
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGas-phase oxidative dehydrogenation of long chain alkenols for the production of key fragrance ingredients: from Rosalva isomers to Costenal analogues†
Jacopo
De Maron
 a,
Tommaso
Tabanelli
*a,
Francesca
Ospitali
a,
Carlos
Lopez Cruz
b,
Paolo
Righi
a and
Fabrizio
Cavani
*a
a,
Tommaso
Tabanelli
*a,
Francesca
Ospitali
a,
Carlos
Lopez Cruz
b,
Paolo
Righi
a and
Fabrizio
Cavani
*a
aDipartimento di Chimica Industriale “Toso Montanari”, Alma Mater Studiorum – Università di Bologna, Viale del Risorgimento 4, 40136 Bologna, Italy. E-mail: tommaso.tabanelli@unibo.it
bIFF Benicarló S.L., Avenida Felipe Klein 2, 12580 Benicarló, Spain
First published on 4th January 2023
Abstract
The continuous-flow, gas-phase oxidative dehydrogenation (ODH) of an actual mixture of decen-1-ol isomers (“Isorosalva” alcohol) towards the corresponding mixture of aldehydes (“Costenal” analogues, valuable ingredients in perfumes formulation) is herein reported for the first time over noble metal-free catalysts. In particular, the optimisation of the reaction conditions over a copper ferrite (Cu/Fe/O), as well as dedicated characterizations and comparisons between the fresh, the post-reaction (reduced) and regenerated (re-oxidised) catalytic material, allowed us to underline the key role of well dispersed copper oxide over a Fe-enriched spinel in promoting the selective ODH of Isorosalva alcohol. The superior catalytic activity and selectivity of CuO/γ-Fe2O3 synthesized ad hoc were attributed to the very high dispersion of Cu over the support as well as to a cooperative effect between Cu and Fe species in promoting the redox cycle.
Introduction
The selective oxidation of complex alcohols (e.g., aromatic, branched or unsaturated fatty alcohols) to the corresponding aldehydes is a fundamental step in the preparation of a wide variety of fragrance ingredients and food additives.1 Unsaturated fatty aldehydes such as neral, geranial, citronellal and decenal, to cite a few, are among the most important ingredients in perfumery and are used, individually or in combination, in the formulation of most perfume types.2 Traditionally, the oxidation of fatty alcohols to the corresponding aldehydes has been carried out in high yield using stoichiometric amounts of oxidants such as pyridinium chlorochromate, activated MnO2, 2-iodobenzoic acid, the Dess–Martin periodinane or the Swern reagent.3 Unfortunately, all of these methods present safety/toxicity issues, produce large amounts of waste and have significant scale-up issues; on top of that, their sustainability is further reduced because apart from Swern-type oxidations that are carried out in dimethyl sulfoxide, the preferred solvent for all the other methods is dichloromethane.Owing to these limitations of stoichiometric oxidants, significant efforts have been made in the past years to develop the aerobic oxidation of saturated/unsaturated fatty alcohols in the liquid phase: as a result, several homogeneous catalytic systems based on complexes or salts of Cu,4–6 Ru,7–12 Pd,13–15 Au,16 Fe17,18 and Co19 were reported to be active and selective under mild reaction conditions for the oxidation of model substrates such as 1-octanol, 1-decanol, citronellol and geraniol to the corresponding aldehydes. Still, the scale up of these processes remains difficult due to the cost of noble metals, the difficult catalyst recovery and recycle on an industrial scale, the use of non-green solvents (with the only exception of ref. 9) and the need for organic co-catalysts (e.g. DBAB,4 TEMPO derivatives,5,6,10,17–19 hydroquinone,7 and amines13–15). These issues prompted the research towards the development of heterogeneous catalysts containing the same active elements immobilized onto a suitable support: the aerobic oxidation of saturated/unsaturated fatty alcohols has been carried out successfully over a number of supported catalysts based on platinum (Pt and Pt/Bi supported onto Al2O3 and active carbon),20 on palladium (Pd supported onto hydrotalcite,21 mesocellular silica foam (MCF),22 γ-Al2O3 (ref. 23) and MgO (ref. 24)), on ruthenium (Ru supported onto hydroxyapatite25 and γ-Al2O3 (ref. 26 and 27) or immobilized into the pores of MCM-41 (ref. 28)), on silver (Ag supported onto SiO2 and promoted by physical mixing with CeO2),29 on gold (Au supported onto CeO2,30 Al2O3–CeO2 mixed oxide31 and TiO2 (ref. 32)), and on vanadium (vanadyl acetylacetonate supported on polyaniline).33 On the other hand, a number of bulk oxides and mixed oxides such as Ru/Co/O,34 Ru–Co–Al-hydrotalcite,35 Ru–Co(OH)2–CeO2,36 MnFe1.5Ru0.35Cu0.15O4 (ref. 37) and Mn3O4 (ref. 38) were reported to be active and selective as well. The oxidation of fatty alcohols in the liquid-phase is usually carried out in batch under mild reaction conditions resulting in good selectivities; moreover, the substrate scope is wide and not limited to vaporizable fatty alcohols. Despite these advantages, the productivity of these liquid-phase oxidations is usually limited by the long reaction times needed and by the need for expensive work up operations.
On the other hand, a gas-phase process can be easily carried out continuously and, with catalyst separation being unnecessary, work up operations are extremely simplified. As an example, light alcohols may be converted into the corresponding aldehydes by means of gas-phase dehydrogenation (DH) with a catalyst such as MgO,39 mixed oxides of Mg and transition metals possessing redox properties (Fe,40 Cr41 or Ga42,43), or V-containing oxides44 such as FeVO4 and V2O5, to cite a few. Meanwhile, the literature dealing with the gas-phase DH of fatty alcohols is less investigated and focused on the transformation of model substrates such as octanol45 and decanol.46 However, DH processes are endothermic and suffer from several limitations, such as the relatively high reaction temperature required to promote dehydrogenations, the presence of a thermodynamic equilibrium that limits the conversion of the substrate and the occurrence of parasitic reactions such as dehydrations47 and oligomerisations, the latter often leading to catalyst deactivation due to coke formation. Another drawback of DH processes depends on the reaction mechanism, involving the dissociation of the alcohol into a proton (H+) and an alkoxide anion (RO−) on the catalyst surface, followed by a hydride (H−) transfer from the alkoxide anion to the vicinal proton. In fact, since the hydride can be transferred also to another reactive hydrogen acceptor such as a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, DH catalysts foster to a certain extent also the so-called catalytic transfer hydrogenation (CHT).48,49 For this reason, DH is less suited than ODH for the preparation of unsaturated fatty aldehydes. The oxidative dehydrogenation (ODH) of light alcohols in the gas-phase over a catalyst consisting of bulk or supported Ag,50 Fe–Mo mixed oxides,51 FeVO4 and V2O5 (ref. 44 and 52) is a well-established technology and represents the prevailing method to produce on a large-scale formaldehyde from methanol53 and, to a lower degree, acetaldehyde from ethanol.54 However, the applicability of gas-phase ODH is not limited to light alcohols: in fact, the BASF citral process (which involves the ODH of prenol to prenal over Au, Ag or Cu as a key intermediate step55) represents a successful application of ODH to the industrial-scale preparation of a valuable unsaturated fatty aldehyde. The co-feeding of a limited amount of oxygen, with respect to DH processes, allows the reaction temperature to be lowered by promoting the oxidation of the co-produced hydrogen to water via a concerted mechanism, eventually shifting the thermodynamic equilibrium towards the product side. However, despite the potential of gas-phase ODH, this reaction remains very challenging, because of the high reactivity of fatty alcohols and aldehydes at the relatively high temperatures required by their vaporization and reaction. As a consequence, the academic literature available on this topic is narrower than that about liquid-phase oxidations, and deals mostly exclusively with octanol (as a model substrate) and catalytic systems based on either gold (Au/SiO2,56 Au–Ni alloy/SiO2,57 Au/Cu-fibres58,59 and Au/Ni fibres60) or silver (bulk Ag,61 Ag supported over a zeolite coated Cu grid,62 Ag/silicon nanowire arrays63 and Ag-Cu/SiC64).
C double bond, DH catalysts foster to a certain extent also the so-called catalytic transfer hydrogenation (CHT).48,49 For this reason, DH is less suited than ODH for the preparation of unsaturated fatty aldehydes. The oxidative dehydrogenation (ODH) of light alcohols in the gas-phase over a catalyst consisting of bulk or supported Ag,50 Fe–Mo mixed oxides,51 FeVO4 and V2O5 (ref. 44 and 52) is a well-established technology and represents the prevailing method to produce on a large-scale formaldehyde from methanol53 and, to a lower degree, acetaldehyde from ethanol.54 However, the applicability of gas-phase ODH is not limited to light alcohols: in fact, the BASF citral process (which involves the ODH of prenol to prenal over Au, Ag or Cu as a key intermediate step55) represents a successful application of ODH to the industrial-scale preparation of a valuable unsaturated fatty aldehyde. The co-feeding of a limited amount of oxygen, with respect to DH processes, allows the reaction temperature to be lowered by promoting the oxidation of the co-produced hydrogen to water via a concerted mechanism, eventually shifting the thermodynamic equilibrium towards the product side. However, despite the potential of gas-phase ODH, this reaction remains very challenging, because of the high reactivity of fatty alcohols and aldehydes at the relatively high temperatures required by their vaporization and reaction. As a consequence, the academic literature available on this topic is narrower than that about liquid-phase oxidations, and deals mostly exclusively with octanol (as a model substrate) and catalytic systems based on either gold (Au/SiO2,56 Au–Ni alloy/SiO2,57 Au/Cu-fibres58,59 and Au/Ni fibres60) or silver (bulk Ag,61 Ag supported over a zeolite coated Cu grid,62 Ag/silicon nanowire arrays63 and Ag-Cu/SiC64).
Among fatty aldehydes, the isomers of decenal are notable ingredients in the formulation of perfumes and other personal care products, not only as single isomers (i.e., dec-9-enal, commercial name “Costenal”)65 but also as a combination of isomers: as an example, mixtures of dec-6-enal, dec-7-enal and dec-8-enal with well-defined compositions were patented as novel ingredients by International Flavors & Fragrances Inc.66 Despite their relevance, very few catalytic processes have been proposed as alternatives to the traditional oxidation of dec-9-en-1-ol (commercially known as “Rosalva”) with stoichiometric reactants such as pyridinium chlorochromate,67 Dess–Martin periodinanes,68 activated MnO2,69 K2S2O8/NiSO4 in the presence of a base70 or oxalyl dichloride, DMSO and alkyl amines in Swern-type oxidations),71 which are not sustainable for large-scale production. To the best of our knowledge, the liquid-phase aerobic oxidation of pure Rosalva to Costenal has been reported only twice: with air and a homogeneous tetrapropylammonium perruthenate catalyst,12 and with pure O2 and a heterogeneous Ru–Co oxide catalyst.34
Similarly, also the gas-phase ODH of Rosalva to Costenal was reported only twice, by Gallezot et al.65 over a 4.5% Ag/SiC catalyst charged into a traditional fixed bed reactor and by Cao et al.72 with an Ag/SiO2 catalyst deposited onto a continuous-flow microreactor. Considering that a pure Rosalva alcohol can be easily isomerised into a selected mixture of its isomers,66 in this study an actual mixture of decen-1-ols (“Isorosalva” alcohols), produced and supplied by International Flavors & Fragrances Inc., was used as the starting material for the continuous-flow, gas-phase ODH aimed at the production of the corresponding mixture of aldehydes (“Opalene” aldehydes), directly exploitable in fragrance formulation. Moreover, herein we report for the first time this peculiar ODH reaction promoted by a cheap, noble metal-free, non-stoichiometric Cu-ferrite catalyst with a Fe-to-Cu ratio equal to 4 (Cu0.6Fe2.4O4.2) and by a supported copper oxide catalyst (6 wt% CuO/γ-Fe2O3). The comparison between our synthetic strategy and the alternatives reported in the literature is depicted in Scheme 1. In particular, dedicated catalytic tests showed that our catalyst performs better, in terms of selectivity, than a typical V2O5–TiO2 benchmark catalyst for the ODH of light alcohols; moreover, analyses of the product distribution and an in-depth characterisation of the catalytic materials allowed us to highlight that the co-presence of both Cu and Fe result in a synergistic effect that boosts the selectivity to the desired Opalene mixture.
 | ||
| Scheme 1 Comparison between a) oxidation of Rosalva to Costenal with traditional stoichiometric oxidants, b) aerobic oxidation of Rosalva with air/O2 in the liquid-phase, c) oxidative dehydrogenation (ODH) of Rosalva in the gas-phase and d) oxidative dehydrogenation (ODH) of an Isorosalva mixture of isomers in the gas-phase. | ||
Experimental
Catalyst preparation
A copper ferrite with a nominal Fe/Cu atomic ratio of 4 (henceforth in the text, Cu/Fe/O) was synthesized adapting a co-precipitation technique from the literature.73 Briefly, 200 mL of a solution containing 0.5 mol L−1 Fe(NO3)3·9 H2O (Sigma-Aldrich, 98%), and 0.125 mol L−1 Cu(NO3)2·2.5 H2O (Sigma-Aldrich, 98%) was slowly added dropwise under vigorous stirring to 300 mL of a solution containing 2 mol L−1 NaOH (Sigma-Aldrich, 98%). At the end of the addition, the resulting brown suspension was aged for 1 h under stirring, filtered over a Buchner funnel, and washed with deionized water until pH 7 to remove hydroxide, nitrate, and sodium ions. The resulting wet solid was dried overnight at 120 °C, ground in an agate mortar, and calcined with a heating rate of 5 °C min−1 up to 450 °C; the final temperature was kept for 4 h. The vanadium oxide supported over titania (V2O5/TiO2) was prepared by means of wet impregnation, adapting the procedure reported here.74 To obtain a wt% of V equal to 3.6, equivalent to a 6 wt% of V2O5, 9.4 g of commercial TiO2 (9.4 g, CristalACTiV DT-51) was suspended under stirring in 30 mL of a hot (≈50 °C) solution containing 0.22 mol L−1 NH4VO3 (Sigma Aldrich, 99%). The suspension was stirred for 1 h, evaporated in a rotavapor and finally dried in an oven at 120 °C overnight. The resulting material was ground in an agate mortar and calcined with a heating rate of 5 °C min−1 up to 500 °C; the final temperature was kept for 5 h. γ-Fe2O3 (maghemite) was prepared from commercial Fe3O4 (magnetite, Sigma-Aldrich, 97%, particle size 50–100 nm) by calcination at 200 °C for 3 h. Upon heating the powder changed its colour from black to brown-reddish and maintained its magnetic properties. The copper oxide supported over silica (CuO/SiO2) was prepared by means of incipient wetness impregnation. To obtain a wt% of Cu(0) equal to 5, equivalent to 6.1 wt% of CuO, 1.83 g of Cu(NO3)2·2.5 H2O (Sigma-Aldrich, 98%) was dissolved in 20 mL of deionized water and added dropwise to 9.5 g of SiO2 (Grace 360) until the mud point was reached (2 mL of solution per gram of support). The resulting wet powder was dried in an oven at 120 °C for 2 h and calcined with a heating rate of 5 °C min−1 up to 450 °C; the final temperature was kept for 4 h. The copper oxide supported over maghemite (CuO/γ-Fe2O3) was prepared by means of incipient wetness impregnation in the same way as CuO/SiO2. In order to obtain a wt% of Cu(0) equal to 5, equivalent to 6.1 wt% of CuO, 1.83 g of Cu(NO3)2·2.5 H2O was dissolved in 5.5 mL of deionized water and added dropwise to 9.5 g of Fe3O4 (magnetite, Sigma-Aldrich, 97%, particle size 50–100 nm) until the mud point was reached (0.55 mL of solution per gram of support). The resulting wet powder was dried in an oven at 120 °C for 2 h and calcined with a heating rate of 5 °C min−1 up to 450 °C; the final temperature was kept for 4 h. All the catalysts were charged into the reactor in the form of pellets with a granulometry between 30 and 60 mesh, which were obtained by grinding a self-sustaining disk (height ≈ 1 mm, diameter ≈ 30 mm) through a 30-mesh sieve placed on top of a 60-mesh sieve and collecting the fraction of pellets trapped between the two sieves.Catalyst characterization
The XRD powder patterns of all the catalysts were collected using a Bragg–Brentano Philips X'Pert diffractometer and Cu Kα radiation (λ = 1.54178 Å, Ni-filtered). The average size of the coherently scattering domain (CSD) of materials was calculated applying the Scherrer equation (eqn (1)):CSD = (K·λ)/(β·cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ϑ) ϑ) | (1) |
Catalytic tests
All catalytic tests were carried out at atmospheric pressure on the gas-phase plant shown in Fig. S1 (ESI†). The reactor was a conventional fixed bed down-flow quartz reactor, and all the catalysts were charged as pellets with size between 30 and 60 mesh.The liquid reactant “Isorosalva Alcohol” (IRA, mixture of isomers of decen-1-ol provided by Iff) was injected with a KD Scientific Legacy 100 volumetric pump into a thin stainless-steel line (1/16 inches of diameter) and directly driven ≈5 cm above the catalytic bed with 10 mL min−1 N2. A second flux of pre-heated (250 °C) air diluted with N2 was driven to the top of the reactor to obtain the desired molar fraction of O2 and the desired total flux. Usually, the catalyst was flowed with the same air/N2 mixture to be used during the catalytic test and heated up to reaction temperature at 20 °C min−1; the final temperature was kept for 30 min before starting to feed the liquid reactant. The effluent from the reactor was bubbled through two cold traps in series: the former was filled with 15 mL of acetonitrile (AcCN, Sigma-Aldrich, 99.8%) and kept at room temperature to absorb the condensable products; the latter was filled with glass spheres and kept at 0 °C by means of an ice bath to condensate the AcCN stripped from the first trap. At regular intervals of time, the reaction mixture was transferred from the first cold trap to a 50 mL flask, together with the AcCN needed to wash the lower portion of the reactor below the catalytic bed and to rinse the cold trap 3 times. Finally, 1 g of dodecane (Sigma-Aldrich, 99%) solution (4 × 10−5 mol g−1) was added as the internal standard. All catalytic tests were carried out for the time required to obtain at least 5 samples (5–6 h) to be analysed by means of GC-FID; the mean values of conversion and selectivities were calculated once steady-state conditions were achieved, usually during the last 3 h of time on stream. The gas mixture exiting the cold traps were driven to an Agilent 8860 GC instrument equipped with an FID and TCD. The quantification of the condensable products was carried out offline with an Agilent HP-5 capillary column (30 m × 0.32 mm × 0.25 μm) connected to the FID. The quantification of the gaseous products (O2, N2, CO, CO2) was carried out online with a set of two parallel columns combining an Agilent CP-Molsieve 5 Å capillary column and an Agilent CP-PoraBOND Q capillary column (“Agilent J&W Select Permanent Gases/CO2”) connected to the TCD. An Agilent Technologies 6890 gas chromatograph equipped with an Agilent HP-5 capillary column (30 m × 250 μm × 1.05 μm) and coupled to an Agilent Technologies 5973 mass analyser (GC-MS) was used to identify unknown products; also, the retention times of unknown products were compared to those of purchased standard reference compounds. The following equations were used to calculate Isorosalva Alcohol conversion (X IRA, eqn (2)), yields (Yi, eqn (3)), selectivities (Si, eqn (4)), sum of yields (Yield Sum, eqn (5)), and carbon balance (C-balance, eqn (6)). All these parameters were calculated in terms of moles of carbon.
| X IRA = (molIRACIN − molIRACOUT/molIRACIN) × 100 | (2) |
| Yi = (moliCOUT/molIRACIN) × 100 | (3) |
| Si = (Yi)/(X IRA) × 100 | (4) |
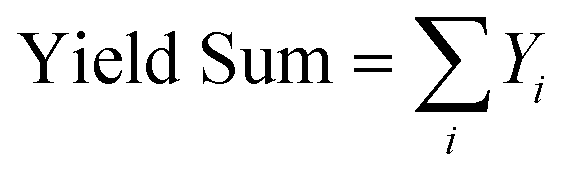 | (5) |
| C-balance = (Yield Sum)/(X IRA) × 100 | (6) |
Results and discussion
Characterization of fresh catalysts
The main physico-chemical features of the catalysts are listed in Table 1. CuO/SiO2 possessed the highest specific surface area (455 m2 g−1) among the materials investigated, followed by the Cu/Fe/O mixed oxide (165 m2 g−1), V2O5/TiO2 (22 m2 g−1), CuO/γ-Fe2O3 (9 m2 g−1) and γ-Fe2O3 (9 m2 g−1). Apart from the precipitated copper ferrite, for all the other materials the final specific surface area was mainly affected by the initial surface area of the oxide support.| SSAa [m2 g−1] | CSD sizeb [nm] | Element or oxide contentc,d [wt%] | |
|---|---|---|---|
| a SSA measured by single-point BET. b CSD size calculated from XRD. c Elemental analysis carried out by SEM-EDS. d Theoretical values are reported within brackets. | |||
| V2O5/TiO2 | 22 | 125 (V2O5) | V2O5 = 6.2 (6) |
| 58 (TiO2) | |||
| Cu/Fe/O | 165 | 5 (Cu-ferrite) | Cu = 14.3 (16) |
| Fe = 57.4 (56) | |||
| CuO/SiO2 | 455 | 35 (CuO) | CuO = 6.9 (6.1) |
| CuO/γ-Fe2O3 | 9 | 81 (γ-Fe2O3) | CuO = 4.8 (6.1) |
| γ-Fe2O3 | 9 | 78 (γ-Fe2O3) | — |
The XRD powder pattern of Cu/Fe/O (Fig. S2a†) was characterized by broad reflections, attributable to a nano-crystalline, defective spinel structure (CSD size = 5 nm, Table 1), with no substantial impurities of segregated CuOx or FeOx. The high defectivity of Cu/Fe/O (Cu0.6Fe2.4O4.2) was attributed to the excess of the trivalent cation Fe(III) with respect to the divalent cation Cu(II) deviating from that of the stoichiometric spinel CuFe2O4. As a result, the material displayed a relatively high specific surface area and a small CSD size. The diffractogram of V2O5/TiO2 (Fig. S2b†), in agreement with the literature,75 arose from the superimposition of the pattern of TiO2 anatase and V2O5, with the former being largely predominant; both phases had high crystallinity. The XRD powder pattern of CuO/SiO2 (Fig. S3a†) was characterized by a very broad band, centred at 2θ = 22° and attributable to amorphous silica, plus a set of sharper reflections that were ascribed to well-developed CuO tenorite crystals (CSD size = 35 nm, Table 1). The pattern of γ-Fe2O3 (Fig. S3b†) was characterized by sharp reflections, attributable to the pattern of the maghemite cubic spinel (CSD size = 78 nm, Table 1). The maghemite pattern was distinguished from that of the parent magnetite (Fe3O4 spinel) because all the reflections of the γ-form were shifted towards higher 2θ values, corresponding to lower d-spacings.76 Finally, the XRD powder pattern of CuO/γ-Fe2O3 is shown in Fig. S4:† unlike CuO/SiO2, in this case a well-defined pattern attributable to CuO was absent: this can mean either that the dispersion of copper was much higher on maghemite than on silica, or that upon calcination Cu(II) was incorporated into the newly formed γ-Fe2O3 structure resulting in a Cu enriched maghemite. Instead, the diffractogram displayed a well-defined pattern of maghemite (CSD size = 81 nm, Table 1), plus a few weak but sharp reflexes attributable to an impurity of hematite (α-Fe2O3, CSD size = 42 nm, Table 1).
The elemental composition of the catalysts was verified by means of SEM-EDS. The experimental wt% of V2O5 on TiO2 measured by EDS was 6.2%, in good agreement with the theoretical value of 6. In the case of Cu/Fe/O, the EDS elemental map of Cu (Fig. S5b†) indicates that it was homogeneously distributed in the sample, which was a true mixed metal solid solution. The electron images and elemental maps of CuO/SiO2, as well as its Raman spectrum (both shown in Fig. S6†) indicated that Cu was concentrated in specific areas, forming flower-like crystals of tenorite (CuO)77 with ≈100 nm diameter. Conversely, the elemental maps of CuO/γ-Fe2O3 suggested that Cu was more homogeneously distributed in the sample (Fig. S7b†).
The high-resolution TEM characterization of Cu/Fe/O is shown in Fig. S8:† the material was formed by small, quasi-spherical and poorly crystalline grains with an average diameter of 5 nm, in agreement with the CSD size calculated from the XRD diffractogram with the Scherrer equation (eqn (1)). On the other hand, the TEM electron images reported in Fig. S9† showed that CuO/γ-Fe2O3 was made of much bigger grains (100–150 nm) and that supported CuO nanoparticles were not systematically present over the support. Nonetheless, the EDS microanalysis carried out over more than 10 grains showed that Cu was always present. In particular, the mass fraction of CuO ranged from 3.7 to 7.1 wt%, with an average value of 5.3 wt%, in good agreement with the value of 4.8 wt% obtained with the SEM elemental analysis, suggesting that Cu-species were extremely dispersed over the support, possibly forming small clusters, overlayers or being partially incorporated into the defects of γ-Fe2O3.
ODH of Isorosalva alcohol over Cu/Fe/O and V2O5/TiO2
A series of preliminary blank runs was carried out in order to optimize the reaction conditions and the feeding system setup. The results of two blank runs carried out at 350 °C reaction temperature, contact time (τ) of 1 s and with a feed mixture containing 5 mol% of Isorosalva Alcohol (IRA), 5 mol% of O2, and 90% of N2 are shown in the ESI† (Fig. S10). Unknown compounds obtained during the catalytic tests were grouped together and are reported in figures under the category “Others”. When IRA was vaporized by injecting the suitable liquid flow rate in a 1/8′′ stainless-steel line heated at 250 °C before the reactor (blank run 1, Fig. S10†), a significant fraction of the reactant decomposed (X IRA = 25%) and the carbon balance was very low (<50%). On the other hand, when IRA was fed directly into the reactor at about 5 cm above the catalytic bed by means of a stainless-steel capillary line, IRA conversion was 7% and the carbon balance became higher than 90% (blank run 2, Fig. S10†). These results indicate that IRA underwent some uncatalyzed homogeneous reactions in the gas-phase when exposed to high temperatures for a relatively long time, as it was in the case of blank run 1; these reactions induced the extensive fouling of the reactor inner walls, both before and after the catalytic bed, as shown in Fig. S11.† However, the extent of these unwanted homogenous reactions can be reduced either by reducing the dead volume of the reactor, or by reducing the time spent by IRA inside the reactor at high temperature. This way, the extent of the fouling was effectively reduced and limited to the reactor walls below the catalytic bed in blank run 2, as shown in Fig. S11.† Therefore, all the following catalytic tests were carried out with the same IRA feeding system used in blank run 2.Initially, the ODH of IRA was carried out over Cu/Fe/O, V2O5/TiO2 and without a catalyst (for comparison) at 300 °C temperature, τ = 1 s and with a feed mixture IRA/O2/N2 = 5/5/90; under these conditions, the O2 in the feed was two-fold the amount required by the stoichiometry of the desired reaction. The results of these catalytic tests are reported as a bar chart in Fig. 1 in terms of conversion of IRA (X IRA), selectivities to products (S), and carbon balance (C-balance); these parameters were calculated in terms of moles of carbon. The conversion of IRA at 300 °C was negligible without a catalyst, therefore the occurrence of homogeneous reactions under these conditions can be ruled out. As expected, V2O5/TiO2 was relatively active in the ODH of IRA: the conversion under these conditions was 34% and O2 was consumed entirely. OPA was the main product in the reaction mixture with a selectivity of 32%, followed by several isomers of decadiene (S DD = 20%) and carbon oxides (S COx = 18%, with CO2 being largely predominant with respect to CO). Decadienes are likely to be formed by the dehydration of IRA over acidic sites,78 as shown in Scheme 2 (reaction b), while carbon oxides are formed by the complete combustion of the reactant or products. Apart from CO and CO2, all the products found in the reaction mixture contained 10 C atoms; therefore, the occurrence of the oxidative cleavage of CC double bonds79 was ruled out. When IRA was fed over Cu/Fe/O, its conversion (23%) was lower than the one obtained over V2O5/TiO2 under the same conditions, but the two materials displayed a similar activity for the ODH, because the OPA yields with V2O5/TiO2 and Cu/Fe/O were 11% and 12%, respectively. The superior selectivity of Cu/Fe/O to the desired products (S OPA = 51%) arose from its negligible activity for the unwanted dehydration of IRA to DD. Similarly, with Cu/Fe/O also the selectivity to COx was higher than that obtained with V2O5/TiO2.
 | ||
| Fig. 1 ODH of IRA over V2O5/TiO2, Cu/Fe/O and without any catalyst. Reaction conditions: volume of catalyst = 1 cm3, temperature = 300 °C, IRA/O2/N2 = 5/5/90 mol%, contact time (τ) = 1 second. Symbols: Isorosalva Alcohol conversion (X IRA, orange), oxygen conversion (X O2, dark red), carbon balance (C-balance, purple), opalene selectivity (S OPA, light blue), decadiene selectivity (S DD, yellow), other by-products' selectivity (S Others, black) and COx selectivity (S COx, grey). Mean values calculated at the steady-state during the last 3 h of time on stream. | ||
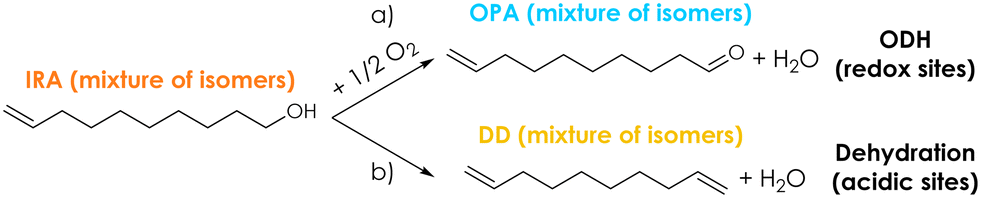 | ||
| Scheme 2 Proposed reaction pathways for a) the desired ODH of IRA towards OPA and b) the parallel parasitic dehydration of IRA towards a mixture of decadiene (DD) isomers. | ||
To better understand the structure–activity relationship governing the reaction scheme, the two catalysts were further characterized by means of NH3- and CO2-TPD to compare their acid–base surface features, and by means of TPR with H2 to compare their reducibility. The NH3- and CO2-TPD profiles for Cu/Fe/O are shown in Fig. S12a and b† respectively, while those for V2O5/TiO2 are reported in Fig. S13a and b† respectively. Also, the density of weak, medium-strength and strong acidic and basic sites measured by deconvolution of the experimental desorption profiles is reported in Table 2 for both materials. Cu/Fe/O was amphoteric and possessed a higher overall density of acidic sites (0.98 μmol m−2 of NH3) than basic sites (0.66 μmol m−2 of CO2). However, the acidic sites of Cu/Fe/O were of medium-strength (two desorption events centred at 223 and 303 °C), while the distribution of basic sites was more complex and several types of sites were shown (four desorption events centred at 117, 242, 310 and 440 °C).
V2O5/TiO2 was amphoteric as well, but it possessed an overall density of both acidic and basic sites higher compared to Cu/Fe/O (3.6 μmol m−2 of NH3 and 3.0 μmol m−2 of CO2). In conclusion, the stronger acidity, in terms of both density and desorption temperature, of V2O5/TiO2 is likely to be responsible for the unwanted dehydration of IRA to DD. The redox properties of Cu/Fe/O and V2O5/TiO2 were characterized by means of TPR with H2 and their reduction profiles as well as a detailed discussion can be found in Chapter S2 and Fig. S14 in the ESI.† The ODH of IRA was further investigated using the Cu/Fe/O catalyst with the aim to increase the yield of OPA. Two catalytic tests were carried out by increasing either the reaction temperature, from 300 to 350 °C (Fig. 2a), or the contact time from 1 to 4.5 s (Fig. 2c) and the results were compared to those of the catalytic tests carried out at 300 °C and τ = 1 s (Fig. 2b). The increase of temperature from 300 °C to 350 °C, while maintaining a contact time of 1 s (Fig. 2a), led to only a moderate increase of IRA conversion (from 23% to 32%). At the same time, OPA selectivity shrank from 51% to 22% because it was consumed by consecutive reactions, such as the formation of 10-nonadecadienones (NDD, red bars, S = 13%) and other unknown by-products (black bars, S = 16%). Also, the carbon balance decreased from 85% at 300 °C down to 70% at 350 °C, suggesting the occurrence of other unwanted reactions (e.g., homogeneous reactions, coking and fouling). The increase of the contact time from 1 s to 4.5 s at 300 °C (Fig. 2c) led to a more significant increment of IRA conversion (from 23% to 52%) but also in this case OPA underwent consecutive reactions that decreased its selectivity from 51% down to 35% and led to the formation of decenoic acids (DA, green bars S = 8%) and 10-dodecatrienals (ODT, blue bars, S = 19%). Remarkably, increasing the contact time did not negatively affect the carbon balance, suggesting that some unwanted reactions (e.g., gas-phase homogeneous reactions, coking and fouling) were fostered mainly by high temperatures, in agreement with the results of the blank runs at 350 °C (Fig. S10 and S11†). Summarising this new information, a more detailed reaction scheme is shown in Scheme 3. The presence of DA among the reaction products was explained by the consecutive oxidation of OPA (Scheme 3, reaction c), which was fostered by harsher reaction conditions. Then, the resulting DA underwent a ketonization reaction80 forming the symmetric C19 ketone NDD (Scheme 3, reaction e); in agreement with previous results43,80 the ketonization was particularly favoured at 350 °C and DA were quantitatively transformed into NDD. Finally, the C20 aldehyde ODT was formed by the aldol condensation of OPA with itself (Scheme 3, reaction d), which was fostered by an increase of the contact time (e.g., 4.5 seconds). Considering the stoichiometry of the selective ODH of IRA to OPA (Scheme 3, reaction a), the O2 fed into the system was always two-fold the one required for the complete conversion of IRA. However, O2 was always quantitatively consumed under all conditions reported in Fig. 2, because it was unselectively converted to overoxidation products (i.e., CO2). Results reported in Fig. 2 showed that an increase of the contact time was the most effective way to improve IRA conversion without fostering its overoxidation to COx. Therefore, we decided to investigate in more depth the behaviour of the Cu/Fe/O catalyst under these conditions (e.g., 300 °C of temperature, τ = 1 s, feed mixture IRA/O2/N2 = 5/5/90 mol%) as a function of time on stream; the results are shown in Fig. 3. During the first hour of reaction, the Cu-ferrite was extremely active, and the conversion of IRA was complete, but the C-balance was lower than 20% and COx were almost the sole products of the reaction. Then, starting from the second hour on stream, the selectivity to condensable products and the C-balance increased, and stabilized after 3 h on stream. At the same time, the conversion of IRA decreased steadily down to half of its initial value during 5 h on stream.
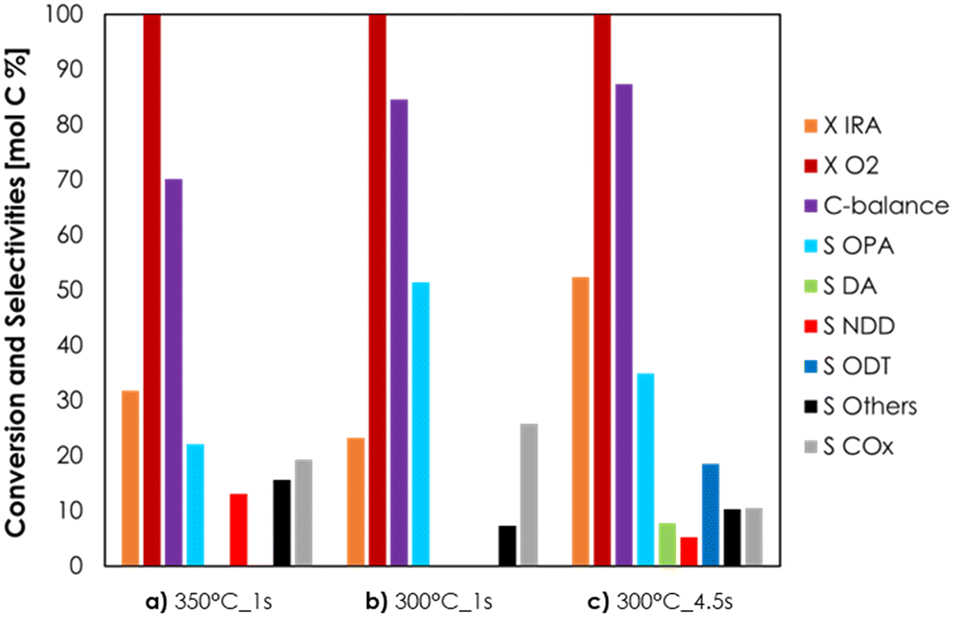 | ||
| Fig. 2 ODH of IRA over Cu/Fe/O as a function of reaction temperature and contact time. Reaction conditions: volume of catalyst = 1 cm3 (τ = 1 second) or 3 cm3 (τ = 4.5 second), IRA/O2/N2 = 5/5/90 mol%. Symbols: Isorosalva Alcohol conversion (X IRA, orange), oxygen conversion (X O2, dark red) carbon balance (C-balance, purple), opalene selectivity (S OPA, light blue), decenoic acid selectivity (S DA, green), 10-nonadecadienone selectivity (S NDD, red), 2-octyldodecatrienal selectivity (S ODT, blue), other by-products' selectivity (S Others, black) and COx selectivity (S COx, grey). Mean values calculated at the steady-state during the last 3 h of time on stream. | ||
 | ||
| Scheme 3 Proposed complete reaction scheme: a) ODH of IRA towards OPA; b) parallel parasitic dehydration of IRA towards DD; c) overoxidation of OPA towards DA; d) aldol condensation of OPA to ODT; e) ketonization of DA to NDD; other reactions include the formation of heavy compounds on the catalyst surface and the total oxidation to COx. | ||
 | ||
| Fig. 3 ODH of IRA over Cu/Fe/O as a function of the time on stream. Reaction conditions: volume of catalyst = 1 cm3, temperature = 300 °C, IRA/O2/N2 = 5/5/90 mol%, contact time (τ) = 4.5 seconds. Symbols: Isorosalva Alcohol conversion (X IRA, orange), oxygen conversion (X O2, dark red) carbon balance (C-balance, purple), opalene selectivity (S OPA, light blue), decenoic acid selectivity (S DA, yellow), 10-nonadecadienone selectivity (S NDD, red), 2-octyldodecatrienal selectivity (S ODT, blue), other by-products' selectivity (S Others, black) and COx selectivity (S COx, grey). | ||
Therefore, we decided to stop the feeding of IRA and regenerate the catalyst in situ by feeding 50 mL min−1 of air diluted with 10 mL min−1 N2 at 400 °C for 3 h. Then, the feeding of IRA was resumed and, surprisingly, a completely different product distribution was obtained: in fact, OPA became the main product of the reaction over the regenerated Cu/Fe/O (S OPA = 70%, X IRA = 45%, values calculated as the average value between the 7th and the 16th hour on stream). At the same time, the obtained C-balance was excellent, and the selectivity to the consecutive products NDD and ODT, which were produced in significant amounts over the fresh Cu/Fe/O, dropped from 18 down to 3% and from 5 to 0%, respectively. Finally, the regenerated Cu/Fe/O catalyst maintained a stable catalytic performance for 10 h. Interestingly, during the very first hour of reaction CO2 was the major product over both the fresh and the freshly regenerated Cu/Fe/O catalyst. The outcome of the catalytic test reported in Fig. 3 indicated that the regeneration in situ after the first run played a key role in improving the catalyst selectivity to OPA.
Characterization of Cu/Fe/O after reaction and after regeneration
The redox mechanism of ODH postulates that, as a consequence of the reduction of a metal cation (e.g., Cu2+ or Fe3+ in the case of Cu/Fe/O) by the organic reactant, the formation of the co-produced water occurs at the expense of the lattice oxygen of the metal oxide. Then, the catalytic cycle is closed by the reoxidation of the reduced metal by the molecular O2 in the gaseous feed. Considering that under all the reaction conditions investigated so far, the conversion of O2 was quantitative, it is likely that the reoxidation of the catalyst was limited by the complete consumption of O2 in the feed, resulting in a catalyst that under reaction conditions was presumably strongly reduced. Upon the ODH of IRA followed by in situ regeneration, the fresh Cu/Fe/O underwent a transformation that significatively enhanced its selectivity to the desired product OPA. Therefore, a small portion of the catalyst employed in the experiment shown in Fig. 3 was withdrawn from the reactor before and after the in situ regeneration; then, these materials were thoroughly characterized. Samples were labelled as follows: Cu/Fe/O-F (fresh), Cu/Fe/O-AR (after reaction) and Cu/Fe/O-R (regenerated).The XRD powder patterns of Cu/Fe/O-F, Cu/Fe/O-AR and Cu/Fe/O-R are shown in Fig. 4a and b. The diffractogram of Cu/Fe/O-AR (Fig. 4a, red) was characterized by the pattern of a spinel phase as it was for Cu/Fe/O-F, plus three peaks attributable to metallic Cu centred at 43, 50 and 74 2θ degrees. Therefore, Cu/Fe/O was reduced during the ODH of IRA leading to the segregation of metallic Cu over a Fe-enriched spinel. The formation of the latter was confirmed by the small shift towards lower 2θ angles of the reflections of the spinel phase (Fig. 4b). Moreover, the reflections attributable to the Fe-enriched spinel-phase were sharper than the ones of Cu/Fe/O-F, indicating that the crystallinity of the catalysts increased during the ODH of IRA; hence, the CSD size of the spinel calculated by the Scherrer equation (eqn (1)) increased from 5 nm to 10 nm and the SSA dropped from 165 m2 g−1 to 63 m2 g−1. In the diffractogram of Cu/Fe/O-R (Fig. 4a, light blue) the reflections of metallic Cu disappeared leaving only a spinel phase, plus a few small reflections attributable to hematite (α-Fe2O3); no reflections attributable to CuO or Cu2O were found. The XRD characterization suggested that upon regeneration all metallic Cu was oxidised to Cu(II) and re-incorporated into the spinel structure of the catalyst, as demonstrated by the shift of the reflections of the spinel phase of Cu/Fe/O-R with respect to Cu/Fe/O-AR (Fig. 4b). However, a partial segregation of very small, well-dispersed, nanoparticles of CuO could not be excluded yet. Also, upon in situ regeneration a small fraction of the Fe-enriched spinel was oxidised to hematite and its crystallinity increased further (spinel CSD size = 15 nm, SSA = 55 m2 g−1).
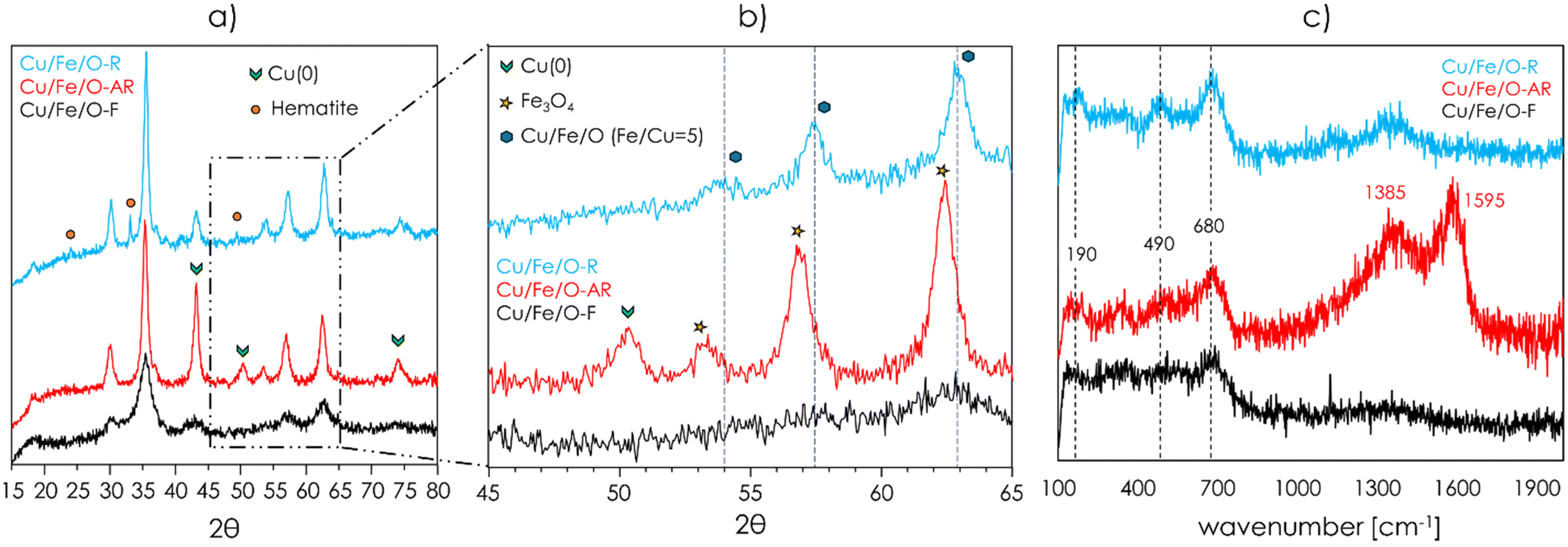 | ||
| Fig. 4 a) and b) XRD powder patterns of Cu/Fe/O-F (fresh, black), Cu/Fe/O-AR (after reaction, red) and Cu/Fe/O-R (regenerated, light blue); c) Raman spectra (excitation source Ar+ laser, 514.5 nm) of Cu/Fe/O-F (fresh, black), Cu/Fe/O-AR (after reaction, red) and Cu/Fe/O-R (regenerated, light blue). | ||
As mentioned previously, the reduction of Cu/Fe/O-F was investigated by means of TPR with H2. The reduction profile (Fig. S14†) showed that the material was reduced in the same temperature range at which the ODH of isorosalva alcohol took place (e.g., maximum of desorption centred at 295 °C). Moreover, the H2 uptake (19.9 mL; 96 mL g−1) calculated from the integrated area of the TPR profiles up to 450 °C corresponded to the amount required to obtain metallic Cu(0) and Fe3O4, in agreement with the outcome of the XRD characterization carried out over Cu/Fe/O-AR shown in Fig. 4a.
The Raman spectra of Cu/Fe/O-F, Cu/Fe/O-AR and Cu/Fe/O-R are reported in Fig. 4c. The spectrum of Cu/Fe/O-F was characterized by broad and poorly defined bands, with the most intense being centred at 680 cm−1 and corresponding to the A1g vibrational mode of ferrite spinels.81 The spectra of Cu/Fe/O-AR and Cu/Fe/O-R were more defined, owing to their higher crystallinity, and displayed also two bands centred at 490 cm−1 and 190 cm−1, corresponding to the vibrational modes F2g(2) and F2g(1) respectively, in agreement with the results of Shebanova et al.81 The presence of two strong bands centred at 1385 and 1595 cm−1 in the Raman spectrum of Cu/Fe/O-AR confirmed that during the ODH of IRA a certain amount of coke was formed over the catalyst surface, as suggested by the low C-balance obtained during the first 6 h of the catalytic tests shown in Fig. 3. The segregation of metallic Cu nanoparticles was clearly observed when high resolution TEM imaging was carried out over Cu/Fe/O-AR (Fig. 5), because the denser Cu appeared black or dark grey with respect to the light grey Fe-enriched spinel. Cu(0) segregated forming nanoparticles with a broad size distribution, ranging from a few nanometres to around 50 nm; the bigger nanoparticles were responsible for the diffraction peaks attributable to Cu(0) in the diffractogram of Cu/Fe/O-AR shown in Fig. 4a. An EDS microanalysis (shown in Fig. S15†) demonstrated that the black/dark grey nanoparticles contained mainly Cu (up to 78 atomic%), while the light grey nanoparticles were enriched in Fe (Fe/Cu atomic ratio = 12.9). The Fe-enriched spinel particles in Cu/Fe/O-AR maintained the quasi-spherical morphology of the parent Cu/Fe/O-F (see Fig. S8†) but their diameter increased from 5 to around 10 nm, in agreement with CSD sizes calculated from XRD. The high-resolution TEM characterization of Cu/Fe/O-R showed that after the in situ regeneration some nanoparticles were still present, suggesting that a fraction of the segregated Cu(0) in the parent Cu/Fe/O-AR was oxidised to CuO without being re-incorporated in the spinel structure of Cu/Fe/O. These nanoparticles of CuO were too small to be detected by means of XRD. Moreover, the light grey spinel particles became more crystalline, their average diameter increased further, and their quasi-spherical shape was lost in favour of a cubic morphology.
 | ||
| Fig. 5 High resolution TEM electron images of a) Cu/Fe/O-AR and b) Cu/Fe/O-R. | ||
ODH of Isorosalva Alcohol over Cu-supported catalysts
The results reported in the previous section suggest that the Cu-ferrite can be considered a precursor of the real catalyst, which was made of small CuO nanoparticles supported over a Fe/Cu spinel, with the latter being formed upon a cycle of reactions followed by in situ regeneration. Starting from this hypothesis, a catalyst consisting of CuO supported over maghemite (CuO/γ-Fe2O3, 6 wt% of CuO) was prepared and its catalytic activity was compared to those of fresh Cu/Fe/O and two other materials containing only Fe (pure γ-Fe2O3) and only Cu (CuO/SiO2, 6 wt% of CuO). The results of a catalyst screening carried out at 300 °C, τ = 1 s and with a feed consisting of IRA/O2/N2 = 5/5/90 mol% are reported in Fig. 6. | ||
| Fig. 6 ODH of IRA over Cu/Fe/O, CuO/γ-Fe2O3, γ-Fe2O3 and CuO/SiO2. Reaction conditions: volume of catalyst = 1 cm3, temperature = 300 °C, IRA/O2/N2 = 5/5/90 mol%, contact time (τ) = 1 second. Symbols: Isorosalva Alcohol conversion (X IRA, orange), oxygen conversion (X O2, dark red) carbon balance (C-balance, purple), opalene selectivity (S OPA, light blue), decadiene selectivity (S DD, yellow), cyclic decene selectivity (S CD, dark blue), other by-products' selectivity (S Others, black) and COx selectivity (S COx, grey). Mean values calculated at the steady-state during the last 3 h of time on stream. | ||
The activity of iron oxide alone (γ-Fe2O3) in the ODH of IRA to OPA was very low because the catalyst underwent a fast deactivation, leading to a negligible IRA conversion after only 4 h on stream, as shown in Fig. S16.† Its lack of activity has been attributed to the absence of Cu, meaning that its presence is crucial for the ODH reaction. The material containing only copper (CuO/SiO2) performed poorly as well, displaying unsatisfactory activity and poor selectivity to OPA (S OPA = 24%, lower than that obtained with V2O5/TiO2). Moreover, this catalyst fostered the dehydration of IRA to DD (S = 25%) and its consecutive cyclization to alkylated cyclic alkenes with 10 carbon atoms (CD, S = 9%) such as butyl-cyclohexene and pentyl-cyclopentene, as shown in Scheme S1 (ESI†). On the other hand, a remarkable cooperative effect between Cu and Fe occurred in the case of CuO/γ-Fe2O3, which displayed enhanced activity and improved selectivity with respect to both γ-Fe2O3 and CuO/SiO2. This material also outperformed Cu/Fe/O: in fact, when Cu was supported over maghemite (CuO/γ-Fe2O3), the conversion of IRA was 33% (compared to 23 for the Cu/Fe/O catalyst), while OPA selectivity was 69% (compared to 51 for Cu/Fe/O); at the same time, the selectivity to COx decreased from 26 to 18% and the selectivity to other unknown compounds remained almost the same compared to Cu/Fe/O. Remarkably, this material was selective to OPA from the very beginning of the reaction, without the need for any “activation” procedure (Fig. S17†).
The synergistic effect between Cu and Fe observed for CuO/γ-Fe2O3 can be ascribed to the extremely high dispersion of Cu over the surface of maghemite as suggested by the TEM and EDS characterization (Fig. S9†). In fact, maghemite seemed to be capable of strongly bonding and stabilizing highly dispersed CuO-species (e.g., small clusters or overlayers) or even incorporating Cu(II) into its lattice, possibly thanks to its surface defects and cationic vacancies.82 As a consequence, both the conversion and the selectivity to OPA were improved, thanks to the formation of isolated active sites. A similar behaviour was observed for the regenerated, fully oxidized Cu/Fe/O-R catalyst, which was characterized by the presence of small nanoparticles of CuO dispersed over its surface, as shown by TEM characterization (Fig. 5b). However, this material displayed a transient activity with very high selectivity to total oxidation products during the first hour of reaction after regeneration (Fig. 3), which then dropped favouring the formation of OPA already during the second hour of reaction. In the literature it has been pointed out that in order to achieve high selectivity in partial oxidations with copper-based catalysts, CuO must be reduced to a certain extent in order to form isolated surface domains with a limited number of active lattice oxygen atoms, in agreement with the “site isolation” principle.83 In line with this concept, some industrially relevant partial oxidations are carried out with catalysts containing Cu(0)55 or Cu(I).84 On the other hand, supported Cu(II)O is well known as a very active and selective catalyst for the total oxidation of methane85 and VOCs (e.g., toluene86), and high activities have been correlated with increasingly high dispersion of CuO on the support by several authors.85,86 Hence, it is likely that the aforementioned change of selectivity of Cu/Fe/O-R from CO2 (total oxidation) to OPA (selective partial oxidation, ODH) was due to the partial reduction of CuO nanoparticles to Cu(0). Although a similar behaviour might also be expected for CuO/γ-Fe2O3, it was not observed in the test reported in Fig. S17,† probably due to the higher dispersion of CuO-species and the lower Cu-loading (5 wt% expressed as metal) with respect to Cu/Fe/O (14.3 wt%).
Characterization of Cu-supported catalysts after reaction
A comparison between the XRD patterns before and after the ODH of IRA for CuO/SiO2, γ-Fe2O3 and CuO/γ-Fe2O3 is reported in Fig. S18–S20,† respectively. The diffractogram of CuO/SiO2 after reaction showed the superimposition of a broad band attributable to silica, the pattern of Cu(0) and that of Cu2O, with no traces of the parent CuO; therefore, Cu(II) was completely reduced to Cu(I) and Cu(0) during the ODH of IRA. Unlike CuO/SiO2, pure γ-Fe2O3 was not reduced during the reaction: in fact, except for the presence of a new set of weak reflections attributable to an impurity of hematite, its diffractogram after reaction was unaffected (Fig. S19b†). Therefore, the scarce activity of γ-Fe2O3 may be a consequence of its lack of reducibility in the reaction environment. Finally, in the diffractogram of CuO/γ-Fe2O3 after reaction all the reflections attributable to the spinel crystal structure of the support were shifted to lower 2θ values (Fig. S20b†), meaning that contrary to what happened with pure maghemite, in this case the γ-Fe2O3 support was reduced to magnetite (Fe3O4), very likely thanks to the presence of Cu.The latter hypothesis was confirmed by means of TPR characterization of CuO/SiO2, pure γ-Fe2O3 and CuO/γ-Fe2O3. The TPR profiles for these materials as well as a detailed discussion can be found in Fig. S21 and in Chapter S3 of the ESI† respectively. Briefly, the reduction of both CuO/SiO2 and γ-Fe2O3 occurred in a single step, the former at a relatively low temperature (peak maximum at 316 °C) and the latter at higher temperatures (peak maximum at 450 °C, incomplete reduction). Meanwhile, the reduction profile of CuO/γ-Fe2O3 was characterized by two maxima centred at 322 °C (CuO) and 441 °C (γ-Fe2O3) respectively. However, the H2 uptake calculated from the integrated area under the first peak centred at 322 °C was much higher than the one required to reduce all CuO to Cu(0), indicating that also a fraction of Fe(III), possibly the iron oxide support closer to the Cu(0)-species, was reduced at lower temperature. These results indicated that CuO was easily reduced at the temperature at which the ODH was investigated (300 °C) but the pure maghemite was not. However, the presence of supported Cu species in CuO/γ-Fe2O3 decreased the temperature needed for the reduction of the γ-Fe2O3 support to Fe3O4, making it feasible under the actual reaction conditions. These results confirmed the hypothesis of a synergy effect between the two metals, derived from the high dispersion of copper species over maghemite.
Conclusions
In this work the continuous-flow, gas-phase ODH of a mixture of decen-1-ol isomers (“Isorosalva”, IRA) towards the corresponding mixture of aldehydes (“Opalene”, OPA) has been reported for the first time.The IRA mixture used in this study was produced and supplied by International Flavors & Fragrances Inc. and the target reaction was investigated in a conventional fixed-bed reactor over a novel, non-noble metal, Cu-ferrite catalyst (Cu0.6Fe2.4O4.2, Cu/Fe/O) with spinel structure. This material displayed higher selectivity to OPA with respect to a traditional V2O5/TiO2 catalyst (a benchmark catalyst for the ODH of light alcohols).
Increasing the contact time over the fresh Cu/Fe/O catalyst fostered several consecutive reactions such as OPA aldol condensation to form C20 branched aldehydes (ODT) and OPA overoxidation to decanoic acids followed by ketonization of the acids to C19 symmetric ketones (NDD).
However, it was found that OPA selectivity could be greatly increased up to 70% at 45% IRA conversion upon in situ regeneration of Cu/Fe/O with air at 400 °C for 3 h. This remarkable result was rationalized by characterizing in-depth the fresh, the spent and the regenerated catalyst. The combined results of XRD, BET, SEM-EDS, TEM and Raman spectroscopy showed that upon reaction the Cu-ferrite underwent a reduction to metallic Cu and a magnetite-like Fe-enriched spinel possessing a higher crystallinity with respect to the parent Cu-ferrite. Upon regeneration, not all the Cu was reincorporated into the Cu-ferrite structure; instead, it segregated forming small and well dispersed CuO nanoparticles over the surface of a Fe-enriched Cu-ferrite, which was the actual selective phase for OPA production.
Starting from these results, an ad hoc prepared catalyst based on 6 wt% of CuO supported on γ-Fe2O3 (maghemite) was prepared and subjected to comparative testing with 6 wt% CuO supported on SiO2, pure maghemite γ-Fe2O3 and Cu/Fe/O. It was found that the performance of CuO/γ-Fe2O3 was superior to that of all the other materials both in terms of activity and selectivity. On the other hand, copper oxide alone (CuO/SiO2) was not selective, while the iron oxide alone (γ-Fe2O3) was not active, hence the superior catalytic activity and selectivity of Cu/Fe/O and CuO/γ-Fe2O3 were attributed to a cooperative effect between Cu and Fe species.
Author contributions
Conceptualization: T. T., F. C. and P. R.; investigation: J. D. M. and F. O.; methodology: J. D. M. and T. T., writing-original draft: J. D. M. and T. T.; writing-review and editing: F. C., P. R., C. L. C.; Supervision: F. C. and C. L. C. Resources: C. L. C. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
IFF Benicarlò S. L. is acknowledged for funding and for the fruitful collaboration.Notes and references
- C. Lucarelli, A. Lolli, A. Giugni, L. Grazia, S. Albonetti, D. Monticelli and A. Vaccari, Efficient and ecofriendly route for the solvent-free synthesis of piperonal and aromatic aldehydes using Au/CeO2 catalyst, Appl. Catal., B, 2017, 203, 314–323 CrossRef CAS.
- J. Panten and H. Sturburg, Flavors and fragrances, 2. Aliphatic compounds, in Ullmann's Encyclopedia of Industrial Chemistry, 2015, pp. 1–55 Search PubMed.
- C. Parmeggiani and F. Cardona, Transition metal based catalysts in the aerobic oxidation of alcohols, Green Chem., 2012, 14, 547–564 RSC.
- I. Markò, A. Gautier, R. Dumeunier, K. Doda, F. Philippart, S. Brown and C. Urch, Efficient, copper-catalyzed, aerobic oxidation of primary alcohols, Angew. Chem., Int. Ed., 2004, 43, 1588–1591 CrossRef PubMed.
- P. Gamez, I. Arends, R. Sheldon and J. Reedijk, Room temperature aerobic copper-catalysed selective oxidation of primary alcohols to aldehydes, Adv. Synth. Catal., 2004, 346, 805–811 CrossRef CAS.
- J. Hoover and S. Stahl, Highly practical copper(I)/TEMPO catalyst system for chemoselective aerobic oxidation of primary alcohols, J. Am. Chem. Soc., 2011, 133, 16901–16910 CrossRef CAS PubMed.
- A. Hanyu, E. Takezawa, S. Sakaguchi and Y. Ishii, Selective aerobic oxidation of primary alcohols catalyzed by a Ru(PPh3)3Cl2 / hydroquinone system, Tetrahedron Lett., 1998, 39, 5557–5560 CrossRef CAS.
- H. Mizoguchi, T. Uchida, K. Ishida and T. Katsuki, Ru(PPh3)(OH)-salen complex: a designer catalyst for chemoselective aerobic oxidation of primary alcohols, Tetrahedron Lett., 2009, 50, 3432–3435 CrossRef CAS.
- N. Komiya, T. Nakae, H. Sato and T. Naota, Water-soluble diruthenium complexes bearing acetate and carbonate bridges: highly efficient catalysts for aerobic oxidation of alcohols in water, Chem. Commun., 2006, 4829–4831 RSC.
- A. Dijksman, A. Marin-Gonzales, A. Mairata i Payeras, I. Arends and R. Sheldon, Efficient and selective aerobic oxidation of alcohols into aldehydes and ketones using ruthenium/TEMPO as the catalytic system, J. Am. Chem. Soc., 2001, 123, 6826–6833 CrossRef CAS PubMed.
- I. Markò, P. Giles, M. Tsukazaki, I. Chellè-Regnaut, C. Urch and S. Brown, Efficient, aerobic, ruthenium-catalyzed oxidation of alcohols into aldehydes and ketones, J. Am. Chem. Soc., 1997, 119, 12661–12662 CrossRef.
- M. Hasan, M. Musawir, P. Davey and I. Kozhevnikov, Oxidation of primary alcohols to aldehydes with oxygen catalysed by tetra-n-propylammonium perruthenate, J. Mol. Catal. A: Chem., 2002, 180, 77–84 CrossRef CAS.
- M. Schultz, S. Hamilton, D. Jensen and M. Sigman, Development and comparison of the substrate scope of Pd-catalysts for the aerobic oxidation of alcohols, J. Org. Chem., 2005, 70, 3343–3352 CrossRef CAS PubMed.
- S. Gowrisankar, H. Neumann, D. Gordes, K. Thurow, H. Jiao and M. Beller, A convenient and selective palladium-catalyzed aerobic oxidation of alcohols, Chem. – Eur. J., 2013, 19, 15979–15984 CrossRef CAS PubMed.
- T. Nishimura, T. Onoue, K. Ohe and S. Uemura, Palladium(II)-catalyzed oxidation of alcohols to aldehydes and ketones by molecular oxygen, J. Org. Chem., 1999, 64, 6750–6755 CrossRef CAS PubMed.
- B. Guan, D. Xing, G. Cai, X. Wan, N. Yu, Z. Fang, L. Yang and Z. Shi, Highly selective aerobic oxidation of alcohol catalyzed by a gold(I) complex with an anionic ligand, J. Am. Chem. Soc., 2005, 127, 18004–18005 CrossRef CAS PubMed.
- X. Jiang, J. Liu and S. Ma, Iron-catalyzed aerobic oxidation of alcohols: lower cost and improved selectivity, Org. Process Res. Dev., 2019, 23, 825–835 CrossRef CAS.
- W. Yin, C. Chu, Q. Lu, J. Tao, X. Liang and R. Liu, Iron chloride/4-acetamido-TEMPO/sodium nitrite-catalyzed aerobic oxidation of primary alcohols to the aldehydes, Adv. Synth. Catal., 2010, 352, 113–118 CrossRef CAS.
- Y. Jing, J. Jiang, B. Yan, S. Lu, J. Jiai, H. Xue, G. Yang and G. Zheng, Activation of dioxygen by cobaloxime and nitric oxide for efficient TEMPO-catalyzed oxidation of alcohols, Adv. Synth. Catal., 2011, 353, 1146–1152 CrossRef CAS.
- R. Anderson, K. Griffin, P. Johnston and P. Alsters, Selective oxidation of alcohols to carbonyl compounds and carboxylic acids with platinum group metal catalysts, Adv. Synth. Catal., 2003, 345, 517–523 CrossRef CAS.
- N. Kakiuchi, Y. Maeda, T. Nishimura and S. Uemura, Pd(II)-hydrotalcite-catalyzed oxidation of alcohols to aldehydes and ketones using atmospheric pressure of air, J. Org. Chem., 2001, 66, 6620–6625 CrossRef CAS PubMed.
- E. Johnston, O. Verho, M. Karkas, M. Shakeri, C. Tai, P. Palmgren, K. Eriksson, S. Oscarsson and J. Backvall, Highly Dispersed Palladium Nanoparticles on Mesocellular Foam: An Efficient and Recyclable Heterogeneous Catalyst for Alcohol Oxidation, Chem. – Eur. J., 2012, 18, 12202–12206 CrossRef CAS PubMed.
- H. Wu, Q. Zhang and Y. Wang, Solvent-free aerobic oxidation of alcohols catalyzed by an efficient and recyclable palladium heterogeneous catalyst, Adv. Synth. Catal., 2005, 347, 1356–1360 CrossRef CAS.
- U. Pillai and E. Sahle-Demessie, Selective oxidation of alcohols by molecular oxygen over a Pd/MgO catalyst in the absence of any additives, Green Chem., 2004, 6, 161–165 RSC.
- K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Creation of a monomeric Ru species on the surface of hydroxyapatite as an efficient heterogeneous catalyst for aerobic alcohol oxidation, J. Am. Chem. Soc., 2000, 122, 7144–7145 CrossRef CAS.
- K. Yamaguchi and N. Mizuno, Supported ruthenium catalyst for the heterogeneous oxidation of alcohols with molecular oxygen, Angew. Chem., Int. Ed., 2002, 41, 4538–4542 CrossRef CAS PubMed.
- T. Stuchinskaya, M. Musawir, E. Kozhevnikova and I. Kozhevnikov, Liquid-phase oxidation of alcohols by oxygen and nitrous oxide catalysed by Ru-Co, J. Catal., 2005, 231, 41–47 CrossRef CAS.
- A. Bleloch, B. Johnson, S. Ley, A. Price, D. Shepard and A. Thomas, Modified mesoporous silicate MCM-41 materials: immobilised perruthenate - a new highly active heterogeneous oxidation catalyst for clean organic synthesis using molecular oxyge, Chem. Commun., 1999, 1907–1908 RSC.
- M. Beier, T. Hansen and J. Grunwaldt, Selective liquid-phase oxidation of alcohols catalyzed by a silver-based catalyst promoted by the presence of ceria, J. Catal., 2009, 266, 302–330 CrossRef.
- A. Abad, C. Almela, A. Corma and H. Garcia, Unique gold chemoselectivity for the aerobic oxidation of allylic alcohols, Chem. Commun., 2006, 3178–3180 RSC.
- V. Cortez Corberan, A. Gomez-Avilez, S. Martinez-Gonzalez, S. Ivanova, M. Dominguez and M. Gonzalez-Perewz, Heterogeneous selective oxidation of fatty alcohols: oxidation of 1-tetradecanol as a model substrate, Catal. Today, 2014, 238, 49–53 CrossRef.
- Y. Kotolevich, E. Kolobova, E. Khramov, M. Farias, Y. Zubavichus, H. Tiznado, S. Martinez-Gonzales, V. Cortes Corberan, J. Mota-Morales, A. Pestryakov and N. Bogdanchikova, n-Octanol oxidation on Au/TiO2 catalysts promoted with La and Ce oxides, J. Mol. Catal. A: Chem., 2017, 427, 1–10 CrossRef CAS.
- S. Reddy, S. Das and T. Punniyamurthy, Polyaniline supported vanadium catalyzed aerobic oxidation of alcohols to aldehydes and ketones, Tetrahedron Lett., 2004, 45, 3561–3564 CrossRef CAS.
- M. Musawir, P. Davey, G. Kelly and I. Kozhevnikov, Highly efficient liquid-phase oxidation of primary alcohols to aldehydes with oxygen catalysed by Ru–Co oxide, Chem. Commun., 2003, 1414–1415 RSC.
- T. Matsushita, K. Ebitani and K. Kaneda, Highly efficient oxidation of alcohols and aromatic compounds catalysed by the Ru-Co-Al hydrotalcite in the presence of molecular oxygen, Chem. Commun., 1999, 265–266 RSC.
- H. Ji, T. Mizugaki, K. Ebitani and K. Kaneda, Highly efficient oxidation of alcohols to carbonyl compounds in the presence of molecular oxygen using a novel heterogeneous ruthenium catalyst, Tetrahedron Lett., 2002, 43, 719–7183 CrossRef.
- H. Ji, K. Ebitani, T. Mizugaki and K. Kaneda, Environmentally friendly alcohol oxidation using heterogeneous catalyst in the presence of air at room temperature, Catal. Commun., 2002, 3, 511–517 CrossRef CAS.
- H. Sun, Q. Hua, F.-f. Guo, Z. Wang and W. Huang, Selective aerobic oxidation of alcohols by using manganese oxide nanoparticles as an efficient heterogeneous catalyst, Adv. Synth. Catal., 2012, 354, 569–573 CrossRef CAS.
- T. Tabanelli, S. Passeri, S. Guidetti, F. Cavani, C. Lucarelli, F. Cargnoni and M. Mella, A cascade mechanism for a simple reaction: the gas-phase methylation of phenol with methanol, J. Catal., 2019, 370, 447–460 CrossRef CAS.
- N. Ballarini, F. Cavani, L. Maselli, A. Montaletti, S. Passeri, D. Scagliarini, C. Flego and C. Perego, The transformations involving methanol in the acid- and base-catalyzed gas-phase methylation of phenol, J. Catal., 2007, 251, 423–436 CrossRef CAS.
- V. Crocellà, G. Cerrato, G. Magnacca, C. Morterra, F. Cavani, S. Cocchi, S. Passeri, D. Scagliarini, C. Flego and C. Perego, The balance of acid, basic and redox sites in Mg/Me-mixed oxides: the effect on catalytic performance in the gas-phase alkylation of m-cresol with methanol, J. Catal., 2010, 270, 125–135 CrossRef.
- T. Tabanelli, S. Cocchi, B. Gumina, L. Izzo, M. Mella, S. Passeri, F. Cavani, C. Lucarelli, J. Schutz, W. Bonrath and T. Netscher, Mg/Ga mixed-oxide catalysts for phenol methylation: outstanding performance in 2,4,6-trimethylphenol synthesis with co-feeding of water, Appl. Catal., A, 2018, 552, 86–97 CrossRef CAS.
- J. De Maron, M. Eberle, F. Cavani, F. Basile, N. Dimitratos, P. Maireles-Torres, E. Rodriguez-Castellon and T. Tabanelli, Continuous-flow methyl methacrylate synthesis over gallium-based bifunctional catalysts, ACS Sustainable Chem. Eng., 2021, 9, 1790–1803 CrossRef CAS.
- A. Malmusi, J. Ochoa Velasquez, T. Tabanelli, F. Basile, C. Lucvarelli, S. Agnoli, F. Carraro, G. Granozzi and F. Cavani, Ethanol aerobic and anaerobic oxidation with FeVO4 and V2O5 catalysts, Appl. Catal., A, 2019, 570, 139–147 CrossRef CAS.
- M. Crivello, C. Perez, S. Mendieta, S. Casuscelli, G. Eimer, V. Elias and E. Herrero, n-Octyl alcohol dehydrogenation over copper catalysts, Catal. Today, 2008, 133–135, 787–792 CrossRef CAS.
- D. Sun, T. Misu, Y. Yamada and S. Sato, Advantages of using Cu/SiO2 catalyst for vapor-phase dehydrogenation of 1-decanol into decanal, Appl. Catal., A, 2019, 582, 117109 CrossRef CAS.
- L. Izzo, T. Tabanelli, F. Cavani, P. Vasquez Blair, C. Lucarelli and M. Mella, The competition between dehydrogenation and dehydration reactions for primary and secondary alcohols over gallia: unravelling the effects of molecular and electronic structure via a two-pronged theoretical/experimental approach, Catal. Sci. Technol., 2020, 10, 3433–3449 RSC.
- L. Grazia, A. Lolli, F. Folco, Y. Zhang, S. Albonetti and F. Cavani, Gas-phase cascade upgrading of furfural to 2-methylfuran using methanol as a H-transfer reactant and MgO based catalysts, Catal. Sci. Technol., 2016, 6, 4418–4427 RSC.
- L. Grazia, D. Bonincontro, A. Lolli, T. Tabanelli, C. Lucarelli, S. Albonetti and F. Cavani, Exploiting H-transfer as a tool for the catalytic reduction of bio-based building blocks: the gas-phase production of 2-methylfurfural using a FeVO4 catalyst, Green Chem., 2017, 19, 4412–4422 RSC.
- A. Nagy and G. Mestl, High temperature partial oxidation reactions over silver catalysts, Appl. Catal., A, 1999, 188, 337–353 CrossRef CAS.
- V. Folliard, G. Postole, L. Marra, J. Dubois and A. Aroux, Sustainable acrolein production from bio-alcohols on spinel catalysts: Influence of magnesium substitution by various transition metals (Fe, Zn, Co, Cu, Mn), Appl. Catal., A, 2020, 608, 117871 CrossRef CAS.
- A. Chieregato, J. L. Nieto and F. Cavani, Mixed-oxide catalysts with vanadium as the key element for gas-phase reactions, Coord. Chem. Rev., 2015, 301–302, 3–23 CrossRef CAS.
- G. Reuss, W. Disteldorf, A. Gamer and A. Hilt, Formaldehyde, Ullmann's Encycl. Ind. Chem., 2000, vol. 15, pp. 735–768 Search PubMed.
- M. Eckert, G. Fleischmann, J. Reinhard, H. Bolt and K. Golka, Acetaldehyde, Ullmann's Encycl. Ind. Chem., 2012, vol. 1, pp. 191–207 Search PubMed.
- W. Aquila, H. Fuchs, O. Worz, W. Ruppel and K. Halbritter, Continous industrial production of unsaturated aliphatic aldehydes in a tube bundle reactor. Brevetto US6013843A, 2000.
- S. Biella and M. Rossi, Gas phase oxidation of alcohols to aldehydes or ketones catalysed by supported gold, Chem. Commun., 2003, 378–379 RSC.
- W. Yi, W. Yuan, Y. Meng, S. Zou, Y. Zhou, W. Hong, J. Che, M. Hao, B. Ye, L. Xiao, Y. Wang, H. Kobayashi and J. Fan, A rational solid-State synthesis of supported Au−Ni bimetallic nanoparticles with enhanced activity for gas-phase selective oxidation of alcohols, ACS Appl. Mater. Interfaces, 2017, 9, 31853–31860 CrossRef CAS PubMed.
- G. Zhao, H. Hu, M. Deng, M. Ling and Y. Lu, Au/Cu-fiber catalyst with enhanced low-temperature activity and heat transfer for the gas-phase oxidation of alcohols, Green Chem., 2011, 13, 55–58 RSC.
- I.-D. Huang, L. Polinski and K. Rao, Oxidative dehydrogenation of alcohols to aldehydes and ketones, Brevetto US4154762, 1979.
- G. Zhao, M. Deng, Y. Jiang, H. Hu, J. Huang and Y. Lu, Microstructured Au/Ni-fiber catalyst: galvanic reaction preparation and catalytic performance for low-temperature gas-phase alcohol oxidation, J. Catal., 2013, 301, 46–53 CrossRef CAS.
- Z. Yang, J. Li, X. Yang, X. Xie and Y. Wu, Gas-phase oxidation of alcohols over silver: The extension of catalytic cycles of oxidation of alcohols in the liquid-phase, J. Mol. Catal. A: Chem., 2005, 241, 15–22 CrossRef CAS.
- J. Shen, W. Shan, Y. Zhang, J. Du, H. Xu, K. Fan, W. Shen and Y. Tang, Gas-phase selective oxidation of alcohols: in situ electrolytic nano-silver/zeolite film/copper grid catalyst, J. Catal., 2006, 237, 94–101 CrossRef CAS.
- C. Zhang, P. Chen, J. Liu, Y. Zhang, W. Shen, H. Xu and Y. Tang, Ag microparticles embedded in Si nanowire arrays: a novel catalyst for gas-phase oxidation of high alcohol to aldehyde, Chem. Commun., 2008, 3290–3292 RSC.
- L. Zhao, L. Kong, C. Liu, Y. Wang and L. Dai, AgCu/SiC-powder: a highly stable and active catalyst for gas-phase selective oxidation of alcohols, Catal. Commun., 2017, 98, 1–4 CrossRef CAS.
- P. Gallezot, L. Ceroni and A. Perrard, Oxidative dehydrogenation of rosalva to costenal on supported silver catalyst, J. Mol. Catal. A: Chem., 1998, 129, L127–L130 CrossRef CAS.
- A. Narula, E. Arruda and F. Schiet, Decenal mixtures and their use in perfume composition, Brevetto US8461100B1, 2013.
- F. Huang, Y. Zhang, Y. Yao, W. Yang and Y. Tao, Synthesis of (4E,6Z,10Z)-hexadeca-4,6,10-trien-1-ol and (4E,6E,10Z)-hexadeca-4,6,10-trien-1-ol, the pheromone components of cocoa pod borer moth Conopomorpha cramerella, RSC Adv., 2017, 57, 35575–35580 RSC.
- K. Lorber, G. Zeh, J. Regler and A. Buettner, Structure-odor relationships of (Z)-3-alken-1-ols, (Z)-3-alkenals, and (Z)-3-alkenoic acids, J. Agric. Food Chem., 2018, 66, 2334–2343 CrossRef CAS PubMed.
- E. Brodl, J. Ivkovic, C. Tabib, R. Breinbauer and P. Macheroux, Synthesis of α,β-unsaturated aldehydes as potential substrates for bacterial luciferases, Bioorg. Med. Chem., 2017, 25, 1487–1495 CrossRef CAS PubMed.
- Y. Shigekazu and Y. Yasuyuki, Nickel-catalyzed oxidation of allylic alcohols with K2S2O8, Chem. Lett., 1989, 18, 1361–1364 CrossRef.
- C. Wulkesch and C. Czekelius, Straightforward synthesis of fluorinated enals via photocatalytic α-perfluoroalkenylation of aldehydes, J. Org. Chem., 2021, 86, 7425–7438 CrossRef CAS PubMed.
- E. Cao, I. Zuburtikudis, N. Al-Rifay, M. Roydhouse and A. Gavriilidis, Enhanced performance of oxidation of rosalva (9-decen-1-ol) to costenal (9-decenal) on porous silicon-supported silver catalyst in a microstructured reactor, Processes, 2014, 2, 141–157 CrossRef.
- C. Trevisanut, O. Vozniuk, M. Mari, S. Arenas Urrea, C. Lorentz, J.-M. Millet and F. Cavani, The chemical-loop reforming of alcohols on spinel-type mixed oxides: comparing Ni, Co, and Fe ferrite vs magnetite performances, Top. Catal., 2016, 59, 1600–1613 CrossRef CAS.
- F. Folco, J. Velasquez Ochoa, F. Cavani, L. Ott and M. Janssen, Ethanol gas-phase ammoxidation to acetonitrile: the reactivity of supported vanadium oxide catalysts, Catal. Sci. Technol., 2017, 7, 200–212 RSC.
- W. Zhang, G. Innocenti, M. Ferbinteanu, E. Ramos-Fernandez, A. Sepulveda-Escribano, H. Wu, F. Cavani, G. Rothemberg and R. Shiju, Understanding the oxidative dehydrogenation of ethyl lactate to ethyl pyruvate over vanadia/titania, Catal. Sci. Technol., 2018, 8, 3707–3978 RSC.
- D. Maity and D. Agrawal, Synthesis of iron oxide nanoparticles under oxidizing environment and their stabilization in aqueous and non-aqueous media, J. Magn. Magn. Mater., 2007, 308, 46–55 CrossRef CAS.
- W. Wang, Q. Zhou, X. Fei, Y. He, P. Zhang, G. Zhang, L. Peng and W. Xie, Synthesis of CuO nano- and micro-structures and their Raman spectroscopic studies, CrystEngComm, 2010, 12, 2232–2237 RSC.
- C. Bandinelli, B. Lambiase, T. Tabanelli, J. De Maron, N. Dimitratos, F. Basile, P. Concepcion, N. J. Lopez and F. Cavani, A study of the oxidehydration of 1,2-propanediol to propanoic acid with bifunctional catalysts, Appl. Catal., A, 2019, 582, 117102 CrossRef CAS.
- A. Vassoi, T. Tabanelli, A. Sacchetti, F. Di Gioia, L. Capuzzi and F. Cavani, The oxidative cleavage of 9,10-dihydroxystearic triglyceride with oxygen and Cu oxide-based heterogeneous catalysts, ChemSusChem, 2021, 14, 2375–2382 CrossRef CAS PubMed.
- J. De Maron, L. Bellotti, A. Baldelli, A. Fasolini, N. Schiaroli, C. Lucarelli, F. Cavani and T. Tabanelli, Evaluation of the catalytic activity of metal phosphates and related oxides in the ketonization of propionic acid, Sustainable Chem., 2022, 3, 58–75 CrossRef CAS.
- O. Shebanova and P. Lazor, Raman spectroscopic study of magnetite (FeFe2O4): a new assignment for the vibrational spectrum, J. Solid State Chem., 2003, 174, 424–430 CrossRef CAS.
- R. Vandenberghe, E. De Grave, C. Landuydt and L. Bowen, Some aspect conceriìning the characterization of iron oxides and hydroxides in soils and clays, Hyperfine Interact., 1990, 53, 175–196 CrossRef CAS.
- R. Grasselli, Site isolation and phase cooperation: two important concepts in selective oxidation catalysis: a retrospective, Catal. Today, 2014, 238, 10–27 CrossRef CAS.
- G. Hearne and M. Adams, Production of unsaturated carbonilic compounds, Brevetto US2451485, 1947.
- M. Marion, E. Garbowski and M. Primet, Physicochemical Properties of Copper Oxide Loaded Alumina in Methane Combustion, J. Chem. Soc., Faraday Trans., 1990, 86, 3027–3032 RSC.
- P. Larsson, A. Andersson, L. Wallenberg and B. Svensson, Combustion of CO and toluene; characterisation of copper oxide supported on titania and activity comparisons with supported cobalt, iron, and manganese oxide, J. Catal., 1996, 163, 279–293 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental information of catalyst characterisation, namely NH3 and CO2-TPD, TPR, XRD, SEM-EDS and TEM analyses. Additional catalytic tests as well as the reactor scheme and blank runs are also reported. See DOI: https://doi.org/10.1039/d2cy01836e |
| This journal is © The Royal Society of Chemistry 2023 |