Native functional group directed distal C(sp3)–H activation of aliphatic systems
Received
4th October 2022
, Accepted 20th October 2022
First published on 20th October 2022
Abstract
Aliphatic systems are ubiquitous in numerous natural products and pharmacoactive compounds. Selective remote C–H functionalization of such aliphatic chains is of great significance and remains a challenge to be addressed. The native functional group assisted C(sp3)–H activation has come up as one of the most practical and simpler approaches to transforming aliphatic compounds as it eliminates the need for an external directing group, thereby making the process more step and atom economic. In this regard, aliphatic acids and amines have gained tremendous attention in the recent past due to the unique coordination abilities of carboxylate and amine groups with transition metals. We have summarized here all the concepts and applications reported in the remote C(sp3)–H functionalization of aliphatic systems in recent times. The systematic presentation here is expected to provide a deeper understanding to beginners as well as experts interested in this exciting search field.
1. Introduction
Aliphatic chains with carboxylic acids and amine functionalities are one of organic chemistry's most ubiquitous and vital moieties. This is due to their presence in an enormous number of highly demanding functional materials, pharmaceuticals, natural products, and other biologically active molecules.1 For this reason, the functionalization and derivatisation of such molecules have captivated organic chemists for a long time – particularly the installation of functional groups on hydrocarbons. Although traditional methods using the inherent reactivity of the amine and carboxylic acids have been developed in due course, such synthetic routes come with severe drawbacks – a) inertness of the unactivated C–H bond and b) low yield due to the copious number of steps involved.2 Intending to mitigate this problem and due to the increasing popularity of metal-induced organic transformations, the late 20th century witnessed an alternative sustainable approach to C–H functionalization aided by transition metals. The direct conversion of simple substrates to more complex molecules without involving the pre-functionalization of the molecules makes this method more efficient, step economic and less time-consuming, as a result of which, it finds its diverse application in both academic and industrial arenas.3
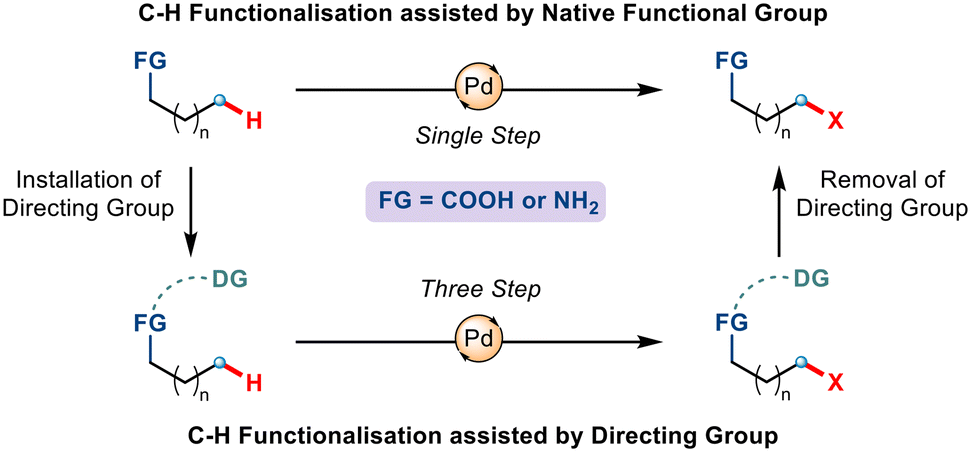
Although metal-catalyzed C–H functionalization has emerged as a powerful tool for the diversification of carboxylic acids and amines, functionalization at one particular position, regioselectively, is the most crucial yet challenging task. This has attracted the implementation of directing groups (DGs) whereby a Lewis basic motif coordinates with the metal centre to dictate the metal to selectively convert a C–H bond to a C–M bond through the formation of a cyclometallated complex.4 Despite being conceptually appealing, the use of such exogenous directing groups requires additional steps of incorporation and removal. Interestingly, if the directing ability of the native functional group already present could be utilized, then the transformation could be achieved in a single and more efficient step (Scheme 1). Among all the functional groups, the carboxylate and amine groups have shown promising results when it comes to coordinating with transition metals and thereby assisting in activating C–H bonds.
 |
| | Scheme 1 Design of native functional groups and pre-installed directing groups in assisting C–H functionalization. | |
Among the various transition metals, palladium salts have been reported to show extraordinary reactivity in cleaving C(sp3)–H bonds either via an inter- or intra-molecular fashion.5 So far, a number of strategies have been reported that demonstrate C–H functionalization at the proximal positions (α and β positions). Whilst functionalization at α and β positions involve a thermodynamically stable five-membered palladacycle, functionalization at γ and δ positions demand the formation of a larger palladacycle which is relatively less stable than smaller palladacycles.6 Nevertheless, there have been a few reports by some groups which exhibit the ability of free carboxylic acids and amines to act as a director for C(sp3)–H functionalization. No other functional group except carboxylate and amine is known yet to activate C(sp3)–H bonds without the usage of extra auxiliaries. A number of multifarious reviews are available that highlight the proximal C(sp3)–H functionalization of aliphatic systems.7 We have focused here on the C(sp3)–H activation at remote sites that are beyond α and β positions.
This review encompasses such reports and highlights the recent advancements and developments of the native functional group-directed remote C–H activation along with mechanistic insights.
2. Native amine directed strategy
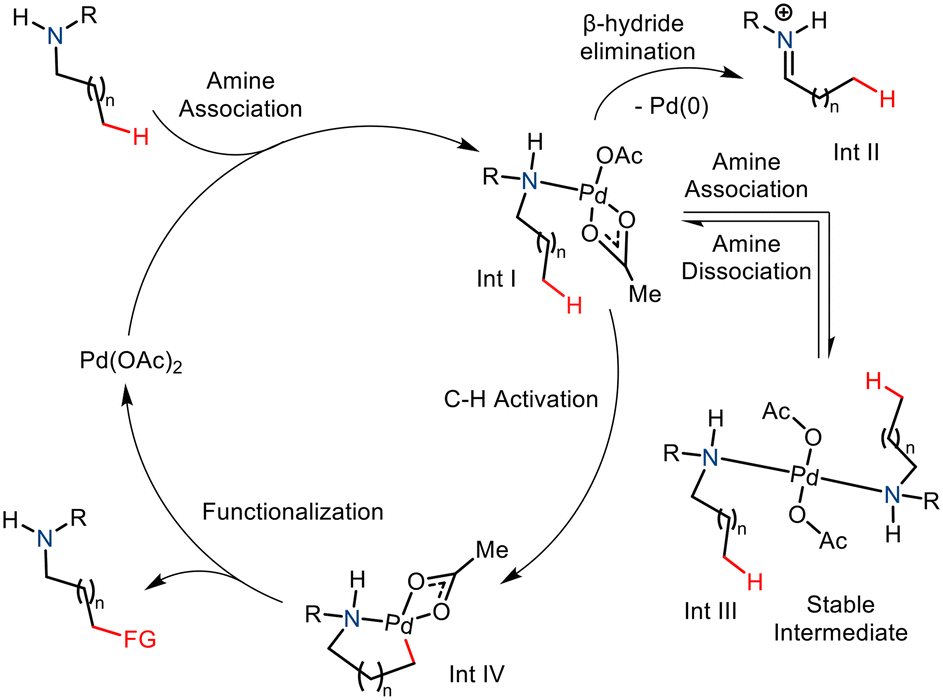
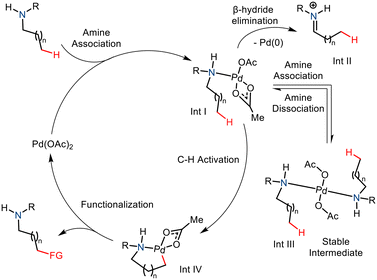
The amine group is an essential constituent in the marketed drug molecules.8 So, in recent years, substantial effort has been made to aid the synthesis of derivatized aliphatic amines.9 Notable methods include directed group (DG) assisted transition metal catalyzed C–H functionalization,10,11 and the use of transient directed groups (TDGs),12 which involves in situ attachment and removal of the directing groups. Usage of static directing group requires addition and removal of the directing group pre- and post-functionalization which ultimately decreases the practical aspect of a methodology. In comparison, transient directing groups need not be attached prior to reaction and removal is straightforward after the reaction. Thus the transient directing group approach offers a better strategy to carry out distal C–H functionalization. Nonetheless, most often an excess catalytic amount of the transient directing group is being utilized in the reactions and only a few specific types of substrates (amine, aldehyde, and ketone) could be employed as substrates in this approach. An efficient and more practical method would involve no directing group whatsoever, thereby eliminating the requirement of harsh conditions to remove the directing groups making it more atom efficient. Here, the innate ability of the amine group to direct and activate the C(sp3)–H bond at the γ and δ positions is explored. The nitrogen atom coordinates electrophilic transition metal complexes such as Pd(II) salts and facilitates C–H activation via cyclometallation (Scheme 2).13 This strategy is currently underdeveloped due to a few challenges in the Pd-catalyzed strategy. One such drawback is the formation of the stable and unreactive bis(amine) Pd(II) complex (Int. III).4a The C–H cleavage requires a coordinatively unsaturated Pd centre and thus cannot proceed due to strong coordination with amine nitrogen. Also, the amine complex may undergo β hydride elimination, thereby leading to oxidative degradation of the substrate and reduction of the palladium (Int. II). Nevertheless, the native amine-directed C–H functionalization strategy has been developed and well explored in recent years. This methodology has proved to be a successful strategy for synthesizing enantiopure N-heterocycles from free amines.
 |
| | Scheme 2 Native amine-directed strategy for C–H functionalization and the associated challenges. | |
2.1 Native amine group directed formation of the azetidine ring
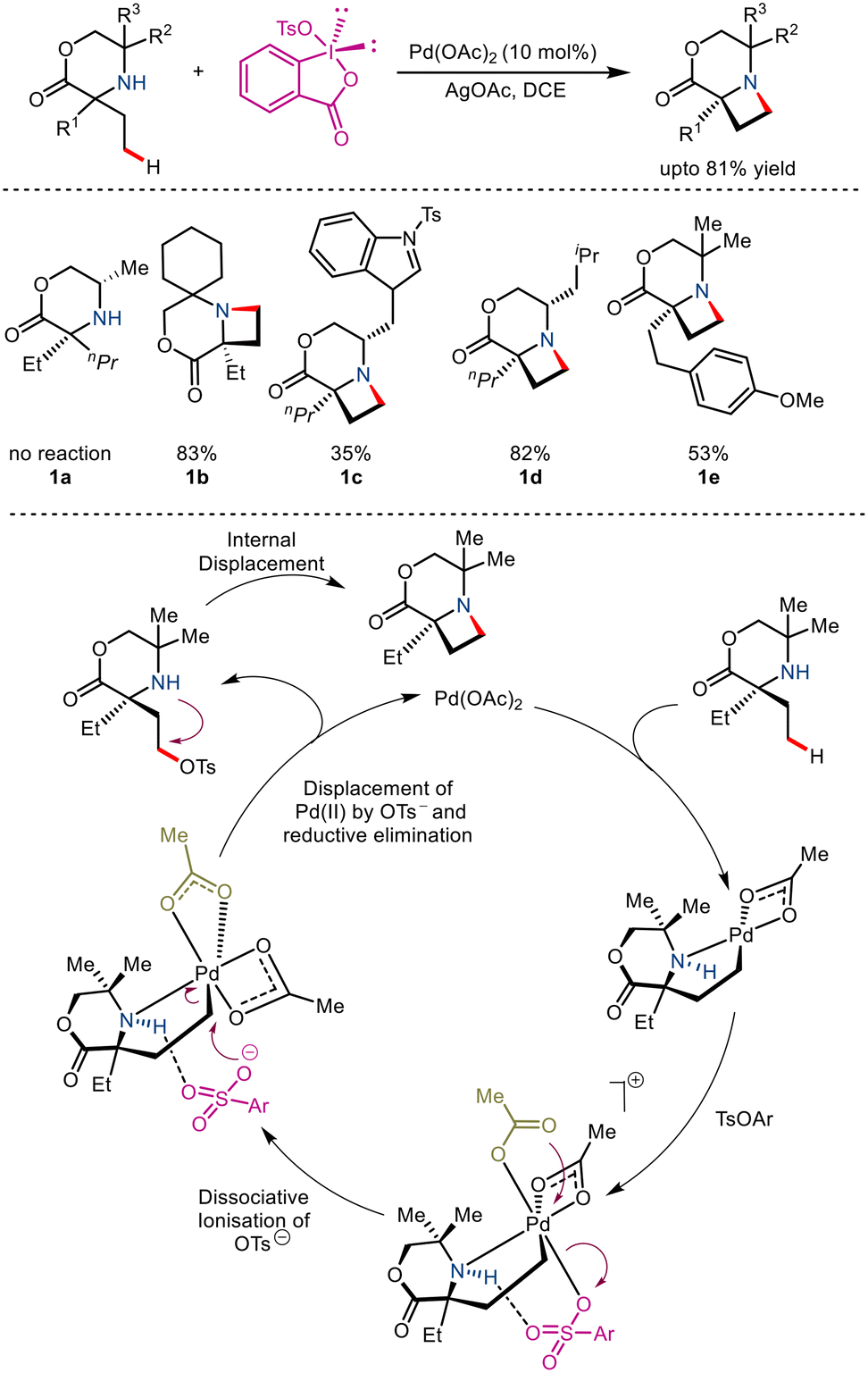
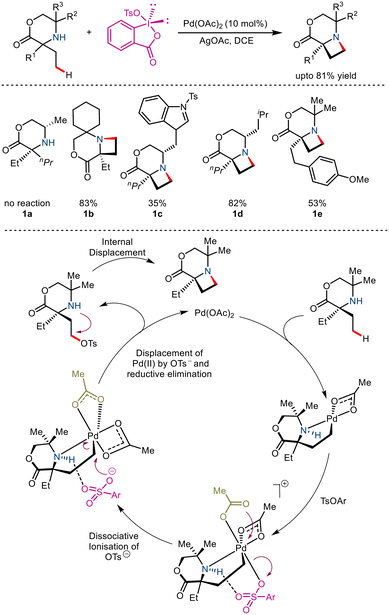
Generation of saturated N-heterocycles from free amines using palladium catalyzed intramolecular C–H amination as the key step has gained immense popularity due to its apparent simplicity. In 2018, Gaunt and co-workers utilized this tactic to report the synthesis of substituted azetidines via Pd(II) catalyzed intramolecular γ-C(sp3)–H amination.14 The primary purpose of this strategy was to target more facile C–O reductive elimination rather than direct C–N reductive elimination to produce a reactive intermediate that undergoes cyclisation (Scheme 3). It was seen that the use of benziodoxole tosylate along with AgOAc and Pd(OAc)2 resulted in good yields. Aided by computational studies, it was proposed to go via a selective reductive elimination pathway which involved dissociative ionisation of the tosylate and anchimeric κ2 carboxylate binding, thereby forming an octahedral aminoalkyl Pd(IV) complex. Attack by the OTs at the nucleofugal carbon atom bearing the Pd metal resulted in the formation of the C–OT bond. Subsequent displacement of this C–OT by the amino group gave access to the azetidine ring.
 |
| | Scheme 3 Pd-catalysed γ-C–H amination to form azetidines. | |
2.2 Native amine directed distal C–H arylation
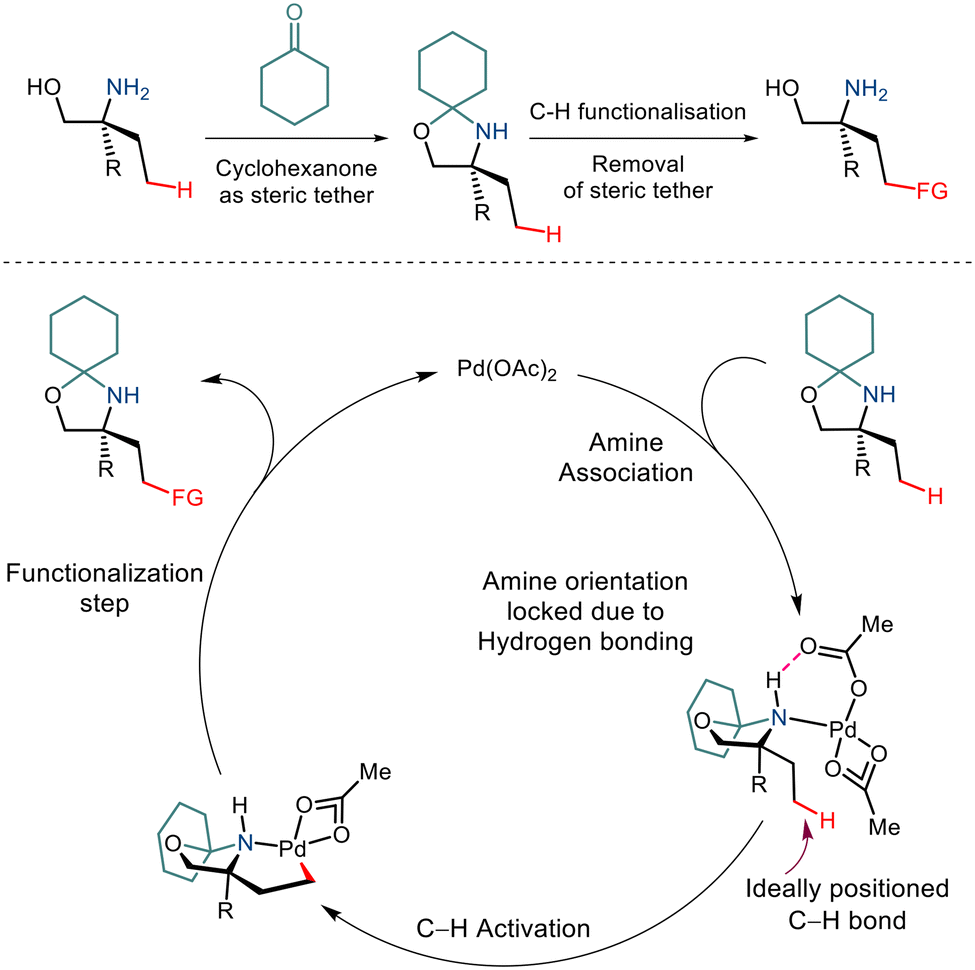
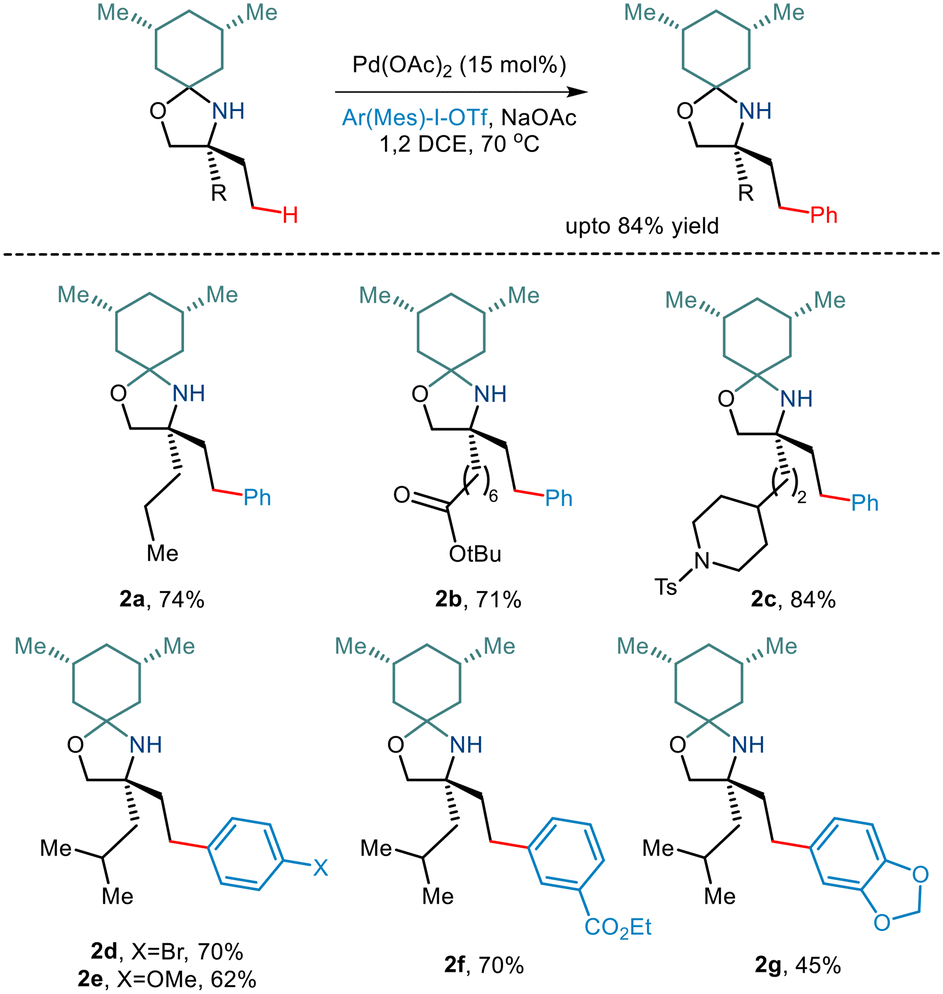
Over the last few years, chemists have exploited the electronic properties of the directing functional groups and ligands to facilitate C–H bond functionalization. But considering the several drawbacks encountered, steric parameters have been utilized to affect the C–H activation reaction. Given the importance of amino alcohols as an essential precursor motif in small drug molecules, Gaunt and co-workers, in 2015, implemented steric parameters to cause effective C–H activation of amino alcohols.15 They used a simple cyclohexanone as a steric tether to form a hindered N,O-ketal motif from amino alcohols (Scheme 4). The use of Pd(OAc)2 along with the hypervalent iodine reagent diphenyliodonium triflate resulted in γ C–H arylation (Scheme 5). The mechanism was proposed to form a lower energy monoamine Pd(II) complex, which involved a hydrogen bond between the Pd-bound amine and the acetate ligand. This intermediate facilitated the activation of the C–H bond and led to the formation of the cyclopalladation complex (Scheme 4). The presence of the triflate counterion resulted in the best outcomes, and the base added helped in quenching the trifluoromethanesulfonic acid formed as a by-product. This C–H arylation strategy has been implemented in synthesising Novartis' billion-dollar drug Gilenya used to treat sclerosis.
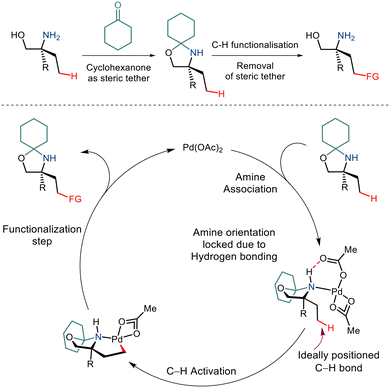 |
| | Scheme 4 A proposed strategy to functionalize the C–H bond in primary amino alcohols using a steric tether. | |
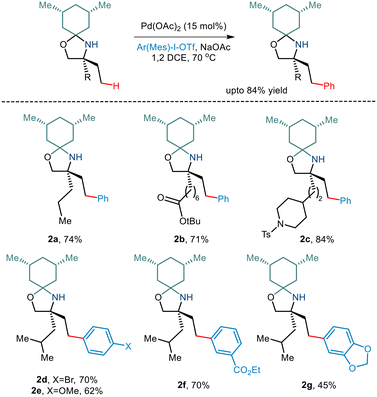 |
| | Scheme 5 Pd catalyzed C–H bond arylation of primary amino alcohols using a steric tether. | |
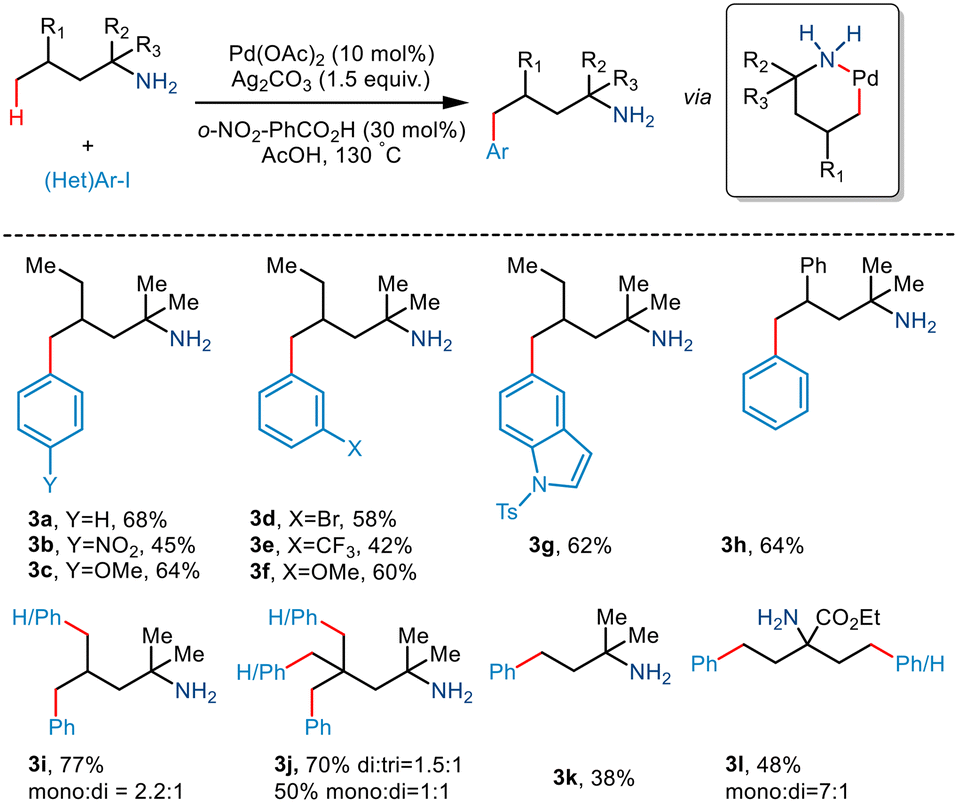
Although γ C–H activation has been well developed in recent years, δ C–H activation is a far less developed field due to the formation of a six-membered palladacycle which is kinetically less favourable.16 Bannister and co-workers, in 2019, reported the δ-C(sp3)–H arylation of primary aliphatic amines,17 albeit, the prevalent challenges. 2,4-Dimethylhexan-2-amine and iodobenzene underwent catalytic conversion in the presence of Pd(OAc)2 and Ag2CO3 (Scheme 6). Weak acid additives like acetic acid and 2-nitrobenzoic acid reportedly prompted dissociation of the complex. Although para-substituted aryl iodides containing electron-withdrawing and electron-donating groups resulted in the converted products, meta and ortho-substituted aryl iodides failed to give the desired yields. Interestingly, substrates containing two equivalent methyl groups gave mono and diarylated products with a ratio of 2.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (3i). Even the triarylated product (3j) was obtained with three equivalent methyl groups in 2,4,4-trimethylpentan-2-amine (di:tri = 1.5:1). However, the group observed that aliphatic amines having a large substituent at the α position resulted in low yields, and in some cases, even γ arylated products were also obtained.
:1 (3i). Even the triarylated product (3j) was obtained with three equivalent methyl groups in 2,4,4-trimethylpentan-2-amine (di:tri = 1.5:1). However, the group observed that aliphatic amines having a large substituent at the α position resulted in low yields, and in some cases, even γ arylated products were also obtained.
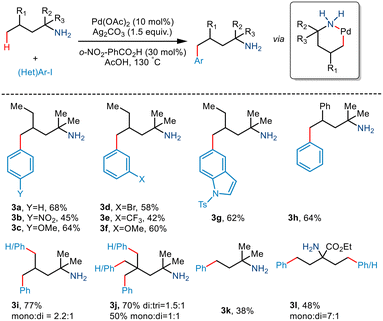 |
| | Scheme 6 Pd catalyzed C–H bond arylation of primary amino alcohols using a steric tether. | |
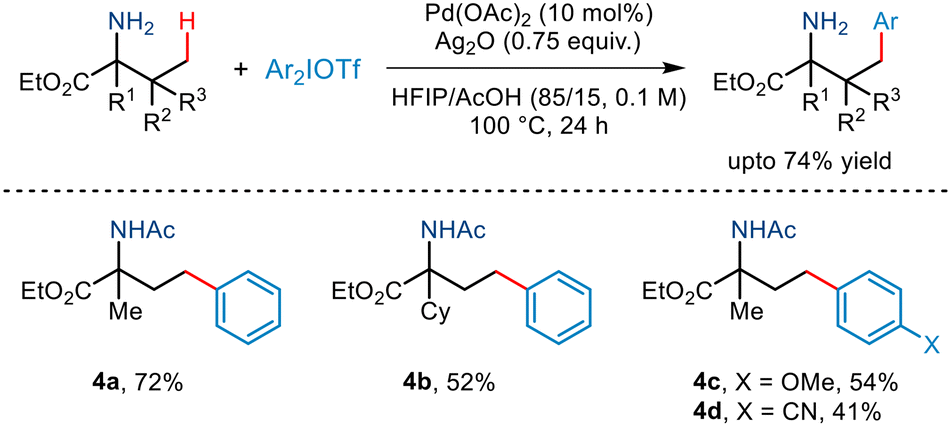
In the same year, Yao demonstrated γ-C(sp3)–H arylation of α-amino esters for the first time (Scheme 7).18 This was realised using diaryliodonium triflates and a Pd(OAc)2 catalyst, Ag2O and the AcOH/HFIP cosolvent system. This free amino group-directed arylation strategy was found to be compatible with a wide range of amino acid derivatives having various functionalities attached to the alkyl chain. Even a wide range of aryl partners successfully furnished the desired arylated amino ester in moderate yields. However, this protocol requires the presence of a tertiary carbon centre at the α site of amino esters.
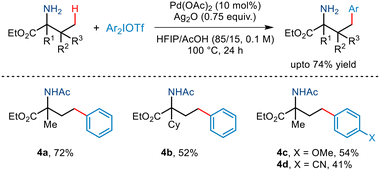 |
| | Scheme 7 Free amino group-directed γ-C(sp3)–H arylation of α-amino esters. | |
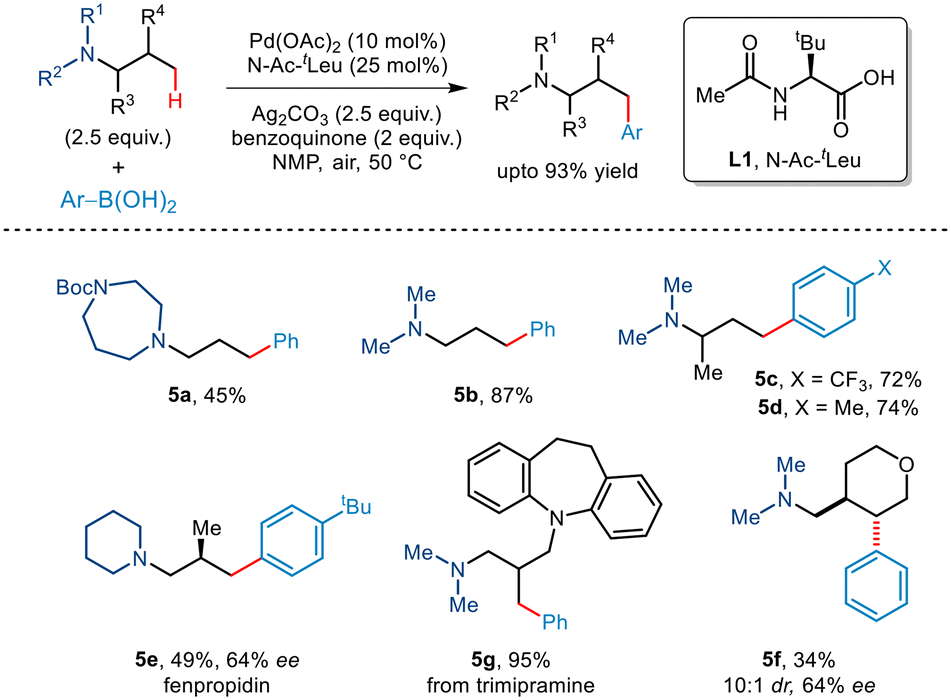
In the subsequent year, Gaunt and co-workers reported the first tertiaryamine-directed C–H arylation.19 While arylboronic acids were used as arylating partners, the strategy required the involvement of the N–Ac–tLeu ligand (Scheme 8). The role of the ligand lies in distorting the coplanarity of the Pd(II)·ligand complex thereby eliminating the chances of deleterious β-hydride elimination and favouring the desired C(sp3)–H activation. Investigations revealed that tert-amines appended with protected piperidines, piperazines and morpholines were well tolerated. In addition, arylboronic acids with a wide range of functionalities participated in this organic transformation to furnish γ-arylated amines in good yields. The protocol was further extended to the late-stage transformation of trimipramine. Interestingly, even substrates having enantiotopic β-methyl groups furnished non-racemic β-methyl–γ-aryl tertiary alkyl amines. For cyclic substrates, activation of methylene C–H showcased trans-selectivity.
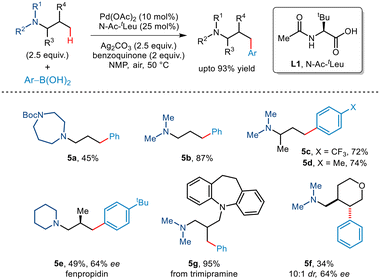 |
| | Scheme 8 γ-C(sp3)–H arylation of tertiary amines. | |
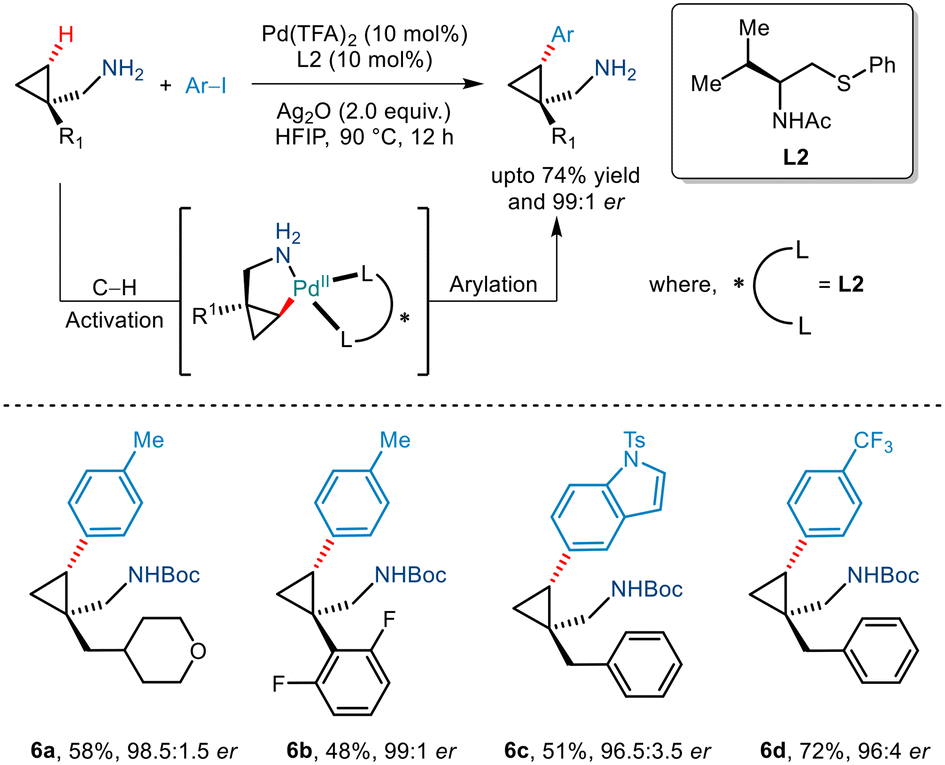
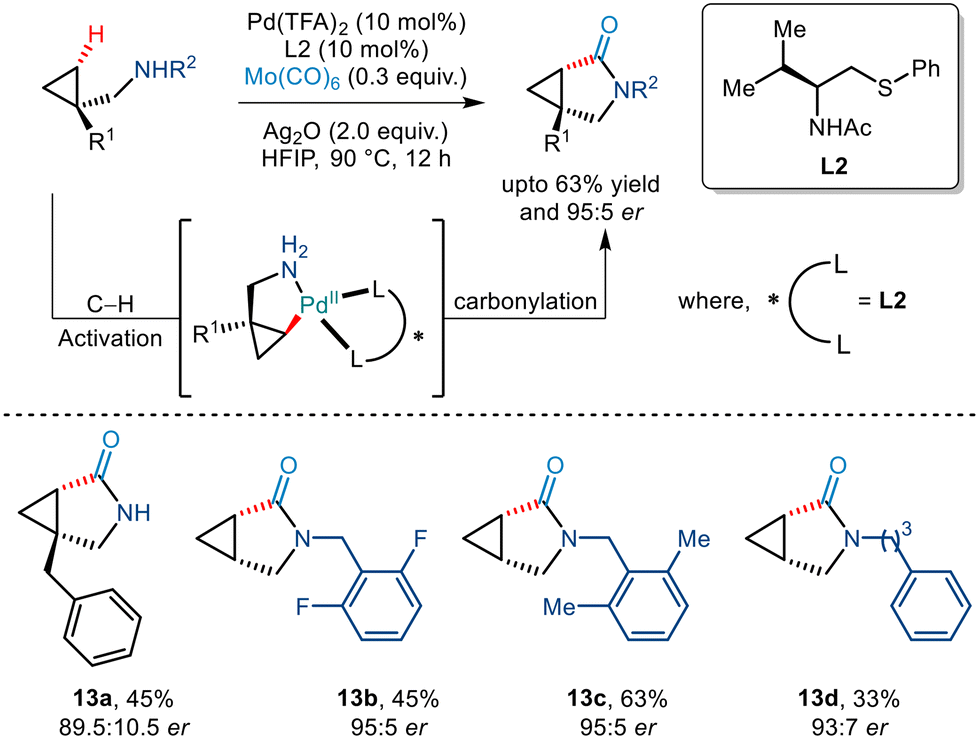
In 2020, Yu et al. demonstrated an efficient strategy for enantioselective γ C(sp3)–H arylation of cyclopropylmethylamines without the involvement of any exogenous directing group (Scheme 9).20 This was achieved by using a bidentate thioether ligand which is said to promote the formation of the mono(amine)–Pd(II) intermediate by dissociating the bis(amine)–Pd(II) complex through its strong σ donor properties. In addition to this, Pd(TFA)2, Ag2O and HFIP were crucial for this Pd(II)/Pd(IV) catalytic conversion. Aryl iodides with a diverse range of functionalities were tolerated to give the desired arylated products in good enantioselectivities of >95% ee. The bidentate properties of the ligand L2 were said to be responsible for the formation of a rigid palladacycle thereby allowing the transfer of chirality to the cyclopropylmethylamines during the C–H activation step. Furthermore, the generality of this protocol was extended to showcase carbonylation and olefination reactions via a Pd(0)/Pd(II) catalytic cycle.
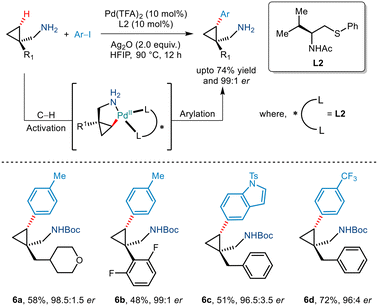 |
| | Scheme 9 Enantioselective γ-C(sp3)–H arylation of free cyclopropylmethylamines. | |
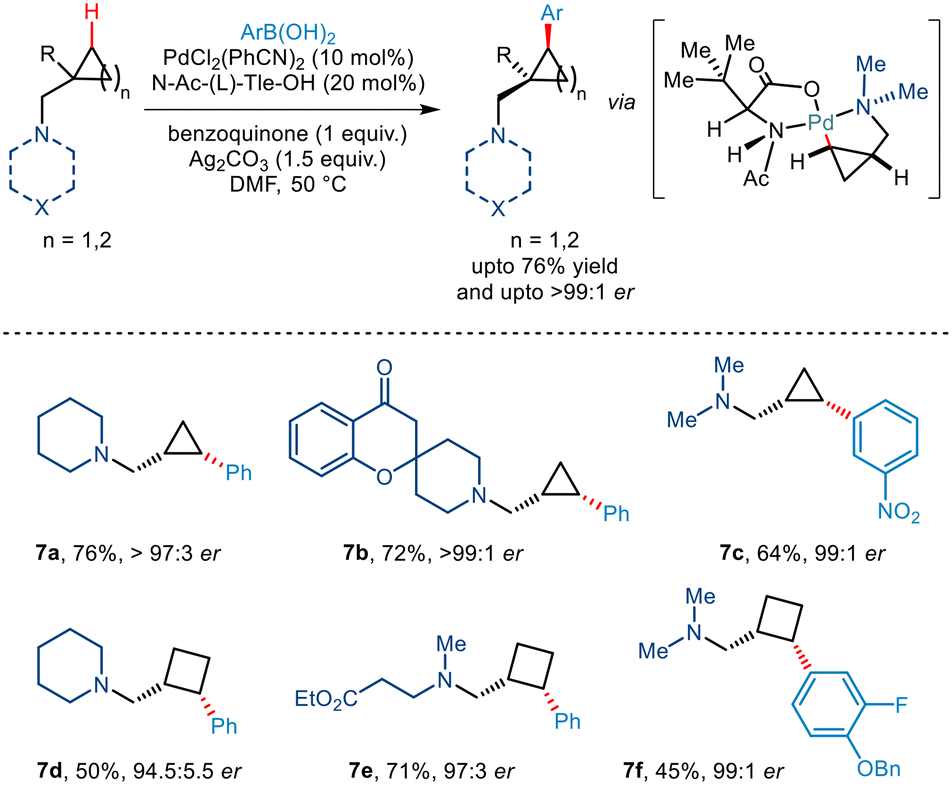
Tertiary alkylamine directed enantioselective γ C(sp3)–H arylation of similar aminomethyl cyclopropane and cyclobutane scaffolds was further illustrated by Gaunt and co-workers, very recently in 2022 (Scheme 10).21 Using PdCl2(PhCN)2 as the catalyst, Ag2CO3 as the silver salt and benzoquinone they observed that the best yield could be obtained in DMF solvent. In addition to this, the use of N-Ac(L)-Tle-OH ligand was found to be very crucial for inducing enantioselectivity through restricted orientation of the acetamide group towards the γ C–H bond in order to form cis diastereomers. Furthermore, the use of this amino acid ligand also promoted the γ C–H activation in place β hydride elimination pathways. N-Heterocycles like piperidines and azetidines along with acyclic tertiary alkyl amines successfully furnished cis arylated cyclopropanes and cyclobutanes in excellent enantioselectivities >95:5.
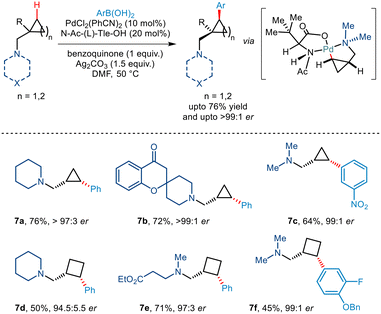 |
| | Scheme 10 Enantioselective C(sp3)–H arylation of aminomethyl–cyclopropanes and cyclobutanes directed by tertiary alkylamine. | |
2.3 Native amine group directed remote acetoxylation
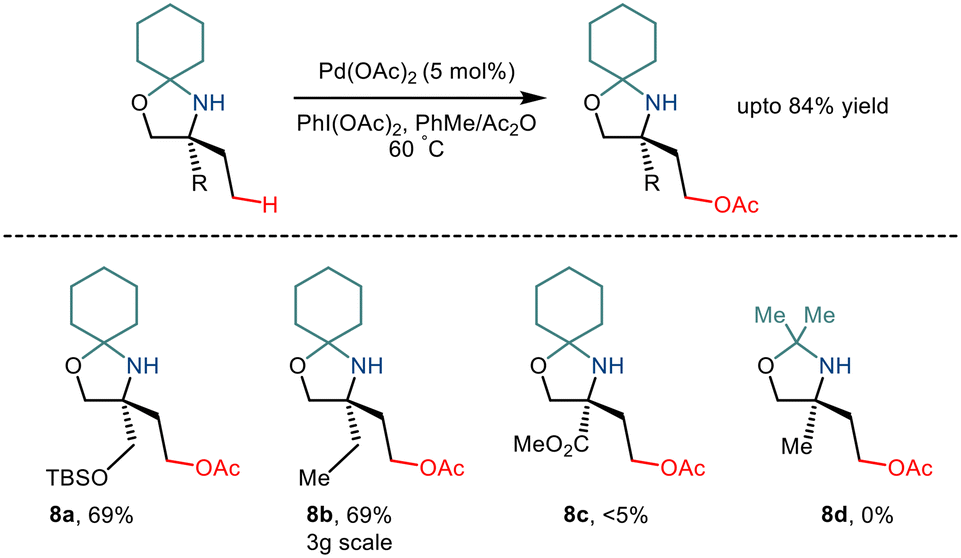
As already discussed, the C–H functionalization of primary aliphatic amines comes with a catch: the formation of the bis(amine) metal complex. Various measures like steric control have been implemented previously to combat this difficulty. In 2015, Gaunt and co-workers implemented steric factors to produce C–H activation of amino alcohols.15 Using Pd(OAc)2 and PhI(OAc)2, the group reported γ C–H acetoxylation on N,O-ketal motifs, formed by using a simple cyclohexanone as a steric tether (Scheme 11). The mechanism was hypothesized to go via the involvement of a high valent palladium intermediate where subsequent reductive elimination led to the formation of the acetoxy product in moderate yields.22 However, the reaction was less efficient for substrates with electron-withdrawing functionality.
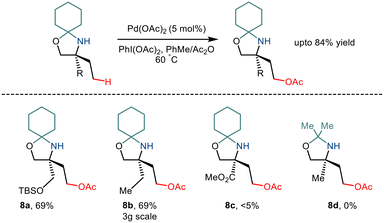 |
| | Scheme 11 Pd catalysed C–H bond acetoxylation of primary amino alcohols using a steric tether. | |
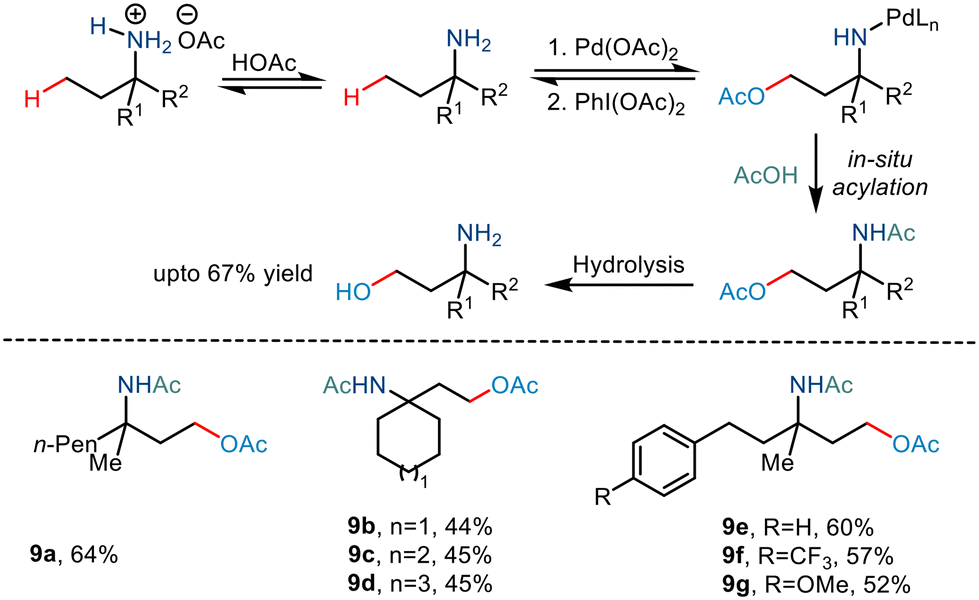
In 2017, Shi and co-workers demonstrated C(sp3)–H acetoxylation23 in weak acids like acetic acid to overcome the associated challenges. Protonation under acidic conditions led to – (i) restriction of the coordinating ability of the nitrogen atom to Pd catalysts and (ii) increased their stability in the presence of electrophilic or oxidative reagents, making the amino group more tolerant to harsh conditions. The conversion was carried out in the presence of Pd(OAc)2, and PhI(OAc)2 led to the formation of a five-membered palladacycle intermediate, which resulted in the C–H functionalization through metal-assisted C–H cleavage (Scheme 12). The C–OAc motif was in situ acylated to hinder the further reaction of the free amino group. This strategy gave a straightforward way to synthesize γ amino alcohols from primary amines.
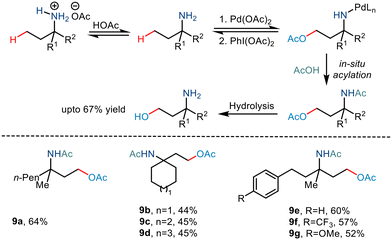 |
| | Scheme 12 The free amino group directed C–H bond acetoxylation via in situ acylation. | |
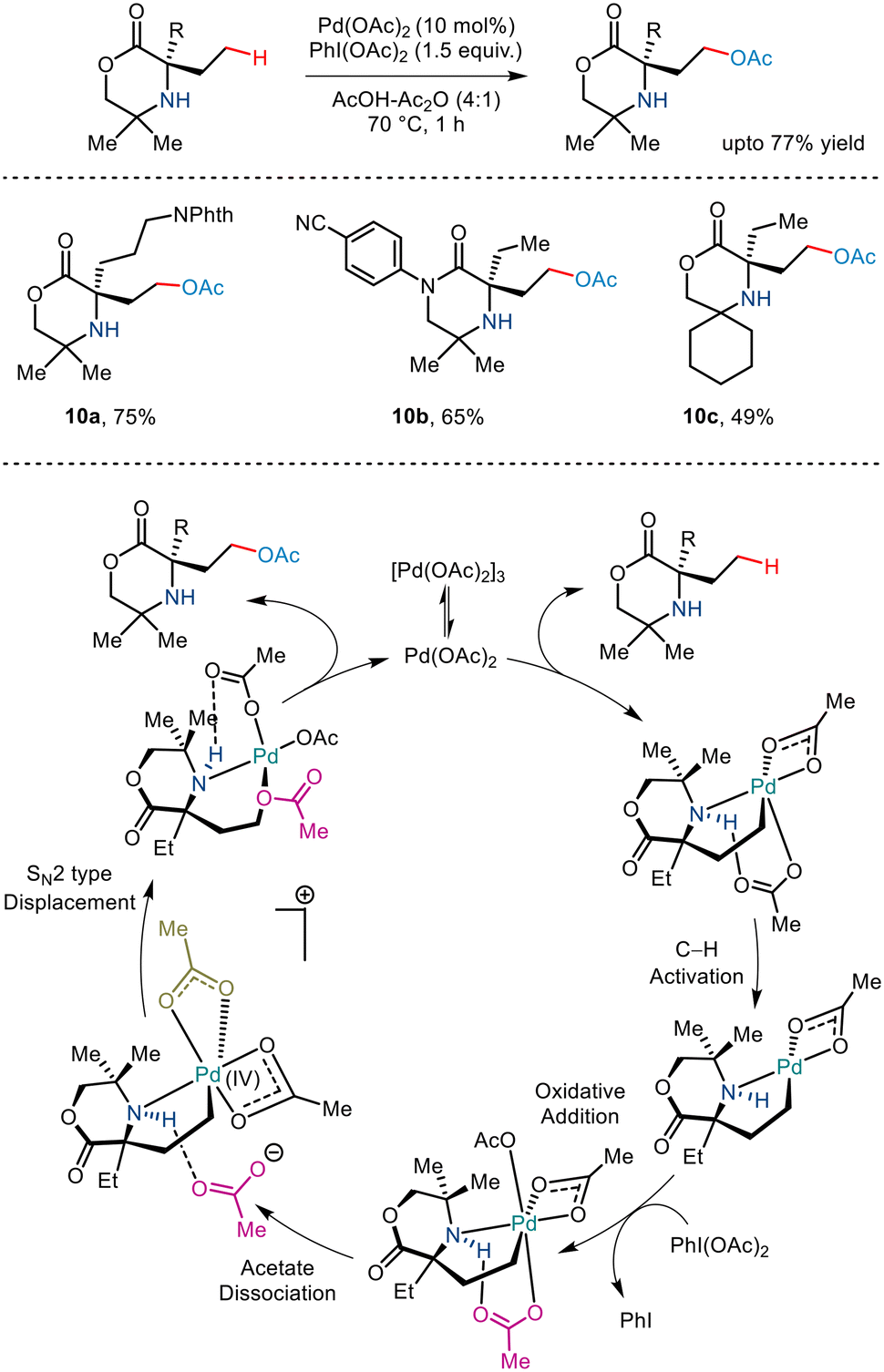
Banking on the strategy developed by the Gaunt group to synthesize azetidine rings,14 the group, in 2019, reported a similar γ C–H acetoxylation of morpholinone derivatives by exploiting the native amine functionality (Scheme 13).24 DFT and kinetic studies revealed that the reaction proceeded via the formation of a five-membered palladacycle where C–H activation formed the rate-limiting step. A similar two-step process of dissociative ionisation/SN2-type reductive elimination paved the way to the C(sp3)–O bond formation. Interestingly, additional use of chiral BINOL–phosphoric acid ligands aided enantioselectivity, thereby forming a rare example of asymmetric C–H acetoxylation.
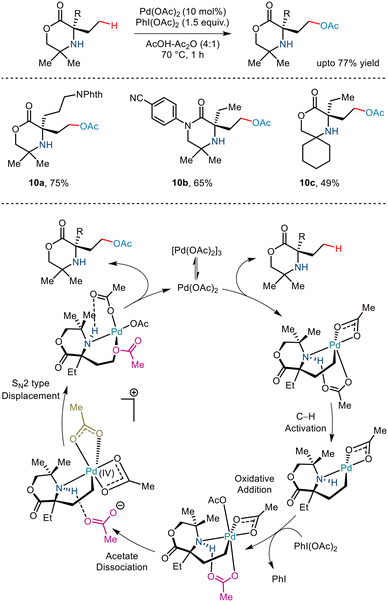 |
| | Scheme 13 γ-C(sp3)–H acetoxylation of morpholinones. | |
2.4 Native amine directed distal carbonylation
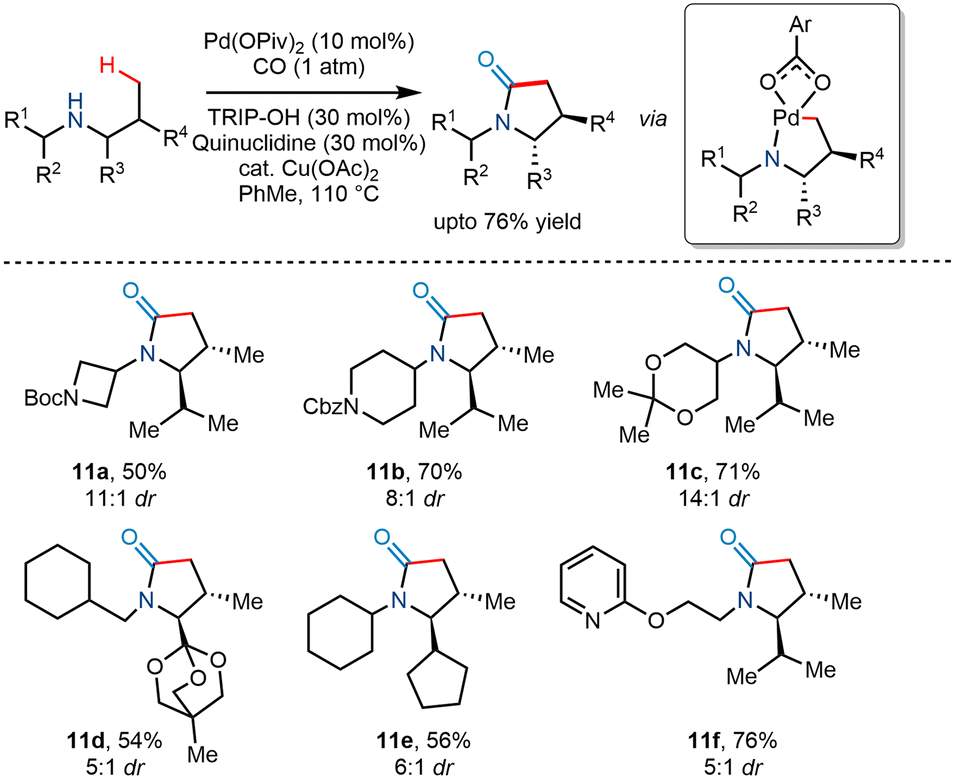
In 2018, Gaunt and co-workers utilized Pd(II) catalyzed C(sp3)–H carbonylation to transform less substituted acyclic secondary amines to trans substituted γ lactams in good yields and high diastereoselectvities ranging from 5:1 to 11:1 (Scheme 14).25 Using Pd(OPiv)2 as a catalyst and Cu(OAc)2 as an oxidant reportedly produced modest yields. The addition of sterically hindered 2,4,6-triisopropylbenzoic acid (TRIP-OH) in the presence of quinuclidine further enhanced the diastereoselectivity and yield. However, it was found that increased carbon monoxide concentration favoured β lactams instead of γ lactams, while the addition of electronically poor benzoate additives favoured γ lactams.
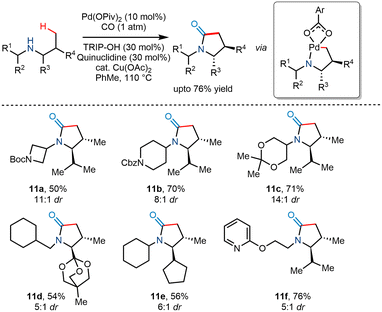 |
| | Scheme 14 Diastereoselective γ-C–H carbonylation to trans-substituted γ-lactams. | |
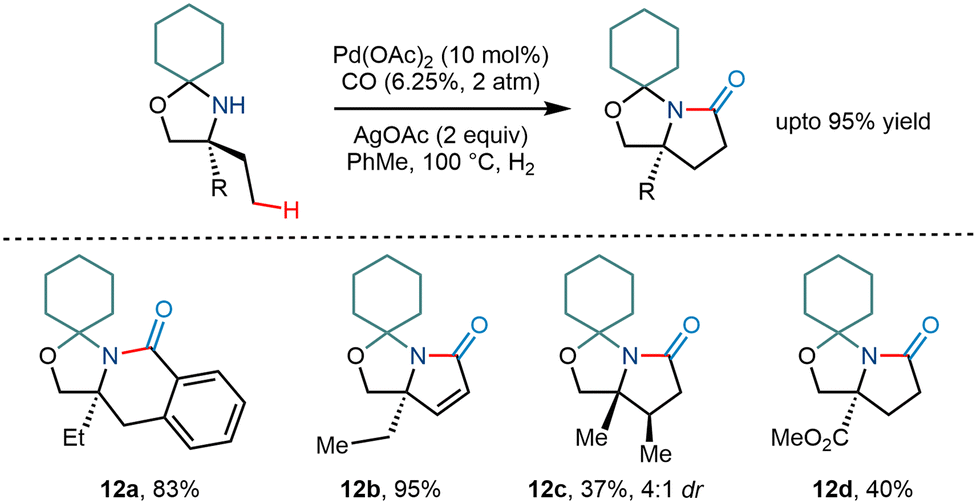
The Gaunt group in 2015 demonstrated the C–H activation of amino alcohols using a cyclohexanone molecule as a steric tether (Scheme 15).15 Although the group mentioned the formation of arylated and acetoxy products, they envisioned that trapping of the cyclopalladation would lead to one carbon homologation and result in subsequent pyrrolidinones, a constituent in many natural products.26 Upon carrying out the reaction, they found that corresponding pyrrolidinone compound derivatives were obtained in modest yields. Silver salts were found to be crucial for the reaction as they – (i) acted as a terminal oxidant and (ii) led to the formation of a bimetallic complex with palladium.27,28 However, the reaction was less efficient and resulted in relatively lower yields when the amine functionality had a robust electron-withdrawing group next to it. Interestingly, β hydride elimination of the cyclopalladation product provided α, β unsaturated pyrrolidinone products in large amounts.
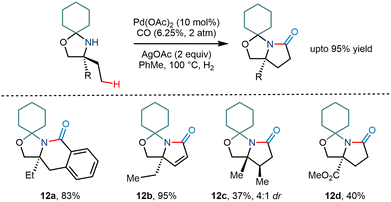 |
| | Scheme 15 Pd catalyzed C–H bond carbonylation of primary amino alcohols using a steric tether. | |
In 2020, the Yu group reported an efficient strategy to functionalize the γ C(sp3)–H bond of cyclopropylmethylamine derivatives (Scheme 16).20 Although it primary focused on enantioselective arylation, the strategy could be extended to demonstrate the directing group free carbonylation reaction in an asymmetric fashion – the first of its kind. Utilizing Mo(CO)6 as a CO source in addition to Pd(OAc)2 and L2, the group successfully achieved bicyclic γ-lactams in acceptable yields.
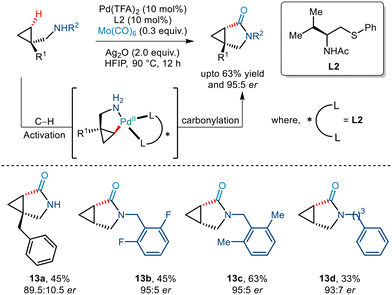 |
| | Scheme 16 Enantioselective γ-C(sp3)–H carbonylation of free cyclopropylmethylamines. | |
2.5 Native amine group-directed distal alkenylation
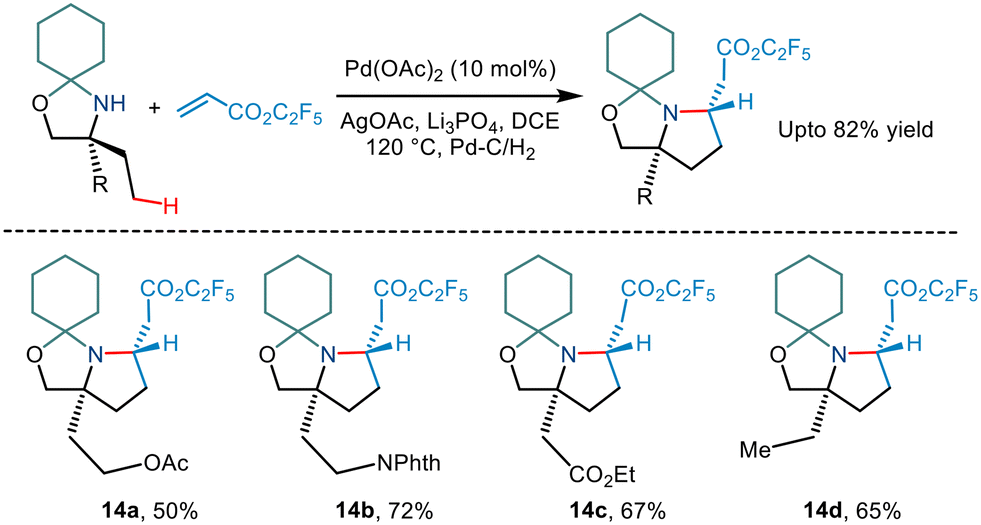
Carbon–carbon coupling especially combining C(sp3)–H bonds with an alkene has been a traditional challenge to chemists. Gaunt's 2015 steric tethering approach to enable C–H activation15 was further extended to form carbon–carbon bonds based on C–H alkenylation (Scheme 17). Treating the N,O-ketal motif with trifluoroethyl acrylate in the presence of Pd(OAc)2, AgOAc and Li3PO4 in 1,2 dichloromethane solvent led to alkenylation. This, followed by an intramolecular aza-Michael reaction furnished pyrrolidine compounds. Cleaving of N,O-ketals under mild conditions gave way to remotely functionalized amino alcohols.
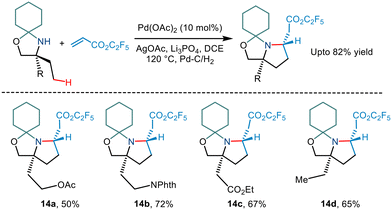 |
| | Scheme 17 Pd catalysed C–H bond alkenylation of primary amino alcohols using a steric tether. | |
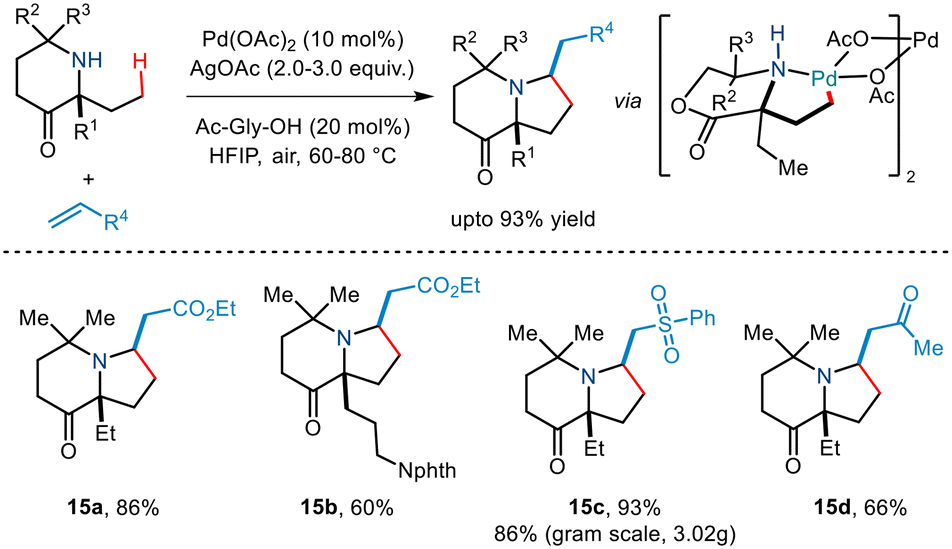
Subsequently, Gaunt and co-workers in 2017 disclosed another example of Pd-catalyzed C–H alkenylation of aliphatic amines.29 Besides using Pd(OAc)2 as a Pd source and AgOAc in HFIP solvent, they utilized an amino acid ligand (Ac-Gly-OH) to realise the reversible C–H bond activation of morpholinone moieties to afford various pyrrolidine scaffolds (Scheme 18). A wide range of α,α′ disubstituted morpholinones underwent alkenylation to deliver the desired product in modest to good yields. Usage of chiral morpholinones enabled the synthesis of enantiopure pyrrolidine derivatives. In the case of alkene coupling partners, various α,β-unsaturated ketones, acrolein and even vinyl sulfones and vinyl phosphonates, etc. were successfully incorporated into the pyrrolidine moiety. The group suggested that this alkenylation proceeded through the formation of a five-membered palladacycle which gives way for alkene insertion and subsequent aza-Michael cyclisation to form a pyrrolidine ring.
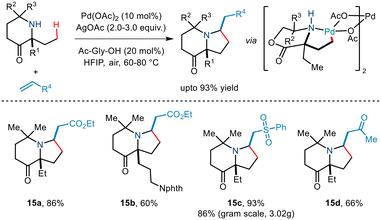 |
| | Scheme 18 C(sp3)–H alkenylation of aliphatic amines for the synthesis of functionalized pyrrolidines. | |
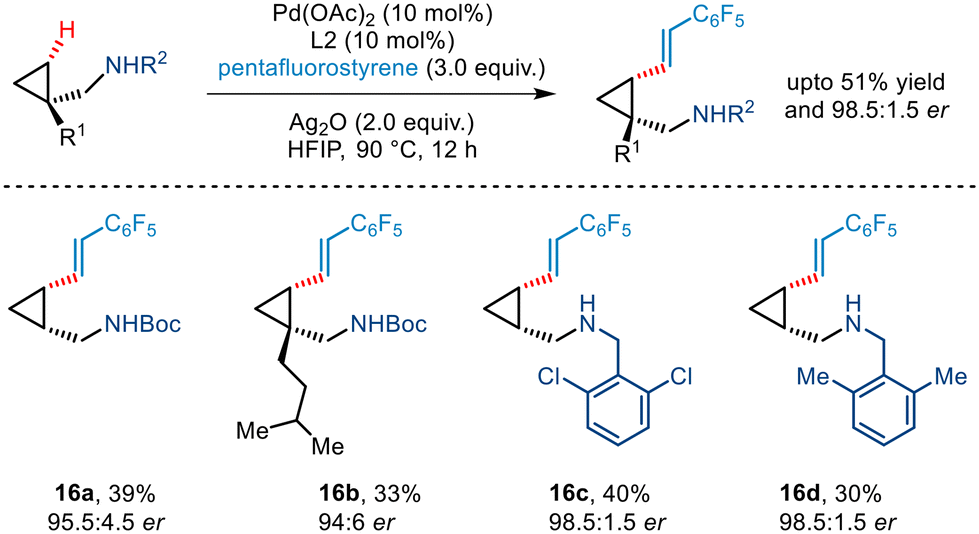
Yu's seminal work on enantioselective arylation of free cyclopropylmethylamines20 was further extended to demonstrate asymmetric olefination involving pentafluorostyrene as the olefin partner (Scheme 19). Various substituted cyclopropyl amines were utilized in the protocol that furnished olefinated products in moderate yield and excellent enantioselectivity.
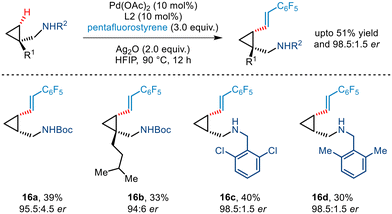 |
| | Scheme 19 Enantioselective γ-C(sp3)–H olefination of free cyclopropylmethylamines. | |
3. Native carboxylic acid group directed strategy
Carboxylic acid is among the most abundant functional groups present in organic chemistry. The importance is escalated due to the broad applicability of these acids in synthetic chemistry30 and biological processes.31 The use of covalently and transiently attached directing groups to aliphatic carboxylic acids led to the development of arylated, alkylated, carbonylated and olefinated derivatives.28,32 However, such transformations come with an additional cost to the atom and step economy. In this regard, there have been quite a few instances where reports of C–H functionalization have been made without involving any exogenous directing groups. Considering the desirability of this strategy,6,33 functionalization of free carboxylic acid has been explored mainly in the proximal and, to some extent, a distal position.
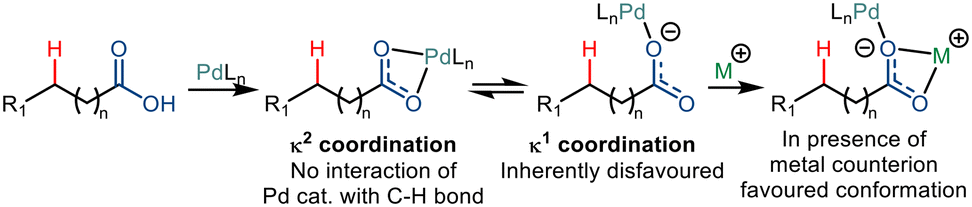
Two different coordination modes of κ1 and κ2 are known for a carboxylate group. In the κ2 binding mode, the metal atom coordinates with both oxygen atoms of a carboxylate. On the other hand, in the κ1 binding mode, the metal binds with one of the carboxyl oxygen atoms. The binding of the carboxylate ligand to the empty coordination site of the transition metal complex in the κ1 mode leads to a spatial conformation that enables the C–H functionalization (Scheme 20).34 However, palladium favours coordination in the κ2 mode. So, the shift from the κ2 mode to κ1 mode has been made in the presence of inorganic and organic counterions like Na+, K+, Cs+, NR4+, etc. These counter ions preferably coordinate with the carboxylate in a κ2 fashion, forcing the palladium metal to bind in the κ1 mode.35,36
 |
| | Scheme 20 Coordination mode of free carboxylic acid. | |
Although these distinctive nature of the carboxylate group has been employed to synthesize functionalized products at the proximal position by various groups, functionalization at the distal part is relatively challenging due to a thermodynamically less favoured six and seven-membered palladacycle, respectively. Nevertheless, selective remote functionalization has been reported by a few groups covered in this review.
3.1 Native carboxylic acid group directed distal arylation
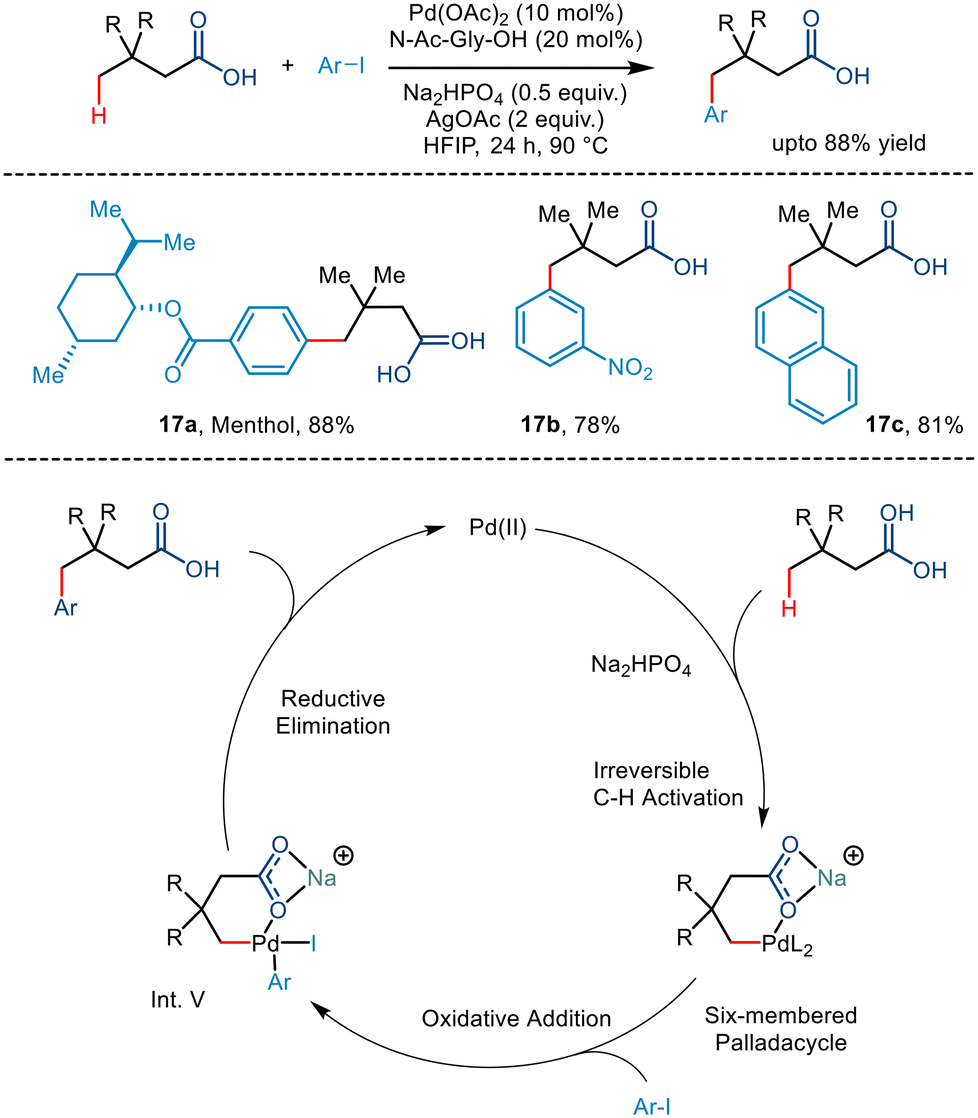
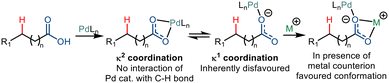
Maiti and co-workers, in 2019, reported the directing group free regioselective C(sp3)–H arylation of aliphatic carboxylic acid for the first time.36 They functionalized tert-butyl acetic acid with various aryl iodides where the N-Ac-Gly-OH ligand was most effective (Scheme 21). The addition of bases like Na2HPO4 and AgOAc favoured the κ1 binding mode rather than the κ2 binding mode, which facilitated C–H functionalization at the distal position in higher yields. The choice of solvent was also crucial to the reaction where 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) stabilized the palladium intermediate due to its strong hydrogen bonding properties. The group proposed that the conversion occurred via a Pd(II)/Pd(IV) catalytic cycle where the acid initially coordinates with the palladium catalyst. A subsequent C–H activation step formed an alkali metal ion stabilized six-membered palladacycle. Upon conducting kinetic isotope studies, the group concluded that the activation step is the rate-determining step. Oxidative addition of the aryl iodide resulted in the formation of the Pd(IV) intermediate (Int. V), which, after C–C forming reductive elimination, yielded the corresponding γ arylated product. Aryl iodides containing both electrons withdrawing and electron-donating groups resulted in significant yields. Structurally complex aryl partners like naphthyl and biphenyl and derivatives of natural products were also tolerant to the reaction conditions. The utility of this γ arylation methodology can be deduced from the fact that derivatives of drug molecules can be utilized to prepare value-added drugs like ketoprofen and ibuprofen and natural products like fenchyl alcohol.
 |
| | Scheme 21 Pd-catalyzed γ-C(sp3)–H arylation of free carboxylic acid. | |
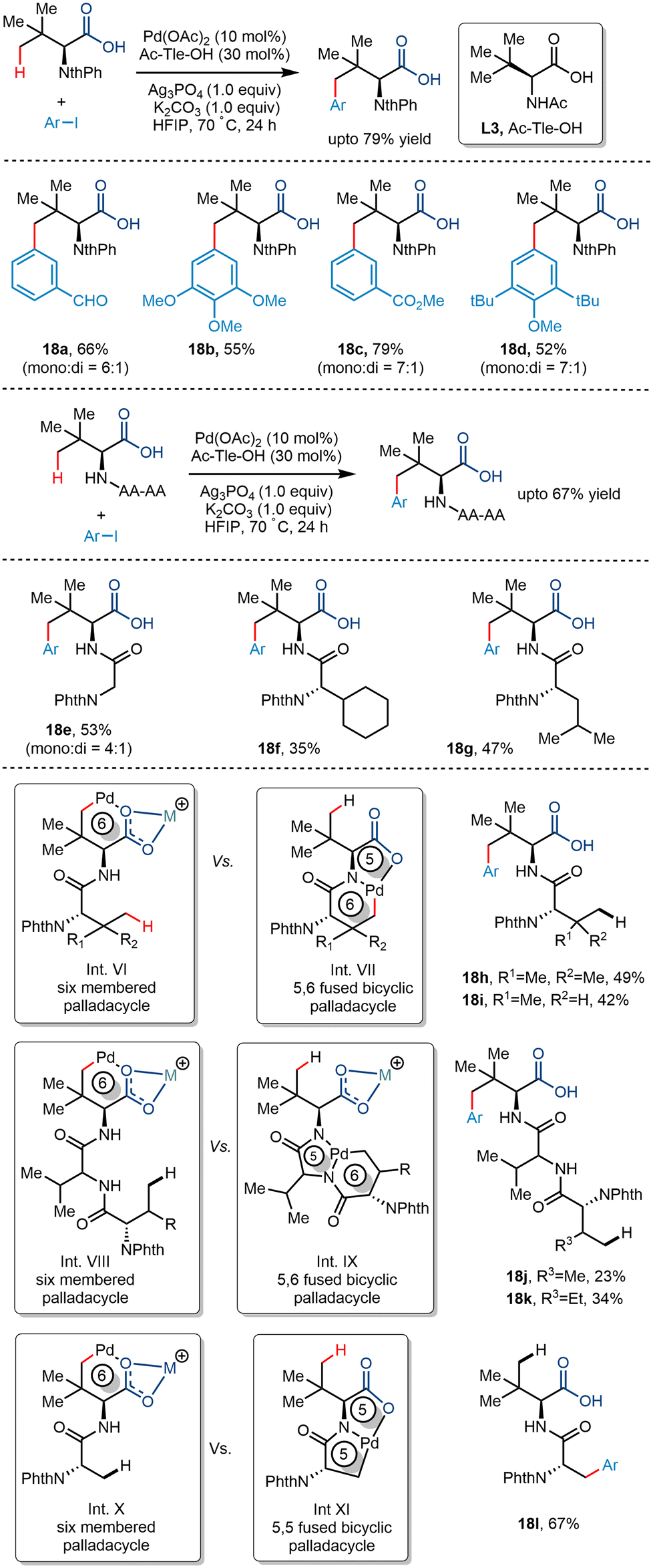
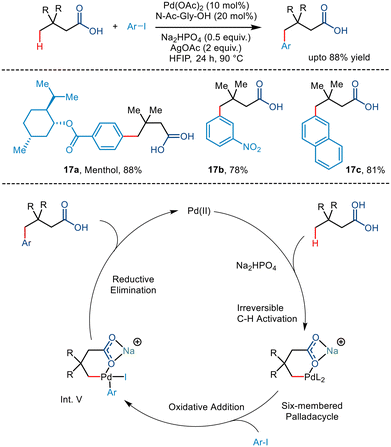
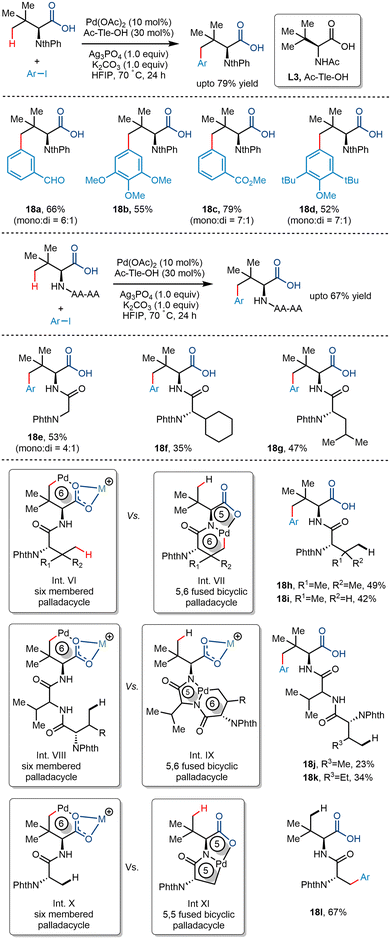
Later, Shi and co-workers utilized the palladium-catalyzed carboxylate-directed γ-C(sp3)–H arylation to synthesize a wide range of sizeable sterically hindered amino acids (Scheme 22).37 The group reported the γ-C(sp3)–H arylation of t-leucine derived peptides using aryl iodides in the presence of Pd(OAc)2 as a catalyst. Using Ac-Tle-OH as a ligand was crucial, where the ligand's bulky side chain was said to promote the reaction. Ag3PO4, used as an additive, played a dual role of a halide scavenger and a heteronuclear active species, thereby enabling the C–H bond cleavage.38 Examination of the substrate scope revealed that aryl partners containing both electron-donating and electron-withdrawing groups resulted in arylated products at moderate yields. Even diarylated products were obtained for a few cases with a mono:di ratio of 7:1 to 4:1 (Scheme 22, 18a, 18c, 18d, 18e). Phthaloyl-t-leucine was also arylated to form the corresponding product, which was previously achieved using a strong bidentate directing group39 and biscoordination of the peptide backbone.40 During a competition of two C(sp3)–H bonds of bi and tripeptides, arylation occurred at the γ position of the Tle moiety (18h–k). It was hypothesized that the formation of a thermodynamically less stable six-membered palladacycle was preferred to the 5,6-fused palladacycle. However, interestingly, the 5,5-fused palladacycle was found to be more durable than the six-membered palladacycle for N-Phth-Ala-Tle-OH (18l) ensuing β methyl arylation in the alanine residue.
 |
| | Scheme 22 Pd-catalyzed γ-C(sp3)–H arylation to synthesize amino acids with bulky side chains. | |
3.2 Native carboxylic acid group-directed distal alkynylation
Palladium catalyzed C–H functionalization has wide application in forming carbon–carbon bonds. In addition to advances in the C(sp3)–C(sp2) coupling, the applicability of C–H functionalization has been extended to C(sp3)–C(sp) coupling. Using free carboxylic acids to direct C(sp3)–H alkynylation yielded compounds having two highly versatile functional groups, thereby enabling the synthesis of value-added drugs and materials.41,42
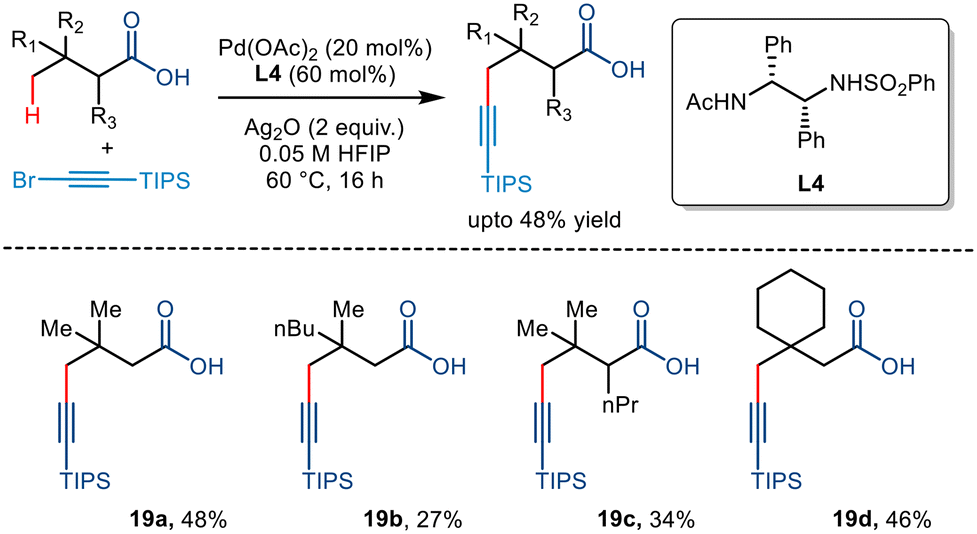
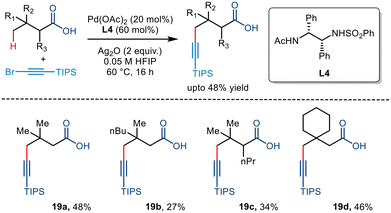
In 2020, the van Gemmeren group reported the directing group free carboxylate directed alkynylation for the first time using 1-bromo-2(triisopropylsilyl)acetylene as the alkyne partner (Scheme 23).43 Although initially, the group reported β alkynylation of the carboxylic acid substrates using diamine-derived ligands, they further expanded this reactivity to the distal γ position using a modified version of the diamine ligand used for the β alkynylation (L4). The strategy involved an increased catalyst loading but required no base for the transformation. Upon further examination, it was found that substrates with varying chain lengths and structural complexities could be transformed with moderate yields, comparable to the previously devised strategies involving directing groups,44,45 proving themselves to be more atom economic.
 |
| | Scheme 23 γ-C(sp3)–H alkynylation of carboxylic acids. | |
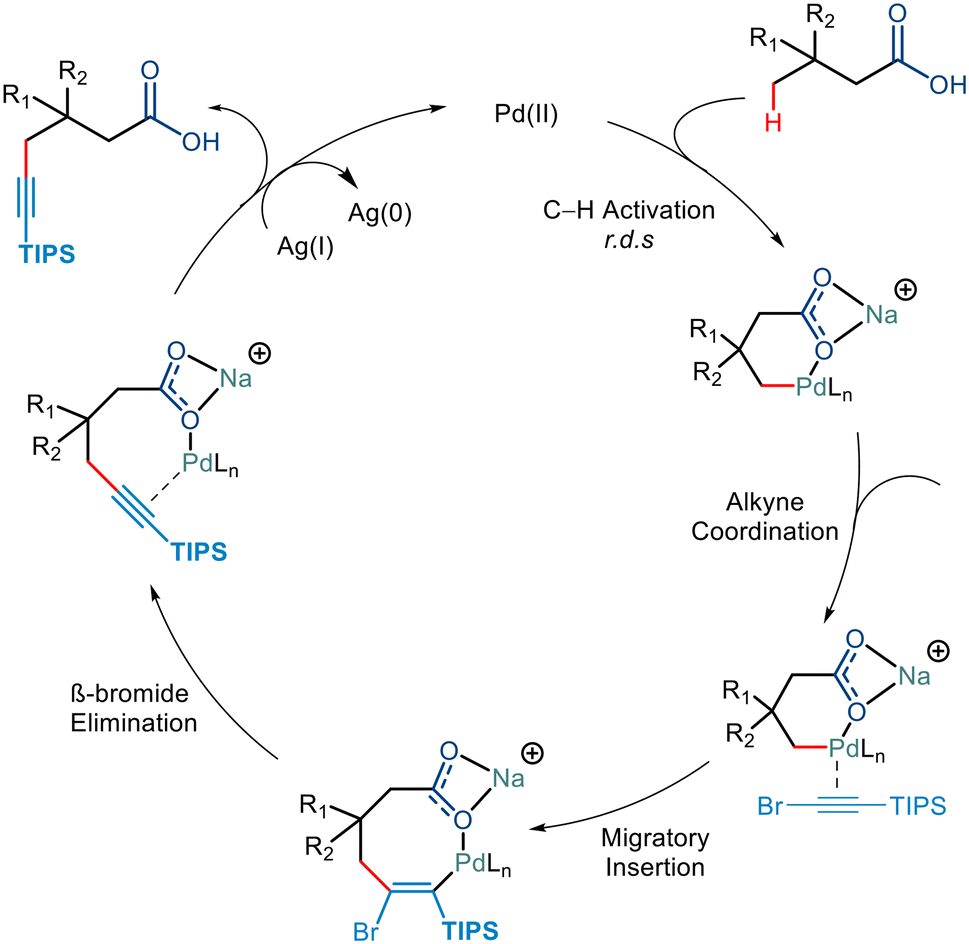
KIE studies revealed that the C–H Activation step was the rate-determining step. Further mechanistic steps in the catalytic cycle were hypothesised to go via either of the following steps. It could either go via the Pd(IV)/Pd(II) cycle, which involved the oxidative addition of the C–Br bond followed by the reductive elimination resulting in the C–C bond formation,42b,46 or it could go via the Pd(II) pathway consisting of alkyne insertion and then a Pd–Br elimination (Scheme 24).47
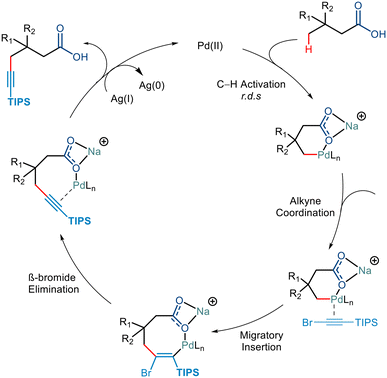 |
| | Scheme 24 Plausible mechanism for γ-alkynylation of aliphatic acid. | |
3.3 Native carboxylic acid group-directed lactone formation via distal C(sp3)–H activation
Lactones are industrially valuable48 moieties, especially the six and seven-membered counterparts, which find broad applicability as flavouring agents. Owing to the extensive functional group interconversions capabilities of carboxylic acid, it is considered an ideal precursor for lactone synthesis.
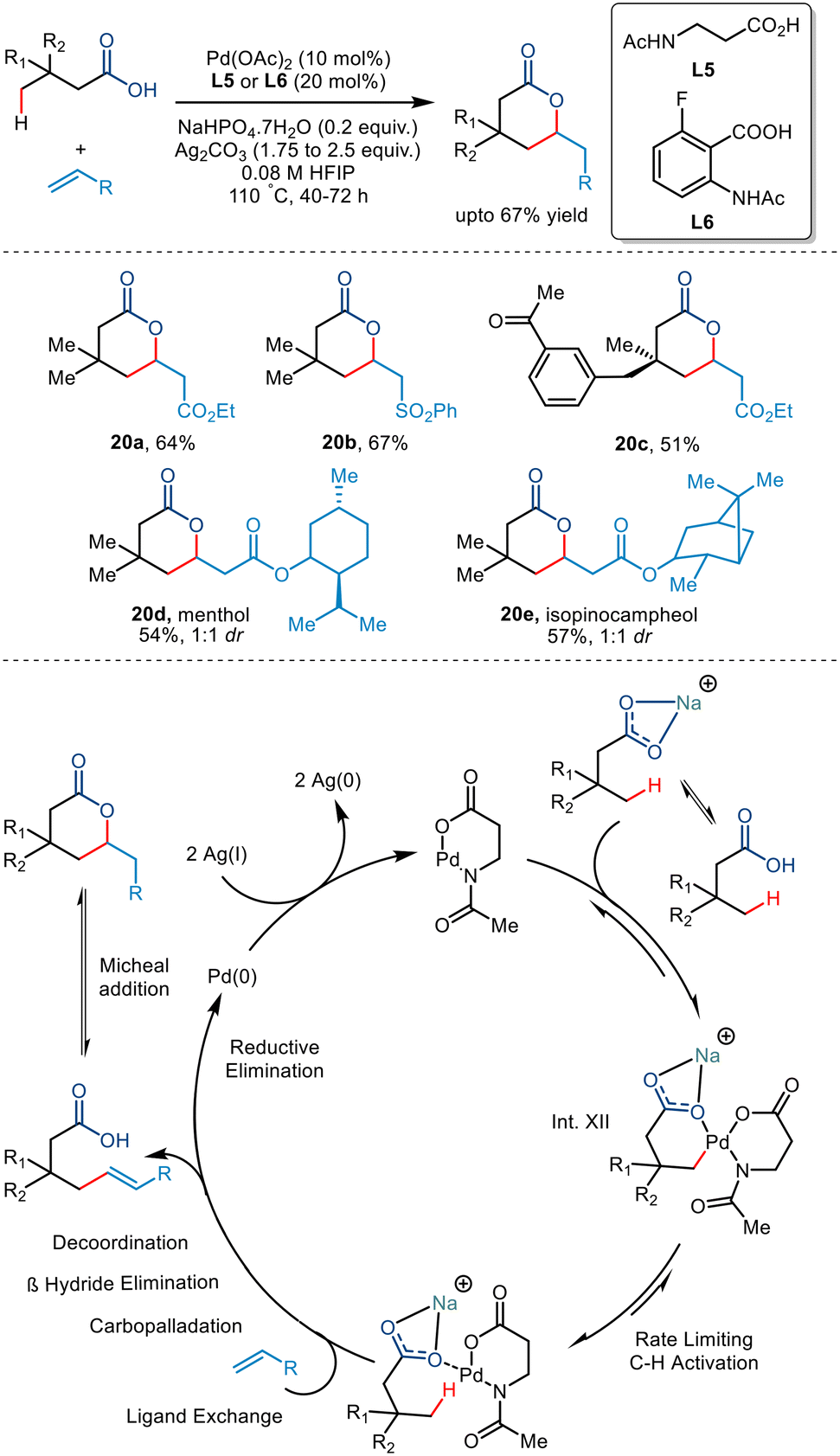
In 2020, the van Gemmeren group reported the synthesis of δ lactones formed via an intramolecular Michael reaction of γ olefinated products (Scheme 25).49 They initially reported the lactonised product developed from 3,3-dimethylbutyric acid and ethyl acrylate using the reactive anthranilic acid ligand (L6). However, the amino acid-derived N-Ac-β alanine ligand (L5) was preferred due to its wider availability. Investigating the substrate scope led to the conclusion that alkyl-substituted acids produced cis diastereomers in good yields. In contrast, structurally complex acids required additional reaction time and acrylate loading to generate modest yields.
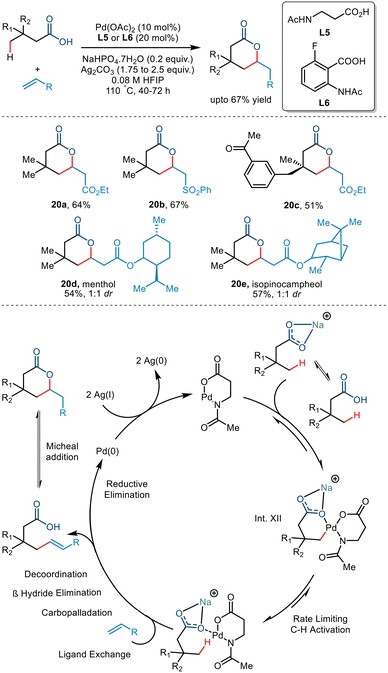 |
| | Scheme 25 γ-C(sp3)–H olefination of carboxylic acids enabled by the anthralinic acid-derived ligand. | |
Interestingly, it was observed that carboxylic acids having no quaternary centre at the β position failed to transform due to the absence of acceleration of the Thorpe–Ingold effect. Use of olefinic partners having electron-withdrawing groups and structurally complex molecules also resulted in moderate yields. Olefins bearing electron-withdrawing groups (20a–d) resulted in high-yielding transformations. The reaction was proposed to go via a Pd(II)/Pd(0) catalytic cycle (Scheme 25). The sodium carboxylate coordinated with the Pd(II) intermediate to give Int. XII. The step involving the olefinic partner was found to have a non-zero order of 0.2, attributed to the reversibility of the rate-determining C–H activation step. This step followed subsequent ligand exchange, carbopalladation, β-H elimination, decoordination and an intramolecular Michael reaction to form a δ lactone. The Pd(II) hydride donor was converted to Pd(0) via reductive elimination, which was again oxidised to Pd(II) by Ag salts. The carboxylic-directed allylic C–H activation also resulted in the formation of a γ lactone as a side product.
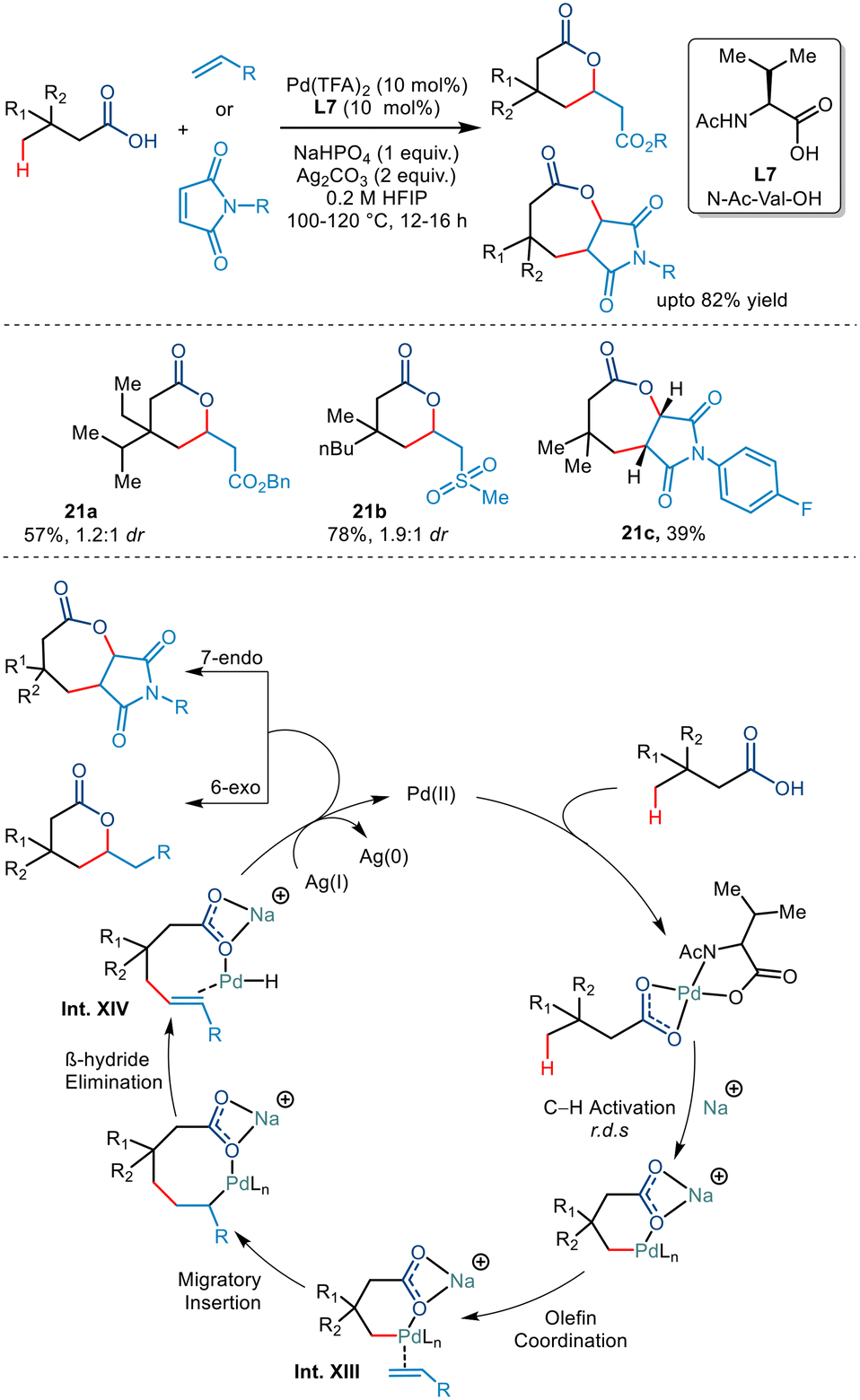
Subsequently at the same time, Maiti et al. synthesized δ and ε lactones via palladium catalyzed γ C(sp3)–H activation of carboxylic acids using suitable olefinic partners (Scheme 26).50 They initiated the reaction using 3,3 dimethyl butanoic acid and ethyl acrylate in Pd(OAc)2, 2-pyridone, Ag2CO3 and Na2CO3. On optimising various palladium salts and ligands, Pd(TFA)2 and N-Ac-Val-OH (L7) found to be the best catalyst and best ligand, respectively. Further optimisation revealed that Na2HPO4 acted as a better base and HFIP as a suitable solvent. Carboxylic acids containing ethyl, propyl, butyl, diethyl and ethyl–isopropyl at the β position successfully transformed with good yields. Besides acrylates, both sulphones and acrylonitriles behaved as suitable olefinic partners. However, the presence of the β quaternary site in the acid was necessary as it provided suitable geometrical alignment (via the Thorpe–Ingold effect) to form a thermodynamically stable six-membered palladacycle. For the first time, the group also demonstrated the formation of seven-membered fused lactones using maleimides as an olefin coupling partner. Both N-alkyl and N-aryl maleimides were found to provide corresponding transformed products. The mechanism was proposed to go via a six-membered palladacycle (Int. XIII). Binding to the olefin coupling partner and subsequent 1,2 migratory insertions resulted in the formation of an eight-membered palladacycle (Int. XIV). The seven-membered lactone was obtained following a subsequent Michael reaction after the β hydride elimination and reductive elimination steps of the eight-membered palladacycle. The catalytic cycle was terminated by Ag(I) salts which oxidised Pd(0) to Pd(II).
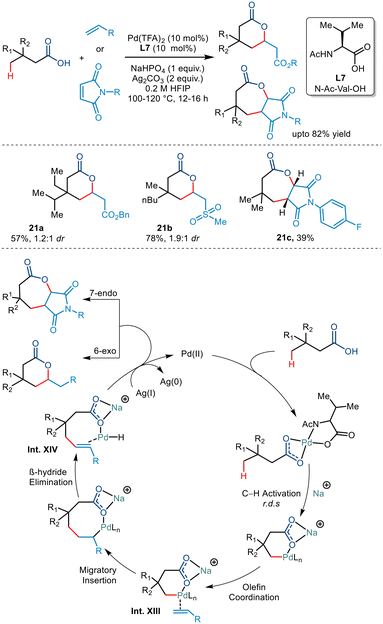 |
| | Scheme 26 γ-C(sp3)–H olefination of carboxylic acids to form δ and ε lactones. | |
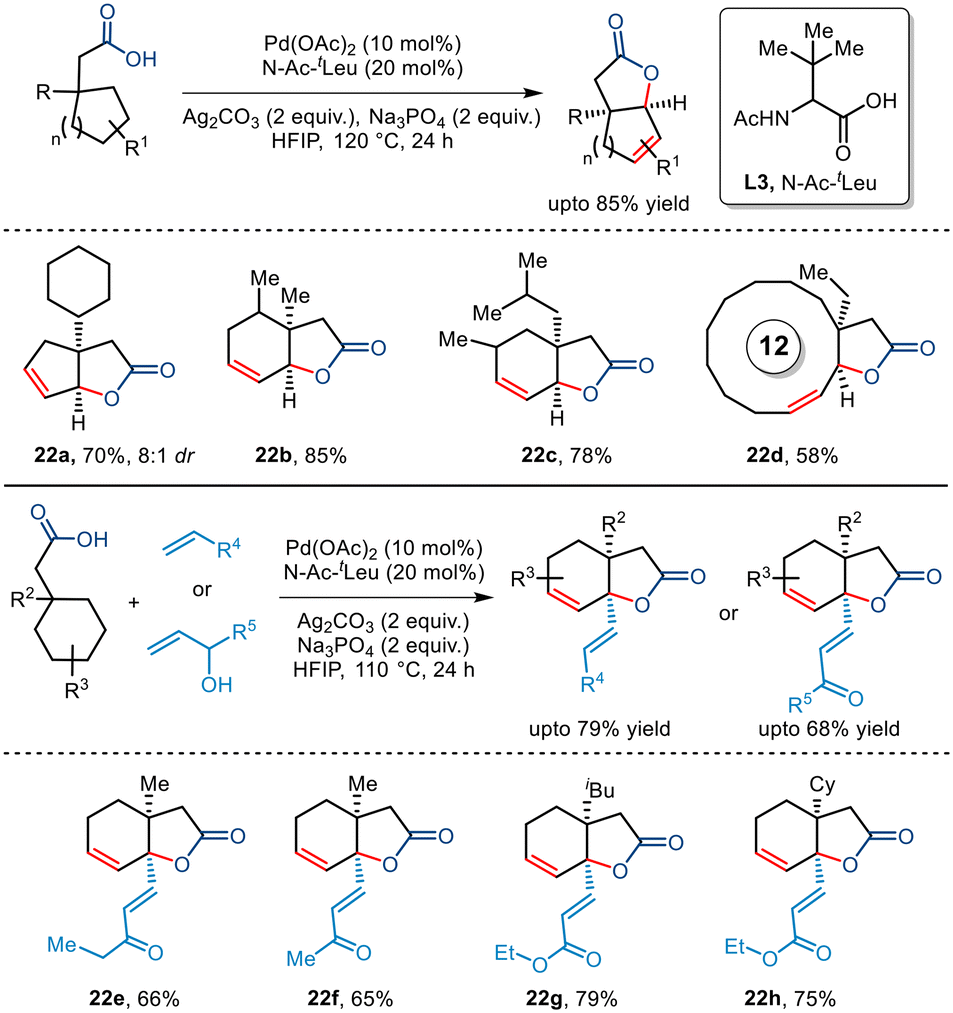
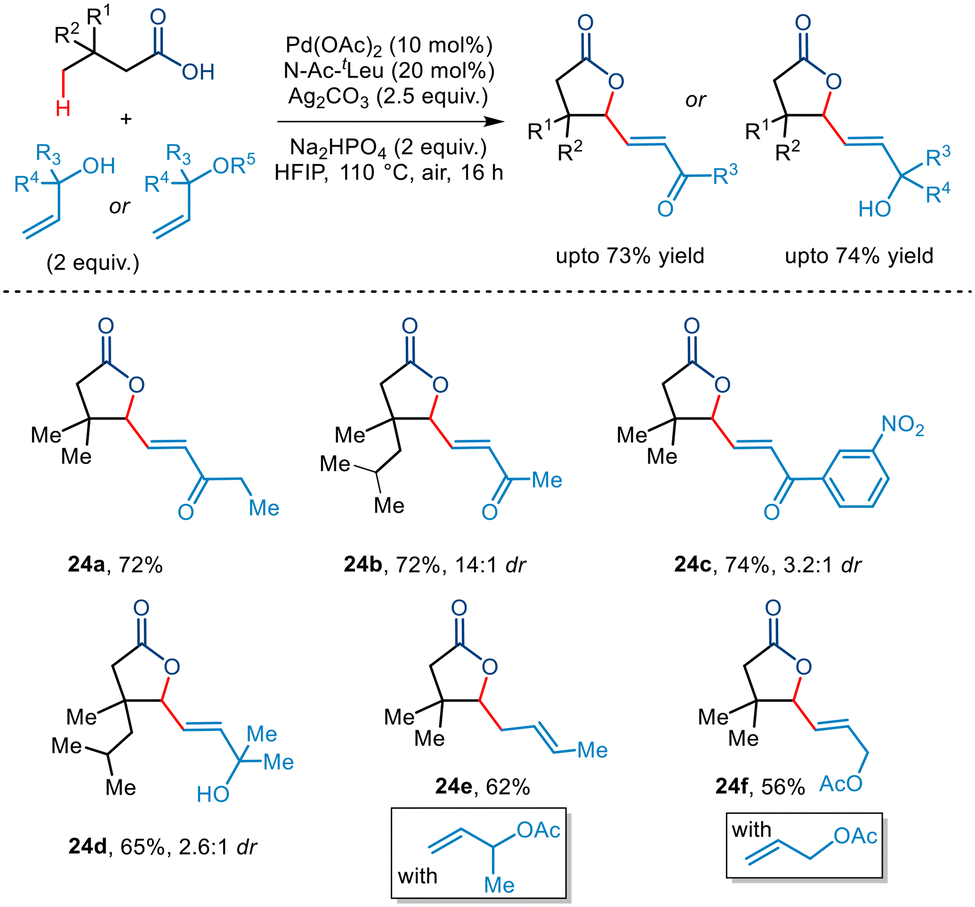
In 2022, Maiti and co-workers disclosed a novel report of free carboxylic acid-directed methylene γ C–H activation (Scheme 27).51 Interestingly, this was achieved by keeping a competing terminal methyl C–H bond intact throughout, thereby showcasing reverse regioselectivity. Aided by the bidentate N-Ac-tLeu ligand, bicyclic unsaturated lactones bearing different functionalities and varied sizes were synthesized including [12,5] and [15,5] fused lactones whose direct synthetic routes present a great challenge. This protocol was further extended to an intermolecular version where a reactive palladacycle upon interception with olefins and allyl alcohols furnished bicyclic unsaturated lactones bearing γ-olefinated products. Thus, this protocol presents an efficient route to transforming the methylene centre into a quaternary centre through tandem one-pot cyclization–dehydrogenation and olefination process.
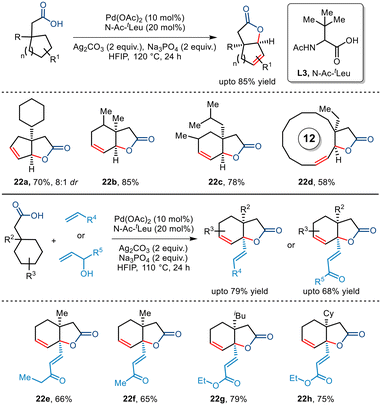 |
| | Scheme 27 Synthesis of unsaturated bicyclic lactones via carboxylic acid-directed methylene γ C–H activation. | |
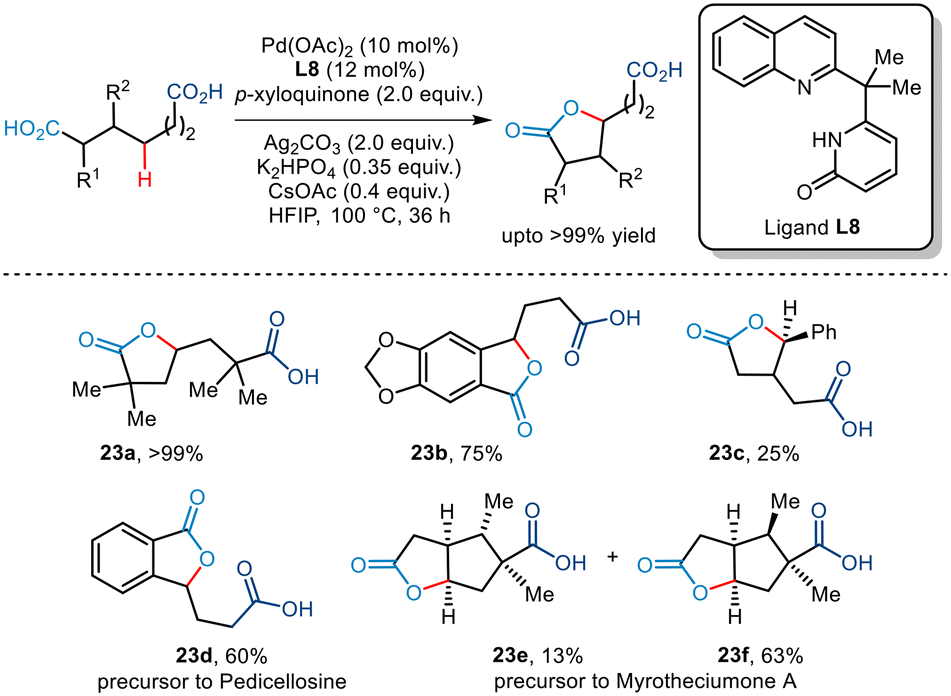
Subsequently, the Yu group established an efficient Pd-catalyzed pathway to achieve site-selective methylene γ C–H lactonization of dicarboxylic acids (Scheme 28).52 This transformation enabled the conversion of symmetrical dicarboxylic acids to unsymmetrical lactones while lactonizing the more substituted fragment in the case of unsymmetrical dicarboxylic acids, thus allowing effective regiocontrol. The use of quinoline–pyridone ligands inhibited bis-carboxylate chelation thereby fostering the C–H activation. In addition, the use of p-xyloquinone as an additive also aided in reductive elimination at the Pd-centre. The group envisaged that the desired tri-functionalized monolactone was furnished via carboxylic acid-directed γ-C–H activation followed by subsequent lactonization by the carboxylic group present at the other end. Substrates bearing substituents at the α, β, and γ positions successfully yielded the desired lactone in acceptable yields, even allowing the synthesis of a range of benzo-fused compounds. Furthermore, the group also highlighted the synthesis of two natural products namely, myrotheciumone A and pedicellosine using this strategy.
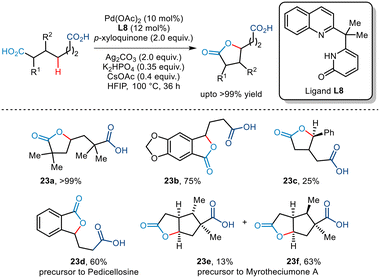 |
| | Scheme 28 Site-selective methylene γ-C–H lactonization of dicarboxylic acids. | |
Very recently, Maiti and co-workers demonstrated the first example of Pd-catalyzed dual-γ-1,1-C(sp3)–H activation of aliphatic carboxylic acids with allyl alcohols as coupling partners to furnish a wide range of multisubstituted γ-lactones (Scheme 29).53 The bidentate N-Ac-tLeu ligand was found to be essential for this transformation. Kinetic studies led to the conclusion that the C(sp3)–H activation step formed the rate-determining step. Further investigation revealed that a variety of primary, secondary, and tertiary allyl alcohols were tolerated. Interestingly, allyl acetates were also found to be compatible with the protocol. Aliphatic acids having various substituents at the β position and longer chain lengths successfully resulted in the five-membered lactone in synthetically acceptable yields. Incorporation of the α,β-unsaturated moiety at the γ position of the lactone allows further synthetic transformation to construct complex molecules.
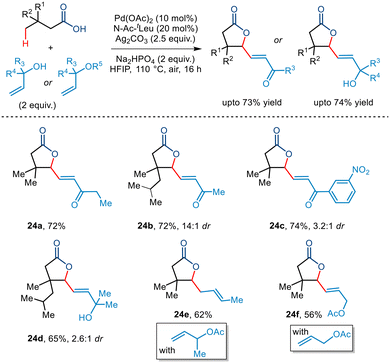 |
| | Scheme 29 Pd-catalyzed dual-γ-1,1-C(sp3)–H activation of aliphatic carboxylic acids with allyl alcohols. | |
Conclusions and outlook
Palladium-catalyzed C–H activation of unactivated aliphatic chains mediated by free carboxylic acids and amines has gained predominance in recent years. Extensive research carried out by various research groups has contributed to the establishment of this research area. Although the efficient and atom economic, i.e. ‘one step’ is viable for activating C(sp3)–H at proximal positions, activation of distal C(sp3)–H bonds suffer from a few limitations. The six or larger palladacycle formed during the process is thermodynamically not favourable, thereby making it highly challenging. Nevertheless, quite a few research groups have been successful in reporting C–H functionalization at distal positions without the requirement of exogenous directing groups. Such reports showcase the inherent ability of amine and carboxylic acid groups to act as directing groups. However, this strategy is still at its inception and requires further refinement for its general application in the industrial setting. For example, enantioselective approaches, less catalyst loadings, usage of abundant 3d metals as catalysts, and access to further remote positions are current challenges yet to be addressed. Such developments will further aid in bridging the gap and add significant value to this emerging field.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Council of Scientific and Industrial Research 01(3033)/21/EMR-II for funding. J. D. thanks IIT Bombay for financial support.
Notes and references
-
(a)
C. Lamberth and J. Dinges, Bioact. Carboxylic Compd. Classes Pharm. Agrochem., 2016, vol. 1 Search PubMed;
(b) L. J. Gooßen, N. Rodríguez and K. Gooßen, Carboxylic Acids as Substrates in Homogeneous Catalysis, Angew. Chem., Int. Ed., 2008, 47, 3100–3120 (
Angew. Chem.
, 2008
, 120
, 3144–3164
) CrossRef;
(c) G. D. Hartman, M. S. Egbertson, W. Halczenko, W. L. Laswell, M. E. Duggan, R. L. Smith, A. M. Naylor, P. D. Manno, R. J. Lynch, G. Zhang, C. T. Chang and R. J. Gould, Non-Peptide Fibrinogen Receptor Antagonists. 1. Discovery and Design of Exosite Inhibitors, J. Med. Chem., 1992, 35, 2–4642 CrossRef PubMed;
(d) V. Vranova, K. Rejsek and P. Formanek, Aliphatic, Cyclic, and Aromatic Organic Acids, Vitamins, and Carbohydrates in Soil: A Review, Sci. World J., 2013, 2013, 524239 Search PubMed;
(e) S. D. Roughley and A. M. Jordan, The Medicinal Chemist's Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
-
(a) B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Selective Intermolecular Carbon-Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution, Acc. Chem. Res., 1995, 28, 154–162 CrossRef CAS;
(b) S. J. Blanksby and G. B. Ellison, Bond Dissociation Energies of Organic Molecules, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed;
(c) H. M. L. Davies and D. J. Morton, Recent Advances in C–H Functionalization, J. Org. Chem., 2016, 81, 343–350 CrossRef CAS.
-
(a) B. M. Trost, Atom Economy-A Challenge for Organic Synthesis: Homogeneous Catalysis Leads the Way, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 (
Angew. Chem.
, 1995
, 107
, 285–307
) CrossRef CAS;
(b) R. G. Bergman, C–H Activation, Nature, 2007, 446, 391–393 CrossRef CAS PubMed;
(c) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Transition metal-catalyzed C–H activation reactions: diastereoselectivity and enantioselectivity, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC;
(d) D. A. Colby, R. G. Bergman and J. A. Ellman, Rhodium-Catalyzed C–C Bond Formation via Heteroatom-Directed C–H Bond Activation, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed;
(e) J. Le Bras and J. Muzart, Intermolecular Dehydrogenative Heck Reactions, Chem. Rev., 2011, 111, 1170–1214 CrossRef CAS PubMed;
(f) C. S. Yeung and V. M. Dong, Catalytic Dehydrogenative Cross-Coupling: Forming Carbon–Carbon Bonds by Oxidizing Two Carbon–Hydrogen Bonds, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS;
(g) J. Li, S. De Sarkar and L. Ackermann, meta- and para-Selective C–H Functionalization by C–H Activation, Top. Organomet. Chem., 2015, 55, 217–257 CrossRef;
(h) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Mild metal-catalyzed C–H activation: examples and concepts, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC;
(i) F. Wang, S. Yu and X. Li, Transition metal-catalysed couplings between arenes and strained or reactive rings: combination of C–H activation and ring scission, Chem. Soc. Rev., 2016, 45, 6462–6477 RSC;
(j) R. H. Crabtree and A. Lei, Introduction: C–H Activation, Chem. Rev., 2017, 117, 8481–8482 CrossRef CAS PubMed;
(k) J. A. Labinger, Platinum-Catalyzed C–H Functionalization, Chem. Rev., 2017, 117, 8483–8496 CrossRef CAS PubMed;
(l) C. G. Newton, S.-G. Wang, C. C. Oliviera and N. Cramer, Catalytic Enantioselective Transformations Involving C–H Bond Cleavage by Transition-Metal Complexes, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS PubMed;
(m) D. L. Davies, S. A. Macgregor and C. L. McMullin, Computational Studies of Carboxylate-Assisted
C–H Activation and Functionalization at Group 8–10 Transition Metal Centers, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed;
(n) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Transition-Metal-Catalyzed C–H Alkylation Using Alkenes, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed;
(o) Y. Wei, P. Hu, M. Zhang and W. Su, Metal-Catalyzed Decarboxylative C–H Functionalization, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed;
(p) X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, The Essential Role of Bond Energetics in C–H Activation/Functionalization, Chem. Rev., 2017, 117, 8622–8648 CrossRef CAS PubMed;
(q) O. Eisenstein, J. Milani and R. N. Perutz, Selectivity of C–H Activation and Competition between C–H and C–F Bond Activation at Fluorocarbons, Chem. Rev., 2017, 117, 8710–8753 CrossRef CAS PubMed;
(r) R. Shang, L. Ilies and E. Nakamura, Iron-Catalyzed C–H Bond Activation, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS;
(s) J. C. K. Chu and T. Rovis, Complementary Strategies for Directed C(sp3)–H Functionalization: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer, Angew. Chem., Int. Ed., 2018, 57, 62–101 (
Angew. Chem.
, 2018
, 130
, 64–105
) CrossRef CAS PubMed;
(t) A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Accessing Remote meta- and para-C(sp2)–H Bonds with Covalently Attached Directing Groups, Angew. Chem., Int. Ed., 2019, 58, 10820–10843 (
Angew. Chem.
, 2019
, 131
, 10934–10958
) CrossRef CAS.
-
(a) A. D. Ryabov, Mechanisms of Intramolecular Activation of C–H Bonds in Transition-Metal Complexes, Chem. Rev., 1990, 90, 403–424 CrossRef CAS;
(b) A. E. Shilov and G. B. Shul'pin, Activation of C–H Bonds by Metal Complexes, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
-
(a) T. W. Lyons and M. S. Sanford, Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed;
(b) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Palladium-Catalyzed Transformations of Alkyl C–H Bonds, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS.
-
(a) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, Palladium-Catalyzed Methylation and Arylation of sp2 and sp3 C–H Bonds in Simple Carboxylic Acids, J. Am. Chem. Soc., 2007, 129, 3510–3511 CrossRef CAS PubMed;
(b) Y. Zhu, X. Chen, C. Yuan, G. Li, J. Zhang and Y. Zhao, Pd-catalysed ligand-enabled carboxylate-directed highly regioselective arylation of aliphatic acids, Nat. Commun., 2017, 8, 14904–14911 CrossRef CAS;
(c) K. K. Ghosh and M. van Gemmeren, Pd-Catalyzed β-C(sp3)–H Arylation of Propionic Acid and Related Aliphatic Acids, Chem. – Eur. J., 2017, 23, 17697–17700 CrossRef CAS PubMed;
(d) Z. Zhuang, C.-B. Yu, G. Chen, Q.-F. Wu, Y. Hsiao, C. L. Joe, J. X. Qiao, M. A. Poss and J.-Q. Yu, Ligand-Enabled β-C(sp3)–H Olefination of Free Carboxylic Acids, J. Am. Chem. Soc., 2018, 140, 10363–10367 CrossRef CAS;
(e) G. Chen, Z. Zhuang, G. C. Li, T. G. Saint-Denis, Y. Hsiao, C. L. Joe and J.-Q. Yu, Ligand-Enabled β-C–H Arylation of α-Amino Acids Without Installing Exogenous Directing Groups, Angew. Chem., Int. Ed., 2017, 56, 1506–1509 (
Angew. Chem.
, 2017
, 129
, 1528–1531
) CrossRef CAS;
(f) P.-X. Shen, L. Hu, Q. Shao, K. Hong and J.-Q. Yu, Pd(II)-Catalyzed Enantioselective C(sp3)–H Arylation of Free Carboxylic Acids, J. Am. Chem. Soc., 2018, 140, 6545–6549 CrossRef CAS;
(g) L. Hu, P. X. Shen, Q. Shao, K. Hong, J. X. Qiao and J.-Q. Yu, Pd(II)-Catalyzed Enantioselective C(sp3)–H Activation/Cross-Coupling Reactions of Free Carboxylic Acids, Angew. Chem., Int. Ed., 2019, 58, 2134–2138 (
Angew. Chem.
, 2019
, 131
, 2156–2160
) CrossRef CAS.
-
(a) J. Das, S. Guin and D. Maiti, Diverse strategies for transition metal catalyzed distal C(sp3)–H functionalizations, Chem. Sci., 2020, 11, 10887–10909 RSC;
(b) J. Das, D. K. Mal, S. Maji and D. Maiti, Recent Advances in External-Directing-Group-Free C–H Functionalization of Carboxylic Acids without Decarboxylation, ACS Catal., 2021, 11, 4205–4229 CrossRef CAS;
(c) C. He, W. G. Whitehurst and M. Gaunt, Palladium-Catalyzed C(sp3)–H Bond Functionalization of Aliphatic Amines, Chem, 2019, 5, 1031–1058 CrossRef CAS.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
-
(a) R. N. Salvatore, C. H. Yoon and K. W. Jung, Synthesis of secondary amines, Tetrahedron, 2001, 57, 7785 CrossRef CAS;
(b) A. Hager, N. Vrielink, D. Hager, J. Lefranc and D. Trauner, Synthetic approaches towards alkaloids bearing α-tertiary amines, Nat. Prod. Rep., 2016, 33, 491–522 RSC.
-
(a) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Palladium(II)-Catalyzed C–H Activation/C–C Cross-Coupling Reactions: Versatility and Practicality, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 (
Angew. Chem.
, 2009
, 121
, 5196–5217
) CrossRef CAS;
(b) O. Baudoin, Transition metal-catalyzed arylation of unactivated C(sp3)–H bonds, Chem. Soc. Rev., 2011, 40, 4902–4911 RSC;
(c) H. Li, B. J. Lia and Z. J. Shi, Challenge and progress: palladium-catalyzed sp3 C–H activation, Catal. Sci. Technol., 2011, 1, 191–206 RSC;
(d) M. C. White, Adding Aliphatic C–H Bond Oxidations to Synthesis, Science, 2012, 335, 807–809 CrossRef CAS;
(e) J. Yamaguchi, A. D. Yamaguchi and K. Itami, C–H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 (
Angew. Chem.
, 2012
, 124
, 9092–9142
) CrossRef CAS;
(f) D. A. Colby, R. G. Bergman and J. A. Ellman, Rhodium-Catalyzed C–C Bond Formation via Heteroatom-Directed C–H Bond Activation, Chem. Rev., 2010, 110, 624–655 CrossRef CAS;
(g) O. Daugulis, J. Roane and L. D. Tran, Bidentate, Monoanionic Auxiliary-Directed Functionalization of Carbon–Hydrogen Bonds, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS;
(h) G. Rouquet and N. Chatani, Catalytic Functionalization of C(sp2)–H and C(sp3)–H Bonds by Using Bidentate Directing Groups, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 (
Angew. Chem.
, 2013
, 125
, 11942–11959
) CrossRef CAS;
(i) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Transition metal-catalyzed C–H bond functionalizations by the use of diverse directing groups, Org. Chem. Front., 2015, 2, 1107–1295 RSC;
(j) G. He, B. Wang, W. A. Nack and G. Chen, Syntheses and Transformations of α-Amino Acids via Palladium-Catalyzed Auxiliary-Directed sp3 C–H Functionalization, Acc. Chem. Res., 2016, 49, 635–645 CrossRef CAS PubMed;
(k) W. J. Yoo and C. J. Li, Cross-Dehydrogenative Coupling Reactions of sp3-Hybridized C–H Bonds, Top. Curr. Chem., 2010, 292, 281–302 CrossRef;
(l) S. A. Girard, T. Knauber and C. J. Li, The Cross-Dehydrogenative Coupling of C–H Bonds: A Versatile Strategy for C–C Bond Formations, Angew. Chem., Int. Ed., 2014, 53, 74–100 (
Angew. Chem.
, 2014
, 126
, 76–103
) CrossRef CAS PubMed.
-
(a) J. E. Spangler, Y. Kobayashi, P. Verma, D. H. Wang and J.-Q. Yu, α-Arylation of Saturated Azacycles and N-Methylamines via Palladium(II)-Catalyzed C(sp3)–H Coupling, J. Am. Chem. Soc., 2015, 137, 11876–11879 CrossRef CAS PubMed;
(b) S. J. Pastine, D. V. Gribkov and D. Sames, sp3 C–H Bond Arylation Directed by Amidine Protecting Group: α-Arylation of Pyrrolidines and Piperidines, J. Am. Chem. Soc., 2006, 128, 14220–14221 CrossRef CAS PubMed;
(c) N. Chatani, T. Asaumi, T. Ikeda, S. Yorimitsu, Y. Ishii, F. Kakiuchi and S. Murai, Carbonylation at sp3 C–H Bonds Adjacent to a Nitrogen Atom in Alkylamines Catalyzed by Rhodium Complexes, J. Am. Chem. Soc., 2000, 122, 12882–12883 CrossRef CAS;
(d) J. J. Topczewski, P. J. Cabrera, N. I. Saper and M. S. Sanford, Palladium-catalysed transannular C–H functionalization of alicyclic amines, Nature, 2016, 531, 220–224 CrossRef CAS PubMed;
(e) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS;
(f) N. Rodríguez, J. A. Romero-Revilla, M. A. Fernμndez-Ibμnez and J. C. Carretero, Palladium-catalyzed N-(2-pyridyl)sulfonyl-directed C(sp3)–H γ-arylation of amino acid derivatives, Chem. Sci., 2013, 4, 175–179 RSC;
(g) K. S. L. Chan, M. Wasa, L. Chu, B. N. Laforteza, M. Miura and J.-Q. Yu, Ligand-enabled cross-coupling of C(sp3)–H bonds with arylboron reagents via Pd(II)/Pd(0) catalysis, Nat. Chem., 2014, 6, 146–150 CrossRef CAS;
(h) Z. Huang, C. Wang and G. Dong, Catalytic C(sp3)–H Arylation of Free Primary Amines with an exo Directing Group Generated In Situ, Angew. Chem., Int. Ed., 2016, 55, 5299–5303 (
Angew. Chem.
, 2016
, 128
, 5385–5389
) CrossRef CAS;
(i) S. Aspin, A.-S. Goutierre, P. Larini, R. Jazzar and O. Baudoin, Synthesis of Aromatic α-Aminoesters: Palladium-Catalyzed Long-Range Arylation of Primary C(sp3)–H Bonds, Angew. Chem., Int. Ed., 2012, 51, 10808–10811 CrossRef CAS;
(j) E. T. Nadres, G. I. F. Santos, D. Shabashov and O. Daugulis, Scope and Limitations of Auxiliary-Assisted, Palladium-Catalyzed Arylation and Alkylation of sp2 and sp3 C–H Bonds, J. Org. Chem., 2013, 78, 9689–9714 CrossRef CAS.
-
(a) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Functionalization of C(sp3)–H bonds using a transient directing group, Science, 2016, 351, 252–256 CrossRef CAS;
(b) Y. Xu, M. C. Young, C. Wang, D. M. Magness and G. Dong, Catalytic C(sp3)–H Arylation of Free Primary Amines with an exo Directing Group Generated In Situ, Angew. Chem., Int. Ed., 2016, 55, 9084–9087 (
Angew. Chem.
, 2016
, 128
, 9230–9233
) CrossRef CAS;
(c) Y. Liu and H. Ge, Site-selective C–H arylation of primary aliphatic amines enabled by a catalytic transient directing group, Nat. Chem., 2017, 9, 26–32 CrossRef CAS;
(d) A. Yada, W. Liao, Y. Sato and M. Murakami, Buttressing Salicylaldehydes: A Multipurpose Directing Group for C(sp3)–H Bond Activation, Angew. Chem., Int. Ed., 2017, 56, 1073–1076 (
Angew. Chem.
, 2017
, 129
, 1093–1096
) CrossRef CAS;
(e) M. Kapoor, D. Liu and M. C. Young, Carbon Dioxide-Mediated C(sp3)–H Arylation of Amine Substrates, J. Am. Chem. Soc., 2018, 140, 6818–6822 CrossRef CAS PubMed;
(f) P. Gandeepan and L. Ackermann, Transient Directing Groups for Transformative C–H Activation by Synergistic Metal Catalysis, Chem, 2018, 4, 199–222 CrossRef CAS;
(g) S. S. J. Campbell and J. A. Bull, Transient imines as ‘next generation’ directing groups for
the catalytic functionalization of C–H bonds in a single operation, Org. Biomol. Chem., 2018, 16, 4582–4595 RSC;
(h) J. I. Higham and J. A. Bull, Transient imine directing groups for the C–H functionalization of aldehydes, ketones and amines: an update 2018–2020, Org. Biomol. Chem., 2020, 18, 7291–7315 RSC;
(i) H. Lin, C. Wang, T. D. Bannister and T. M. Kamenecka, Site-Selective γ-C(sp3)–H and γ-C(sp2)–H Arylation of Free Amino Esters Promoted by a Catalytic Transient Directing Group, Chem. – Eur. J., 2018, 24, 9535–9541 CrossRef CAS PubMed;
(j) S. St John-Campbell, A. K. Ou and J. A. Bull, Palladium-Catalyzed C(sp3)–H Arylation of Primary Amines Using a Catalytic Alkyl Acetal to Form a Transient Directing Group, Chem. – Eur. J., 2018, 24, 17838–17843 CrossRef CAS;
(k) N. Goswami, T. Bhattacharya and D. Maiti, Transient directing ligands for selective metal-catalysed C–H activation, Nat. Rev. Chem., 2021, 5, 646–659 CrossRef CAS.
- D. Willcox, B. G. N. Chappell, K. F. Hogg, J. Calleja, A. P. Smalley and M. J. Gaunt, A general catalytic β-C–H carbonylation of aliphatic amines to β-lactams, Science, 2016, 354, 851–857 CrossRef CAS PubMed.
- M. Nappi, C. He, W. G. Whitehurst, B. G. N. Chappell and M. J. Gaunt, Selective Reductive Elimination at Alkyl Palladium(IV) by Dissociative Ligand Ionization: Catalytic C(sp3)–H Amination to Azetidines, Angew. Chem., Int. Ed., 2018, 57, 3178–3182 (
Angew. Chem.
, 2018
, 130
, 3232–3236
) CrossRef CAS PubMed.
- J. Calleja, D. Pla, T. W. Gorman, V. Domingo, B. Haffemayer and M. J. Gaunt, A steric tethering approach enables palladium-catalysed C–H activation of primary amino alcohols, Nat. Chem., 2015, 7, 1009–1016 CrossRef CAS PubMed.
-
(a) G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, Highly Efficient Syntheses of Azetidines, Pyrrolidines, and Indolines via Palladium Catalyzed Intramolecular Amination of C(sp3)–H and C(sp2)–H Bonds at γ and δ Positions, J. Am. Chem. Soc., 2012, 134, 3–6 CrossRef CAS PubMed;
(b) E. T. Nadres and O. Daugulis, Heterocycle Synthesis via Direct C–H/N–H Coupling, J. Am. Chem. Soc., 2012, 134, 7–10 CrossRef CAS PubMed;
(c) J.-W. Xu, Z.-Z. Zhang, W.-H. Rao and B.-F. Shi, Site-Selective Alkenylation of δ-C(sp3)–H Bonds with Alkynes via a Six-Membered Palladacycle, J. Am. Chem. Soc., 2016, 138, 10750–10753 CrossRef CAS;
(d) B. B. Zhan, Y. Li, J. W. Xu, X. L. Nie, J. Fan, L. Jin and B. F. Shi, Site-Selective δ-C(sp3)–H Alkylation of Amino Acids and Peptides with Maleimides via a Six-Membered Palladacycle, Angew. Chem., Int. Ed., 2018, 57, 5858–5862 (
Angew. Chem.
, 2018
, 130
, 5960–5964
) CrossRef CAS;
(e) W. Cui, S. Chen, J.-Q. Wu, X. Zhao, W. Hu and H.-G. Wang, Palladium-Catalyzed Remote C(sp3)–H Arylation of 3-Pinanamine, Org. Lett., 2014, 16, 4288–4291 CrossRef CAS PubMed.
- H. Lin, X. Pan, A. L. Barsamian, T. M. Kamenecka and T. D. Bannister, Native Directed Site-Selective δ-C(sp3)–H and δ-C(sp2)–H Arylation of Primary Amines, ACS Catal., 2019, 9, 4887–4891 CrossRef CAS.
- P. K. Pramanick, Z. Zhou, Z.-L. Hou and B. Yao, Free Amino Group-Directed γ-C(sp3)–H Arylation of α-Amino Esters with Diaryliodonium Triflates by Palladium Catalysis, J. Org. Chem., 2019, 84, 5684–5694 CrossRef CAS PubMed.
- J. Rodrigalvarez, M. Nappi, H. Azuma, N. J. Flodén, M. E. Burns and M. J. Gaunt, Catalytic C(sp3)–H bond activation in tertiary alkylamines, Nat. Chem., 2020, 12, 76–81 CrossRef CAS PubMed.
- Z. Zhuang and J.-Q. Yu, Pd(II)-Catalyzed Enantioselective γ-C(sp3)–H Functionalizations of Free Cyclopropylmethylamines, J. Am. Chem. Soc., 2020, 142, 12015–12019 CrossRef CAS.
- J. Rodrigalvarez, L. A. Reeve, J. Miró and M. J. Gaunt, J. Am. Chem. Soc., 2022, 144, 3939–3948 CrossRef CAS PubMed.
- A. J. Hickman and M. S. Sanford, High-valent organometallic copper and palladium in catalysis, Nature, 2012, 484, 177–185 CrossRef CAS.
- K. Chen, D. Wang, Z.-W. Li, Z. Liu, F. Pan, Y.-F. Zhang and Z.-J. Shi, Palladium catalyzed C(sp3)–H acetoxylation of aliphatic primary amines to γ-amino alcohol derivatives, Org. Chem. Front., 2017, 4, 2097–2101 RSC.
- C. S. Buettner, D. Willcox, B. G. N. Chappell and M. J. Gaunt, Mechanistic investigation into the C(sp3)–H acetoxylation of morpholinone, Chem. Sci., 2019, 10, 83–89 RSC.
- Z. M. Png, J. R. Cabrera-Pardo, J. Peiró Cada-hía and M. J. Gaunt, Diastereoselective C–H carbonylative annulation of aliphatic amines: a rapid route to functionalized γ-lactams, Chem. Sci., 2018, 9, 7628–7633 RSC.
- T. J. Donohoe, C. J. R. Bataille and G. H. Churchill, Highlights of natural product synthesis, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2006, 102, 98–122 RSC.
- N. Y. Kozitsyna, Y. A. Nefedov, D. I. Kochubey, T. S. Zyubina, A. E. Gekhman, M. N. Vargaftik and I. I. Moiseev, Novel heterometallic palladium–silver complex, Inorg. Chim. Acta, 2011, 370, 382–387 CrossRef CAS.
- S. Li, G. Chen, C.-G. Feng, W. Gong and J.-Q. Yu, Ligand-Enabled γ-C–H Olefination and Carbonylation: Construction of β-Quaternary Carbon Centers, J. Am. Chem. Soc., 2014, 136, 5267–5270 CrossRef CAS.
- C. He and M. J. Gaunt, Ligand-assisted palladium-catalyzed C–H alkenylation of aliphatic amines for the synthesis of functionalized pyrrolidines, Chem. Sci., 2017, 8, 3586–3592 RSC.
-
H. G. Korth and R. Sustman, Carboxylic Acids and Carboxylic Acid Derivatives, Thieme, Stuttgart, 1985, 4th edn, pp. 193–469 Search PubMed.
- G. D. Hartman, M. S. Egbertson, W. Halczenko, W. L. Laswell, M. E. Duggan, R. L. Smith, A. M. Naylor, P. D. Manno, R. J. Lynch, G. Zhang, C. T.-C. Chang and R. J. Gould, Non-Peptide Fibrinogen Receptor Antagonists. 1. Discovery and Design of Exosite Inhibitors 1, J. Med. Chem., 1992, 35, 4640–4642 CrossRef CAS.
-
(a) A. Dey, S. Pimparkar, A. Deb, S. Guin and D. Maiti, Chelation-Assisted Palladium-Catalyzed γ-Arylation of Aliphatic Carboxylic Acid Derivatives, Adv. Synth. Catal., 2017, 359, 1301–1307 CrossRef CAS;
(b) S. KC, R. K. Dhungana, B. Shrestha, S. Thapa, N. Khanal, P. Basnet, R. W. Lebrun and R. Giri, Ni-Catalyzed Regioselective Alkylarylation of Vinylarenes via C(sp3)–C(sp3)/C(sp3)–C(sp2) Bond Formation and Mechanistic Studies, J. Am. Chem. Soc., 2018, 140, 9801–9805 CrossRef CAS;
(c) H. G. Lee, G. Lautrette, B. L. Pentelute and S. L. Buchwald, Palladium-Mediated Arylation of Lysine in Unprotected Peptides, Angew. Chem., Int. Ed., 2017, 56, 3177–3181 (
Angew. Chem.
, 2017
, 129
, 3225–3229
) CrossRef CAS PubMed;
(d) Z. J. Cai, C. X. Liu, Q. Gu and S. L. You, Thioketone-Directed Palladium(II)-Catalyzed C–H Arylation of Ferrocenes with Aryl Boronic Acids, Angew. Chem., Int. Ed., 2018, 57, 1296–1299 (
Angew. Chem.
, 2018
, 130
, 1310–1313
) CrossRef CAS;
(e) J. C. Lewis, A. M. Berman, R. G. Bergman and J. A. Ellman, Rh(I)-Catalyzed Arylation of Heterocycles via C–H Bond Activation: Expanded Scope through Mechanistic Insight, J. Am. Chem. Soc., 2008, 130, 2493–2500 CrossRef CAS;
(f) B. Wang, W. A. Nack, G. He, S.-Y. Zhang and G. Chen, Palladium-catalyzed trifluoroacetate-promoted mono-arylation of the β-methyl group of alanine at room temperature: synthesis of β-arylated α-amino acids through sequential C–H functionalization, Chem. Sci., 2014, 5, 3952–3957 RSC;
(g) Y. Aihara and N. Chatani, Nickel-Catalyzed Direct Arylation of C(sp3)–H Bonds in Aliphatic Amides via Bidentate-Chelation Assistance, J. Am. Chem. Soc., 2014, 136, 898–901 CrossRef CAS;
(h) R. Giri, J. Liang, J. G. Lei, J. J. Li, D. H. Wang, X. Chen, I. C. Naggar, C. Guo, B. M. Foxman and J.-Q. Yu, Pd-Catalyzed Stereoselective Oxidation of Methyl Groups by Inexpensive Oxidants under Mild Conditions: A Dual Role for Carboxylic Anhydrides in Catalytic C–H Bond Oxidation, Angew. Chem., Int. Ed., 2005, 44, 7420–7424 (
Angew. Chem.
, 2005
, 117
, 7586–7590
) CrossRef CAS PubMed;
(i) R.-Y. Zhu, L.-Y. Liu and J.-Q. Yu, Highly Versatile β-C(sp3)–H Iodination of Ketones Using a Practical Auxiliary, J. Am. Chem. Soc., 2017, 139, 12394–12397 CrossRef CAS;
(j) X. Wu, Y. Zhao and H. Ge, Nickel-Catalyzed Site-Selective Alkylation of Unactivated C(sp3)–H Bonds, J. Am. Chem. Soc., 2014, 136, 1789–1792 CrossRef CAS PubMed;
(k) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, Stereoselective Synthesis of β-Alkylated α-Amino Acids via Palladium-Catalyzed Alkylation of Unactivated Methylene C(sp3)–H Bonds with Primary Alkyl Halides, J. Am. Chem. Soc., 2013, 135, 12135–12141 CrossRef CAS PubMed;
(l) A. Deb, S. Singh, K. Seth, S. Pimparkar, B. Bhaskararao, S. Guin, R. B. Sunoj and D. Maiti, Experimental and Computational Studies on Remote γ-C(sp3)–H Silylation and Germanylation of Aliphatic Carboxamides, ACS Catal., 2017, 7, 8171–8175 CrossRef CAS;
(m) S. Guin, A. Deb, P. Dolui, S. Chakraborty, V. K. Singh and D. Maiti, Promoting Highly Diastereoselective γ-C–H Chalcogenation of α-Amino Acids and Aliphatic Carboxylic Acids, ACS Catal., 2018, 8, 2664–2669 CrossRef CAS;
(n) S. Li, G. Chen, C.-G. Feng, W. Gong and J.-Q. Yu, Ligand-Enabled γ-C–H Olefination and Carbonylation: Construction of β-Quaternary Carbon Centers, J. Am. Chem. Soc., 2014, 136, 5267–5270 CrossRef CAS;
(o) G. Liao, X.-S. Yin, K. Chen, Q. Zhang, S.-Q. Zhang and B.-F. Shi, Stereoselective alkoxycarbonylation of unactivated C(sp3)–H bonds with alkyl chloroformates via Pd(II)/Pd(IV) catalysis, Nat. Commun., 2016, 7, 12901–12909 CrossRef PubMed;
(p) Y. Shen, Y. Gu and R. Martin, sp3 C–H Arylation and Alkylation Enabled by the Synergy of Triplet Excited Ketones and Nickel Catalysts, J. Am. Chem. Soc., 2018, 140, 12200–12209 CrossRef CAS;
(q) G. Laudadio, S. Govaerts, Y. Wang, D. Ravelli, H. F. Koolman, M. Fagnoni, S. W. Djuric and T. Noël, Selective C(sp3)–H Aerobic Oxidation Enabled by Decatungstate Photocatalysis in Flow, Angew. Chem., Int. Ed., 2018, 57, 4078–4082 (
Angew. Chem.
, 2018
, 130
, 4142–4146
) CrossRef CAS;
(r) J. Pedroni, M. Boghi, T. Saget and N. Cramer, Access to β-Lactams by Enantioselective Palladium(0)-Catalyzed C(sp3)–H Alkylation, Angew. Chem., Int. Ed., 2014, 53, 9064–9067 (
Angew. Chem.
, 2014
, 126
, 9210–9213
) CrossRef CAS PubMed;
(s) J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Photoredox-Controlled Mono- and Di-Multifluoroarylation of C(sp3)–H Bonds with Aryl Fluorides, Angew. Chem., Int. Ed., 2017, 56, 7266–7270 (
Angew. Chem.
, 2017
, 129
, 7372–7376
) CrossRef CAS;
(t) S. Jerhaoui, J.-P. Djukic, J. Wencel-Delord and F. Colobert, Asymmetric, Nearly Barrierless C(sp3)–H Activation Promoted by Easily-Accessible N-Protected Aminosulfoxides as New Chiral Ligands, ACS Catal., 2019, 9, 2532–2542 CrossRef CAS;
(u) M.-Z. Lu, X.-R. Chen, H. Xu, H.-X. Dai and J.-Q. Yu, Ligand-enabled ortho-C–H olefination of phenylacetic amides with unactivated alkenes, Chem. Sci., 2018, 9, 1311–1316 RSC.
- Y. Zhu, X. Chen, C. Yuan, G. Li, J. Zhang and Y. Zhao, Pd-catalysed ligand-enabled carboxylate-directed highly regioselective arylation of aliphatic acids, Nat. Commun., 2017, 8, 1–8 Search PubMed.
-
(a) T. A. Stephenson, S. M. Morehouse, A. R. Powell, J. P. Heffer and G. Wilkinson, Carboxylates of Palladium, Platinum, and Rhodium, and their Adducts, J. Am. Chem. Soc., 1965, 3632–3640 RSC;
(b) G. Shi and Y. Zhang, Carboxylate-Directed C–H Functionalization, Adv. Synth. Catal., 2014, 356, 1419–1442 CrossRef CAS;
(c) S. Li, H. Wang, Y. Weng and G. Li, Carboxy Group as a Remote and Selective Chelating Group for C–H Activation of Arenes, Angew. Chem., 2019, 131, 18673–18678 CrossRef;
(d) L. Cai, S. Li, C. Zhou and G. Li, Carboxyl-Assisted meta-Selective C–H Functionalizations of Benzylsulfonamides, Org. Lett., 2020, 22, 7791–7796 CrossRef CAS PubMed;
(e) A. Uttry and M. van Gemmeren, Direct C(sp3)–H Activation of Carboxylic Acids, Synthesis, 2020, 52, 479–488 CrossRef CAS.
- R. Giri and J.-Q. Yu, Synthesis of 1,2- and 1,3-Dicarboxylic Acids via Pd(II)-Catalyzed Carboxylation of Aryl and Vinyl C–H Bonds, J. Am. Chem. Soc., 2008, 130, 14082–14083 CrossRef CAS.
- P. Dolui, J. Das, H. B. Chandrashekar, S. S. Anjana and D. Maiti, Ligand-Enabled Pd(II)-Catalyzed Iterative γ-C(sp3)–H Arylation of Free Aliphatic Acid, Angew. Chem., Int. Ed., 2019, 58, 13773–13777 (
Angew. Chem.
, 2019
, 131
, 13911–13914
) CrossRef CAS PubMed.
- L. Liu, Y.-H. Liu and B.-F. Shi, Synthesis of amino acids and peptides with bulky side chains via ligand-enabled carboxylate-directed γ-C(sp3)–H arylation, Chem. Sci., 2020, 11, 290–294 RSC.
- K. L. Bay, Y. F. Yang and K. N. Houk, Multiple roles of silver salts in palladium-catalyzed C–H activations, J. Organomet. Chem., 2018, 864, 19–25 CrossRef CAS.
-
(a) E. Hernando, J. Villalva, A. M. Martínez, I. Alonso, N. Rodríguez, R. Arrayas Gomez and J. C. Carretero, Palladium-Catalyzed Carbonylative Cyclization of Amines via γ-C(sp3)–H Activation: Late-Stage Diversification of Amino Acids and Peptides, ACS Catal., 2016, 6, 6868–6882 CrossRef CAS;
(b) X. Zhang, G. Lu, M. Sun, M. Mahankali, Y. Ma, M. Zhang, W. Hua, Y. Hu, Q. Wang, J. Chen and G. He, A general strategy for synthesis of cyclophane-braced peptide macrocycles via palladium-catalysed intramolecular sp3 C–H arylation, Nat. Chem., 2018, 10, 540–548 CrossRef CAS PubMed.
-
(a) J. Tang, Y. He, H. Chen, W. Sheng and H. Wang, Synthesis of bioactive and stabilized cyclic peptides by macrocyclization using C(sp3)–H activation, Chem. Sci., 2017, 8, 4565–4570 RSC;
(b) W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, Site-Selective C(sp3)–H Functionalization of Di-, Tri-, and Tetrapeptides at the N-Terminus, J. Am. Chem. Soc., 2014, 136, 16940–16946 CrossRef CAS.
-
(a)
P. J. Stang, in Modern Acetylene Chemistry, ed. P. J. Stang and F. Diederich, VCH, Weinheim, 1995, pp. 67–98 CrossRef;
(b)
R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley-VCH, Weinheim, 1999 Search PubMed;
(c)
J. Gorzynski Smith, Org. Chem., McGraw-Hill Education, 2008 Search PubMed;
(d) J. F. Lutz and Z. Zarafshani, Efficient construction of therapeutics, bioconjugates, biomaterials and bioactive surfaces using azide–alkyne “click” chemistry, Adv. Drug Delivery Rev., 2008, 60, 958–970 CrossRef CAS.
-
(a) J. P. Brand and J. Waser, Electrophilic alkynylation: the dark side of acetylene chemistry, Chem. Soc. Rev., 2012, 41, 4165–4179 RSC;
(b) L. D. Caspers and B. J. Nachtsheim, Directing-Group-mediated C–H-Alkynylations, Chem. – Asian J., 2018, 13, 1231–1247 CrossRef CAS.
- F. Ghiringhelli, A. Uttry, K. K. Gosh and M. van Gemmeren, Direct β- and γ-C(sp3)–H Alkynylation of Free Carboxylic Acids, Angew. Chem., Int. Ed., 2020, 59, 23127–23131 (
Angew. Chem.
, 2020
, 132
, 23327–23331
) CrossRef CAS PubMed.
- J. He, Q. Shao, Q. Wu and J.-Q. Yu, Pd(II)-Catalyzed Enantioselective C(sp3)–H Borylation, J. Am. Chem. Soc., 2017, 139, 3344–3347 CrossRef CAS.
- H. Fu, P.-X. Shen, J. He, F. Zhang, S. Li, P. Wang, T. Liu and J.-Q. Yu, Ligand-Enabled Alkynylation of C(sp3)–H Bonds with Palladium(II) Catalysts, Angew. Chem., Int. Ed., 2017, 56, 1873–1876 (
Angew. Chem.
, 2017
, 129
, 1899–1902
) CrossRef CAS.
- J. P. Brand, C. Chevalley and J. Waser, Beilstein, One-pot gold-catalyzed synthesis of 3-silylethynyl indoles from unprotected o-alkynylanilines, J. Org. Chem., 2011, 7, 565–569 CAS.
-
(a) M. Tobisu, Y. Ano and N. Chatani, Palladium-Catalyzed Direct Alkynylation of C–H Bonds in Benzenes, Org. Lett., 2009, 11, 3250–3252 CrossRef CAS;
(b) A. S. Dudnik and V. Gevorgyan, Formal Inverse Sonogashira Reaction: Direct Alkynylation of Arenes and Heterocycles with Alkynyl Halides, Angew. Chem., Int. Ed., 2010, 49, 2096–2098 (
Angew. Chem.
, 2010
, 122
, 2140
) CrossRef CAS PubMed;
(c) A. Mondal, H. Chen, L. Flämig, P. Wedi and M. van Gemmeren, Sterically Controlled Late-Stage C–H Alkynylation of Arenes, J. Am. Chem. Soc., 2019, 141, 18662–18667 CrossRef CAS.
-
Natural Lactones and Lactams: Synthesis Occurrence and Biological Activity, ed. T. Janecki, Wiley-VCH, Weinheim, 2013 Search PubMed.
- K. K. Ghosh, A. Uttry, A. Mondal, F. Ghiringhelli, P. Wedi and M. van Gemmeren, Ligand-Enabled γ-C(sp3)–H Olefination of Free Carboxylic Acids, Angew. Chem., Int. Ed., 2020, 59, 12848–12852 (
Angew. Chem.
, 2020
, 132
, 12948–12952
) CrossRef CAS PubMed.
- J. Das, P. Dolui, W. Ali, J. P. Biswas, H. B. Chandrashekar, G. Prakash and D. Maiti, A direct route to six and seven membered lactones via γ-C(sp3)–H activation: a simple protocol to build molecular complexity, Chem. Sci., 2020, 11, 9697–9702 RSC.
-
D. Maiti, J. Das, W. Ali, A. Ghosh, T. Pal, A. Mandal, C. Teja, S. Dutta, R. Pothikumar, H. Ge and X. Zhang, Access to Unsaturated Bicyclic Lactones by Overriding Conventional C(sp3)-H Site Selectivity, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-4brs3.
- H. S. S. Chan, J.-M. Yang and J.-Q. Yu, Catalyst-controlled site-selective methylene C–H lactonization of dicarboxylic acids, Science, 2022, 376, 1481–1487 CrossRef CAS.
- J. Das, T. Pal, W. Ali, S. R. Sahoo and D. Maiti, Pd-Catalyzed Dual-γ-1,1-C(sp3)–H Activation of Free Aliphatic Acids with Allyl–O Moieties, ACS Catal., 2022, 12, 11169–11176 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Shaeel Ahmed
Al-Thabaiti
c,
Soha M.
Albukhari
c,
Qana A.
Alsulami
b,
Shaeel Ahmed
Al-Thabaiti
c,
Soha M.
Albukhari
c,
Qana A.
Alsulami