
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of structural changes in CaCu3Ti4O12 on high energy ball milling and their effect on photocatalytic performance†
Andrea
Bartoletti
ab,
Angela
Gondolini
*a,
Nicola
Sangiorgi
 a,
Matteo
Aramini
c,
Matteo
Ardit
d,
Marzio
Rancan
e,
Lidia
Armelao
bf,
Simon A.
Kondrat
*g and
Alessandra
Sanson
a
a,
Matteo
Aramini
c,
Matteo
Ardit
d,
Marzio
Rancan
e,
Lidia
Armelao
bf,
Simon A.
Kondrat
*g and
Alessandra
Sanson
a
aInstitute of Science and Technology for Ceramics (ISTEC) of the National Research Council (CNR), Via Granarolo 64, I-48018 Faenza (RA), Italy. E-mail: angela.gondolini@istec.cnr.it
bDepartment of Chemical Sciences, Università degli Studi di Padova, Via Marzolo 1, 35131, Padova, Italy
cDiamond Light Source, Harwell Science and Innovation Campus, Chilton, Didcot, OX11 0DE, UK
dDepartment of Physics and Earth Sciences, University of Ferrara, Via Saragat 1, Ferrara 44122, Italy
eInstitute of Condensed Matter Chemistry and Technologies for Energy (ICMATE), National Research Council (CNR), c/o Department of Chemistry, University of Padova, Via F. Marzolo 1, 35131 Padova, Italy
fDepartment of Chemical Sciences and Materials Technologies (DSCTM), National Research Council (CNR), Italy
gDepartment of Chemistry, Loughborough University, Loughborough, Leicestershire, LE113TU, UK. E-mail: s.kondrat@lboro.ac.uk
First published on 21st December 2022
Abstract
The perovskite CaCu3Ti4O12 is known for its ability to photocatalytically degrade model dye molecules using visible light. The influence of ball milling preformed CaCu3Ti4O12 on the catalysts structure and performance in the degradation of rhodamine B and the antihistamine cetirizine hydrochloride, which does not absorb light in the visible region, was investigated. The surface area of CaCu3Ti4O12 increased from 1 m2 g−1 to >80 m2 g−1 on milling with a retention of 96% CaCu3Ti4O12 phase purity, as determined by X-ray diffraction and extended X-ray absorption fine structure analysis. Multiple characterisation techniques showed an increase in structural defects on milling, including, for the first time, X-ray absorption near edge spectroscopy which showed changes in the local electronic structure from the perspective of Cu and Ti. Photocatalytic degradation was notably higher with the milled sample than that observed for the as-synthesized sample, even after normalisation for surface area, with a doubling of surface normalised rate constant from 4.91 × 10−4 to 9.11 × 10−4 L min−1 m2 for rhodamine B degradation and a tripling for cetirizine hydrochloride degradation from 2.64 × 10−4 to 7.92 × 10−4 L min −1 m−2. The improvement in catalytic performance can be correlated to the defects observed by X-ray absorption spectroscopy.
1. Introduction
The harnessing and converting of solar energy using semiconducting inorganic materials remains an area of fundamental scientific interest with significant potential to contribute to a more sustainable society. Photocatalytic reactions for organic transformations, sustainable fuel production, and pollutant degradation present low carbon and energetically mild options towards such sustainable chemistry. Yet, early work on photocatalytic metal oxides predominantly focused on harnessing UV light, which only accounts for approximately 4% of solar irradiance.1 To access visible light, which accounts for ca. 50% of solar energy, materials with a reduced band gap relative to UV-active materials are required.To find an alternative strategy to the inherently flawed doping of UV-active TiO2 with coloured transition metals, ordered mixed metal oxides with multiple photoactive cations have been employed for visible light driven photocatalysis.2,3 Rosseinsky and co-workers highlighted the potential of this strategy with a CaCu3Ti4O12 double-perovskite catalyst for visible light photooxidation of model pollutants.4 CaCu3Ti4O12 is unusual, in that in addition to Ca, it has square planer Cu2+ on the A site, accommodated by the significant tilting of the vertex sharing TiO6 octahedra. The material is well known for its dielectric properties,5 photoluminescence6 and antiferromagnetism.7 Its potential as a visible light photocatalyst being due to photoexcitation from occupied CuO states at the top of the valance band to a Ti based conduction band, reducing the band gap relative to TiO2 alone. However, the photoexcitation process is partially hindered by localised Cu(3d)-O(2p) σ-antibonding states within the band gap, that limit the mobility of Cu+ carriers thus facilitating carrier recombination.4 Further, the conventional synthesis of perovskites (e.g., solid-state reaction synthesis) commonly produces compounds with low surface area (ca. <5 m2 g−1), and therefore a low number of surface-active sites.
Since this seminal work, several reports have focused on synthesising the CaCu3Ti4O12 photocatalyst and applying it to a host of other organic substrates for photodegradation. Attempts to overcome recombination and surface area issues have been made by producing hybrid catalysts, including incorporation of Pt nanoparticles,4 polymers,8,9 carbon nanotubes,10 carbon nitrides11 and additional metal oxides (zeolites, SiO2 and TiO2).12,13 Other work has utilised alternative sol–gel and molten salt routes to improve surface area, control morphology and induce defects in the structure.14–17
The presence of point or planar defects such as oxygen nonstoichiometry, metal site vacancies or antisite disorder, are often reported and hypothesised to influence CaCu3Ti4O12 properties. For example, the unexpectedly high dielectric constant of CaCu3Ti4O12, relative to that predicted by DFT, has been hypothesized as being due to planar defects or Ca–Cu antisite defects.18,19 While, photoluminescence effects are ascribed to [TiO5VO]–[TiO6] clusters from oxygen vacancies.6 Photocatalytic properties for organic substrate decomposition have been stated to be improved by bulk and surface defects, as determined by photoluminescence measurements, EPR and XPS. Hailili et al.14–16 stated that the photocatalytic performance of CaCu3Ti4O12 could be correlated with EPR line broadening, which has been attributed to oxygen vacancies and other defects, such as the formation of Cu+[CuO4]′ and Ti3+[TiO5VO]′ species. Complimentary XPS analysis further showing additional features in the Cu 2p and O 1s that were attributed to the presence of Cu1+ on the catalyst surface and oxygen vacancies. These defects are theorised to alter carrier mobility and act as electron scavenger sites to generate superoxide radicals to the benefit of catalytic activity.
While molten salt synthesis routes have produced defined morphologies with structural defects, surface areas remain below 30 m2 g−1.16 Variation of Cu–Ca ratios in conventional oxide synthetic routes has also been used to create defective CaCu3Ti4O12 but at the expense of phase purity (CuO and CaTiO3 being formed) and poor surface area. Mechanical milling of CaCu3Ti4O12 represents an appealing strategy to increase surface area substantially, retain phase purity (if the process is sufficiently controlled), and also induce structural defects. The latter point being highlighted in a series of papers by Modi and co-workers,20,21 where on milling, positron annihilation spectroscopy and photoluminescence measurements confirmed an increase in structural defects, UV-vis spectroscopy that mechano-chromism can be induced, and lastly that antiferromagnetic properties are disrupted.
Therefore, the impact of the simple process of milling on the photocatalytic properties of CaCu3Ti4O12 requires further investigation. The prospect of a simple and affordable process to high surface area mixed metal oxide photocatalysts being appealing. Thus, this study describes the mechanochemical processing of CaCu3Ti4O12, which provides the highest reported surface area of this material to date and exceptional mass and surface area normalised photodegradation rate of rhodamine B and cetirizine hydrochloride (Ctz). These substrates have been chosen, as they have been frequently employed for studying the photocatalytic properties of metal oxides, including titania and also CaCu3Ti4O12, allowing for effective comparison with other synthesis methodologies within the literature. The issue of rhodamine B dye sensitization in its photodegradation masking or complicating structure–function relationships, which is frequently not discussed, has been mitigated through the use of the second Ctz substrate that does not absorb light in the viable region. Structural defects, which are important to catalytic performance, have been studied by X-ray absorption spectroscopy and supported by theoretical simulations.
2. Materials and method
2.1 Synthesis of CCTO powder
Calcium copper titanate (CCTO–CaCu3Ti4O12) was synthesized by sol–gel method22 using Ca(NO3)2·4H2O (Carlo Erba, 99.0%), Cu(NO3)2·2.5H2O (Sigma Aldrich, 98.0%) and Ti(OC4H9)4 (Sigma Aldrich, 97.0%) as starting materials. In a typical preparation, stoichiometric amount of Ca(NO3)2·4H2O and Cu(NO3)2·2.5H2O (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) were dissolved in deionized water and absolute ethanol (3:1 vol%) to produce a 2.0 M nitrate solution. Meanwhile, stoichiometric amount of Ti(OC4H9)4 was dissolved in ethanol to obtain a 0.5 M solution. Once the complete precursor dissolution occurs the as-produced solutions were mixed until the formation of a sky-blue gel. The as-obtained gel was heated up to 60 °C for 4 h in a water bath and then at 100 °C for 24 h to obtain the dried precursors. The produced powder was homogenised for 30 minutes in agate mortar, calcined at 1100 °C for 2 h in air and sieved at 64 μm to obtain the final CaCu3Ti4O12 powder (CCTO). Part of the obtained powder was milled in a high energy planetary mill (Fritsch, Pulverisette 6) using zirconia beads as grinding media and absolute ethanol as solvent. The milling speed was kept at 400 rpm, using a powder to beads ratio of 1:10 until a cumulative kinetic energy of 4.6 × 104 kJ g−1 was conferred to the system (the description of the calculation method is reported in ESI,† Section S1, Table S1). The milled powder was dried at room temperature and sieved at 64 μm to obtain the so-labelled CCTO-GM sample.
:3) were dissolved in deionized water and absolute ethanol (3:1 vol%) to produce a 2.0 M nitrate solution. Meanwhile, stoichiometric amount of Ti(OC4H9)4 was dissolved in ethanol to obtain a 0.5 M solution. Once the complete precursor dissolution occurs the as-produced solutions were mixed until the formation of a sky-blue gel. The as-obtained gel was heated up to 60 °C for 4 h in a water bath and then at 100 °C for 24 h to obtain the dried precursors. The produced powder was homogenised for 30 minutes in agate mortar, calcined at 1100 °C for 2 h in air and sieved at 64 μm to obtain the final CaCu3Ti4O12 powder (CCTO). Part of the obtained powder was milled in a high energy planetary mill (Fritsch, Pulverisette 6) using zirconia beads as grinding media and absolute ethanol as solvent. The milling speed was kept at 400 rpm, using a powder to beads ratio of 1:10 until a cumulative kinetic energy of 4.6 × 104 kJ g−1 was conferred to the system (the description of the calculation method is reported in ESI,† Section S1, Table S1). The milled powder was dried at room temperature and sieved at 64 μm to obtain the so-labelled CCTO-GM sample.
2.2 CCTO characterization
X-ray powder diffraction (XRPD) on CCTO and CCTO-GM samples were collected at room-temperature in a D8 Advance Da Vinci diffractometer (Bruker) working in Bragg–Brentano geometry, and equipped with a Cu-anode X-ray tube, Ni-filter to suppress CuKβ component, and a LynxEye XE silicon strip detector (angular range of the detector window size = 2.585° 2θ) set to discriminate CuKα1,2 radiation. The powder of each sample was loaded in a 0.5 mm-deep cavity in a poly(methyl methacrylate) specimen holder and scanned in a continuous mode from 5 to 135° 2θ with step size of 0.015° 2θ and a counting time of 1.5 s per step. In order to minimize preferred orientation phenomena and increase crystallites statistic, samples were spun at 25 rpm, and a knife perpendicular to the sample holder was placed at a suitable distance from the sample surface to reduce the air-induced scattering. Qualitative phase analysis was performed by means of the Bruker AXS EVA software (v.5), and each collected X-ray powder diffraction pattern was Rietveld refined by means of the fundamental-parameter approach (TOPAS v.5.0). Identified phases were modelled by carrying out multiphase refinements in which a nine-terms Chebyshev polynomial to model the background, the scale factor and unit-cell parameters were varied. Known instrumental parameters (e.g., goniometer radius, slit sizes, geometrical parameters of the X-ray tube, etc.) were used to calculate the instrumental contribution to the peak profile, and specimen-related Lorentzian crystallite size and microstrain broadening information were extracted from the observed profiles.Specific surface area of powders was obtained from Brunauer, Emmett, and Teller analysis of nitrogen adsorption isotherms in vacuum with a Sorpty 1750 (Carlo Erba Strumentazione). Before analysis, all the samples were degassed at 100 °C for 1 h. Morphology of the as-prepared and milled samples was investigated using a ∑IGMA SEM-FEG(Zeiss).
XPS analyses were performed with a Perkin-Elmer Φ 5600-ci spectrometer using Mg Kα radiation. The sample analysis area was 800 μm in diameter. The standard deviation for the BEs values was ± 0.2 eV (further details are reported in ESI,† Section S2). It cannot be excluded that Cu+ in CCTO can derive from X-ray irradiation or vacuum effects during XPS analysis.23,24 For these reasons, particular attention has been posed to assure the same experimental conditions for the two samples (i.e., time in vacuum, X-ray source power, and X-ray exposure time).
Photoluminescence (PL) analysis where performed using a Horiba JobinYvon Fluorolog-3 spectrofluorimeter in front-face acquisition geometry exciting samples at 380 nm.
X-ray absorption spectroscopy was performed on the B18 beamline at Diamond Lightsource. Measurements were performed using the Si (111) monochromator in both transmission and fluorescence mode. Samples were diluted in cellulose and pressed into pellets to achieve the optimal edge step. All data was calibrated using appropriate Cu and Ti foils. X-ray absorption spectroscopy and EXAFS processing was performed using Athena and Artemis software. Amplitude reductor factors were determined from known CuO and TiO2 reference compounds. Aside from 1st shell M–O paths, all coordination numbers were fixed at those expected from the crystallographic data and mean squared displacement parameters floated during fits.
Infrared spectroscopy experiments were carried out on investigated powders afters photocatalytic measurements using a Nicolet™ iS™ 5 FTIR spectrometer (Thermo Fischer instrument) equipped with an internal reflection element (ATR crystal) i.e., a diamond crystal. Collected spectra, obtained by means of 16 scans in the range of 400–4000 cm−1, have a resolution of 4 cm−1.
In the photolysis process, the electronic band structure of each sample has to be considered. The position of the conduction (ECB) and the valence (EVB) bands of a semiconductor can be calculated from eqn (1) and (2) respectively:14,15
 | (1) |
| EVB = Eg + ECB | (2) |
Transmission electron microscopy (TEM) was performed on a JEOL JEM 2100 LaB6 operating at 200 kV. Samples were dry dispersed onto holey carbon film 300 mesh copper grids.
2.3 Photocatalytic measurements
Photocatalytic properties were investigated monitoring the decrease in concentration of an organic dye aqueous solution under visible light. The experimental procedure was carried out using 50 mL solution of 2.5 mg L−1 rhodamine B (RhB). Initially, 100 mg of CaCu3Ti4O12 catalyst were dispersed in the dye solution and kept in dark under stirring for 60 minutes to allow stabilization and pre-adsorption of RhB molecules on the catalyst surface. Photocatalytic degradation test was conducted exposing the reaction vessel under illumination at 1000 W m−2 AM 1.5 (calibrated with a Silicon reference cell) using a solar simulator SUN 2000 (ABET Technologies) and keeping the reactor at room temperature throughout the whole experiment. During the analysis, the suspension was collected at different time, and the powder was separated by centrifugation at 5500 rpm. The dye concentration was determined by UV-vis spectrometry Lambda 35 (PerkinElmer) before and after degradation, in the wavelength range of 650–450 nm. The degradation efficiency (DE) was calculated using eqn (3) | (3) |
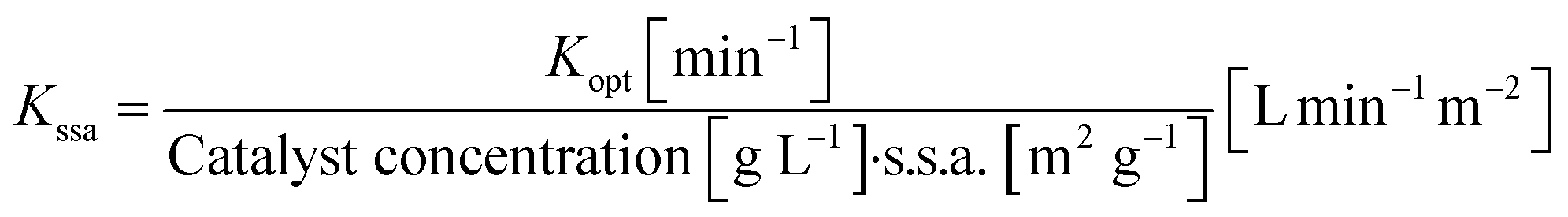 | (4) |
2.4 Density functional theory simulations
The density functional theory (DFT) calculations presented in this work were performed using the plane-wave pseudopotential DFT method available within the code CASTEP29 using the meta-generalised-gradient approximation for the exchange–correlation energies in the form of the strongly constrained and appropriately normed (SCAN) functional.30 Self-consistently generated, ultrasoft pseudopotentials were used in the calculations. A kinetic energy cutoff of 825 eV for the wave function, together with a (8 × 8 × 8) Monkhorst–Pack k-point grid were determined as parameters for converged calculations. All the structures were relaxed up to an energy change of 5 × 10−6 eV per atom.An extended (10 × 10 × 10) k-point grid was instead used for the calculation of the X-ray absorption spectra. Self-consistent calculations were performed to a energy threshold of 1 × 10−7 eV. Due to the isolated nature of the core–hole excitation, a 2 × 2 × 2 supercell bearing P1 symmetry was used for the calculation of the excited state, using the same convergence parameters as found for the ground-state calculation, after evaluating that the convergence criteria were still respected, and that the choice of larger values was not influencing both the energy and the simulated spectra.
An energy shift of 8979 eV was applied to the simulated spectra, in order to overlap with the experimental data, and computational results were normalised through trapezoidal integration of simulated spectrum. Transition broadening as a consequence of instrumental resolution (Gaussian) and core-lifetime effects (Lorentzian) was applied as 0.2 and 1.25 eV FWHM respectively.
3. Results and discussion
3.1 Structural, compositional and physical properties of CCTO and CCTO-GM
XRPD revealed that CaCu3Ti4O12 cubic phase (s.g. Im-3) was retained on milling (>95 wt%), with small fractions of CaTiO3 (orthorhombic, s.g. Pbnm) and CuO tenorite (monoclinic, s.g. C2/c) detected as minor associated phases (Fig. 1 and Table 1). The concentration of CuO was found to have decreased on milling, but was compensated by a corresponding increase in CaTiO3, such that the total concentration of associated phases was relatively constant. It is worth noting that Raval et al.20 observed a significant increase in CaTiO3 and CuO bi-phases with extended milling above 16 h. The lack of bi-phases in the present study demonstrates that milling conditions, such as rotation speed, milling material hardness and sample to milling ball ratio can significantly influence product phase evolution. The possible occurrence of nanocrystalline or amorphous impurities were ruled out from EXAFS analysis (vide infra). Elemental analysis (Table S2†) showed that the concentration of Ca, Cu and Ti were similar to that expected from the stoichiometry, i.e., with an atomic ratio of 1:3:4. As expected but often not reported, high energy milling resulted in the sample contaminated with 3 ppm of Zr from the mill media, which was considered sufficiently low in concentration to have minimal influence on catalytic properties.
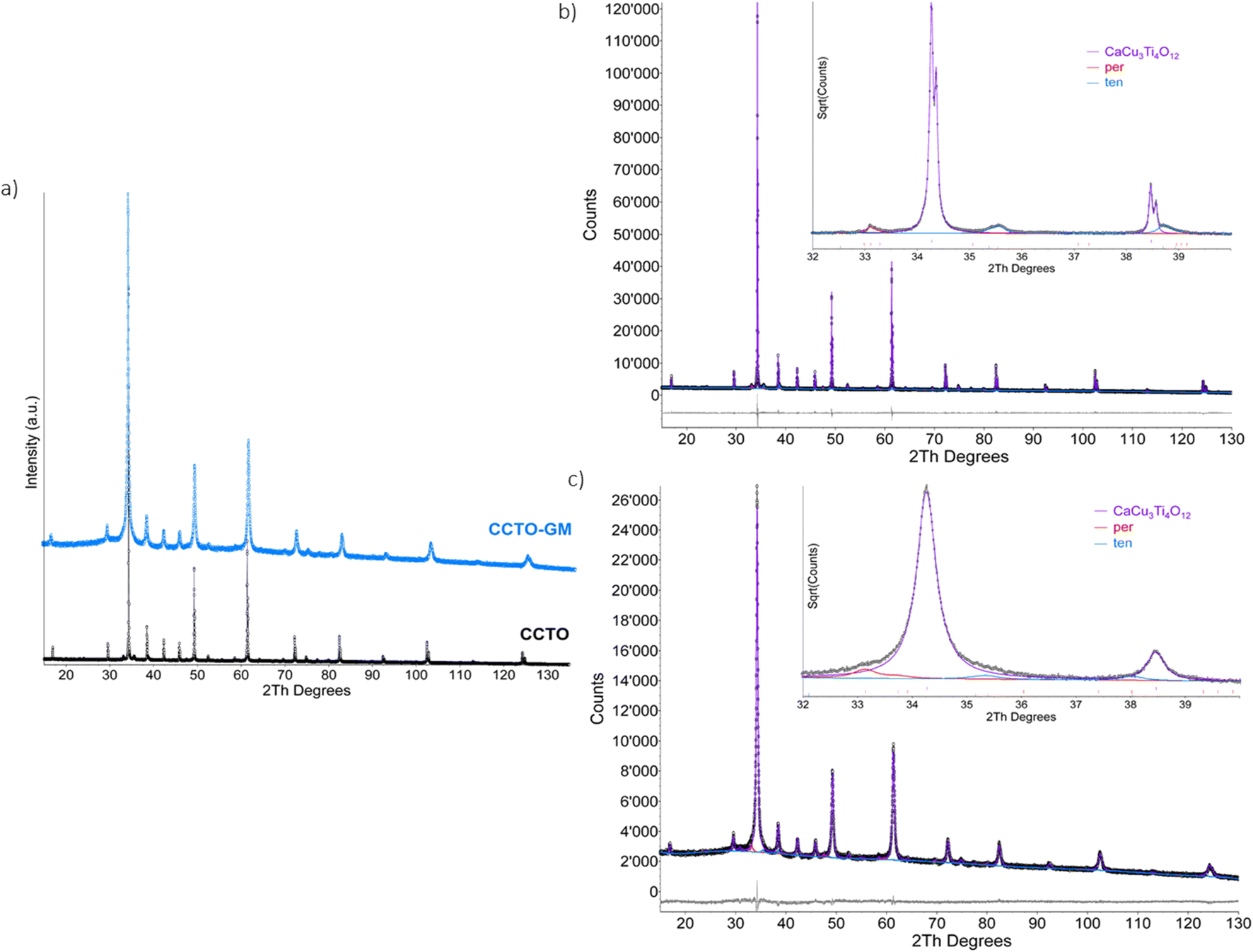 | ||
| Fig. 1 X-ray power diffraction patterns a) and Rietveld refinement plots for CCTO b) and CCTO-GM c). Refinement plot legend: experimental profile is represented by black dots, phases contribution for CaCu3Ti4O12, CaTiO3 perovskite (per), and CuO tenorite (ten) are in purple, red, and blue continuous lines respectively. | ||
| CaCu3Ti4O12 | prv | ten | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R exp | R wp | G.O.F | wt% | a (Å) | V (Å3) | D XRD (nm) | Strain × 10−4 | wt% | wt% | |
| CCTO | 0.022 | 0.034 | 0.015 | 95.6(1) | 7.393(2) | 404.02(3) | 263(4) | 0.5(1) | 1.9(1) | 2.5(1) |
| CCTO-GM | 0.022 | 0.033 | 0.015 | 95.7(1) | 7.395(1) | 404.32(2) | 22(2) | 1.8(1) | 3.2(1) | 1.1(1) |
A notable effect of milling was a reduction in CaCu3Ti4O12 crystallite sized determined from XRD, from 260 to 20 nm for the CCTO and CCTO-GM, with the associated reduction in particle size being confirmed by SEM and TEM analysis (Fig. 2 and S1†). TEM analysis shows the presence of large crystallites in the CCTO sample (Fig. 2a and b) with the CCTO-GM displaying agglomerations of smaller crystallites without a distinct morphology after milling (Fig. 2c and d). The inset selected area electron diffraction highlights the transformation from large single crystals to a polycrystalline material. Such a reduction in particle size on milling is reflected in a significant increase in surface area of the powder from 1.4(1) m2 g−1 to 80.1(4) m2 g−1. The reported surface area of CaCu3Ti4O12 after milling is, to the authors knowledge, the highest reported in the literature and significantly greater than those reported using sol–gel and molten salts synthetic routes (<30 m2 g−1).4,14–16 It is anticipated that such an increase in surface area will significantly enhance photocatalytic performance through increasing the number of adsorption sites (vide infra).
 | ||
| Fig. 2 Transmission electron micrographs of CCTO (a and b) and CCTO-GM (c and d). Inset: selected area electron diffraction patterns of the areas shown. | ||
While the CaCu3Ti4O12 phase is retained on milling, the observed lattice parameter from XRPD increased from 7.393 (CCTO) to 7.395 Å (CCTO-GM) and it was coupled with an increase in crystallite microstrain. The latter situation has previously been observed by Raval et al.,20 and was attributed to milling induced defects and lattice disorder at grain boundaries. As stated above, the presence of defects has previously been reported to influence the photocatalytic performance of CaCu3Ti4O12 when prepared using molten salts. Therefore, it is important to further understand the nature of defects induced by milling and how they influence the intrinsic catalytic performance of CaCu3Ti4O12 in addition to the potential benefit from an increase in surface area.
3.2 Optical properties of CCTO and CCTO-GM
The photocatalytic potential of CCTO and CCTO-GM can be inferred from their optical properties and band gap energies, as determined by diffuse reflectance spectroscopy (DRS). Fig. 3 shows the DRS spectra converted to absorbance (K/S) by Kubelka–Munk equation: K/S = (1 − R∞)2 × (2R∞)−1, and Tauc plots for the two samples. Table 2 provides the wavelength and energy of each transition. The absorption spectrum of CCTO matches well with that reported in the literature for CaCu3Ti4O12. Assignment of the observed bands has previously been attributed to simple electronic transitions of square planar Cu2+ and to more complex transitions between band structures.4,21,31 Specifically, the low energy feature (α) is attributed to d–d electronic transitions from occupied Cu(3d)–O(2p) states to the unoccupied localised α*Cu(3d)–O(2p) states within the band gap. Features (β) and (γ) can be attributed to electronic transitions from the valance band to the Ti (3d) conduction band. Further, the charge transfer band within the UV region and the excitation of TiO6 octahedral orbitals O(2p6)–Ti(3d0), as seen in both CaCu3Ti4O12 and CaTiO3, complicated the spectra in the (γ) region.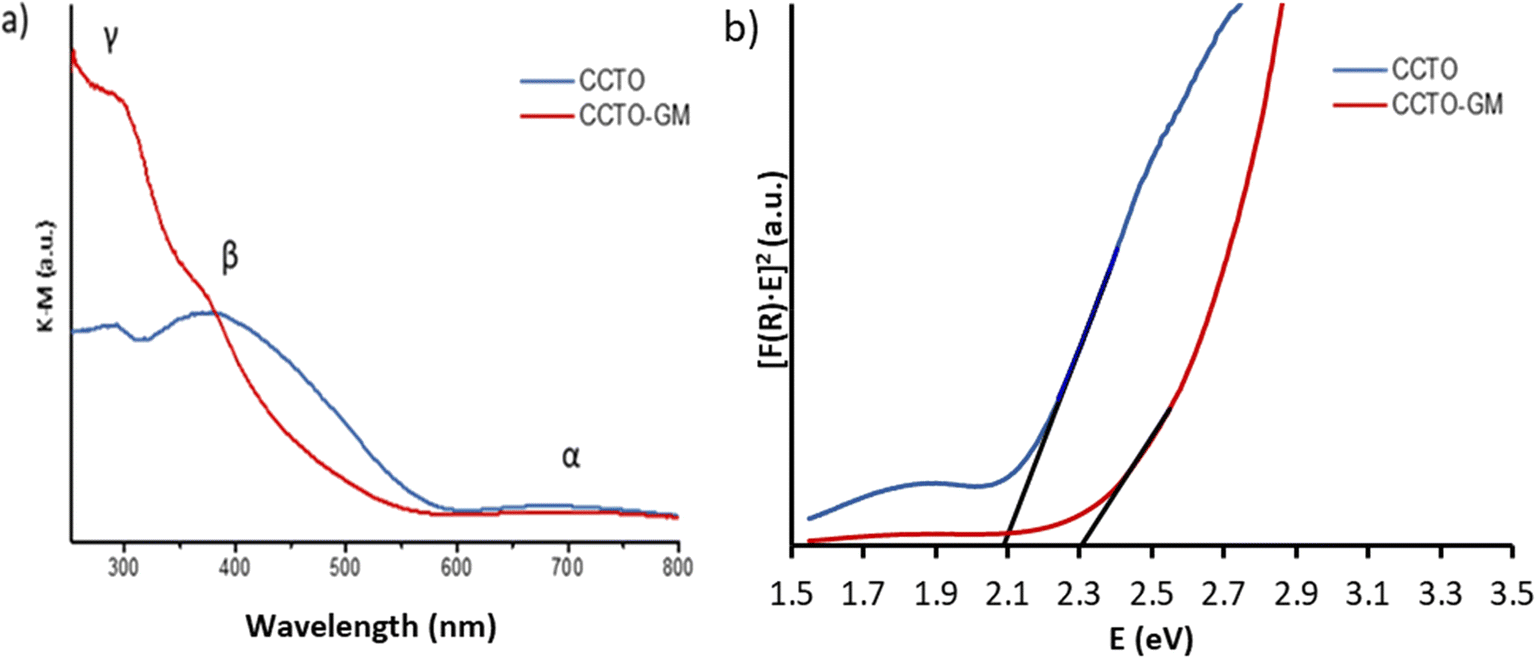 | ||
| Fig. 3 K.M. function for CCTO and CCTO-GM (a); Tauc plots for band gap determination (b). | ||
| CCTO | ||||
|---|---|---|---|---|
| Transition | Energy (eV) | Max. band wavelength (nm) | E CB (V vs. NHE) | E VB (V vs. NHE) |
| α | 1.8 | 681 | 0.068 | 2.15 |
| β | 2.1 | 596 | ||
| γ | 4.2 | 293 | ||
| CCTO-GM | ||||
|---|---|---|---|---|
| Transition | Energy (eV) | Max band wavelength (nm) | ECB (V vs. NHE) | EVB (V vs. NHE) |
| α | 1.8 | 692 | −0.044 | 2.26 |
| β | 2.3 | 538 | ||
| γ | 4.2 | 298 | ||
Milling of CaCu3Ti4O12 (CCTO-GM) resulted in an appreciable blue shift and increase in intensity of the band related to transition β. Further, the intensity of feature γ significantly increased on milling. Band gaps from direct transitions of 2.1(1) eV and 2.3 (1) eV are derived from Tauc plots (Fig. 3b) for CCTO and CCTO-GM samples, respectively. It is well-known that the optical and electronic properties of materials heavily depend on the synthesis method and subsequent powder treatments, as well as on the physical state of the samples under examination (powder, pellets, films). Comparable blue shift and relative intensity changes have been observed by P. Y. Raval et al. on the milling of CaCu3Ti4O12, which were attributed to a lowering in the point symmetry of [CuO4] units from D2h (square planar coordination) to D2 (highly distorted tetrahedral coordination), combined with a reduction in particle size.21 Yet, no other evidence is provided to support such a change in Cu local coordination (note such as change is not supported from Cu XANES vide infra). Hailili et al. also saw significant changes in spectra for CaCu3Ti4O12 prepared using different molten salt synthesis techniques.14,15 The intensity of β features were associated with metal site defects (i.e., Ti3+ and Cu–O vacancies), supported by EPR and XPS analysis. However, the observed changes in UV-vis spectra cannot easily be attributed solely to defects and can be influenced by a number of other parameters, such as particle size and impurities.
3.3 XPS analysis of surface properties
XPS was used to investigate the effect of milling on the surface of CaCu3Ti4O12. Fitting of the Ti2p region (Fig. 4) revealed that only Ti4+ centres are present on the CCTO surface (BE of Ti2p3/2: 458.2 eV), while in CCTO-GM there is evidence of a small Ti3+ component, in addition to that from Ti4+ (BEs of Ti2p3/2: 458.2 eV and 456.1 for Ti4+ and Ti3+, respectively) at a Ti3+:Ti4+ ratio of 1:10. The fitting of the Cu2p3/2 region, along with its shake-up feature, has been performed using three peaks to model the Cu2+ component, with a single Cu+ contribution in the main peak as previously reported.32,33 As seen in Fig. 4, both samples have Cu2+ and Cu+ contributions, with the latter having comparable BEs of 932.1 eV to that reported previously for other CCTO samples.32 However, CCTO-GM surface had a higher concentration of Cu+ than the unmilled CCTO, with a Cu2+:Cu+ ratio of 3:1 and 2:1 in CCTO and CCTO-GM, respectively. Increased signals fitted as Cu+ and Ti3+ from XPS have, as discussed above, previously been used to validate the hypothesis of increasing oxygen defects within CaCu3Ti4O12. Our observations concur that the increase of structural defects, identifiable from changes in the optical properties, paired with the increase of crystallite microstrain and the lattice parameter, can be correlated with the increase of Cu+ and Ti3+ as detected from XPS analysis. Yet, several points of caution require highlighting, namely: (i) XPS is a surface technique and an over-extrapolation to bulk properties must be avoided. For instance, structural features at the perovskite surface are known to significantly differ to that of the bulk.34 (ii) Analysis of Cu oxidation state from Cu2p region is challenging due to small shifts in binding energy coupled with the broadness of the Cu2+ 2p3/2 peak.
 | ||
| Fig. 4 XPS of CCTO and CCTOM-GM. Top row; Ti2p region for CCTO (left) and CCTO-GM (right) with associated data fitting. Second row; Cu 2p3/2 region for CCTO (left) and CCTO-GM (right) and fittings. Bottom row; Zr 3d region for CCTO-GM. | ||
It is also worth noting that the CCTO-GM XPS spectrum revealed the presence of Zr with a Zr/Ti ratio of 0.24. Notably, the latter value is significantly higher than that of the bulk, where a Zr/Ti ratio of 0.03(1) was found, meaning that Zr species are not well dispersed within the bulk of CCTO-GM, but they preferentially concentrate on the CCTO surface as small particles. The Zr 3d5/2 binding energy was observed at 182.3 eV and suggested that the Zr was present in an oxide environment such as ZrO2,35i.e. fragments of milling media.
3.4 EXAFS analysis of the short-range structure of CCTO and CCTO-GM
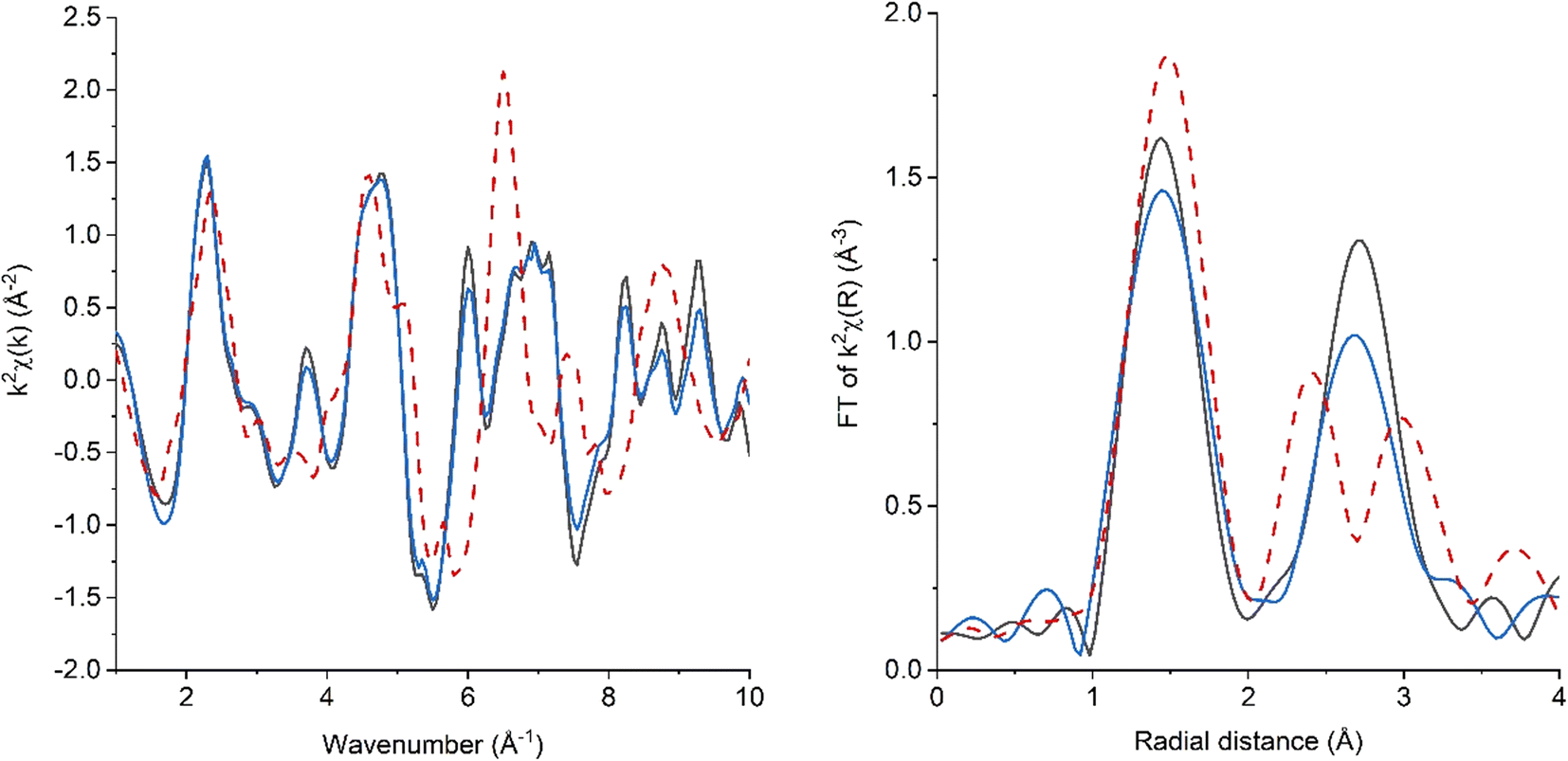
The study of the local structure, including low crystalline materials, can be performed by Cu K-edge EXAFS. The fitting of both the CCTO and CCTO-GM EXAFS data (Fig. 5, Table 3), using a structural model (space group: Im-3 (204), a = 7.391 Å), was successful. These good fitting parameters indicated that both samples were predominantly comprised of CaCu3Ti4O12 in agreement with XRD analysis. Significant quantities of phase separated CuO or Cu2O forming on milling can therefore be discounted. For both samples, most paths could be fitted with a positive symmetric expansion coefficient, with minimal expansion (within error) being observable for CCTO, and a more significant expansion for CCTO-GM. Such expansion agrees with the unit cell expansion seen by XRD. However, it is interesting to note that the 1st Cu–O shells, for both samples, were best fitted with a shorter distance than expected (1.953 Å vs. the literature value of 1.971 Å). Such a contraction of 0.02 Å is far greater than the error of the fit (ca. 0.003 Å) and can be attributed to a proportion of the Cu being present in a different coordination environment/oxidation state (given that EXAFS gives a global average path lengths). Molecular dynamic simulations of CuO and TiO2 nanostructures do report reduced metal–oxygen distances, but at particle sizes way below those observed for the investigated samples (i.e., <5 nm in contrast to the 260 nm and 20 nm crystallite sizes for CCTO and CCTO-GM).36 It could be speculated that a significant concentration of CuO clusters (bond length from EXAFS of 1.948(9) Å) are present, or alternatively that a proportion of the Cu is Cu+ with notably shorter bond length. The former is considered unlikely, as there is no evidence of CuO exceeding 2.5 wt% of the sample (as determined from XRD). Therefore, the contraction is likely attributable to a proportion of Cu+ being present within the sample. It is notable that the 1st Cu–O path length did not change on milling (within error), so if the path length was attributable to Cu+, its concentration within the bulk of the sample remained unchanged on milling (within error). The increase in Cu+ from XPS would then be attributed to an increase in surface Cu+ species vs. a significant change to bulk concentration. A further detail regarding the 1st shell Cu–O is that, when modelled, the coordination numbers were 3.5(2) Å and 4.3(2) Å for CCTO and CCTO-GM respectively. Given the error observed in CNs from EXAFS it is important not to overinterpreted the apparent increase in CN on milling GM, but to simply conclude that there is not clear evidence for any significant increase in oxygen non-stoichiometry on milling around the Cu centre from the 1st shell fit. | ||
| Fig. 5 Cu K-edge k3χ data EXAFS and the associated Fourier transform of CCTO (top) and CCTO-GM (bottom). (Black line): experimental data, (red dashed line) sum of fitting. Fitting parameters: 3 < k < 12 and 1.1 < R < 3.7. | ||
| Sample | Path | Coordination number(i) | Symmetric expansion | Path length (Å) | 2σ2 (Å2) | ΔE (eV) | R-Factor |
|---|---|---|---|---|---|---|---|
| Fitting parameters: 3 < k < 12 and 1.1 < R < 3.7. (i) S02 values (fitted from CuO) fixed at 0.85. (ii) Cu–O(1) increase to 3.7(1) if 2σ2 fixed at 0.0048. (iii) 2σ2 defined as 2σ2 for Cu–O(2) × 1.17. (iv) 2σ2 defined as 2σ2 for Cu–O(2) × 3.6. Multiples defined by factor of increased path length vs. path length of calculated 2σ2. | |||||||
| CCTO | Cu–O(1) | 3.5(2)(ii) | n/a | 1.953(3) | 0.0042(1) | −0.2(6) | 0.0034 |
| Cu–O(2) | 4 | 0.0025(20) | 2.788 | 0.0184(36) | |||
| Cu–Ti | 8 | 3.208 | 0.0080(4) | ||||
| Cu–O(3) | 4 | 3.271 | 0.0216(iii) | ||||
| Cu–O–Ti–Cu (MS) | 16 | 3.573 | 0.0204(iv) | ||||
| Cu–Cu | 4 | 3.705 | 0.0096(14) | ||||
| Cu–Ca | 2 | 3.705 | 0.0111(47) | ||||
| CCTO-GM | Cu–O(1) | 4.3(2) | n/a | 1.955(2) | 0.0048(6) | 0.3(5) | 0.0026 |
| Cu–O(2) | 4 | 0.0057(15) | 2.797 | 0.0179(26) | |||
| Cu–Ti | 8 | 3.218 | 0.0093(4) | ||||
| Cu–O(3) | 4 | 3.281 | 0.0209(iii) | ||||
| Cu–O–Ti–Cu (MS) | 16 | 3.584 | 0.0149(iv) | ||||
| Cu–Cu | 4 | 3.716 | 0.0107(13) | ||||
| Cu–Ca | 2 | 3.716 | 0.0166(64) | ||||
Considering further shells, differences can be observed between samples in the magnitude of the Fourier Transform of the k3 weighted EXAFS, with a notable decrease in the feature just below 3 Å (not phase corrected). This feature is dominated by the Cu–Ti path with contributions from a Cu–O–Ti–Cu multiple scattering path and the Cu–Cu/Cu–Ca paths. The fitting was performed with fixed CN values with the 2σ2 values being used as an indicator of structural disorder (thermal disorder being constant). A clear increase in 2σ2 can be determined for the Cu–Ti path on milling, demonstrating a reduction in local structural order/CN. Reduced coordination is often seen for metal–metal paths in nanostructured oxides, due to the strong contribution of undercoordinated surface sites, however this is normally observed for particles <5 nm. Therefore, the observed particles are considered too large from an EXAFS perspective to see significant particle size affects. Numerous studies have suggested that oxygen vacancies and the formation of distorted [TiO5VOz] square pyramidal geometries can be accommodated in CaCu3Ti4O12. Whangbo and Subramanian hypothesized that sterically crowded CuO6 units, formed by twinning parallel to the (100) plane, could relax to form CuO4 units through oxygen vacancy formation on the connected distorted TiO6.18 The electrons left to the plane defect then facilitate Cu+ and Ti3+ formation. The increased structural disorder in the Cu–Ti path on milling may reflect such planer defects being formed. Increases in 2σ2 were also observed on milling for both Cu–Cu and Cu–Ca paths. Zheng et al. demonstrated that Cu–Ca antisite defects are present in CaCu3Ti4O12, through a combination of techniques including Cu K-edge EXAFS.19 Relatively high 2σ2 values for these paths (0.0096 and 0.0111 Å2) could be attributed as evidence for antisite defects. However, the error in fitted 2σ2 values makes change in the contribution of these defects on milling difficult to verified.
Ti K-edge EXAFS (Fig. 6) proved challenging to fit, with high errors in the fit preventing clear distinction between CCTO and CCTO-GM (Fig. S2, S3 and Table S3†). A clear difference in phasing and the magnitude of the FT between CCTO and TiO2 showed that there are no significant amounts of TiO2 in either CaCu3Ti4O12 sample. Further, both samples could be fitted to a CaCu3Ti4O12 model, although Ti–O distances were slightly shorter than predicted (1.94(1) Å vs. an expected 1.96 Å), potentially demonstrating reduced Ti–O coordination as reported in simulations of TiO2 nanostructures. Between CCTO and CCTO-GM differences are observed in the intensity of features. In particular, the feature at 2.8 Å in the magnitude of the FT, which has contributions from Ti–Cu/Ca paths (in addition to Ti–O multiple scattering, Ti–O (2nd shell) and Ti–Ti paths), is dampened in CCTO-GM. The 2σ2 values for the Ti–Cu/Ca paths were higher for CCTO-GM at 0.009(2)Å2vs. 0.006(2) Å2 seen for CCTO. Broadly, and without over interpreting the K-edge EXAFS data, this correlates with the observations from the Cu K-edge data, in that the A–B (Cu–Ti) site path is more disordered after milling.
 | ||
| Fig. 6 Ti K-edge k2χ data EXAFS (left) and the associated magnitude of the Fourier transform (right) of CCTO, CCTO-GM and rutile TiO2. (Black) CCTO; (blue) CCTO-GM; (dashed red) rutile TiO2. | ||
3.5 XANES analysis of the electronic environment of Cu and Ti in CCTO and CCTO-GM
Lastly, the X-ray absorption near edge structure (XANES) from the Cu and Ti K-edges can be considered. In the Cu k edge XANES (Fig. 7), the CaCu3Ti4O12 samples and the standard CuO all comprise of square planer Cu(II) species and contain 5 notable features. The dipole forbidden 1s → 3d transition (feature A) can be observed as a quadrupole transition, which is sensitive to coordination geometry and changes in centrosymmetry.37 The 1s → 4p transition for planer Cu compounds is rich with information, with 1s → 4pπ (feature B) and 1s → 4pσ transitions (feature C) being clearly distinct. Further, these transitions are strongly modified by the electronic environment of the Cu with 2 electron shakedown transitions (denoted as *) associated with ligand to metal charge transfer (L → 3d; 3d10 L−1). Each transition is therefore split into a lower energy well screened transition and a poorly screened higher energy transition. The notable difference between CCTO and CCTO-GM is the peak intensities of the 1s → 4p features. Specifically, in CCTO-GM the B* feature has reduced intensity relative to CCTO, while features B and C/C* are higher in CCTO-GM. Notably, an absence of change in the 1s → 3d transition (feature A) on milling, does however suggest no significant deviation in local geometry from square planar on milling. | ||
| Fig. 7 Cu K-edge XANES of CCTO and CuO. (Black) CCTO, (blue) CCTO-GM and (red dashed) CuO. (A) 1s → 3d, (B) 1s → 4pπ, (C) 1s → 4pσ, (*) shakedown feature. | ||
Solomon and co-workers attributed the relative intensity of peak B*/B with the degree of bond covalency,38 while Heald et al. noted lower than expected B feature in La1.85Sr0.15CuO4, and attributed this to the presence of holes in the O 2p band.39 Hence, simplistically it can state that milling has created vacancies or reduced covalency of the Cu–O bond, which limits the probability of ligand to metal transfer and thus the screening of this transition. To shed further light on this, ab initio simulations were performed to provide XANES spectral positions, intensities and the wave-function associated with the orbital response of any given band. Fig. 8a shows that the Cu k edge XANES could be simulated with good agreement to experiment. From simulation, feature B* at 8.988 keV has an excited state with O hybridised with Ti d states, while B is localised on the solely Cu–O hybridised orbitals. Lastly, peak C at 8998.5 eV, responsible for the strongest feature in the spectra, is the result of the collective contribution of p-symmetry states of two neighbouring Cu sites, linked together by O p states. With this additional information, the reduction of the B* feature on milling suggests a reduction in the interaction between Cu states and Ti(d)–O(p) hybridised states, potentially through disruption of Cu–Ti centres ordering as seen from EXAFS.
 | ||
| Fig. 8 Simulation of Cu K-edge XANES of Cu3CaTi4O12. (a) Comparison of experimental and simulated XANES. (b) Graphical representations of the bands responsible for features at; (i) 8.988 keV – 1s → 4pπ shakedown feature, (ii) 8.994 keV – 1s → 4pπ feature, and (iii) 8.999 keV – 1s → 4pσ shakedown. | ||
Ti K-edge XANES, shown in Fig. 9, provides analogues evidence, from the perspective of Ti of a decrease in local ordering. Detailed comparison between theory and experiment allows for a correlation of Ti coordination number with band position and intensity (increasing intensity and lower energy position = lower coordination).6,40 The complex pre-edge features, seen in all samples, correlate with those seen by Oliveira et al.6 and were assigned as; (A) 1s → t2g of [TiO6] octahedra quadrupole excitations, (B) 1s → eg of [TiO6] octahedra and (C) dipole excitation 1s → eg of neighbouring [TiO6] octahedra. Notably, on milling, the position of feature B shifts from 4971.2 eV to 4970.8 eV, while the normalised peak area slightly increases. Using the correlation determined by Farges and co-workers it can be estimated that the average Ti coordination number decreased on milling.40 Oliveira et al. attributed this to the formation of oxygen vacancies as [TiO5VOz], where Z denotes different charged vacancies, and therefore the coordination number can be expressed as a [TiO6]:[TiO5VOz] ratio. The ratio for CCTO can be determined as being 4.2:1 [TiO6]:[TiO5VOz], which then decreases on milling (CCTO-GM) to 1.2:1. In addition to the change in feature B, the intensity of features C decrease on milling, which suggests that the interaction between adjacent vertex shared [TiO6] has changed. The change can be hypothesised as being a subtle alteration in the tilting angle of the octahedra or due to vacancies, leading to a reduced excitation 1s → eg of neighbouring [TiO6] octahedra. These findings are supported by changes in the photoluminescence spectra (Fig. S4†) of the two samples, with a splitting of the predominant peak at 400 nm and an decrease in intensity on milling. These changes are interpreted as an increase in the concentration of oxygen vacancies and a change in their nature within complex [TiO6–TiO5VOz] clusters on milling, although changes in scattering on milling could also contribute to intensity changes. Lastly, Farges and co-workers noted that the intensity of main features (M1 and M2) broaden and reduce in intensity with medium range disorder (i.e. glassy vs. crystalline states).40 Interestingly, the normalised intensity of these features didn't decrease on CCTO milling but marginally increased, which suggests a retention, or moderate increase, of medium range order.
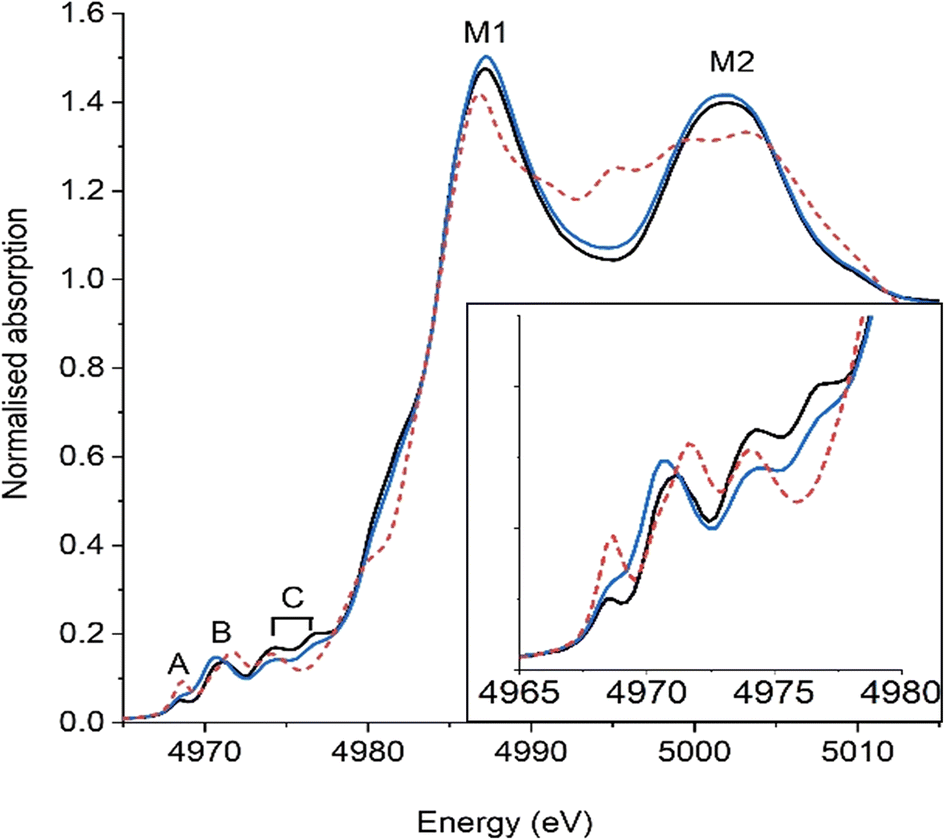 | ||
| Fig. 9 Ti K-edge XANES of CCTO and rutile TiO2. (Black) CCTO, (blue) CCTO-GM and (red dashed) TiO2. Pre edge features: (A) 1s → t2g of [TiO6], (B) 1s → eg of [TiO6], (C) 1s → eg of neighbouring [TiO6], inset shows this region expanded. M1 and M2 main features in CCTO. | ||
In summary, detailed characterisation shows that CaCu3Ti4O12 retains medium and long-range order on milling with an increase in crystallite strain coupled with an increase in surface area due to a reduction in crystallite/particle size. EXAFS supports the retention of the CaCu3Ti4O12 phase but with a decrease in ordering between A and B centres. Cu K-edge XANES showed that milling resulted in a change in the electronic structure, with a reduced probability of transitions to an excited state with significant Ti d state character. An increase in [TiO5VOz] concentration, coupled with a change in short range [TiO6] octahedral interactions is observed. Currently this is hypothesised as being due to an increase in plane defects and proportion of surface structures on milling. These defects are responsible for the observed change in optical properties with little evidence for a significant change in Cu coordination environment.
3.6 Photocatalytic activity for rhodamine B degradation
The ability of CCTO samples to remove a model organic dye through adsorption and subsequent photocatalytic degradation, was assessed initially using an aqueous rhodamine B solution. After optimisation of conditions, the degradation of Ctz was performed. Given that the determination of a photochemical rate constant is complicated by quantum yields, light path lengths and related catalyst concentrations, a simple comparison of performance normalised by catalyst mass or surface area is insufficient (results are shown in Fig. S5†). Therefore, both catalysts have been tested at a range of concentrations (Fig. S6†) through two different regimes. The 1st being that if a linear response between conversion and catalyst mass, and the latter a point at which this proportionality is lost. Kisch and Bahnemann state that the inflection point between these two regimes represents the optimal conditions to test the catalysts, as light absorption is at its maximum.28 The optimal concentrations for CCTO and CCTO-GM were found to be 4 g L−1 and 0.14 g L−1 respectively.Fig. 10 shows the conversion of rhodamine B and the related first order kinetic decay plots for both catalysts under their respective ideal conditions. As a control, rhodamine solution was illuminated using visible light irradiation in absence of photocatalysts with no change in concentration being observed, indicating that degradation takes place by the photocatalytic action rather than by self-photosensitization.4,8,22 CCTO and CCTO-GM samples show notable adsorption capacity towards rhodamine B before exposure to sunlight, as previously reported by K. V. Ivanov et al.41 After this initial absorption no further reactivity was observed, with blank experiments in dark conditions show no remarkable rhodamine B conversion between 60 and 140 minutes. Under solar irradiation the rate constants for the two catalysts were distinctly different at 2.75 × 10−3 min−1 and 1.02 × 10−2 min−1 for CCTO and CCTO-GM respectively. As anticipated the significant increase in surface area on milling was beneficial to catalytic performance. Further, when normalised to surface area and catalyst concentration the rate of the milled CCTO-GM was almost twice that of the original perovskite at 9.11 × 10−4 L min −1 m−2 and 4.91 × 10−4 respectively. Therefore, milling intrinsically increased catalytic activity, which can be ascribed to the influence of structural defects, as identified by changes to XRD determined unit cell parameters, Cu–Ti path disorder from Cu K-edge EXAFS and changes in the Cu and Ti K-edge pre edge XANES. These are considered to be line or point defects but may also be associated with a higher proportion of high index surface terminations on milling. A further consideration is that an intrinsic particle size effect is responsible for improved reactivity, with CCTO-GM having significantly smaller particle size than CCTO. i.e. that the formation of nanoparticles has resulted in a change in band structure and hence photocatalytic performance. However, it should be considered that the average crystallite size of CCTO-GM from XRD is 20 nm, which is considerably larger than that expected for such changes in band structure.
 | ||
| Fig. 10 Rhodamine B removal under solar light irradiation and in dark condition, using optimal concentrations of CCTO and CCTO-GM. (a) Time on line data; (b) first order kinetic decay plot related to photodegradation tests under sun light irradiation. | ||
To better understand the dye adsorption behaviour on the catalyst's surface, Z potential analysis were carried out at pH 7 (pH of the RhB solutions before degradation). At this pH the dye can exist in a zwitterionic form [29] and can interact with the powders through the negatively (COO−) or positively (NR4+) charged functionalities. CCTO and CCTO-GM samples show both negative surface charge of −82(3) mV and −31(9) mV, respectively and, therefore, can interact with the cationic part of the dye molecules. Thus, the adsorption difference found between the two samples are probably linked to the surface area of the powders combined with the surface chemistry, that are both greatly influenced by the milling treatment.
3.7 Post reaction FTIR characterisation
To understand these differences between CCTO and CCTO-GM samples during photodegradation, FT-IR spectroscopy was performed (Fig. 11). Bare CCTO had no absorption peaks, whereas after one hour in “dark” conditions with the substrate, features are observed at 1091 cm−1, 1189 cm−1 and 1400 cm−1 (Gaussian fitting shown in ESI,† Section S4, Fig. S9). These features are assigned to C–O–H stretching of the carboxylic functionality, C–H stretching of the ammino group and bending of C–H bonds in the group![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+(C2H6) of the rhodamine B structure (see ESI† Section S3, Fig. S8, Table S4). No changes were seen in this adsorbed species after photocatalytic reaction.
N+(C2H6) of the rhodamine B structure (see ESI† Section S3, Fig. S8, Table S4). No changes were seen in this adsorbed species after photocatalytic reaction.
 | ||
| Fig. 11 IR spectra of CCTO (a) and CCTO-GM (b) samples as-prepared, after an hour of stirring with rhodamine B in dark and after photodegradation. | ||
CCTO-GM in contrast to CCTO had clear features in the as-prepared sample, which could be assigned to surface bound carbonate (1396 cm−1 and 1556 cm−1) and adsorbed water (1650 cm−1 and 3400 cm−1). The presence of carbonate in CCTO-GM was also confirmed from the XPS C1s signal (Fig. S7†). The significant splitting of the ν(CO) mode indicates a significant loss of symmetry from the free CO32− anion (D3h) and that the carbonate is strongly bound to surface metal sites. After exposure to the substrate in the dark, CCTO-GM had two absorption bands at 1149 and 1225 cm−1 ascribable to C–O–H stretching of carboxylic group,8 and C–N stretching vibration of the ammino group presents in RhB. These two peaks were shifted to higher wavenumbers compared to the as prepared sample indicating the formation of weaker interactions through the milled sample and RhB molecules. This can be related to the different surface chemical environment (presence of carbonates and zirconium, higher Cu+/Cu2+ and Ti3+/Ti4+ ratios and a more positive surface charge) of CCTO-GM compared to the as prepared sample. Moreover, the bending of C–H bond in the group N+(C2H6) (visible at 1400 cm−1 in the CCTO spectra) cannot be appreciated in CCTO-GM probably due to the overlap with the carbonate's modes centred at 1396 cm−1. FTIR results suggest that rhodamine B molecules are firstly adsorbed onto calcium copper titanate's surface (a schematic rapresentation and description is presented in ESI† Section 5 Fig. S10) and subsequently degraded under sun light irradiation through the destruction of the chromophore group.8
3.8 Mechanistic studies of rhodamine B dye degradation using CCTO and CCTO-GM
To investigate the active species in rhodamine B photocatalytic degradation, radical scavengers, and interfering species were introduced in the system before photodegradation (1 mM concentration) as trapping agents for CCTO-GM sample. Since the degradation kinetics of the CCTO sample is extremely slow (Fig. 10) the addition of radical scavengers did not reveal any differences in the photodegradation rate. In the case of CCTO-GM sample isopropanol (IPA) and potassium iodide (KI) were employed as OH˙ (ref. 12 and 42) and h+ scavenger43 respectively, p-benzoquinone (pBQ) for O2˙− quenching12 and dimethyl sulfoxide (DMSO) as photoelectron trapping agent.11,44 The results about CCTO-GM radical trapping experiments are reported in Fig. 12. Using IPA or KI the degradation reaction is strongly quenched indicating that h+ and OH˙ have a key role in photolysis. When KI is added, RhB concentration decreases in dark (≈10% lower) probably due to the competitive adsorption of K+ ions on the negatively charged CCTO-GM surface. | ||
| Fig. 12 Results for radical trapping experiments in the presence of CCTO-GM photocatalysts. | ||
Moreover, the addition of pBQ seems to reduce the photodegradation rate, suggesting that photoproduced O2˙− radicals are involved in dye degradation mechanism. The presence of DMSO does not have a great influence on the photodegradation reaction, indicating that photoproduced electron are not the main reactive species during dye degradation. Using the band edge positions calculated with eqn (1) and (2) (Table 2), a schematic illustration of the possible photodegradation mechanism is reported in Fig. 13. All standard potentials are taken at pH 7.
 | ||
| Fig. 13 Schematic representation of solar light driven degradation mechanisms of rhodamine B by a) CCTO, b) CCTO-GM. | ||
Both samples show the edge of the valence band more negative than E°(H2O/OH˙) (2.730 V vs. NHE)45 but more positive than E°(OH−/OH˙) (1.902 V vs. NHE).45 Thus, the formation of hydroxyl radicals is thermodynamically favourable from the oxidation of adsorbed OH− groups, but not directly from water molecules. OH˙ and h+ photogenerated are the main active species during RhB photodegradation in CCTO-based compounds12,46 as generally reported for photodegradation of dyes and organic pollutants.47 The position of the conduction band is more positive than E°(O2(aq)/O2˙−) (−0.160 V vs. NHE)45,48 in both CCTO and CCTO-GM, therefore it would be difficult for these radicals to participate in the degradation of pollutant molecules. However, the presence of Ti3+ species in CCTO-band gap,4,14,15 as a consequence of the reaction of the electron transferred between Ti4+/Ti3+ (E° = −0.498 V vs. NHE) and the oxygen molecules adsorbed on the material surface, to give O2˙− species following the scheme:
| Ti3+ + O2 → Ti4+ + O2˙− |
Importantly, but often negated, is the possible importance of dye sensitization process in photocatalytic dye degradation. The energy of the highest occupied molecular orbital and lowest unoccupied molecular orbital (HOMO and LUMO) in rhodamine B are 1.1 and −1.0 V vs. NHE.51 Thus, a photo-excited electron can be transferred from the LUMO of RhB* to the more positive conduction band of CCTO-type catalyst, lowering the photoproduced charge recombination rate and enhancing dye degradation.
3.9 Photocatalytic activity for cetirizine hydrochloride degradation
Given the contribution of direct visible light photoexcitation in rhodamine B degradation, an alternative substrate, which does not absorb light within the visible region, was considered to provide evidence of actual photocatalytic performance for CCTO and CCTO-GM. The antihistamine Ctz was chosen, with photocatalytic degradation results using the prior determined optimised masses (200 and 7 mg for CCTO and CCTO-GM respectively) being reported in Fig. 14. It was noted that CCTO and CCTO-GM samples had poor adsorption capacity towards Ctz before exposure to sunlight, as at a pH of 6.5 Ctz is electrically neutral and exist in molecular form. Under solar irradiation the rate constant for the two catalysts were distinctly different, at 1.48 × 10−3 min−1 and 8.88 × 10−3 min−1 for CCTO and CCTO-GM respectively. The retention of an improved degradation rate for CCTO-GM using a molecule without absorption in the visible region strengthens the conclusion that milling does increase photocatalytic performance. Further, the tripling of surface area normalised rates with CCTO-GM vs. CCTO, at 7.92 × 10−4 L min −1 m−2 and 2.64 × 10−4 L min −1 m−2 respectively, confirmed that this improved activity can be attributed to changes in structure on milling. | ||
| Fig. 14 Cetirizine hydrochloride removal under solar light irradiation and in dark condition, using optimal concentrations of CCTO and CCTO-GM. (Left) Time online data; (Right) first order kinetic decay plot related to photodegradation tests under sun light irradiation. | ||
4. Conclusions
The photocatalytically active CaCu3Ti4O12 perovskite catalyst is known to have an intrinsically poor surface area, which is detrimental to catalytic performance. Further, structural defects have an important influence on optical and catalytic properties. The mechanochemical ball milling of sol–gel synthesised CaCu3Ti4O12 was found to increase its surface area from 1 to 80 m2g−1, with no net increase in phase separated CuO, CaTiO3or TiO2 species, i.e. 96% phase purity of CaCu3Ti4O12 as determined by XRD and EXAFS. The observed surface area on milling of CaCu3Ti4O12 is the highest reported to date. Changes to the UV-vis spectra, an increase in unit cell size and strain from XRD, and an observed decrease in Cu–Ti path length structural order from EXAFS, demonstrated that milling induced structural defects within CaCu3Ti4O12. XANES showed clear changes to the local environment of Ti on milling with an increase in [TiO5VOz] species, which in turn influenced the electronic properties of Cu, seen by XANES and supported by relevant simulations. Specifically, the changes in Cu K-edge XANES on milling were a decrease in the intensity of shakedown features associated with delocalised excited states. These changes are interpreted as being indicative of structural defects within CaCu3Ti4O12. Optimised photodegradation studies of rhodamine B showed that milling increased catalytic activity by almost a factor of 5, due to an increase in active surface area. Further, when the results are normalised by surface area, milling resulted in a ×2 increase in CaCu3Ti4O12 performance which can be attributed to the creation of point and line defects during the milling process. Comparable improvement in catalytic activity were also seen for cetirizine hydrochloride degradation, which unlike rhodamine B does not absorb visible light, with a ×3 increase in CaCu3Ti4O12 surface area normalised photocatalytic activity. Thereby confirming that structural changes, in the form of defect formation, enhance photocatalytic degradation performance of CaCu3Ti4O12.Author contributions
Simon Kondrat: conceptualization, formal analysis, investigation, writing-original draft, writing review & editing, funding acquisition. Andrea Bartoletti: formal analysis, investigation, writing-original draft, writing review & editing, visualization. Angela Gondolini: conceptualization, investigation, writing-original draft, writing review & editing. Nicola Sangiorgi: conceptualization, formal analysis, writing-original draft, writing review & editing. Alessandra Sanson: resources, supervision, project administration, funding acquisition. Marzio Rancan: investigation, writing review & editing. Lidia Armelao: resources, writing review & editing. Matteo Ardit: investigation, data curation, writing review & editing, visualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank The Royal Society for funding through the International Exchanges grant (IES\R3\170381). Further, the UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC grant: EP/R026939/1, EP/R026815/1, EP/R026645/1, EP/R027129/1. XAFS measurements were performed at Diamond Light Source on beamline B18 through the UK Catalysis BAG (SP15151-10). We thank Dr. Diego Gianolio and Dr. Martin Wilding for their support. TEM analysis was performed by Dr. Thomas Davies (Cardiff Catalysis Institute), who would like to thank the European Regional Development Fund, the Welsh European Funding Office and The Wolfson Foundation for funding the CCI-Electron Microscopy Facility. Experimental data is available at https://doi.org/10.17028/rd.lboro.21316503.References
- G. Thuillier, M. Hersé, D. Labs, T. Foujols, W. Peetermans, D. Gillotay, P. C. Simon and H. Mandel, The solar sectral irradiance from 200 to 2400 nm as measured by the solspec spectrometer from the atlas and eureca missions, Sol. Phys., 2003, 214, 1–22 CrossRef.
- G. Zhang, G. Liu, L. Wang and J. T. S. Irvine, Inorganic perovskite photocatalysts for solar energy utilization, Chem. Soc. Rev., 2016, 45, 5951–5984 RSC.
- A. Kumar, A. Kumar and V. Krishnan, Perovskite Oxide Based Materials for Energy and Environment-Oriented Photocatalysis, ACS Catal., 2020, 10, 10253–10315 CrossRef CAS.
- J. H. Clark, M. S. Dyer, R. G. Palgrave, C. P. Ireland, J. R. Darwent, J. B. Claridge and M. J. Rosseinsky, Visible Light Photo-oxidation of Model Pollutants Using CaCu3Ti4O12: An Experimental and Theoretical Study of Optical Properties, Electronic Structure, and Selectivity, J. Am. Chem. Soc., 2011, 133, 1016–1032 CrossRef CAS PubMed.
- M. A. Subramanian, D. Li, N. Duan, B. A. Reisner and A. W. Sleight, High Dielectric Constant in ACu3Ti4O12 and ACu3Ti3FeO12 Phases, J. Solid State Chem., 2000, 151, 323–325 CrossRef CAS.
- L. H. Oliveira, E. C. Paris, W. Avansi, M. A. Ramirez, V. R. Mastelaro, E. Longo and J. A. Varela, Correlation Between Photoluminescence and Structural Defects in Ca1+xCu3−xTi4O12 Systems, J. Am. Ceram. Soc., 2013, 96, 209–217 CrossRef CAS.
- A. Koitzsch, G. Blumberg, A. Gozar, B. Dennis, A. P. Ramirez, S. Trebst and S. Wakimoto, Antiferromagnetism in CaCu 3 Ti 4 O 12 studied by magnetic Raman spectroscopy, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 052406 CrossRef.
- T. A. Otitoju, D. Jiang, Y. Ouyang, M. A. M. Elamin and S. Li, Photocatalytic degradation of Rhodamine B using CaCu3Ti4O12 embedded polyethersulfone hollow fiber membrane, J. Ind. Eng. Chem., 2020, 83, 145–152 CrossRef CAS.
- H. S. Kushwaha, P. Thomas and R. Vaish, Polyaniline/CaCu 3 Ti 4 O 12 nanofiber composite with a synergistic effect on visible light photocatalysis, RSC Adv., 2015, 5, 87241–87250 RSC.
- M. Ahmadipour, A. A. Hamzah, A. L. Pang, A. Thi Le, S.-L. Chiam, Z. A. Ahmad, B. Rajitha and S. Y. Pung, Photodegradation of rhodamine B-dye pollutant using CaCu3Ti4O12-multiwall carbon nanotube nanocomposites, Journal of Environmental, Chem. Eng., 2021, 9, 105185 CAS.
- K. Pal, A. Mondal, R. Jana, P. P. Ray and A. Gayen, Domestic LED light driven methylene blue degradation by g-C3N4-CaCu3Ti4O12 composite, Appl. Surf. Sci., 2019, 467–468, 543–553 CrossRef CAS.
- M. Ahmadipour, M. Arjmand, M. Z. A. Thirmizir, A. T. Le, S. L. Chiam and S.-Y. Pung, Synthesis of core/shell-structured CaCu3Ti4O12/SiO2 composites for effective degradation of rhodamine B under ultraviolet light, J. Mater. Sci.: Mater. Electron., 2020, 31, 19587–19598 CrossRef.
- N. U. Saqib, R. Adnan, I. Shah and I. Hussain, Sequential uptake of cadmium and methylene blue from binary solution using ZEOLITE-TIO2 modified porous CACU3TI4O12 photocatalyst, Environ. Prog. Sustainable Energy, 2020, 40 DOI:10.1002/ep.13595.
- R. Hailili, Z.-Q. Wang, X.-Q. Gong and C. Wang, Octahedral-shaped perovskite CaCu3Ti4O12 with dual defects and coexposed {(001), (111)} facets for visible-light photocatalysis, Appl. Catal., B, 2019, 254, 86–97 CrossRef CAS.
- R. Hailili, Z.-Q. Wang, Y. Li, Y. Wang, V. K. Sharma, X.-Q. Gong and C. Wang, Oxygen vacancies induced visible-light photocatalytic activities of CaCu3Ti4O12 with controllable morphologies for antibiotic degradation, Appl. Catal., B, 2018, 221, 422–432 CrossRef CAS.
- R. Hailili, X. Yuan and C. Wang, A systematic investigation on morphology tailoring, defect tuning and visible-light photocatalytic functionality of Ti-based perovskite nanostructures, Catal. Today, 2019, 335, 591–598 CrossRef CAS.
- P. Saikia, H. J. Sarmah, S. Ahmed, S. Lahkar, J. P. Das and S. K. Dolui, Synthesis of CaxCu3-xTi4O12 Perovskite Materials and House-Hold LED Light Mediated Degradation of Rhodamine Blue Dye, J. Inorg. Organomet. Polym., 2021, 31, 2161–2167 CrossRef CAS.
- M.-H. Whangbo and M. A. Subramanian, Structural Model of Planar Defects in CaCu3Ti4O12 Exhibiting a Giant Dielectric Constant, Chem. Mater., 2006, 18, 3257–3260 CrossRef CAS.
- J.-C. Zheng, A. I. Frenkel, L. Wu, J. Hanson, W. Ku, E. S. Božin, S. J. L. Billinge and Y. Zhu, Nanoscale disorder and local electronic properties of ${\text{CaCu}}_{3}{\text{Ti}}_{4}{\text{O}}_{12}$: An integrated study of electron, neutron, and x-ray diffraction, x-ray absorption fine structure, and first-principles calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 144203 CrossRef.
- P. Y. Raval, P. R. Pansara, N. H. Vasoya, K. B. Modi and P. M. G. Nambissan, Positron annihilation spectroscopic investigation of high energy ball – milling engendered defects in CaCu3Ti4O12, Ceram. Int., 2018, 44, 15887–15895 CrossRef CAS.
- P. Y. Raval, P. R. Pansara, N. H. Vasoya, K. Punia, S. N. Dolia, K. B. Modi and S. Kumar, First observation of reversible mechanochromism and chromaticity study on calcium–copper–titanate, J. Am. Ceram. Soc., 2019, 102, 6872–6881 CrossRef CAS.
- J. Zhao, J. Liu and G. Ma, Preparation, characterization and dielectric properties of CaCu3Ti4O12 ceramics, Ceram. Int., 2012, 38, 1221–1225 CrossRef CAS.
- B. Wallbank, C. E. Johnson and I. G. Main, Surface reduction of solid 3d transition metal compounds during x-ray photoelectron spectroscopy, J. Electron Spectrosc. Relat. Phenom., 1974, 4, 263–269 CrossRef CAS.
- J. P. Tobin, W. Hirschwald and J. Cunningham, XPS and XAES studies of transient enhancement of Cu1 at CuO surfaces during vacuum outgassing, Appl. Surf. Sci., 1983, 16, 441–452 CrossRef CAS.
- Q. Liu, Y. Zhou, J. Kou, X. Chen, Z. Tian, J. Gao, S. Yan and Z. Zou, High-Yield Synthesis of Ultralong and Ultrathin Zn2GeO4 Nanoribbons toward Improved Photocatalytic Reduction of CO2 into Renewable Hydrocarbon Fuel, J. Am. Chem. Soc., 2010, 132, 14385–14387 CrossRef CAS PubMed.
- R. Beranek, (Photo)electrochemical Methods for the Determination of the Band Edge Positions of TiO2-Based Nanomaterials, Adv. Phys. Chem., 2011, 2011, 1–20 CrossRef.
- L. Kavan, M. Grätzel, S. E. Gilbert, C. Klemenz and H. J. Scheel, Electrochemical and Photoelectrochemical Investigation of Single-Crystal Anatase, J. Am. Chem. Soc., 1996, 118, 6716–6723 CrossRef CAS.
- H. Kisch and D. Bahnemann, Best Practice in Photocatalysis: Comparing Rates or Apparent Quantum Yields?, J. Phys. Chem. Lett., 2015, 6, 1907–1910 CrossRef CAS PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, First principles methods using CASTEP, Z. Kristallogr. - Cryst. Mater., 2005, 220, 567–570 CrossRef CAS.
- A. P. Bartók and J. R. Yates, Regularized SCAN functional, J. Chem. Phys., 2019, 150, 161101 CrossRef PubMed.
- P. Thomas, K. Dwarakanath, K. B. R. Varma and T. R. N. Kutty, Nanoparticles of the giant dielectric material, CaCu3Ti4O12 from a precursor route, J. Phys. Chem. Solids, 2008, 69, 2594–2604 CrossRef CAS.
- T. T. Fang and Y. H. Wang, Reassessment of Copper and Titanium Valences and Excess Holes in Oxygen 2p Levels of CaCu3Ti4O12, J. Electrochem. Soc., 2011, 158, G207–G210 CrossRef CAS.
- R. P. Gupta and S. K. Sen, Calculation of multiplet structure of core $p$-vacancy levels. II, Phys. Rev. B: Solid State, 1975, 12, 15–19 CrossRef CAS.
- D. R. Inns, A. J. Mayer, V. Skukauskas, T. E. Davies, J. Callison and S. A. Kondrat, Evaluating the Activity and Stability of Perovskite LaMO3-Based Pt Catalysts in the Aqueous Phase Reforming of Glycerol, Top. Catal., 2021, 64, 992–1009 CrossRef CAS.
- J. F. Moulder and J. Chastain, Handbook of x-ray photoelectron spectroscopy: a reference book of standard spectra for identification and interpretation of XPS data, Physical Electronics Division, Perkin-Elmer Corp., Eden Prairie, Minn., 1992 Search PubMed.
- P. K. Naicker, P. T. Cummings, H. Zhang and J. F. Banfield, Characterization of Titanium Dioxide Nanoparticles Using Molecular Dynamics Simulations, J. Phys. Chem. B, 2005, 109, 15243–15249 CrossRef CAS PubMed.
- T. Lais, L. Lukashuk, L. van de Water, T. I. Hyde, M. Aramini and G. Sankar, Elucidation of copper environment in a Cu–Cr–Fe oxide catalyst through in situ high-resolution XANES investigation, Phys. Chem. Chem. Phys., 2021, 23, 5888–5896 RSC.
- S. DeBeer, D. W. Randall, A. M. Nersissian, J. S. Valentine, B. Hedman, K. O. Hodgson and E. I. Solomon, X-ray Absorption Edge and EXAFS Studies of the Blue Copper Site in Stellacyanin: Effects of Axial Amide Coordination, J. Phys. Chem. B, 2000, 104, 10814–10819 CrossRef CAS.
- S. M. Heald, J. M. Tranquada, A. R. Moodenbaugh and Y. Xu, Orientation-dependent x-ray-absorption near-edge studies of high-${T}_{c}$ superconductors, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 761–764 CrossRef CAS PubMed.
- F. Farges, G. E. Brown and J. J. Rehr, Ti $K$-edge XANES studies of Ti coordination and disorder in oxide compounds: Comparison between theory and experiment, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1809–1819 CrossRef CAS.
- K. V. Ivanov, O. V. Alekseeva and A. V. Agafonov, Synthesis of CaCu3Ti4O12, Study of Physicochemical and Photocatalytic Properties, Russ. J. Inorg. Chem., 2020, 65, 1541–1546 CrossRef CAS.
- F. Grieser, Free radical formation and scavenging by solutes in the sonolysis of aqueous solutions, Proc. Meet. Acoust., 2013, 19, 045093 Search PubMed.
- J. T. Schneider, D. S. Firak, R. R. Ribeiro and P. Peralta-Zamora, Use of scavenger agents in heterogeneous photocatalysis: truths, half-truths, and misinterpretations, Phys. Chem. Chem. Phys., 2020, 22, 15723–15733 RSC.
- G. Liu, M. Liao, Z. Zhang, H. Wang, D. Chen and Y. Feng, Enhanced photodegradation performance of Rhodamine B with g-C3N4 modified by carbon nanotubes, Sep. Purif. Technol., 2020, 244, 116618 CrossRef CAS.
- D. A. Armstrong, R. E. Huie, W. H. Koppenol, S. V. Lymar, G. Merényi, P. Neta, B. Ruscic, D. M. Stanbury, S. Steenken and P. Wardman, Standard electrode potentials involving radicals in aqueous solution: inorganic radicals (IUPAC Technical Report), Pure Appl. Chem., 2015, 87, 1139–1150 CrossRef CAS.
- M. Ahmadipour, M. Arjmand, Z. A. Ahmad and S.-Y. Pung, Photocatalytic Degradation of Organic Dye by Sol–Gel-Synthesized CaCu3Ti4O12 Powder, J. Mater. Eng. Perform., 2020, 29, 2006–2014 CrossRef CAS.
- J. Duan, Y. Liu, X. Pan, Y. Zhang, J. Yu, K. Nakajim and H. Taniguchi, High photodegradation efficiency of Rhodamine B catalyzed by bismuth silicate nanoparticles, Catal. Commun., 2013, 39, 65–69 CrossRef CAS.
- D. A. Armstrong, R. E. Huie, S. Lymar, W. H. Koppenol, G. Merényi, P. Neta, D. M. Stanbury, S. Steenken and P. Wardman, Standard electrode potentials involving radicals in aqueous solution: inorganic radicals, BioInorg. React. Mech., 2014, 9 DOI:10.1515/irm-2013-0005.
- P. M. Wood, The potential diagram for oxygen at pH 7, Biochem. J., 1988, 253, 287–289 CrossRef CAS PubMed.
- J. Li, W. Ma, P. Lei and J. Zhao, Detection of intermediates in the TiO2-assisted photodegradation of Rhodamine B under visible light irradiation, J. Environ. Sci., 2007, 19, 892–896 CrossRef CAS PubMed.
- F. Collin, Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases, Int. J. Mol. Sci., 2019, 20, 2407 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy01299e |
| This journal is © The Royal Society of Chemistry 2023 |