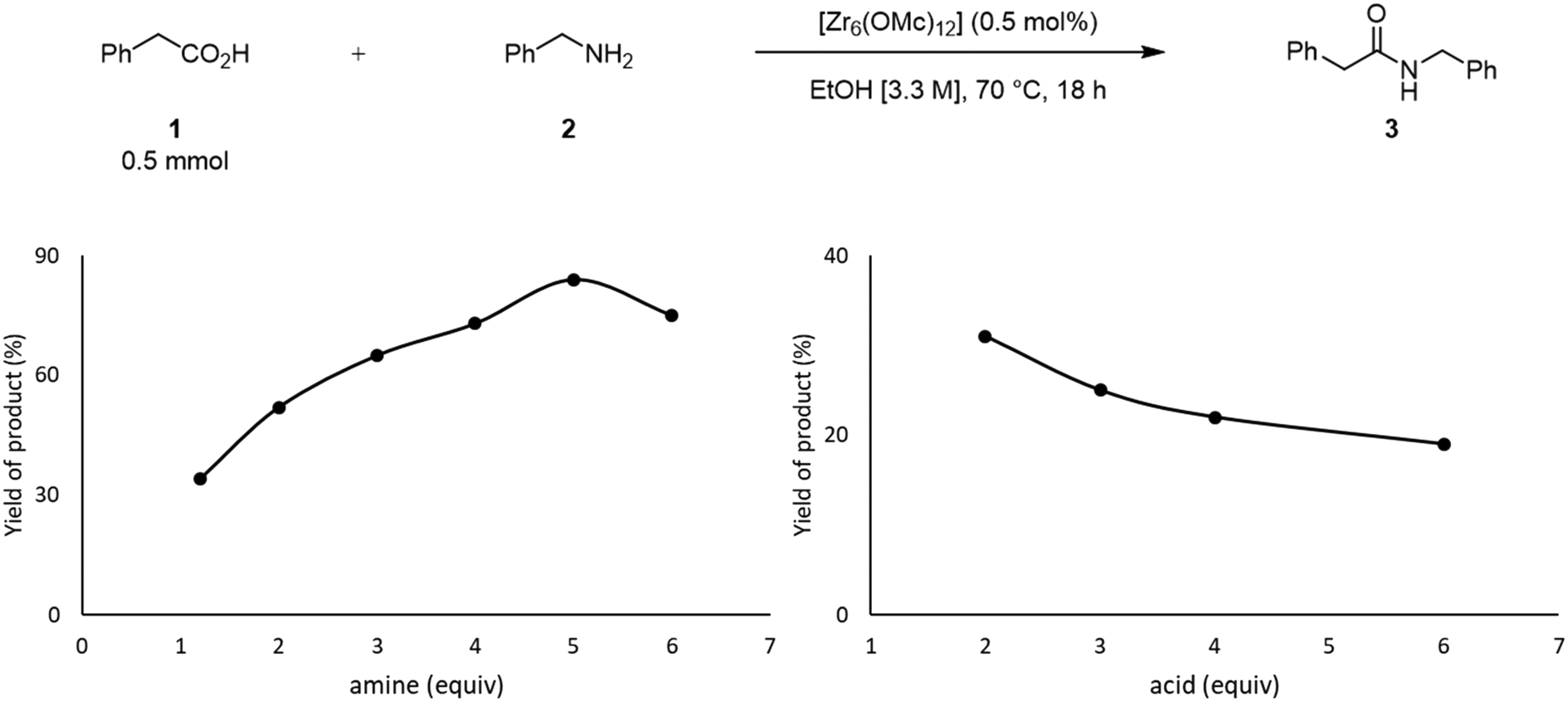 | ||
| Fig. 3 Effect of stoichiometry on the yield of product 3. | ||
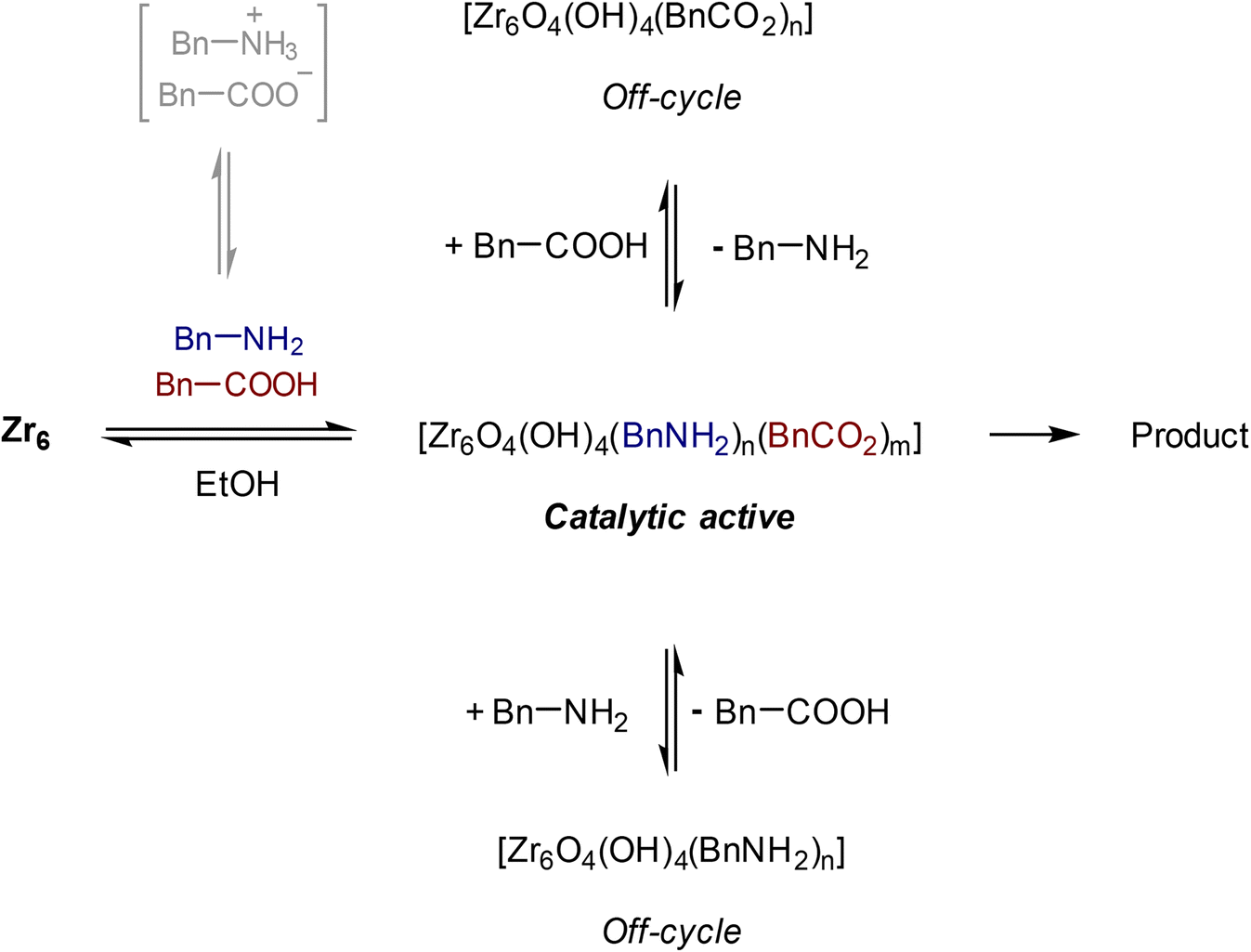 | ||
| Scheme 1 Reaction yield is maximized when amine, and carboxylic acid interact with Zr6. | ||
The stoichiometric balance boost in the reactivity is consistent with 1H NMR observations that both phenylacetic acid and benzylamine can coordinate to the cluster, and that formation of amide product 4 is only observed in the presence of an excess of amine. The effect of acid 1 and amine 2 on the 1H NMR spectrum of Zr6 were evaluated under the above reaction conditions to better understand its ligand exchange dynamics (Fig. S5 and S6†). In general, both substrates turned the Zr6 originally broad resonances into three narrow peaks consistent with methacrylic acid, suggesting the release of methacrylate ligands in solution (see ESI† for the full results). Accordingly, coordination of 1 and 2 to Zr6 was evident by the broadening of benzylic CH2 peaks between 3.00 and 4.00 ppm (3.30 ppm for 1, and 3.80–3.90 ppm for 2).50 In addition, results point to 1 and 2 coordinated with Zr6 to be in equilibrium with their counterparts free in solution. Besides the single component experiments, a solution containing both 1 and 2 was also probed (Fig. S7†). Notably, formation of product 3 was only detected when amine 2 was added in excess, consistent with the beneficial effect of increasing the amount of amine for the yield of 3.21,22,49
Interestingly, two new 1H NMR peaks appeared in the region of methacrylate ligands upon addition of amine to a Zr6 solution (Fig. 4A), suggesting that a change in the coordination mode of carboxylate ligands happened.51 Presumably, some of original bridging carboxylates converted to either chelating or monodentate binding modes in order to vacate sites for the coordination of amine (Fig. 4B). This hypothesis is consistent with a previously reported Zr6 structure in which a bridging carboxylate became monodentate to allow for the coordination of a propanol molecule.52 In addition, a similar coordination environment is also slightly more stable state in UiO-66 MOFs.53,54 Together with reactivity data pointing that reaction happens only if both reactants bind to the cluster (Scheme 1), this change in the coordination mode shows that acid 1, and amine 2 are coordinated to sites nearby each other. This suggests an ‘inner sphere mechanism’ might be operational for the C–N bond formation step.20
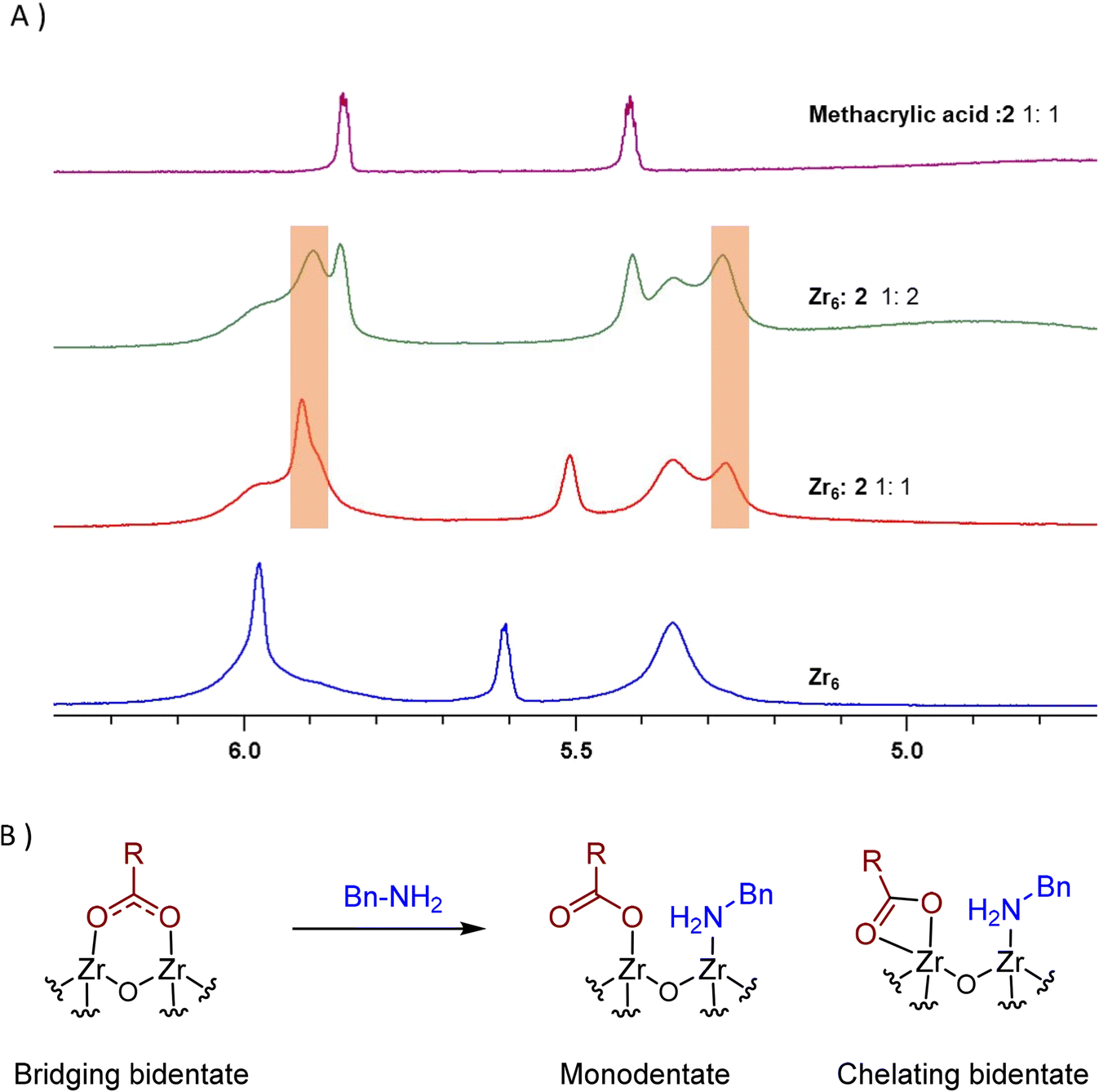 | ||
Fig. 4 New peaks (orange boxes) appearing upon addition of benzylamine (2) to Zr6 solution evidence potential change in the coordination of 1 to Zr centers of Zr6 cluster, suggesting 1 and 2 bind the cluster at proximal sites. (A) 1H NMR of a Zr6 cluster solution (0.02 M) equivalent to the amidation reaction using a catalyst loading of 2 mol% titrated with increasing amounts of 2. At the top, a mixture of methacrylic acid/benzylamine 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 is shown for comparison. (B) Changes in the ligand-cluster interactions upon amine addition hypothesized on the basis of the 1H NMR results, and literature precedents.20,52–54 :1 is shown for comparison. (B) Changes in the ligand-cluster interactions upon amine addition hypothesized on the basis of the 1H NMR results, and literature precedents.20,52–54 | ||
The large excess of amine needed to afford high yields prompted a detailed screening of conditions by varying the amount of amine, reaction temperature and time to better understand how these parameters relate to each other regarding the reaction efficiency (Fig. 5). Using ethanol as a solvent at 70 °C and a catalyst loading of 0.5 mol%, different amounts of amine and reaction times resulted in different yields. For 1.2 equivalent of amine, increasing the reaction time from 18 to 48 hours, increased the yield of product 3 from 34% to 51%, but no additional product formation was observed after one week. Similarly, in the case of 3 equivalents of amine, an increase of the reaction time from 18 hours to 48 hours raised the yield from 65% to 85%, but little improvement was obtained by extending the reaction time to one week. However, using 2 equivalents of amine, a consistent increase of the yield upon increasing the reaction time from 18 hours to 1 week was observed. Similar trends were obtained for the experiments conducted at 60 °C, while a steady increase in yield upon extension of the reaction time up to one week was observed for all amine equivalents at 50 °C. Therefore, these results show that both temperature and time will affect the reaction efficiency. Notably, even though higher yield of 3 is observed at 60 °C than at 50 °C in the first 48 h, both temperatures afford comparable yield after one week showing that the cluster remains active after one week. This is consistent with the previously reported resilience of Zr6 core structure in solution.24,55,56 Moreover, it also shows that the benefit of the temperature is largely kinetic in nature, and can be compensated by a longer reaction time. Finally, a greater impact than temperature and reaction time was observed for the amount of amine used, as a larger excess of amine clearly resulted in higher yields regardless of the reaction time. This suggests that an excess of amine is important to push reaction equilibrium towards the amide formation, presumably by favoring ligand exchanges, and the amine addition step of the reaction pathway.
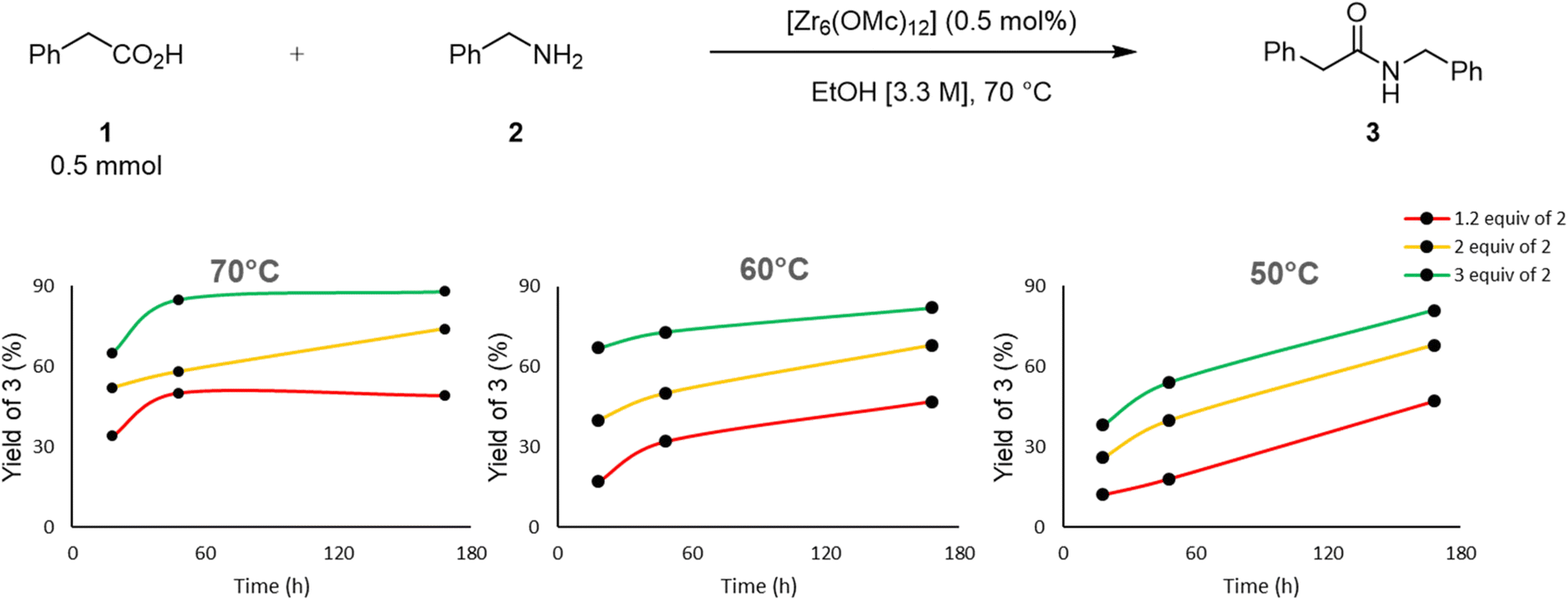 | ||
| Fig. 5 Influence of temperature and amount of amine on the yield of amide 3. | ||
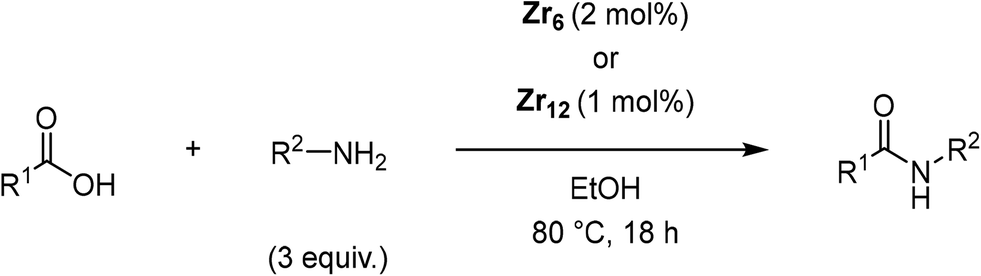
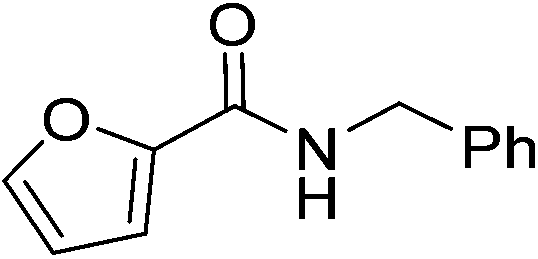
Once main reactivity trends were identified, the generality of Zr6 catalytic activity was probed using several carboxylic acids and amines (Table 2). In general, several substrates can be successfully used, but the reaction yields were significantly affected by the nature of substrates, requiring harsher conditions to provide yields comparable to those obtained with model substrates 1 and 2. For example, substituted benzylamines performed similar to simple benzylamine (2) using 0.5 mol% of Zr6, providing ca. 60% of desired amides (6–8). However, while the yield with 2 could be easily improved by increasing the Zr6 loading to 1 mol%, other amines required higher catalyst loading, temperature, and reaction time. Representative optimization carried out using 2-methoxybenzylamine (8) showed that good yields could only be obtained after using a much larger excess of amine at 80 °C for 48 h. Similar scenario was observed when using 2 as amine while varying the carboxylic acids, revealing that aliphatic acids (68–92% yields) performed better than heteroaromatic ones (32–50% yields). This unexpected susceptibility of Zr6 to the substrates' structure was also observed in 1,4-dioxane (results not shown). In addition, it is quite distinct from what we observed with the dodecanuclear Zr12 cluster, for which good to excellent yields could be obtained for a broader variety of substrates with comparatively minimal modifications to the optimized reaction conditions. This suggests that the irregular performance of Zr6 likely derives from its intrinsic reactivity rather than other factors related to the reaction conditions. For example, the decrease of Zr6 reactivity using more sterically hindered substrates might result directly from its smaller size compared to Zr12, which would slow key ligand exchanges, and subsequent affect product formation. In line with this hypothesis, the sluggish reactivity observed with aromatic acids is consistent with the low yielding synthesis reported for the Zr6 cluster bearing benzoic acid ligands.52
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Product | Equiv. amine | Catalyst (mol%) | T (°C) | Time (h) | Yield (%) | |
| 1 |
|
(3) | 3 | 0.5 | 70 | 24 | 65 |
| 3 | 1.0 | 70 | 48 | 85 | |||
| 2 |
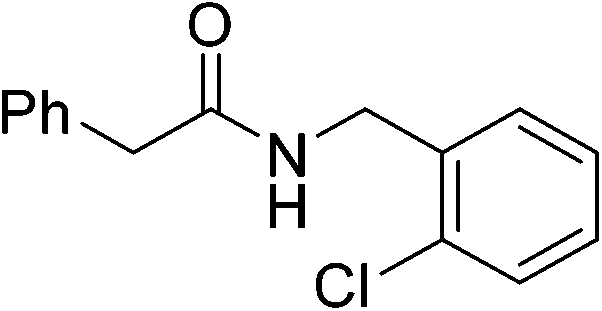
|
(6) | 3 | 0.5 | 70 | 24 | 60 |
| 3 |
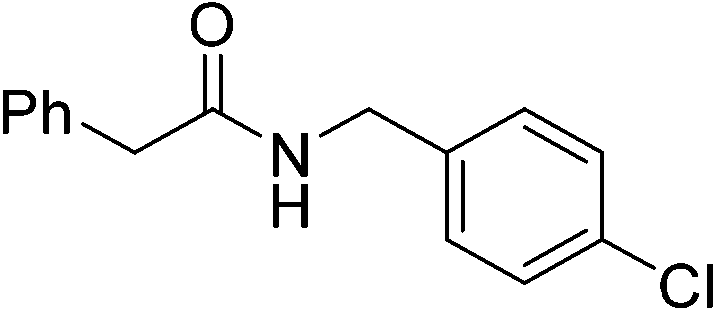
|
(7) | 3 | 0.5 | 70 | 24 | 50 |
| 3 | 2.0 | 80 | 24 | 69 | |||
| 4 |
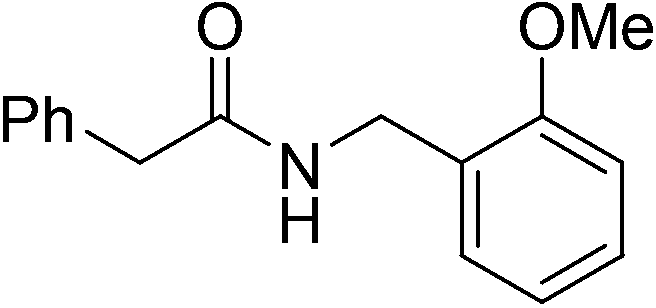
|
(8) | 3 | 0.5 | 70 | 24 | 56 |
| 3 | 2.0 | 80 | 24 | 57 | |||
| 5 | 2.0 | 80 | 48 | 89 | |||
| 5 |
|
(9) | 3 | 0.5 | 70 | 24 | 82 |
| 6 |
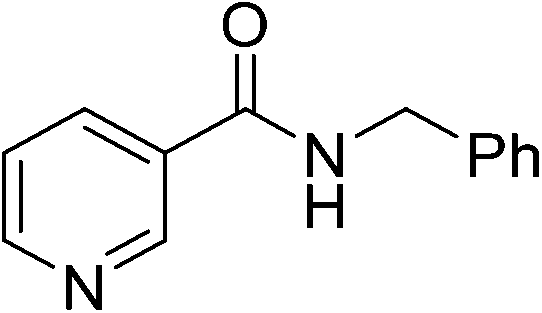
|
(10) | 3 | 2.0 | 80 | 24 | 30 |
| 5 | 2.0 | 80 | 26 | 49 | |||
| 7 |
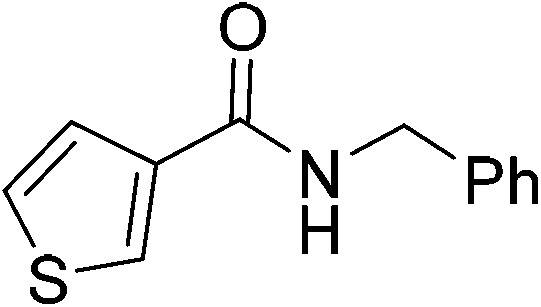
|
(11) | 5 | 2.0 | 80 | 26 | 50 |
| 8 |
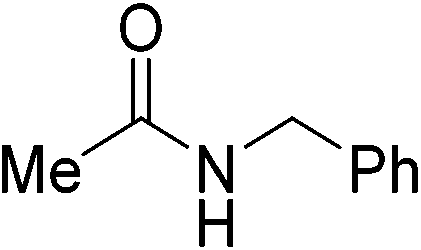
|
(12) | 3 | 1.0 | 80 | 24 | 54 |
| 3 | 2.0 | 80 | 24 | 58 | |||
| 5 | 2.0 | 80 | 26 | 92 | |||
| 9 |
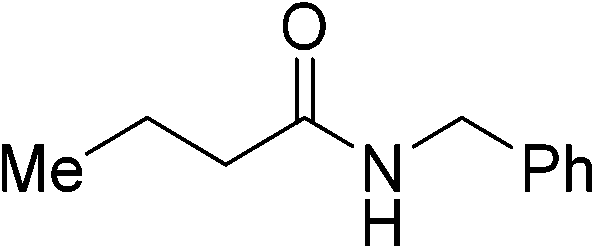
|
(13) | 3 | 2.0 | 80 | 24 | 37 |
| 5 | 2.0 | 80 | 26 | 61 | |||
| 5 | 2.0 | 80 | 48 | 85 | |||
| 10 |
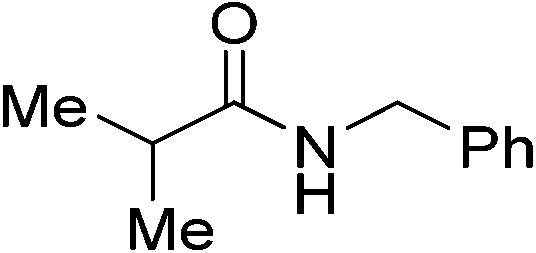
|
(14) | 5 | 2.0 | 80 | 26 | 49 |
| 5 | 2.0 | 80 | 48 | 68 | |||
| 11 |
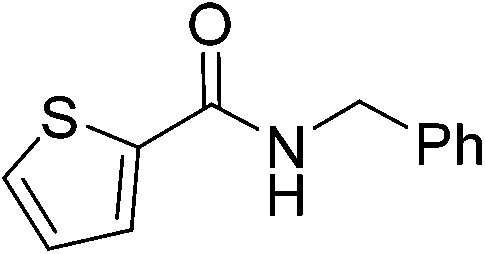
|
(15) | 5 | 2.0 | 80 | 48 | 50 |
| 12 |
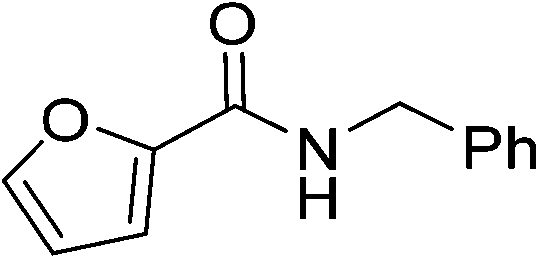
|
(16) | 3 | 2.0 | 80 | 18 | 30 |
Intrigued by the fickle reactivity of Zr6, we compared it with the Zr12 cluster studied previously ([Zr6(OH)4O4(OAcr)12]2, OAcr = acrylate) (Fig. 1).24 Specifically, Zr6 and Zr12 clusters were compared as catalysts using representative substrates under the same conditions. Importantly, an equivalent load of Zr (1 mol% Zr12vs. 2 mol% Zr6) was used to avoid yield differences to be caused by an excess of catalytic sites available in the reaction (see Table S1† for control experiments using equivalent loadings of different Zr salts). For the reaction between 2-furoic acid and benzylamine (2), similar performances were observed (Table 3). Given that the only cluster bearing aromatic carboxylate ligands reported so far is a hexazirconium one,52 this similarity suggests that both clusters are generating the same active hexanuclear ZrOC species in situ. Although the dissociation of a dodecanuclear cluster into two independent hexanuclear units contradicts previous findings, this hypothesis would explain the systematically lower yields obtained with aromatic acid substrates compared with aliphatic ones (Table 2).57 In contrast, Zr12 provided a 29% higher yield than Zr6 (98% vs. 69%, respectively) for the reaction between 1 and 4-chlorobenzylamine (7). As both reactions were carried out with the same load of Zr, these results clearly indicate the reactions were catalyzed by different species, and that Zr12 is a more efficient catalyst in this case. Ultimately, these data collectively shows Zr12 is more reactive than Zr6.56
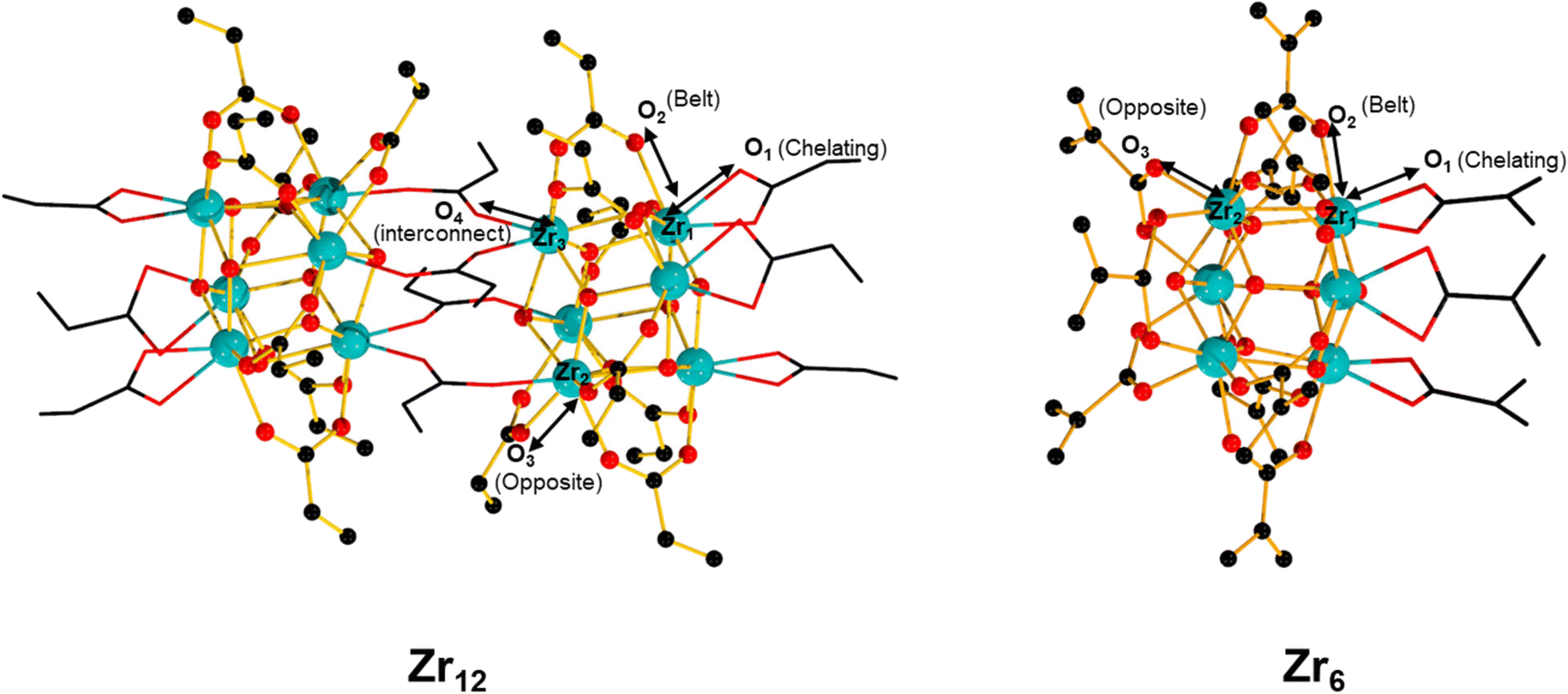
A possible explanation for the lower efficiency of Zr6 compared to Zr12 may arise from its stronger metal–ligand interactions, which would retard key ligand exchanges needed for the reaction. Comparison of Zr–O bond lengths for similarly coordinated carboxylates in Zr6 (ref. 58) and Zr12 (ref. 52) crystal structures evidences the bonds in Zr12 cluster are generally longer than in the Zr6 ones, which indicates carboxylates are more strongly attached to the Zr6O8 inorganic core in Zr6 than in Zr12 (Table 4). This could impact the reactivity of Zr6 in two distinct ways: i) slower ligand exchange kinetics, delaying the formation of product; ii) less favored coordination of the amine in comparison to the carboxylic acid substrate, which agrees with the large excess of amine needed to obtain the products in good yield. Alternatively, ligand exchange differences could also arise from different steric demands around the cluster, given the probable associative mechanism of exchange in this case.59 Thus, further studies on the ligand exchange of these clusters that succeed in identifying ways to streamline ligand exchange steps could enhance the catalytic activity of Zr6 clusters. These studies could include, for example, the use of Lewis basic additives that mimic the effect of amine excess or engineering ligands based on functional groups other than a carboxylate group as a binding moiety, whose distinct affinity for the cluster would facilitate ligand exchange steps.60
|
|
||
|---|---|---|
| Bond | Zr 12 (Å) | Zr 6 (Å) |
| a Ball and stick representation of Zr6 and Zr12. The chelating and the interconnected bridging carboxylate are represented by two-color wires. Color code: Zr (teal), O (red), C (black). Hydrogen atoms omitted for clarity. | ||
| Zr1–O1(chelating) | 2.2707–2.3191 | 2.3297–2.3298 |
| Zr2–O2(belt) | 2.1916–2.2318 | 2.1293–2.1935 |
| Zr2–O3(opposite) | 2.2134–2.2184 | 2.1004–2.1005 |
| Zr3–O4(interconnect) | 2.1595–2.2103 | — |
| Zr–O(core) | 2.0260–2.4087 | 2.0504–2.4109 |
Finally, the reactivity disclosed here towards the formation of intermolecular amide bonds has important similarities to the one observed using Zr-MOF UiO-66.20 Both catalysts required three equivalents of amine to provide the product in good yield, and performed well in 1,4-dioxane. This indicates the reactivity of both systems is relatable, and greater understanding of the reactivity of discrete clusters could enlighten the development of more efficient MOF catalysts. Notably, the low efficiency observed for UiO-66 in EtOH clearly indicates that factors other than the intrinsic reactivity of [Zr6O4(OH)4], e.g. the diffusion of products and substrates throughout the porous network, also play an important role in the reaction outcome. However, comparing the discrete clusters used in this work with representative Zr-MOFs (UiO-66, MOF-808, and NU-1000) indicates that subtle structural features might contribute to the reactivity difference. In Zr-MOFs, the lability of coordinated water and formate ligands bound to the Zr6O8 core plays a crucial role into their reactivity.53 Comparison of Zr–O(H2O/formate) bond lengths with the ones in Zr6 suggests that at least some of the carboxylates in Zr6 are more easily exchanged, which could result in a faster reaction as discussed above (Table S2†). Although other factors might influence the ligand exchanges in Zr6 embedded in MOFs,59 this comparison indicates once again that finding ways to streamline ligand exchange steps could enhance the catalytic activity of Zr6 clusters, and by extension boost the reactivity of Zr-MOF catalysts as well.
Conclusion
In summary, this study on the intrinsic reactivity of hexanuclear discrete zirconium oxo clusters showcases their value as archetype structures for the development of novel Zr-based catalysts with enhanced robustness and utility. Here, the cluster [Zr6(OH)4O4(OMc)12] (OMc = methacrylate) (Zr6), which is isostructural to Zr6O8 nodes found in a large family of Zr-MOFs, was shown to efficiently catalyze the formation of amide bonds without requiring anhydrous conditions, and water scavenging techniques. Remarkably, the cluster hydrolytic stability rendered the reaction feasible in ethanol, a green solvent previously inaccessible for other metal catalysts.More importantly, a comparative analysis of the structural and reactivity features of Zr6 with Zr12 cluster and the UiO-66 Zr-MOF, both used previously as catalysts for similar amide formations, revealed that the reactivity of these three systems is relatable. Thus, discrete ZrOC provide a rather unique insight into the chemistry of broadly useful Zr-MOFs catalysts. Specifically, the soluble and discrete nature of Zr6 allowed us to obtain molecular level insight into factors influencing a challenging bimolecular reaction between substrates of completely different nature, a scenario scarcely evaluated with MOF catalysts, and otherwise challenging to be performed in heterogeneous systems. In summary, such analysis revealed the following: i) for a bimolecular reaction such as the amide bond formation probed here, an optimal stoichiometric ratio that allows both substrates to interact with the cluster surface is of paramount importance for the reaction yield; ii) Zr6 reactivity was largely affected by the structure of substrates, in line with our previous study using UiO-66 MOF; iii) under the same conditions, Zr6 was less reactive than Zr12, but more reactive than UiO-66 MOFs, indicating that intrinsic features of the cluster, and of the material network play a role in the reactivity; and iv) comparison of Zr–O bond lengths in all systems, strongly suggests that reactivity is related to the strength of ligand coordination, presumably due to its influence on key ligand exchanges. Thus, the slightly slower ligand exchange in Zr6 due to stronger [Zr6O8]-ligand interactions is an intrinsic feature of these clusters that, if addressed properly, should result in more efficient ZrOC, and Zr-MOF catalysts.
Combined with the various previous studies on MOF structure–activity relationships, these findings underline molecular-level design elements that should be considered in the development of ZrOC-based catalysts. Further studies on the fundamental reactivity of discrete ZrOC should contribute to a deeper molecular understanding of Zr-MOF family of catalysts based on ZrOC building blocks.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank KU Leuven and Research Foundation Flanders (FWO) for generous financial support. Y. Z. thanks the Chinese Scholar Council (CSC) for doctoral fellowship (201804910511). F. d. A. thanks the FWO for fellowship (195931/1281921N). I. Y. K. thanks The Scientific and Technological Research Council of Turkey (TUBITAK) for fellowship (1059B052001345), and Bilkent University for additional grant support.References
- Y. Zhang, F. de Azambuja and T. N. Parac-Vogt, The forgotten chemistry of group(IV) metals: A survey on the synthesis, structure, and properties of discrete Zr(IV), Hf(IV), and Ti(IV) oxo clusters, Coord. Chem. Rev., 2021, 438, 213886, DOI:10.1016/j.ccr.2021.213886.
- D. Van den Eynden, R. Pokratath and J. De Roo, Nonaqueous Chemistry of Group 4 Oxo Clusters and Colloidal Metal Oxide Nanocrystals, Chem. Rev., 2022, 122, 10538–10572, DOI:10.1021/acs.chemrev.1c01008.
- U. Schubert, Polymers Reinforced by Covalently Bonded Inorganic Clusters, Chem. Mater., 2001, 13, 3487–3494, DOI:10.1021/cm001258r.
- U. Schubert, Cluster-based inorganic–organic hybrid materials, Chem. Soc. Rev., 2011, 40, 575–582, 10.1039/C0CS00009D.
- M. Carraro and S. Gross, Hybrid Materials Based on the Embedding of Organically Modified Transition Metal Oxoclusters or Polyoxometalates into Polymers for Functional Applications: A Review, Materials, 2014, 7, 3956–3989 CrossRef , https://www.mdpi.com/1996-1944/7/5/3956.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability, J. Am. Chem. Soc., 2008, 130, 13850–13851, DOI:10.1021/ja8057953.
- V. Guillerm, S. Gross, C. Serre, T. Devic, M. Bauer and G. Férey, A zirconium methacrylate oxocluster as precursor for the low-temperature synthesis of porous zirconium(iv) dicarboxylates, Chem. Commun., 2010, 46, 767–769, 10.1039/B914919H.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Zr-based metal–organic frameworks: design, synthesis, structure, and applications, Chem. Soc. Rev., 2016, 45, 2327–2367, 10.1039/C5CS00837A.
- S. Dai, C. Simms, I. Dovgaliuk, G. Patriarche, A. Tissot, T. N. Parac-Vogt and C. Serre, Monodispersed MOF-808 Nanocrystals Synthesized via a Scalable Room-Temperature Approach for Efficient Heterogeneous Peptide Bond Hydrolysis, Chem. Mater., 2021, 33, 7057–7066, DOI:10.1021/acs.chemmater.1c02174.
- X. Feng, H. S. Jena, C. Krishnaraj, K. Leus, G. Wang, H. Chen, C. Jia and P. Van Der Voort, Generating Catalytic Sites in UiO-66 through Defect Engineering, ACS Appl. Mater. Interfaces, 2021, 13, 60715–60735, DOI:10.1021/acsami.1c13525.
- M. Rimoldi, A. J. Howarth, M. R. DeStefano, L. Lin, S. Goswami, P. Li, J. T. Hupp and O. K. Farha, Catalytic Zirconium/Hafnium-Based Metal–Organic Frameworks, ACS Catal., 2017, 7, 997–1014, DOI:10.1021/acscatal.6b02923.
- K. O. Kirlikovali, Z. Chen, T. Islamoglu, J. T. Hupp and O. K. Farha, Zirconium-Based Metal–Organic Frameworks for the Catalytic Hydrolysis of Organophosphorus Nerve Agents, ACS Appl. Mater. Interfaces, 2020, 12, 14702–14720, DOI:10.1021/acsami.9b20154.
- H. G. T. Ly, G. Fu, A. Kondinski, B. Bueken, D. De Vos and T. N. Parac-Vogt, Superactivity of MOF-808 toward Peptide Bond Hydrolysis, J. Am. Chem. Soc., 2018, 140, 6325–6335, DOI:10.1021/jacs.8b01902.
- A. Loosen, F. de Azambuja, S. Smolders, J. Moons, C. Simms, D. De Vos and T. N. Parac-Vogt, Interplay between structural parameters and reactivity of Zr6-based MOFs as artificial proteases, Chem. Sci., 2020, 11, 6662–6669, 10.1039/D0SC02136A.
- H. G. T. Ly, G. Fu, F. de Azambuja, D. E. De Vos and T. N. Parac-Vogt, Nanozymatic Activity of UiO-66 Metal-Organic Frameworks: Tuning the Nanopore Environment Enhances Hydrolytic Activity toward Peptide Bonds, ACS Appl. Nano Mater., 2020, 3, 8931–8938, DOI:10.1021/acsanm.0c01688.
- J. Moons, A. Loosen, C. Simms, F. de Azambuja and T. Parac-Vogt, Heterogeneous nanozymatic activity of Hf oxo-clusters embedded in a metal-organic framework towards peptide bond hydrolysis, Nanoscale, 2021, 13, 12298–12305, 10.1039/D1NR01790J.
- C. Simms, F. de Azambuja and T. N. Parac-Vogt, Enhancing the Catalytic Activity of MOF-808 Towards Peptide Bond Hydrolysis through Synthetic Modulations, Chem. – Eur. J., 2021, 27, 17230–17239, DOI:10.1002/chem.202103102.
- S. Wang, H. G. T. Ly, M. Wahiduzzaman, C. Simms, I. Dovgaliuk, A. Tissot, G. Maurin, T. N. Parac-Vogt and C. Serre, A zirconium metal-organic framework with SOC topological net for catalytic peptide bond hydrolysis, Nat. Commun., 2022, 13, 1284, DOI:10.1038/s41467-022-28886-5.
- C. Simms, N. Savić, K. De Winter and T. N. Parac-Vogt, Understanding the role of surfactants in the interaction and hydrolysis of myoglobin by Zr-MOF-808, Eur. J. Inorg. Chem., 2022, e202200145, DOI:10.1002/ejic.202200145.
- F. de Azambuja, A. Loosen, D. Conic, M. van den Besselaar, J. N. Harvey and T. N. Parac-Vogt, En Route to a Heterogeneous Catalytic Direct Peptide Bond Formation by Zr-Based Metal–Organic Framework Catalysts, ACS Catal., 2021, 11, 7647–7658, DOI:10.1021/acscatal.1c01782.
- F. de Azambuja and T. N. Parac-Vogt, Water-Tolerant and Atom Economical Amide Bond Formation by Metal-Substituted Polyoxometalate Catalysts, ACS Catal., 2019, 9, 10245–10252, DOI:10.1021/acscatal.9b03415.
- F. de Azambuja, J. Lenie and T. N. Parac-Vogt, Homogeneous Metal Catalysts with Inorganic Ligands: Probing Ligand Effects in Lewis Acid Catalyzed Direct Amide Bond Formation, ACS Catal., 2021, 11, 271–277, DOI:10.1021/acscatal.0c04189.
- F. de Azambuja, J. Moons and T. N. Parac-Vogt, The Dawn of Metal-Oxo Clusters as Artificial Proteases: From Discovery to the Present and Beyond, Acc. Chem. Res., 2021, 54, 1673–1684, DOI:10.1021/acs.accounts.0c00666.
- Y. Zhang, F. de Azambuja and T. N. Parac-Vogt, Zirconium oxo clusters as discrete molecular catalysts for the direct amide bond formation, Catal. Sci. Technol., 2022, 12, 3190–3201, 10.1039/D2CY00421F.
- D. Conic, K. Pierloot, T. N. Parac-Vogt and J. N. Harvey, Mechanism of the highly effective peptide bond hydrolysis by MOF-808 catalyst under biologically relevant conditions, Phys. Chem. Chem. Phys., 2020, 22, 25136–25145, 10.1039/D0CP04775A.
- V. R. Pattabiraman and J. W. Bode, Rethinking Amide Bond Synthesis, Nature, 2011, 480, 471, DOI:10.1038/nature10702.
- X. Wang, Challenges and Outlook for Catalytic Direct Amidation Reactions, Nat. Catal., 2019, 2, 98–102, DOI:10.1038/s41929-018-0215-1.
- A. El-Faham and F. Albericio, Peptide Coupling Reagents, More than a Letter Soup, Chem. Rev., 2011, 111, 6557–6602, DOI:10.1021/cr100048w.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Large-Scale Applications of Amide Coupling Reagents for the Synthesis of Pharmaceuticals, Org. Process Res. Dev., 2016, 20, 140–177, DOI:10.1021/op500305s.
- C. L. Allen and J. M. J. Williams, Metal-Catalysed Approaches to Amide Bond Formation, Chem. Soc. Rev., 2011, 40, 3405–3415, 10.1039/C0CS00196A.
- R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Nonclassical Routes for Amide Bond Formation, Chem. Rev., 2016, 116, 12029–12122, DOI:10.1021/acs.chemrev.6b00237.
- E. Valeur and M. Bradley, Amide Bond Formation: Beyond the Myth of Coupling Reagents, Chem. Soc. Rev., 2009, 38, 606–631, 10.1039/B701677H.
- H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Catalytic Amide Formation from Non-Activated Carboxylic Acids and Amines, Chem. Soc. Rev., 2014, 43, 2714–2742, 10.1039/C3CS60345H.
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, A Green Chemistry Perspective on Catalytic Amide Bond Formation, Nat. Catal., 2019, 2, 10–17, DOI:10.1038/s41929-018-0211-5.
- D. Yang and B. C. Gates, Catalysis by Metal Organic Frameworks: Perspective and Suggestions for Future Research, ACS Catal., 2019, 9, 1779–1798, DOI:10.1021/acscatal.8b04515.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Metal–Organic Frameworks in Heterogeneous Catalysis: Recent Progress, New Trends, and Future Perspectives, Chem. Rev., 2020, 120, 8468–8535, DOI:10.1021/acs.chemrev.9b00685.
- V. Pascanu, G. González Miera, A. K. Inge and B. Martín-Matute, Metal–Organic Frameworks as Catalysts for Organic Synthesis: A Critical Perspective, J. Am. Chem. Soc., 2019, 141, 7223–7234, DOI:10.1021/jacs.9b00733.
- G. Kickelbick and U. Schubert, Oxozirconium Methacrylate Clusters: Zr6(OH)4O4(OMc)12 and Zr4O2(OMc)12 (OMc = Methacrylate), Chem. Ber., 1997, 130, 473–478, DOI:10.1002/cber.19971300406.
- F. R. Kogler, M. Jupa, M. Puchberger and U. Schubert, Control of the ratio of functional and non-functional ligands in clusters of the type Zr6O4(OH)4(carboxylate)12 for their use as building blocks for inorganic–organic hybrid polymers, J. Mater. Chem., 2004, 14, 3133–3138, 10.1039/B405769D.
- K. Epp, A. L. Semrau, M. Cokoja and R. A. Fischer, Dual Site Lewis-Acid Metal-Organic Framework Catalysts for CO2 Fixation: Counteracting Effects of Node Connectivity, Defects and Linker Metalation, ChemCatChem, 2018, 10, 3506–3512, DOI:10.1002/cctc.201800336.
- C. L. Allen, A. R. Chhatwal and J. M. J. Williams, Direct Amide Formation from Unactivated Carboxylic Acids and Amines, Chem. Commun., 2012, 48, 666–668, 10.1039/C1CC15210F.
- H. Lundberg, F. Tinnis and H. Adolfsson, Direct Amide Coupling of Non-activated Carboxylic Acids and Amines Catalysed by Zirconium(IV) Chloride, Chem. – Eur. J., 2012, 18, 3822–3826, DOI:10.1002/chem.201104055.
- H. Lundberg, F. Tinnis and H. Adolfsson, Titanium(IV) Isopropoxide as an Efficient Catalyst for Direct Amidation of Nonactivated Carboxylic Acids, Synlett, 2012, 23, 2201–2204 CrossRef CAS.
- F. Tinnis, H. Lundberg and H. Adolfsson, Direct Catalytic Formation of Primary and Tertiary Amides from Non-Activated Carboxylic Acids, Employing Carbamates as Amine Source, Adv. Synth. Catal., 2012, 354, 2531–2536, DOI:10.1002/adsc.201200436.
- H. Lundberg and H. Adolfsson, Hafnium-Catalyzed Direct Amide Formation at Room Temperature, ACS Catal., 2015, 5, 3271–3277, DOI:10.1021/acscatal.5b00385.
- N. Li, L. Wang, L. Zhang, W. Zhao, J. Qiao, X. Xu and Z. Liang, Air-stable Bis(pentamethylcyclopentadienyl) Zirconium Perfluorooctanesulfonate as an Efficient and Recyclable Catalyst for the Synthesis of N-substituted Amides, ChemCatChem, 2018, 10, 3532–3538, DOI:10.1002/cctc.201800590.
- H. Lundberg, F. Tinnis and H. Adolfsson, Zirconium catalyzed amide formation without water scavenging, Appl. Organomet. Chem., 2019, 33, e5062, DOI:10.1002/aoc.5062.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Green and Sustainable Solvents in Chemical Processes, Chem. Rev., 2018, 118, 747–800, DOI:10.1021/acs.chemrev.7b00571.
- H. Lundberg, F. Tinnis, J. Zhang, A. G. Algarra, F. Himo and H. Adolfsson, Mechanistic Elucidation of Zirconium-Catalyzed Direct Amidation, J. Am. Chem. Soc., 2017, 139, 2286–2295, DOI:10.1021/jacs.6b10973.
- J. De Roo, N. Yazdani, E. Drijvers, A. Lauria, J. Maes, J. S. Owen, I. Van Driessche, M. Niederberger, V. Wood, J. C. Martins, I. Infante and Z. Hens, Probing Solvent–Ligand Interactions in Colloidal Nanocrystals by the NMR Line Broadening, Chem. Mater., 2018, 30, 5485–5492, DOI:10.1021/acs.chemmater.8b02523.
- M. S. Refat, S. A. El-Korashy, D. N. Kumar and A. S. Ahmed, FTIR, magnetic, 1H NMR spectral and thermal studies of some chelates of caproic acid: Inhibitory effect on different kinds of bacteria, Spectrochim. Acta, Part A, 2008, 70, 217–233, DOI:10.1016/j.saa.2007.07.036.
- G. Kickelbick, P. Wiede and U. Schubert, Variations in capping the Zr6O4(OH)4 cluster core: X-ray structure analyses of [Zr6(OH)4O4(OOC–CH=CH2)10]2(μ-OOC–CH=CH2)4 and Zr6(OH)4O4(OOCR)12(PrOH) (R=Ph, CMe=CH2), Inorg. Chim. Acta, 1999, 284, 1–7, DOI:10.1016/S0020-1693(98)00251-5.
- K. Tan, H. Pandey, H. Wang, E. Velasco, K.-Y. Wang, H.-C. Zhou, J. Li and T. Thonhauser, Defect Termination in the UiO-66 Family of Metal–Organic Frameworks: The Role of Water and Modulator, J. Am. Chem. Soc., 2021, 143, 6328–6332, DOI:10.1021/jacs.1c01408.
- Y. Fu, Z. Kang, J. Yin, W. Cao, Y. Tu, Q. Wang and X. Kong, Duet of Acetate and Water at the Defects of Metal–Organic Frameworks, Nano Lett., 2019, 19, 1618–1624, DOI:10.1021/acs.nanolett.8b04518.
- J. Kreutzer, M. Puchberger, C. Artner and U. Schubert, Retention of the Cluster Core Structure during Ligand Exchange Reactions of Carboxylato-Substituted Metal Oxo Clusters, Eur. J. Inorg. Chem., 2015, 2015, 2145–2151, DOI:10.1002/ejic.201403209.
- F. Faccioli, M. Bauer, D. Pedron, A. Sorarù, M. Carraro and S. Gross, Hydrolytic Stability and Hydrogen Peroxide Activation of Zirconium-Based Oxoclusters, Eur. J. Inorg. Chem., 2015, 2015, 210–225, DOI:10.1002/ejic.201402767.
- M. Puchberger, F. R. Kogler, M. Jupa, S. Gross, H. Fric, G. Kickelbick and U. Schubert, Can the Clusters Zr6O4(OH)4(OOCR)12 and [Zr6O4(OH)4(OOCR)12]2 Be Converted into Each Other?, Eur. J. Inorg. Chem., 2006, 2006, 3283–3293, DOI:10.1002/ejic.200600348.
- G. Kickelbick and U. Schubert, Oxozirconium Methacrylate Clusters: Zr6(OH)4O4(OMc) and Zr4O2(OMc)12 (OMc = Methacrylate), Chem. Ber., 1997, 130, 473–478, DOI:10.1002/cber.19971300406.
- C.-C. Chiu, F.-K. Shieh and H.-H. G. Tsai, Ligand Exchange in the Synthesis of Metal–Organic Frameworks Occurs Through Acid-Catalyzed Associative Substitution, Inorg. Chem., 2019, 58, 14457–14466, DOI:10.1021/acs.inorgchem.9b01947.
- X. Feng, Y. Song and W. Lin, Dimensional Reduction of Lewis Acidic Metal–Organic Frameworks for Multicomponent Reactions, J. Am. Chem. Soc., 2021, 143, 8184–8192, DOI:10.1021/jacs.1c03561.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplementary experiments, copy of NMR spectra (PDF). See DOI: https://doi.org/10.1039/d2cy01706g |
| This journal is © The Royal Society of Chemistry 2023 |