
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBreaking linear scaling relationships with transition metal carbides†
Hector
Prats
 * and
Michail
Stamatakis
* and
Michail
Stamatakis
Department of Chemical Engineering, University College London, Roberts Building, Torrington Place, London WC1E 7JE, UK. E-mail: h.garcia@ucl.ac.uk
First published on 13th July 2023
Abstract
Transition metal carbides (TMCs) are proposed as catalysts and supports for small metal particles to replace expensive late transition metals as heterogeneous catalysts. High-throughput screening based on density functional theory shows that TMCs break the limitations that the linear scaling relations impose to transition metals.
It is well known that the adsorption energies of numerous species on transition metals (TM) scale linearly with each other.1 Such dependencies arise from bond order conservation theory, electron counting rules, and local coordination numbers, and lower the degrees of freedom in complex reactions with several steps, greatly simplifying computational screening. Moreover, the activation barriers can be traced back to the energy of one or more intermediates in a linear form, as illustrated by the Brønsted–Evans–Polanyi (BEP) relationships.2 Then, by focusing on a few descriptors, typically the formation energies of one or two reaction intermediates,2,3 it is possible to derive volcano plots, which provide a systematic way for catalytic optimisation and have become the gold standard in catalyst design.1 Note that such relations are not externally imposed constraints or restrictions on the chemistry, but rather, they arise naturally from the underlying physical laws governing the interactions between nuclei and electrons that give rise to chemistry. The downside of the existence of such linear scaling (LS) relations is that they limit the efficiency of the reactions (i.e., the top of the volcano plots). For instance, it is thought that the scaling between the adsorption energies of *OOH and *OH limits the activity of oxygen reduction and oxygen evolution reactions.4 As the need to go beyond the top of these volcanoes becomes more pressing,5,6 new strategies are explored to break the limitations imposed by the LS relations which are based on increasing the complexity of the catalyst.
One way to increase the complexity of transition metal catalysts is by incorporating carbon atoms into the interstitial sites of the parent metals, leading to transition metal carbides (TMC). The bonding in TMCs involves a metallic, a covalent, and an ionic contribution,7 resulting in TMCs displaying properties characteristic of these three different classes of materials, and they have been shown to exhibit catalytic properties similar to those of Pt-group metals for a wide range of reactions.8,9 The most stable facet of cubic fcc TMCs is the (001),10 which contains 50% C atoms and 50% metal atoms in the surface layer, thereby exhibiting a higher variety of adsorption sites compared to e.g. the (111) surface of fcc TMs (Fig. 1). In addition, surface C atoms are negatively charged while surface metal atoms are positively charged,11 meaning that the possible adsorption sites might interact in a very different way depending on the nature of the adsorbed species, being more likely to break LS relations.
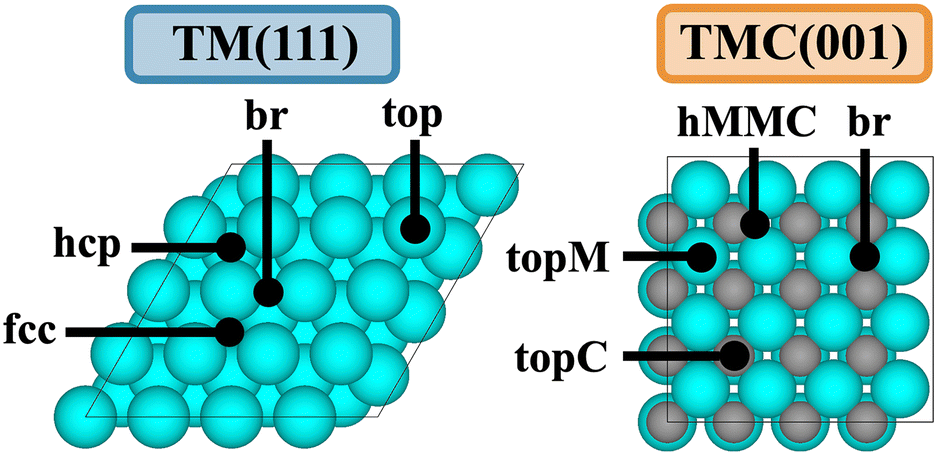 | ||
| Fig. 1 Top views of the (111) facet of a fcc TM and the (001) facet of a fcc TMC. Black dots show the positions of the different adsorption sites. | ||
An additional level of complexity can be achieved by supporting small clusters of TMs on TMCs (namely TM@TMC). In the last decade, TMCs have been shown to also be excellent substrates to disperse metallic particles, since they polarise the electron density of the supported particles in such a way that their catalytic activity can be much superior to those dispersed on more traditional oxide supports.11–14 For instance, small Au, Cu and Ni particles supported on TiC display a very high activity for CO2 hydrogenation,15 orders of magnitude higher compared to Au(100), Cu(100), or Ni(100) extended surfaces. The diversity of adsorption sites in TM@TMCs skyrockets due to their much more complex structure.
In this article, we present a DFT-based study screening study in which we assess the formation energies of an assortment of catalytically relevant species on TMC surfaces and TM@TMCs. These TM@TMCs consist of all combinations between 7 metal nanoclusters (Rh, Pd, Pt, Au, Co, Ni and Cu) supported on the most stable surfaces of 8 fcc TMCs (TiC(001), ZrC(001), HfC(001), VC(001), NbC(001), TaC(001), cMoC(001) and cWC(001)) as well as 3 hcp TMCs (hMoC(0001)-Mo, hMoC(0001)-C and hWC(0001)-W) (77 materials in total). Note that the cubic and hexagonal phases for MoC and WC are referred to as cTMC and hTMC, respectively, and that there exist two possible terminations for the (0001) facet of hexagonal TMC. The (001) facet of fcc TMCs, apart from being the most stable one (and therefore predominant in large TMC particles), is expected to be more active compared to other low Miller index surfaces, such as the (011) or the (111), as the latter interact too strongly with adsorbate species,16 especially with O atoms, and therefore can be more easily oxidised.
Additional details on the computational methods, the chosen dataset, and the calculated formation energy (Ef) values for all systems are provided in the ESI† (sections S1–S4). We analyse the generated database to study the validity of LS relations in TMCs and TM@TMCs, and unravel trends in the adsorptive properties of these materials.
The subset of adsorbates considered in this study includes 4 stable molecules (CH4, CO2, H2O and CO) and 7 catalytically relevant molecular fragments (C, CH, CH2, CH3, O, OH and H). The main reason for not considering other adsorbates such as HCO or COOH, which are relevant in e.g., carbon dioxide reduction in TMCs,16 is simply to keep time and computational resources within reasonable limits, while taking care to explore adsorbed species that are representative and relevant to practical applications. Thus, we limit the species considered to only those having two elements.
A general observation from the calculated formation energies displayed in Fig. 2a is that adsorbed species bind more strongly to TMCs than TMs. This can be explained in part by the ionic character of the TMC(001) facets, the higher diversity of adsorption sites, which can adapt better to the electronic properties of each adsorbate, and also to the alteration of the d-band of the carbide metal atoms due to the mixing with the s–p orbitals of C atoms, which results in an increase of the density of states (DOS) near the Fermi level compared to the parent metal17 and hence an increased binding strength. This trend is particularly pronounced for CO2. As opposed to the weakly physisorbed configuration adopted in extended TM surfaces, anionic CO2δ− species with bent geometry are formed on TMC surfaces. Thus, while the average adsorption energy for CO2 on TMs is about −0.2 eV, it increases by a factor of about 6 on average on TMCs. In fact, the good performance of TMCs towards capture, storage and activation of CO2 has already been reported by Kunkel et al.18 However, the interaction of the highly stable CH4 molecule and TMCs remains weak, except for two outliers (cMoC and cWC). In fact, porous MXenes, a novel family of 2D TMCs, have been proposed as very promising sorbent materials for CO2 separation from CH4, a critical step in biogas upgrading, due to their strong versus weak interactions with CO2versus CH4, respectively.19Fig. 2a also illustrates what is probably the main problem of TMCs in catalysts, which is that they are easily oxidised in air, forming surface oxide structures known as oxycarbides.20 The average Ef decreases from ∼−0.3 eV in TMs to ∼−1.6 eV in TMCs, with hWC-W and hMoC-Mo exhibiting Ef ∼ −3.4 eV. Oxygen is regarded as a poison for TMCs, since, for example, oxycarbides are known to undermine the Mo2C catalytic performance for the water-gas shift reaction, where O moieties are created from H2O decomposition.21
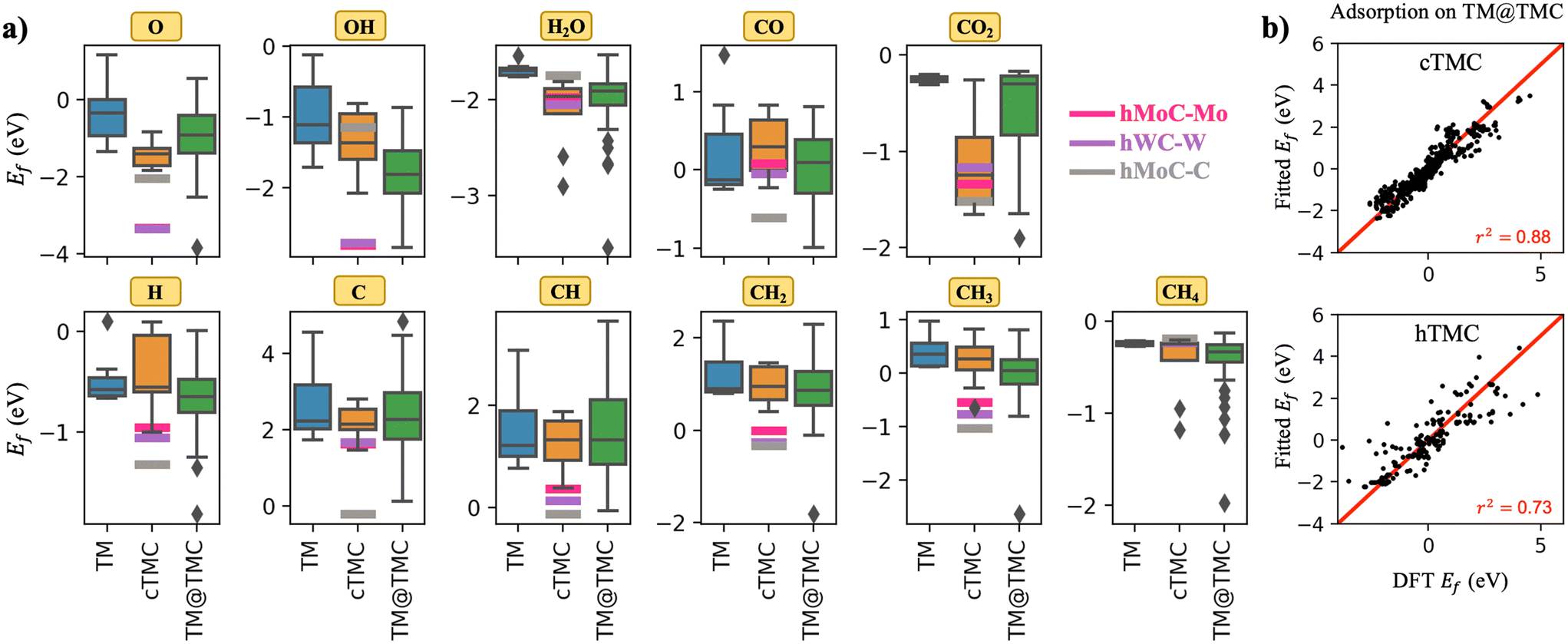 | ||
| Fig. 2 a) Box plots showing the distribution of formation energies for all adsorbed species on TMs (blue), cubic TMCs (orange) and TM@TMCs (green). The box limits correspond to the interquartile range (IQR), formed between the 1st and 3rd quartiles (Q1 and Q3), and the whiskers (black lines) extend to Q1 − 1.5·IQR and Q3 + 1.5·IQR. Outliers are shown as black rhombi. For comparison, the formation energies on hexagonal TMCs are plotted as thick, straight, horizontal lines. b) Parity plot of formation energies on TM@TMC (fitted versus computed by DFT) excluding those where the cluster displaces, deforms, or breaks, see details in Table S7.† | ||
Shifting our attention to the different types of TMC slabs, Fig. 2a shows that hexagonal TMCs (i.e., magenta, purple and grey lines) are potentially much more reactive than cubic TMCs. This trend is attributed to a shift of the d-band centre towards higher energies (Fig. S2†). In fact, the stronger adsorption of hexagonal TMCs compared to cubic TMCs is not only limited to chemical species, but also to supported metal clusters, which bind much stronger in the former.11 Regarding the two possible terminations of hexagonal TMCs, note how all species binding through O atoms (O, OH and H2O) favour the M-termination while all species binding through C atoms (C, CO, CO2, CH, CH2 and CH3) prefer the C-termination. This trend can be rationalised by considering that the surface layer is positively charged in the M-termination and negatively charged in the C-termination11 because of the different electronegativities of C and metal atoms, and the partial ionic character of the C-metal bonds in TMCs. All species that bind to the surface via O carry a negative partial charge. Thus, it is expected, from an electrostatic perspective, that the positively charged M-termination would bind these species more strongly than the C-termination. On the other hand, CO and CO2 bind to the surface via C, which carries a positive partial charge, and therefore prefer the C-termination. For CHx species (x = 0–3) which also bind to the surface via C, the preference for the C-termination is attributed to the formation of strong covalent C–C bonds, as shown in Fig. S3.†
Next, we analyse the interaction of TMCs with CHx (i.e., C, CH, CH2 and CH3) and O-containing (O, OH, H2O and CO) chemisorbed species (Fig. S4†). Most formation energies are positive (weaker interaction) for CHx species and negative (stronger interaction) for O-containing species, in agreement with the known resistance of TMCs against coke formation22 but weak resistance against oxidation.20 Interestingly, TMCs of group 6 chemisorb CHx species more strongly than TMCs of groups 4 and 5, with no significant differences within the TMCs of the same group (Fig. S4a†). On the other hand, the interaction strength with O-containing species is similar for TMCs of groups 4, 5 and 6, but increases when going down a group (Fig. S4b†), with period VI TMCs (HfC, TaC and WC) being more likely to become poisoned by O.
So far, we have discussed the adsorption behaviour of extended TM and TMC facets. Now we proceed to analyse the case of TMC-supported TM clusters. As shown by the green boxes in Fig. 2a, the adsorption strength of most species on TM@TMCs covers a much broader range of formation energies than on the corresponding extended TM and TMC surfaces. Thus, while some TM@TMCs interact with adsorbed species as weakly as Au(111) (i.e., the least reactive metal towards atoms or molecules),23 other supported clusters strongly chemisorb most adsorbates with formation energies even more negative than those of hexagonal TMCs. The case of gas-phase CO2 and CH4 molecules is particularly interesting from a catalysis perspective because of these species' high thermodynamic stability, which makes the activation of the C–H bond in CH4 and the C–O bond in CO2 a great challenge. Earlier, we showed that extended TM slabs can barely activate either of these molecules due to their weak interaction, while TMCs can easily adsorb and dissociate CO2 but are mostly inactive for CH4. TM@TMCs, on the other hand, can in principle activate both molecules with the right combination of cluster and support, as shown by the wide range of formation energies for CO2 and CH4 in Fig. 2a. This was shown experimentally for small Ni clusters supported on TiC, which were able to adsorb and dissociate CH4 at room temperature.14
We have discussed above some trends in the interaction of TMCs with CHx and O-containing chemisorbed species along the groups and periods of the periodic table. When TMCs are used as supports, however, most of these trends disappear, especially regarding the nature of the TMC support (Fig. S4†). To assess if the formation energies of adsorbates on TMs and TMCs can be used to predict their formation energies on TM@TMCs, we have performed multilinear regression using both parameters as descriptors. The parameters of the regression are given in Table S7.†Fig. 2b shows the parity plots for fcc (left) and hcp (right) TMCs. The formation energy on TM@TMC is reproduced with mean absolute errors of 0.33 eV and 0.55 eV for cubic and hexagonal TMCs, respectively. The agreement is not good enough for a quantitative analysis, especially in the case of the hexagonal carbides, where the charged nature of the cluster is not captured by either of the two descriptors. However, it allows to easily predict a narrow range of formation energies that the adsorbate can have on the supported cluster. The analysis of the coefficients indicates that Ef on the TM has a greater weight than Ef on the TMC, which is also concluded from the single-parameter regressions (Fig. S5†).
Following the original discovery of LS on transition metals24 and later in transition metal oxide, sulphide, and nitride surfaces,25 Lewis acid zeolites,26 and single atom alloys,27 we investigate if such relations also hold for transition metal carbides. LS relations are mainly observed when the two adsorbates are chemically similar (i.e., bind to the metal through the same atom, such as CH and C). Fig. 3 shows that, while LS relations for the selected pair of similar adsorbates hold well for TMs, they are in general not obeyed by TMCs. This is a consequence of the higher diversity of adsorption sites on TMCs and the more ionic nature of the interactions. Note that, in fcc TM(111) facets, all surface atoms are equivalent, charge-neutral, and the preferred adsorption sites tend to follow simple valency rules.24 The fcc TMC(001) facets, however, contain negatively charged C atoms and positively charged metal atoms (Fig. 1), which invalidates simple valency rules and leads to a greater diversification of the preferred adsorption sites (Fig. S6†). For instance, the preferred adsorption site for C, CH and CH2 species correlates with the atomic charge of surface atoms, following the sequence br → hMMC → topC as the charge of surface C (metal) atoms becomes more negative (positive), see Table S8.†
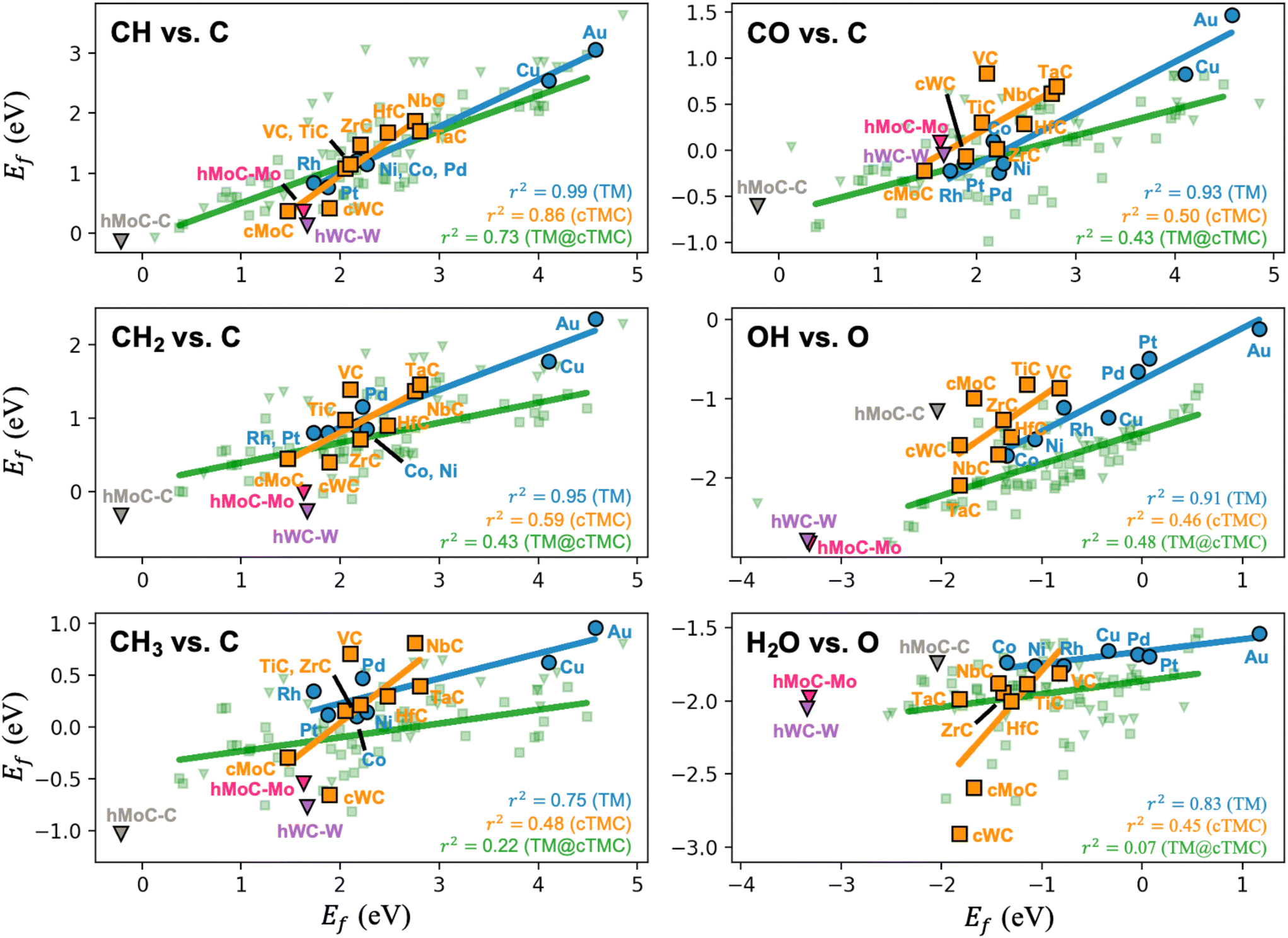 | ||
| Fig. 3 Linear scaling plots between the formation energies of different adsorbates on TMs, TMCs and TMn@TMCs. Trend lines and R-squared coefficients are included. Note that hexagonal TMCs (shown as triangles) have been excluded from the TMC trend lines, as they exhibit different adsorption sites compared to cubic TMCs. All other linear scaling plots for TM and TMC are shown in Fig. S7 and S8,† respectively. | ||
The only decent LS on TMCs is observed for the formation energies of CH and C species, which adsorb on the same site in all TMCs except NbC. In fact, when the two species bind (on a given family of materials) through the same atom and on the same adsorption site, it is more likely that their formation energies will correlate. This is illustrated in Fig. S9 and S10† for TMs and TMCs, respectively. The most representative case is the LS of CHx species with C in TMCs. If we only consider the formation energies on topM sites instead of the most stable site, the correlation between Ef for both species is much stronger (i.e., r2 increases from 0.86 to 0.97 for CH vs. C, from 0.59 to 0.88 for CH2vs. C, and from 0.48 to 0.94 for CH3vs. C, see Fig. S10†). Note that this “same-site” argument does not hold when the selected site is not very stable. TM@TMCs expose a highly diverse set of adsorption sites due to their complex structure, so LS relations are even poorer (Fig. 3). The lack of LS in the formation energies of adsorbate species in TMCs and TM@TMCs makes computational screening more costly but allows these materials to overcome the limitations imposed by such relations, especially for the case of TM@TMCs, as they can be designed in such a way that they favour the adsorption of some key species over others.
Conclusions
TMCs can represent a cheap and attractive alternative to traditional metal-based catalysts, especially in the context of CO2 conversion, since most TMCs are able to activate stable CO2 molecules by charge transfer. Within the set of the TMCs investigated, hexagonal carbides interact more strongly than cubic carbides with supported metal clusters as well as with most reaction intermediates, due to a shift of the d-band centre towards higher energies. Unlike TMs, the interaction between TMCs and reaction intermediates is highly influenced by the electrostatic interactions. For instance, the preferred adsorption site does not follow simple valency rules as in metals, but in some cases correlates with the atomic charge of surface atoms. Moreover, in hexagonal TMCs, species that bind to the surface via O, which carries a negative partial charge, always prefer binding to the positively charged metal termination, while species that bind to the surface via C always prefer binding to the negatively charged C termination. We also show that TMCs interact much more strongly with O-containing species than with CHx species, and the degree of interaction depends on the position of the TMC metal atom in the periodic table. These trends disappear when the TMC is used as a support of small metal particles, where the binding strength of the adsorbed species to the supported cluster depends on the specific combination of TM and TMC instead. Most importantly, the use of TMCs as supports (i.e., TM@TMC) opens up a plethora of opportunities for catalyst design, as these systems cover a broad range of formation energies for the different adsorbates and transcend the limitations imposed by the LS relations, enabling tuning of the adsorption strength of key species.Author contributions
H. P.: conceptualisation, data curation, formal analysis, funding acquisition, investigation, methodology, validation, visualisation, writing – original draft. M. S.: conceptualisation, formal analysis, funding acquisition, project administration, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 891756. We gratefully acknowledge the use of the UCL High Performance Computing Facility Kathleen@UCL in the completion of the simulations of this work. We are grateful to the UK Materials and Molecular Modelling Hub for computational resources, which is partially funded by EPSRC (EP/T022213/1, EP/W032260/1 and EP/P020194/1).Notes and references
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 1999, 57, 6 Search PubMed.
- J. K. Nørskov, F. Studt, F. Abild-Pedersen and T. Bligaard, Fundamental concepts in heterogeneous catalysis, Wiley, 2014 Search PubMed.
- I. C. Man, H. Su, F. Calle-vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- M. Busch, N. B. Halck, U. I. Kramm, S. Siahrostami, P. Krtil and J. Rossmeisl, Nano Energy, 2016, 29, 126–135 CrossRef CAS.
- N. B. Halck, V. Petrykin, P. Krtil and J. Rossmeisl, Phys. Chem. Chem. Phys., 2014, 16, 13682–13688 RSC.
- J. G. Chen, Chem. Rev., 1996, 96, 1477–1498 CrossRef CAS PubMed.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CrossRef CAS PubMed.
- J. A. Rodriguez, P. Liu, J. Dvorak, J. Gomes, Y. Takahashi and K. Nakamura, Surf. Sci., 2003, 543, L675–L682 CrossRef CAS.
- M. G. Quesne, A. Roldan, N. H. de Leeuw and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2018, 20, 6905–6916 RSC.
- H. Prats and M. Stamatakis, J. Mater. Chem. A, 2022, 10, 1522–1534 RSC.
- J. A. Rodriguez, F. Viñes, F. Illas, P. Liu, Y. Takahashi and K. Nakamura, J. Chem. Phys., 2007, 127, 211102 CrossRef CAS PubMed.
- H. Prats and M. Stamatakis, Nanoscale Adv., 2023, 5, 3214–3224 RSC.
- H. Prats, R. A. Gutiérrez, J. J. Piñero, F. Viñes, S. T. Bromley, P. J. Ramírez, J. A. Rodriguez and F. Illas, J. Am. Chem. Soc., 2019, 141, 5303–5313 CrossRef CAS PubMed.
- J. A. Rodriguez, J. Evans, L. Feria, A. B. Vidal, P. Liu, K. Nakamura and F. Illas, J. Catal., 2013, 307, 162–169 CrossRef CAS.
- F. Silveri, M. G. Quesne, F. Viñes, F. Illas, C. R. A. Catlow and N. H. de Leeuw, J. Phys. Chem. C, 2022, 126, 5138–5150 CrossRef CAS PubMed.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Catal. Today, 2005, 105, 66–73 CrossRef CAS.
- C. Kunkel, F. Viñes and F. Illas, Energy Environ. Sci., 2016, 9, 141–144 RSC.
- H. Prats, H. McAloone, F. Viñes and F. Illas, J. Mater. Chem. A, 2020, 8, 12296 RSC.
- J. A. Rodriguez, P. Liu, J. Gomes, K. Nakamura, F. Viñes, C. Sousa and F. Illas, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 75427 CrossRef.
- P. Liu and J. A. Rodriguez, J. Phys. Chem. B, 2006, 110, 19418–19425 CrossRef CAS PubMed.
- A. P. E. York, J. B. Claridge, A. J. Brungs, S. C. Tsang and M. L. H. Green, Chem. Commun., 1997, 39–40 RSC.
- B. Hammer and J. K. Nørskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skúlason, T. Bligaard and J. K. Nørskov, Phys. Rev. Lett., 2007, 99, 016105 CrossRef CAS PubMed.
- E. M. Fernández, P. G. Moses, A. Toftelund, H. A. Hansen, J. I. Martínez, F. Abild-Pedersen, J. Kleis, B. Hinnemann, J. Rossmeisl, T. Bligaard and J. K. Nørskov, Angew. Chem., Int. Ed., 2008, 47, 4683–4686 Search PubMed.
- B. C. Bukowski and J. Greeley, J. Phys. Chem. C, 2016, 120, 6714–6722 CrossRef CAS.
- M. T. Darby, R. Réocreux, E. C. H. Sykes, A. Michaelides and M. Stamatakis, ACS Catal., 2018, 8, 5038–5050 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00863k |
| This journal is © The Royal Society of Chemistry 2023 |