
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Definition and exploration of realistic chemical spaces using the connectivity and cyclic features of ChEMBL and ZINC
Thomas
Cauchy
 *a,
Jules
Leguy
b and
Benoit
Da Mota
*b
*a,
Jules
Leguy
b and
Benoit
Da Mota
*b
aUniv Angers, CNRS, MOLTECH-ANJOU, SFR MATRIX, F-49000 Angers, France. E-mail: thomas.cauchy@univ-angers.fr
bUniv Angers, LERIA, SFR MATHSTIC, F-49000 Angers, France. E-mail: benoit.damota@univ-angers.fr
First published on 3rd April 2023
Abstract
Discovering an efficient new molecule can have a huge impact on the chemical research field. For several problems, the current knowledge is too scarce to train robust deep learning models. An exploratory approach can be a solution. However, when we consider several types of atoms, a phenomenal amount of combinations are possible even for small molecules. Many of these combinations contain very exotic associations. In addition to connectivity feature filtering (based on ECFP4), we introduce and stress the importance of a new filter based on cyclic features. In this article, we show that whitelists including all connectivity and cyclic features of either ChEMBL or ChEMBL and ZINC allow for the definition of large realistic chemical spaces. An enumeration dataset, Evo10, has been built with more than 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules having 10 or fewer heavy atoms (C, N, O, F, and S). Starting only from a methane molecule, we were able to navigate through the chemical space of those realistic molecules and rediscover all molecules passing these same filters from the reference datasets which are here ChEMBL, ZINC, QM9, PC9, GDB11, and GDBChEMBL. Unlike previously published scores, SAscores and CLscores, which are based on similarity averages on the most common chemical environments, the method proposed here excludes any molecule with an ECFP and cyclic feature that is absent from the lists. The visualisation of the proposed top solutions, that pass all the filters, for the optimisation of the QED or HOMO and LUMO energies, convinces us of the relevance of this approach for the systematic de novo generation of realistic solutions.
000 molecules having 10 or fewer heavy atoms (C, N, O, F, and S). Starting only from a methane molecule, we were able to navigate through the chemical space of those realistic molecules and rediscover all molecules passing these same filters from the reference datasets which are here ChEMBL, ZINC, QM9, PC9, GDB11, and GDBChEMBL. Unlike previously published scores, SAscores and CLscores, which are based on similarity averages on the most common chemical environments, the method proposed here excludes any molecule with an ECFP and cyclic feature that is absent from the lists. The visualisation of the proposed top solutions, that pass all the filters, for the optimisation of the QED or HOMO and LUMO energies, convinces us of the relevance of this approach for the systematic de novo generation of realistic solutions.
In many chemistry domains, the discovery of new molecules is often the result of an intensification of an already known effective compound through chemical reactions (addition, substitutions, …) in order to improve its properties. The emergence of a truly new molecule is a rarer phenomenon, but one that can pave the way for further intensification and profound transformations of this domain. This is precisely around this objective that an intense research has been developed on the topic of de novo generation of molecules possessing sought properties, especially for drug and material discovery.1–8 Among the challenges of this field of research, we can mention the difficulty of correctly formalising the specifications.9–12 Another crucial challenge is to generate molecules that could be synthesised.8,10,13–17
In the case of organic molecular materials, the chemical space definition will be different from the one for drug discovery with different constraints on toxicity, organic solvent solubility, and intermolecular interactions (H bonding, π stacking, …). Furthermore, the number of already known efficient scaffolds can be quite limited depending on the application. As an example, Harris et al. discuss the handful of molecular scaffolds used as molecular photoswitches such as diarylethene, hydrazone, azo, and hemithioindigo.18 Indeed, in many problems there is not enough data on which one could train a deep learning model that could be sufficiently robust to allow for good generalizability for a large chemical space exploration.
One solution is to use molecular generators based on an evolutionary algorithm.19–21 They are often characterised by a great freedom of construction and thus of exploration of the chemical space, followed by an efficient intensification around high scoring solutions. However, they are criticised for very easily constructing molecules that seem to be silly while they respect the valency rules. This limitation of evolutionary generators is a long-lasting lock that is crucial to overcome for their practical use for de novo generation of molecules.
However, assessing the synthesizability of a molecule on the basis of a structural formula is a very difficult problem. Distributors sell molecules that can be very complex, some being extracted compounds from nature. In 2009, Peter Ertl et al. proposed the synthetic accessibility score (SAscore) based on the most common chemical fragments of 1 M compounds of the PubChem penalised by the numbers of stereocenters, spiro atoms, macrocycles, and large cycles in general.13 The SAscore has a value between 1 (the most accessible) and 10 (the least accessible). In the original article, the bulk of the catalogue molecules have a score lower than 4. But there is no obvious threshold value since realistic natural products have SAscore values between 5 and 8.13 In 2020, the ChEMBL-Likeness Score (CLscore) has been proposed in order to select in the huge GDB17, a small portion called GDBChEMBL, that could be more feasible based on an average occurrence of ChEMBL chemical fragments.22 This time, the higher the CLscore, the more common fragments in the ChEMBL the molecule contains on average. Again, there is no obvious threshold value to establish a classification between realistic or not. The authors have used a cut-off value of 3.3.22 Also in 2020, the SYBA score was developed.23 This score is based on a Bayesian classifier trained on ZINC15 molecules and unrealistic de novo generated compounds. Finally, recent work proposes to evaluate solutions based on deep learning approaches (RAscore) whose domain of validity is associated with the environments present in the ChEMBL and GDBChEMBL.17 The purpose of the RAscore is to classify what is synthesizable (a value close to 1) from what is not (a value close to 0). Unfortunately, this score is not explanatory. And allowing new chemical environments requires new training of the deep learning method. The common idea behind all these studies is that a molecule that is similar to millions of known molecules is more realistic. The similarity is assessed based on molecular fragments.
These scores are useful for generating molecules that are visibly more pleasing to the eye of the chemist but are not infallible.21 Two limits could be observed in this approach. On the one hand, not all combinations of known chemical functions are possible when these functions are defined in a short range. And, on the other hand, a single exotic combination of atoms is enough to make the molecule non-synthesizable. A score based on an average may not penalise enough such solutions. As an example, we could use ribavirin and its analog as discussed by Gao and Coley.15 In both cases, the SAscore and CLscore seem to indicate a realistic molecule, see Fig. 1. However, the synthesis of the analog is expected to be much harder. Indeed, one of its connectivity features does not exist in either ChEMBL or ZINC. The RAscore is indeed lower for the second molecule. But the RAscore can be 0.0 for a commercial molecule because it contains an unknown ECFP6 in its training set, see Fig. 2.
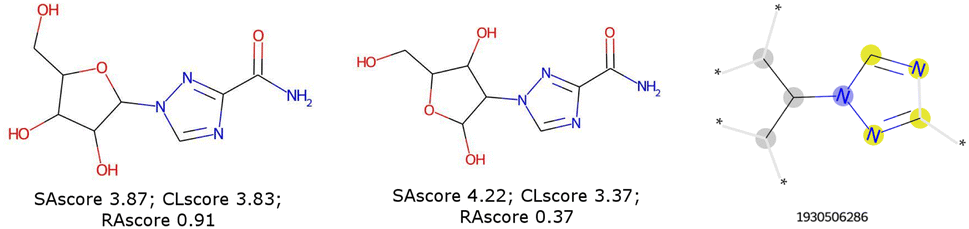 | ||
| Fig. 1 Ribavirin (left) and its analog (center) possess a good SAscore and CLscore. But the analog does not exist and possesses a connectivity feature (right) unknown in ChEMBL and ZINC. | ||
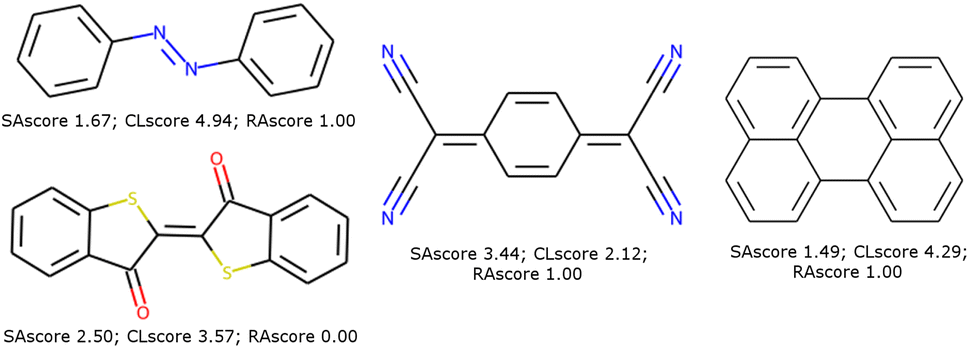 | ||
| Fig. 2 Some molecules used in organic electronic materials that pass our filters with their respective SAscore, CLscore, and RAscore. | ||
Thus, we provide here an approach based on whitelist filtering that can be easily adapted to the specific chemistry of certain applications and allow for some explainability. In this paper, we propose to define realistic chemical spaces of molecules that are constituted of only chemical environments, up to 2 neighbours, of ChEMBL25 and ZINC20.24–26 Prohibiting the creation of new chemical environments at any moment of the optimisation means that the whole areas of chemical combinations are forbidden. Nevertheless, we show that limiting the generator to molecules with only known chemical environments still allows for very large search spaces. We will compare here a whitelist extracted from the whole ChEMBL25 and one including the ChEMBL25 and ZINC20. In addition, we propose a refinement method based on a filter on the cyclic features present in those datasets. This new descriptor fills a lack of information on this subject in the extended-connectivity fingerprints (ECFP). The importance of the cyclic features is demonstrated in the first objective focused on the optimisation of the QED. Since it is legitimate to ask whether it is still possible to navigate between the different possible molecules with an atom-centred evolutionary algorithm under constraints, we will then present the results of rediscovery objectives starting only from methane. The chemical diversity generated using an enumeration objective will be assessed and compared to reference datasets. The last objective is associated with a goal-directed generation of molecular electronic properties.
The connectivity feature filtering has been implemented in our molecular generator called EvoMol.21 It is open source and freely available. Furthermore, the lists of connectivity and cycle features used to define the realistic chemical spaces are also available to be used in any other molecular generators. We believe that it is not only evolutionary algorithms that can benefit from this filtering. In addition, a JSON file is provided. It describes the generated dataset named Evo10 that includes 676875 realistic molecules with up to ten heavy atoms (C, N, O, F, S) with their SMILES and their connectivity filter scores. Similar JSON files of the molecules in reference datasets are also available to ensure the reproducibility of the results presented in the article.
1 Methodology
1.1 Data
To define the ECFP whitelists, we used two datasets. One corresponds to the substances of ChEMBL25 as downloaded in September 2019 with 1817766 unique molecules. It is a large curated database of bioactive molecules. The second one is based on ZINC. ZINC is larger than ChEMBL and is based on commercially available compounds and is not restricted to bioactive molecules. It encompasses in proportion more inorganic and organometallic compounds than ChEMBL. We have used the already prepared version ZINC20-ML by Artem Cherkasov and Francesco Gentile with all the 1006651037 ZINC20 molecules as of early March 2021. ZINC20-ML is available at https://files.docking.org/zinc20-ML/.
In order to compare the various chemical spaces, several other published datasets were also employed. We have built a standardization procedure so that we may compare the chemical spaces with the one produced by the EvoMol program, see specifications below. After converting the SMILES to the RDKit molecular graph object, the stereochemical information is removed. Then, only non-radical neutral compounds were retained whose SMILES writing does not contain formal charges. These last criteria remove only 63498 molecules from the ChEMBL dataset. With this procedure, tautomers should have different SMILES/graphs and correspond to different molecules. Because we needed the cyclic features that are associated with the neutral compounds, the whitelists of cyclic features were built on these subsets of ChEMBL and ZINC. The final filter will be the list of allowed chemical elements and the heavy atom count (HAC, i.e. Z > 1) limit, depending on the objective. The total number of molecules in these subsets is detailed in Table 1.
| Dataset | Atoms | HAC limit | Total | Passing filter 1 | Passing filter 1 ∪ GCF1 | Passing filter 2 | Passing filter 2 ∪ GCF2 |
|---|---|---|---|---|---|---|---|
| QM9 ∪ PC9 | CNOF | 9 | 190300 |
27831 |
27082 |
42791 |
41845 |
| ChEMBL | CNOF | 76 | 804366 |
804366 |
804366 |
804366 |
804366 |
| ChEMBL | CNOFS | 76 | 1191453 |
1191453 |
1191453 |
1191453 |
1191453 |
| GDBChEMBL | CNOF | 17 | 3385555 |
325670 |
298126 |
541409 |
506247 |
| GDBChEMBL | CNOFS | 17 | 3786315 |
355684 |
326053 |
597840 |
561113 |
| GDB11 | CNOF | 11 | 26413375 |
605080 |
582982 |
1002268 |
970519 |
| ZINC | CNOF | 78 | 199278637 |
128284725 |
— | 199278637 |
199278637 |
| ZINC | CNOFS | 78 | 288467281 |
186106225 |
— | 288467281 |
288467281 |
| Evo10 | CNOF | 10 | 491145 |
263630 |
234942 |
491145 |
439701 |
| Evo10 | CNOFS | 10 | 676897 |
348484 |
315335 |
676897 |
614980 |
The selected datasets are QM9 (ref. 27) and PC9 (ref. 28) as defined in the OD9 publication.29 These two sets contain molecules ranging from HAC 1 up to 9 that can be either carbon, nitrogen, oxygen, or fluorine. QM9 is a subset of molecules of the GDB enumerations.30 QM9 was built from a constrained combinatorial approach while PC9 is a subset taken from the PubChemQC dataset with the same number of heavy atom limits and chemical element constraints.28,31 Once filtered the union of these two sets contains 190300 different molecules. Two other combinatorial reference datasets, GDBChEMBL and GDB11, have been also filtered in the same way. The GDB datasets can be downloaded at https://gdb.unibe.ch/downloads/. The total number of molecules in these filtered subsets is detailed in Table 1.
1.2 Whitelist definitions
The definition of whitelist filtering based on connectivity has been inspired by the silly walks program of Patrick Walters.32 The idea is to be able to highlight the chemical environments that are unknown in a reference dataset. To list the connectivity features for each molecular graph, we have used the GetMorganFingerprint function of the RDKit program.33 Considering a medium size radius, i.e. up to 2 bonds for the extended-connectivity fingerprints ECFP4 one could expect that if such chemical environments have never been described, it could be associated with a synthetic challenge.34 The ECFP4 of each molecular graph is composed of all the connectivity features centred on each atom up to 2 bonds. All the connectivity features found in the reference datasets form the allowed whitelist. Patrick Walter used a selection of drugs of the ChEMBL as a reference. Such restrictions can be useful to drug likeness. However, for organic molecular materials such restrictions could be too harsh. So, we propose two different filters. Filter 1 is based on the full ChEMBL25 compounds as a reference dataset. This corresponds to 556187 unique connectivity features. Filter 2 is based on both ChEMBL25 and ZINC20. ZINC20 encompasses approximately 800k unique features, and the union of both encompasses 1156416 unique connectivity features of many atom types.
To illustrate such filters, we have tested several molecules studied in the MOLTECH-Anjou laboratory in molecular materials for electronics and photonics. The azobenzene, thioindigo, tetracyanoquinone, and perylene molecules are composed of only connectivity features present in ChEMBL and thus pass both filters, see Fig. 2. However, the most iconic electron donor molecule, the tetrathiafulvalene presents two connectivity features that do not exist in ChEMBL25, see Fig. 3. Since this molecule exists in the ZINC20 dataset, it passes filter 2.
 | ||
| Fig. 3 The tetrathiafulvalene molecule used in organic electronic materials does not pass filter 1. To the right, the missing ECFP in ChEMBL. | ||
It is worth pointing out here how the ECFP works. An ECFP2 takes into account the central atom, its bonds, and the type of bonds of its first neighbours. Potentially in the case of a carbon bonded to four atoms which are themselves bonded to three other atoms, the largest connectivity feature in an ECFP2 would be defined by 17 atoms as in 2,2-dimethylpropane. With the ECFP4 filtering, we take also into account the chemical nature of the second neighbours and their bonds. Theoretically, it can include implicitly up to 53 atoms. In the whitelists, the ECFP4 max radius has been considered to ensure a short and medium range filtering. However, as it will appear soon in the results, the connectivity features keep the information of atoms being in a cycle but not its size. The evolutionary algorithm can therefore propose highly constrained unsaturated cycles while respecting this filtering. The use of ECPF6 might have limited the use of a whitelist based on cyclic properties, but ECFP6 includes explicitly the atoms up to three of the central atom. This descriptor is therefore very specific. For small molecules, filtering by ECFP6 is almost equivalent to having a list of allowed SMILES.
In order to further refine our realistic chemical space, a second filtering based on generic cyclic features (denoted GCF) is proposed. The process is illustrated on the ribavirin molecule in Fig. 4. Using the NetworkX python library,35 the vertices (bonds) that do not belong to a cycle are deleted (Fig. 4a). RDKit is then used to compute the Murcko scaffold on each remaining subgraph that includes a cycle (Fig. 4b). At this point, the cyclic feature contains information on the bond and atom types. In order to work with more generic cyclic features, all atoms with a coordination number of 4 or less are converted to carbon atoms. Since hypervalent carbon produces RDKit errors, hypervalent atoms are left unchanged. Contrary to the connectivity features that include all atom types and are based on the whole datasets, the GCF lists have been built on the CNOFS chemical subspace of ChEMBL (GCF1) or ChEMBL and ZINC (GCF2). Therefore, the only hypervalent case corresponds to sulphur with a coordination of 5 or 6. To avoid unstable fused small cycles with double and triple bonds, we have decided to keep the bond type information (Fig. 4c). During this procedure, SMILES writing cleaning steps are performed to produce clean cycles. The GCF generation program and lists are available on GitHub at the following address https://github.com/BenoitDamota/gcf.
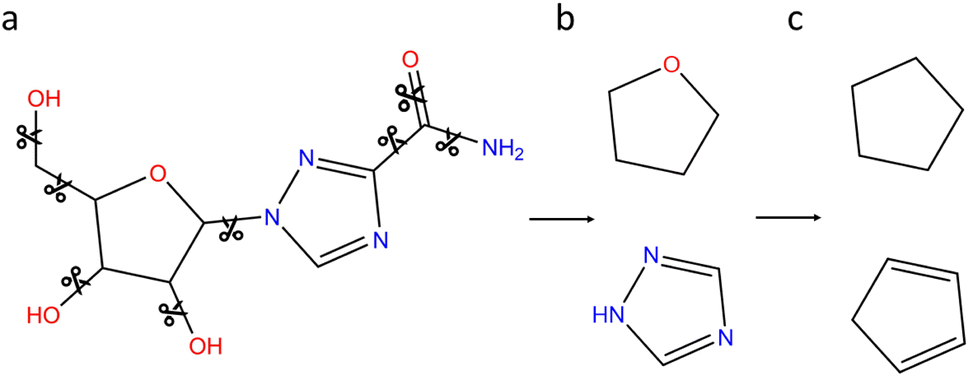 | ||
| Fig. 4 Generation of the generic cyclic features (GCF) of ribavirin. (a) Acyclic bonds are deleted. (b) Murcko scaffolds of remaining subgraphs. (c) Generic cyclic features. | ||
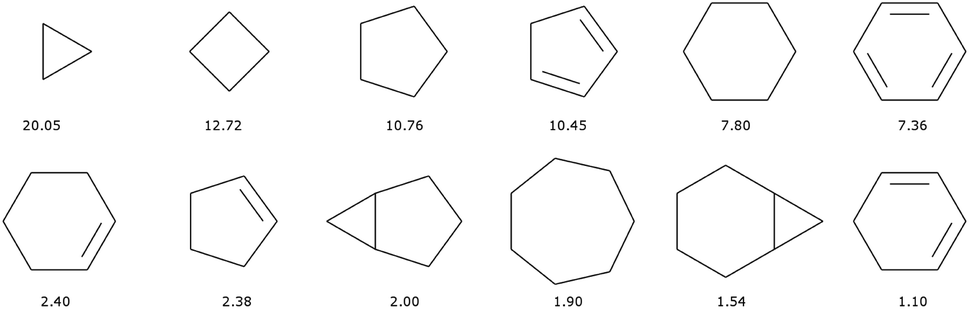
There are 11013 generic cyclic features in the ChEMBL subset that is formed by only H, C, N, O, F, and S atoms. Only eleven of those represent more than 1% of the total number of cycles, see Fig. 5. In fact, the distribution is very uneven with the 6-membered aromatic ring accounting for more than 44% of the ring features, followed by the 5-membered rings (10%) that are probably heteroaromatic. The top trio is completed by a 6-membered ring without unsaturation (10%). Although our method of generation is somewhat different we find similar results to a previous analysis of CAS scaffolds.36 We can also note that the azobenzene, thioindigo, and tetrathaifulvalene of Fig. 2 and 3 are composed of common GCF in ChEMBL. If we consider the ChEMBL and ZINC subsets that are formed by only H, C, N, O, F, and S atoms, the total number of generic cyclic features is 15431.
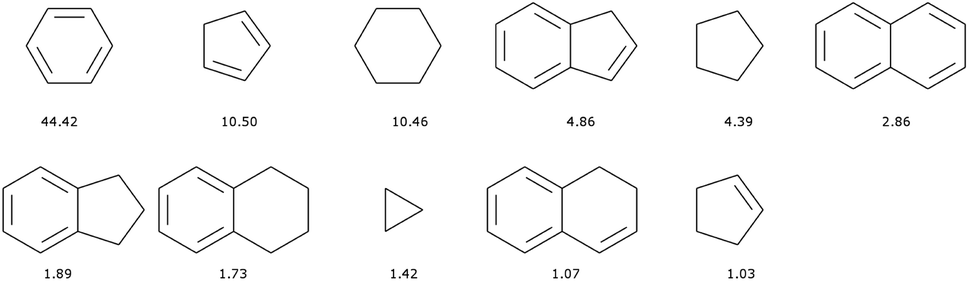 | ||
| Fig. 5 Most common (over 1%) generic cyclic features (GCF) in the ChEMBL dataset. Heteroatoms have been converted to C. Legend: percentage of occurrence of the cyclic features over all cyclic features. | ||
Preliminary tests comparing connectivity features used as filters or in the objective function showed that including even a small proportion of novel chemical environments greatly increased the chemical search space. Moreover, it only takes one unstable chemical environment to drastically change the synthetic accessibility of the entire molecule. Therefore, in this article, the proportion of known connectivity features in the generated molecules is set as a strict conservative filter and equal to 100%. That means that after mutation of the molecular graph, any solution including at least one unknown connectivity feature is discarded before evaluation. So this filtering acts as a chemical space limiter. The cost of calculating the scores of these whitelists was estimated on a simple laptop computer with a sample of 100000 randomly selected molecules. The evaluation of the ECFP features is around 0.33 ms per molecule. The evaluation of the cyclic features is higher, around 3.3 ms per molecule due to the operations on the graph.
1.3 Molecule generation
The use of whitelists based on connectivity and cycle properties is not limited to a single method of molecule generation. The available chemical space is however dependent on this method. We used here EvoMol, an evolutionary algorithm based on RDKit graph objects.21 EvoMol is available at https://github.com/jules-leguy/EvoMol. In EvoMol, hydrogen atoms are treated implicitly and the bond orders are integers. The valence of the atoms is used as a reference to place the hydrogen atoms. The generator does not generate radicals and charged atoms. Ions and zwitterions have therefore been left out of the datasets during the chemical space comparison part. In this representation, the nitro function is considered as a zwitterion. Moreover, EvoMol does not take into account stereochemistry.The actions on the molecular graphs in EvoMol are mainly atom-centred. The list includes append atom, remove atom, change bond, substitute atom type, insert carbon, cut atom, and move group. EvoMol is very flexible and has shown very good performances in optimisation and in chemical diversity generation.21,29 This flexibility of actions can bring the generator to places of the chemical space that seem to be unrealistic or at least not desired according to the problem definition.
In this article, for all objectives we have set the initial population to only a methane molecule in order to start without any prior knowledge and test the chemical space exploration ability under constraints. The action space of EvoMol is also set with a limit on the heavy atoms count (Z > 1) and with the list of allowed chemical elements. For the first objective of the QED optimisation, the search space is set to contain molecules with up to 38 heavy atoms among C, N, O, F, P, S, Cl, and Br. The population size is set to 1000 and the optimisation is run for 1000 steps. At each optimisation step, 10 individuals are replaced. One mutation consists in applying up to two operations on the molecular graph. For the rediscovery objective of the reference datasets, the limit on the HAC has been set to 9 (for QM9 ∪ PC9) and then 10 heavy atoms for the other reference datasets. H, C, N, O, F, and S atoms form the chemical elements list of our chemical space search. At each optimisation step, 10 random individuals are replaced.
1.4 DFT computational details
For the evaluation of molecular electronic properties, it is important to note here that EvoMol operates on molecular graphs but the interface with the ab initio calculation is done using a SMILES representation. From the SMILES, the three-dimensional coordinates are generated by Openbabel (version 3.1) and then optimised by RDkit (version 2021.09.4) with the MMFF94 force field.37–39 For each molecule, the initial MMFF94 geometry is optimised by deactivating the symmetry. Then a density functional theory (DFT) method was chosen for the evaluation of the electronic properties. All DFT calculations were performed with the Gaussian09 software.40 The hybrid functional B3LYP was chosen.41 In order to limit the computational cost, the 3-21G* Pople-type basis set was used.2 Results
2.1 QED optimisation: the importance of cyclic features
As a first qualitative experiment, we propose to assess visually the impact of the connectivity feature filtering on the optimisation of the QED with EvoMol. This multiobjective rewards the presence of some hydrogen bond donors and acceptors, some aromatic cycles, and a medium logP. The best solutions obtained without any filtering are reported in Fig. 6. The RDkit detection of an aromatic ring according to Hückel's rule was exploited by EvoMol to form small heteroatomic fused cycles, where the non-bonding doublets are considered conjugated. In addition, those cycles allow for a better score with hydrogen bond donors and acceptors on the same ring at the same time.
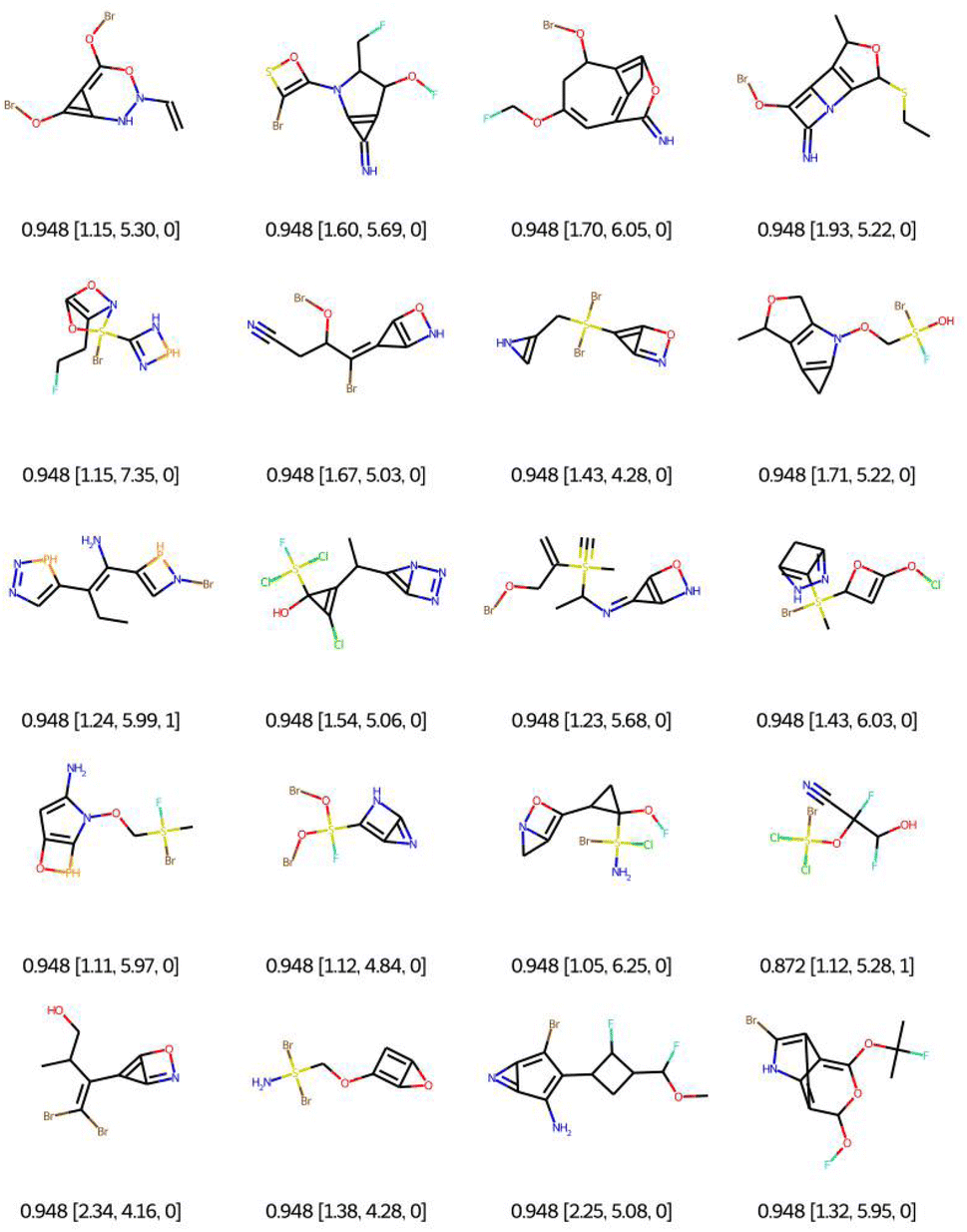 | ||
| Fig. 6 Bests of 20 QED optimisations without any filtering. Legend: QED [CLScore, SAScore, and GCFscore]. | ||
When using filter 1 (the connectivity features of ChEMBL) during optimisation, EvoMol still manages to get top scores, see Fig. 7. Visually the obtained solutions are overall much more pleasant. The combinations of hetero-elements seem more reasonable. However, we observe that there is some assembly of cycles of very different sizes that seem peculiar. This is due to the fact that the ECFP does not encode the cycle size information. For example, in the first row, there is a molecule with a 5-membered ring fused with a 3-membered ring which includes a double bond. This combination is highly constrained and probably not very stable. In order to fill this gap in the connectivity features, we propose to add a second filtering, for the moment a posteriori, based on the cyclic features present in ChEMBL for GCF1. That means that a generic cyclic feature score (GCFscore) will be 1 only if all contained GCF exist in ChEMBL. Keep in mind that this whitelist of cyclic features was set to contain information concerning double and triple bonds since there are saturated fused rings of many kinds. On the 20 experiments, only 9 pass the cyclic feature filtering with a score of 1, highlighted in red. We can observe three different molecules with a top score of 0.948. Interestingly, a derivative of commercially available scaffolds has been found twice, the thieno[3,2-d]pyrimidine-2,4(1H,3H)-dione, PubChem CID number 7059273. This result suggests the relevance of this approach.
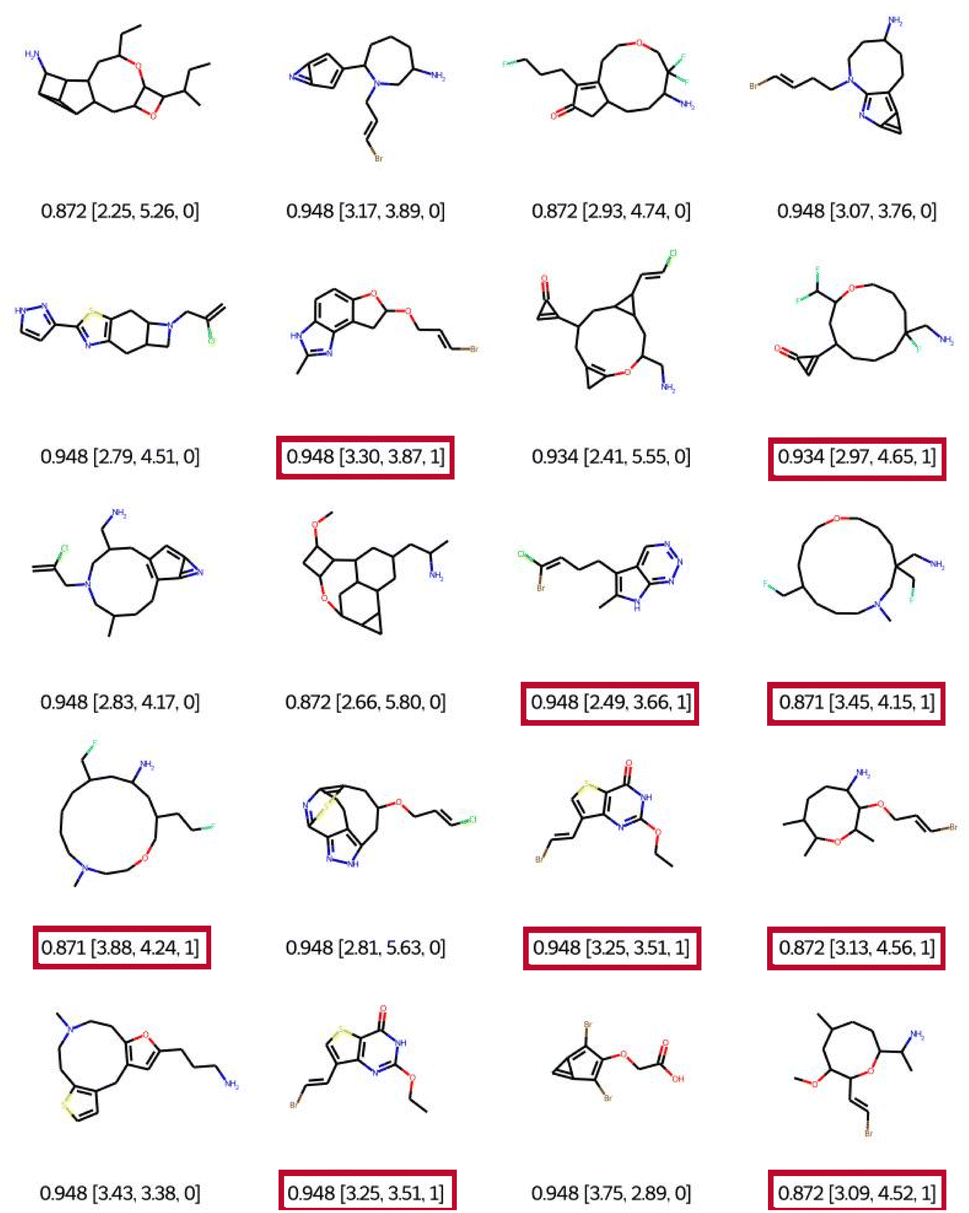 | ||
| Fig. 7 Best of 20 QED optimisations with ChEMBL connectivity feature filtering (filter 1). Legend: QED [CLScore, SAScore, and GCFscore]. | ||
In the 9 solutions that pass the filter of cyclic features, we can also notice the presence of heterocycles of sizes 8, 11, and 14. Indeed, we find in the ChEMBL database heterocyclic derivatives of large sizes such as oxacyclooctane (PubChem CID 12677196, CHEMBL148748) and oxacycloundecane (PubChem CID 20080726). The compound 1-oxa-4-azacyclooctane-3,8-dione is commercial (PubChem CID 55299436). The advantage of the filtering method presented here is that it is possible to trace the connectivity or cycle properties of the proposed solutions and then find these properties in the reference datasets. Therefore we can consider these solutions as realistic. However, this chemistry appears to be quite specific. Depending on the chemistry developed by a given laboratory, it is quite easy to adapt the cyclic property whitelist by eliminating specific cycle sizes.
2.2 Chemical space exploration under constraints
The connectivity feature filtering can be a promising approach to define a realistic chemical space if the exploration is not too much hampered. Considering the quite complex solutions obtained in the QED optimisations, one can expect an atom-centred evolutionary algorithm like EvoMol to always be able to jump from a whitelisted connectivity feature up to another if enough actions are allowed. Therefore, we have tested the exploration capacity of the generator under the constraint of connectivity feature filtering.We start with the objective of rediscovering a set of molecules. The objective set corresponds to the union of QM9 (ref. 27) and PC9 (ref. 28 and 31) as defined in the OD9 publication.29 These two sets contain molecules ranging from HAC 1 up to 9 that can be either carbon, nitrogen, oxygen, or fluorine. QM9 is a subset of molecules of the GDB enumerations.30 QM9 was built from a constrained combinatorial approach while PC9 is a subset taken from PubChem data with the same number of heavy atom limits and chemical element constraints. Once the radicals, the stereochemical information, and the duplicates are removed, the union of these two sets contains 190300 different molecules. Depending on the filter used, it can be seen from Table 1 that the number of molecules that contain only known connectivity features in ChEMBL25 (column passing filter 1) or even the ChEMBL25 and ZINC20 (column passing filter 2) is a minority of this set of molecules. The impact of adding the ZINC connectivity features is evident. For other combinatorial reference datasets like GDBChEMBL and GDB11, the filtering is also quite drastic removing at least 84% of the molecules while accepting all the ChEMBL25 and ZINC20 connectivity features. Using filter 1 instead of filter 2 results in the loss of almost half of the molecules that were left. Applying filter 1 (ChEMBL) to ZINC has a noticeable but comparatively smaller effect. 64% of ZINC correspond to molecules that only possess connectivity features present in ChEMBL. That leaves more than 100000000 unique neutral molecules with just only four types of heavy atoms.
It is important to make it clear here that we are not claiming that all molecules that do not pass the filters are unrealistic. We have opted for a conservative approach, excluding any unknown environment in order to see the impact of severe filtering on the search space. Similarly, we do not claim that all the molecules that pass the filters are synthesisable. But as they only present known chemical environments up to two bonds, they can be considered realistic, especially after the second filter of the cyclic features.
Starting from only a methane molecule, the generator randomly mutates the molecules of its population and if the solution belongs to the QM9 ∪ PC9 dataset, the score for this solution is 1 and if not, it is 0. It is thus a pure enumeration study based on random actions on randomly chosen individuals. The important thing here is to check whether the limitation to certain connectivity features as defined in the ECFP2 and ECFP4 still allows navigation in chemical space. For each heavy atom limit and each filter, ten experiments were performed. We also tested the impact of the number of actions on the molecular graphs for each mutation. As an example, for a depth of four actions, the generator can chain up to four operations such as adding or removing an atom (see the Methodology section) before filtering. We report the results of this experiment in the rest of this paragraph. For molecules up to HAC 4 in size and both filters, the generator is able to systematically find all the molecules by performing only one action each time. At HAC 5, the limit must be increased to two actions, and then to 3 actions at up to HAC 9. Allowing for more actions is therefore mandatory for a few cases, but it increases also the overall enumeration cost. During this task, we found that all actions defined in EvoMol have been used. On average across all these experiments, the most important actions are the two building actions append atom (34%) and insert carbon (30%), followed by change bond (25%), substitute atom type (23%) and move group (14%). Finally, the two destructive actions are for this task the least used with remove atom (9%) and cut atom (6%).
In view of the generation flexibility of EvoMol, we took the opportunity to also test the impact of adding a new chemical element, sulphur, onto the search space. In Table 1, the numbers of unique molecules in the reference datasets and in the enumerations are reported depending on the heavy atoms allowed and the filter. Even considering filter 2 which presents more connectivity features, the expansion induced by the addition of the sulphur atom is not overwhelming. If we look at the numbers in ChEMBL and ZINC, we can expect that adding sulphur represents a size increase of around 45% of the chemical space. If this increase seems important, it can be put in perspective with the huge combinatorial that exists without filters. An enumeration by EvoMol without constraints other than the valence rules managed to generate 5433 different SMILES with C, N, O, and F having HAC 5 or less. Adding sulphur allows us to generate 139689 SMILES with the same size limit. So, filters on connectivity features have a clear and major impact on the definition of the search space and strongly limit exotic combinations.
Rather confident in our ability to enumerate a realistic chemical space of reasonable size, we transformed the objective of random generation into a systematic enumeration of all neighbouring solutions of an initial dataset. With a limit of HAC of 10 and a maximum of 2 actions on the molecular graph, it takes a few days of computation to go over the 600 thousand or so molecules already generated. However, increasing the number of actions to 3 multiplies the combinatorial by several orders of magnitude and makes this approach unreasonable even for datasets of small molecules. All the molecules generated under constraints (either from filter 1 or filter 2) with the two lists of atoms considered, CNOF and CNOFS, were gathered in a single set called Evo10. We propose here to take advantage of this rather large enumeration of realistic chemical spaces associated with the connectivity properties of ChEMBL and ZINC and to compare it to the same size molecules of reference datasets listed in Table 1.
The cumulative evolution of the number of SMILES as a function of the HAC is plotted in Fig. 8 for the CNOF search space. Similar tendencies are observed for the CNOFS search space. We have also added the enumeration tests without filters (denoted as Evo10 no filter) and it can be seen that thanks to the filters the chemical space is several orders of magnitude smaller. The Evo10 dataset is larger than the filtered GDB11 which has the most molecules passing the filters. It can also be seen that QM9 ∪ PC9 contains only a small part of what is possible with realistic filters. Despite the impressive size of ZINC, it contains only a very small part of the chemical space defined by these connectivity features. A logarithmic regression based on Evo10 (filter 2 ∪ GCF2) allows us to estimate a size of 5 × 109 molecules with CNOFS and a HAC limit of 17 and several 1022 molecules with CNOFS and a HAC limit of 40 still excluding radicals, stereoisomers, ions, and zwitterions.
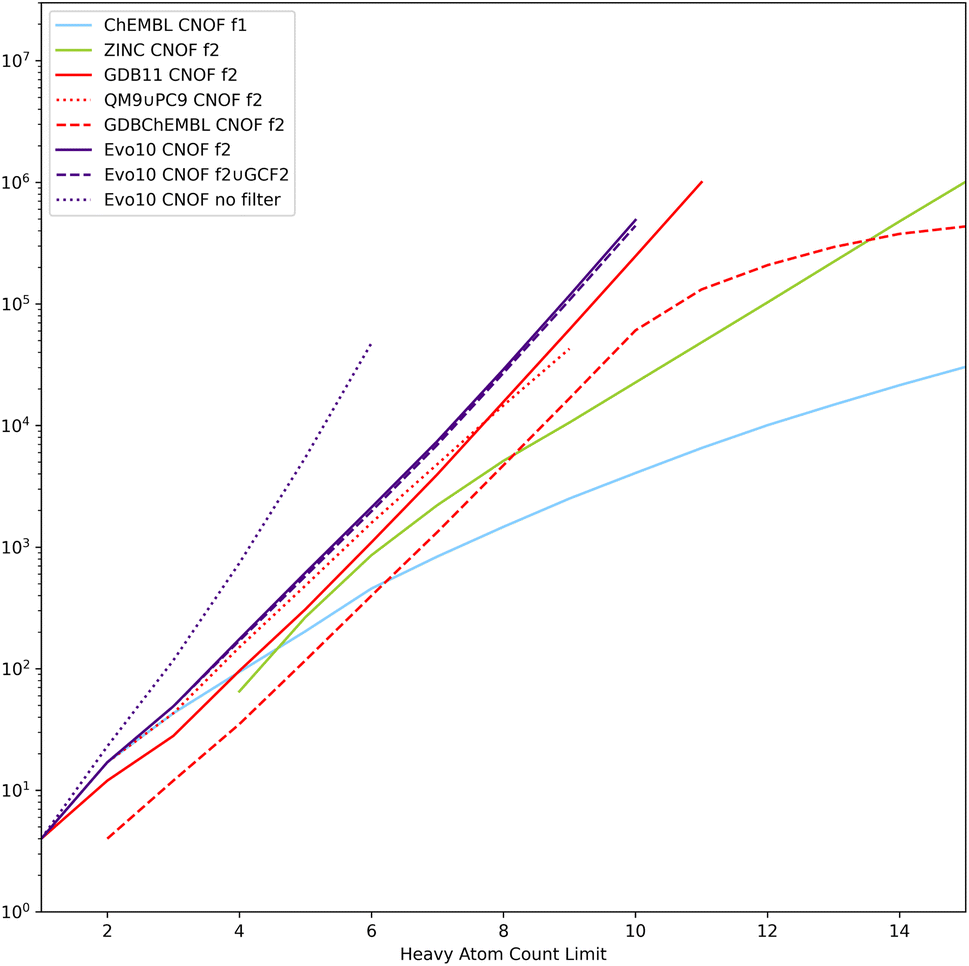 | ||
| Fig. 8 Cumulative plots in the log scale of the number of SMILES by the HAC limit in the filtered reference datasets and enumerated by EvoMol. f1 and f2 correspond to the number of SMILES passing the connectivity filters 1 and 2. | ||
With the connectivity filters, we managed to enumerate more compounds respecting the chemistry of either ChEMBL or ChEMBL plus ZINC. Yet, one could ask if we managed to rediscover every molecule up to HAC 10 present in these reference datasets. In Table 2, the few SMILES that were not found have been reported. Considering the molecules of the reference datasets that pass filter 2 in the CNOFS search space, EvoMol managed to recover all molecules of GDBChEMBL and GDB11. Only 8 molecules have not been found with two actions mutations. Two belong to ChEMBL and 6 to ZINC. The reported SMILES present a large number of heteroelements and correspond probably to connectivity features that are quite isolated. If we enumerate the neighbours at three actions of the missing SMILES, we find that only one of them cannot be mutated to another Evo10 molecule with 3 actions. This peculiar bicycle is commercially available on demand. In the end, it is the only molecule of the filtered reference datasets that we would not find with a mutation depth set at 3 actions. Searching for its neighbours at 4 actions on the molecular graph leads to 17 neighbours present in Evo10. However, just this search took several hours. It is fair to say that there are probably only very few chemical compounds isolated at more than 3 actions away from the rest of the filtered chemical space. Furthermore, for specific problems, it might be relevant to select an initial population that is fitted for the task.
| SMILE | Dataset | Neighbours |
|---|---|---|
CC1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NOC(C)(O)C1NO NOC(C)(O)C1NO |
ChEMBL | 103 |
| ONN(O)S(O)(O)O |
ChEMBL | 20 |
| CN1ON(C)ON(C)O1 | QM9 ∪ PC9, ZINC | 4 |
| c1coc2occoc2o1 |
ZINC | 65 |
| CSC1NNC(SC)NN1 |
ZINC | 10 |
| Sc1[nH]ssc2nnc12 |
ZINC | 8 |
| N1NC(C2NNNN2)NN1 |
ZINC | 1 |
| N1SNC2C1NSN2 |
ZINC | 0 |
Having demonstrated that filters reduce the search space but do not visibly create unreachable compounds, it would be interesting to study the chemical diversity associated with these filters. We have already shown in the past that the chemistry of QM9 and PC9 is somewhat different and that some chemical functions are missing in QM9.28 This is a problem for the generalizability of machine learning methods using QM9 as a training set. However, QM9, GDB11, and GDBChEMBL by their combinatorial construction present a more important diversity in specific associations like the combinations of alkenes with other functions. We can compare the chemical environments that exist in datasets with those that pass the filters. We can consider here that the number of connectivity features as defined in the ECFP2 is a measure of the small range of chemical environment diversity. The ECFP2 takes into account the central atom and its first neighbours and their bond types. We have chosen the ECFP2 since the ECFP4 can include up to 53 atoms and can be very specific. In Table 3, the number of connectivity features before and after filtering is reported. There are initially between 7 and 10 thousand different connectivity features as defined in the ECFP2. Most of them in QM9 ∪ PC9, GDBChEMBL, and GBD11 do not belong to ChEMBL and or ZINC. Thanks to the EvoMol enumeration, we can also see in Table 3 that small sizes datasets cannot reproduce the full range of possible coordinations even at the scale of the ECFP2 since we just managed to generate half of the connectivity features of ChEMBL.
| Dataset | Atoms | Total | Passing filter 1 | Passing filter 2 |
|---|---|---|---|---|
| QM9 ∪ PC9 | CNOF | 7403 | 2247 | 3019 |
| ChEMBL | CNOF | 7174 | 7174 | 7174 |
| ChEMBL | CNOFS | 9947 | 9947 | 9947 |
| GDBChEMBL | CNOF | 7756 | 2341 | 2834 |
| GDBChEMBL | CNOFS | 9247 | 2611 | 3195 |
| GDB11 | CNOF | 8870 | 3077 | 3761 |
| ZINC | CNOF | 6960 | 4138 | 6960 |
| ZINC | CNOFS | 10041 |
5528 | 10041 |
| Evo10 | CNOF | 4648 | 3527 | 4648 |
| Evo10 | CNOFS | 6375 | 4618 | 6375 |
Let us now discuss the impact on the chemical diversity of a posteriori filtering by cyclic features. If we look at the most common cyclic features of the QM9 dataset, see Fig. 9, we can immediately notice a very different distribution from those of ChEMBL where the phenyl group was predominant (see Fig. 5). Table 1 shows that in terms of the number of molecules, filtering removes on average around 10% of the molecules passing the filters. However, if we look at the number of cyclic features in each of the datasets before and after the filters, see Table 4, we can see a drastic reduction in the number of cycles (column fx compared to column fx ∪ GCFx). The 10% or so of molecules removed are associated with the majority of the GCF. The qualitative results on the QED indicated that it was easy for an evolutionary algorithm to construct exotic tangles of nested cycles while respecting the connectivity filters. Again, the correct definition of extensive whitelists can be discussed. But it should be noted that GCF2 includes all the cyclic features of hundreds of millions of known compounds. In total, we have counted 15431 cyclic features. Two-thirds of them concern more than 17 atoms. Small molecule datasets will not be able to represent the topological diversity of ChEMBL and ZINC.
 | ||
| Fig. 9 Most common (over 1%) cyclic features in the QM9 ∪ PC9 dataset. Heteroatoms have been converted to C. Legend: percentage of occurrence of the cyclic features over all cyclic features. | ||
| Dataset | Atoms | No filters | f1 | f1 ∪ GCF1 | Theoretical max 1 | f2 | f2 ∪ GCF2 | Theoretical max 2 |
|---|---|---|---|---|---|---|---|---|
| QM9 ∪ PC9 | CNOF | 858 | 372 | 162 | 248 | 513 | 280 | 393 |
| ChEMBL | CNOFS | 11013 |
11013 |
11013 |
11013 |
11013 |
11013 |
15431 |
| GDBChEMBL | CNOFS | 32268 |
6110 | 1070 | 5255 | 8806 | 1640 | 7493 |
| GDB11 | CNOF | 6663 | 1875 | 469 | 752 | 2636 | 743 | 1168 |
| ZINC | CNOFS | 7685 | — | — | 11013 |
7685 | 7685 | 7685 |
| Evo10 | CNOFS | — | 3063 | 320 | 456 | 4961 | 582 | 718 |
To conclude the study of the chemical diversity passing the filters, we have calculated the SAscore, CLscore, and RAscore for all molecules of the Evo10 dataset according to the chosen filter. In Fig. 10, the comparative distributions between these two scores for the filters allowing the most compounds to pass, i.e. filter 2 and GCF2, show a concentrated area around 2.5 to 4.5 in the SAscore and 2.5 and 4 in the CLscore. A low SAscore should denote better synthesisability and in the SYBA article, the authors propose a threshold value of 4.4 in the SAscore above which the molecule would be too complex to be synthesised.13,23 According to these approaches, the vast majority of Evo10 could become reality.
 | ||
| Fig. 10 Compared distributions of the SAscore and CLscore of all molecules of Evo10 passing the connectivity and cyclic feature filters of ChEMBL and ZINC (filter 2 and GCF2). | ||
In fact, these distributions resemble those of ChEMBL and ZINC.22 Evo10 only contains connectivity and cyclic features existing in these datasets while the two scores favour the most popular environments. Thus, there is a trail of points with a high CLscore and a low SAscore as expected. A past optimisation of these scores showed us that this corresponds to simple molecules consisting only of alkyl and aryl environments with very few cycles.21
The RAscore is a score between 0 and 1 based on a neural network designed as a pre-screening tool to avoid a retrosynthetic analysis of all generated compounds.17 Values near 1 should indicate a compound possessing a synthetic route. The RAscore proportions for all compounds of Evo10 are reported in Table 5. The vast majority of the compounds in this dataset have a score greater than or equal to 0.99. The amount of molecules that the classifier considers without any synthetic route is very low, below 1%. To be fair, it should be noted here that the descriptors used to define the RAscore in the neural network are ECFP6 and that it was trained on a portion of ChEMBL. The two approaches are different but based on similar descriptors and the reference dataset. It is therefore not very surprising that the RAscore rates favourably a large proportion of Evo10.
| RAscore value | Passing f1 ∪ GCF1 | Passing f2 ∪ GCF2 |
|---|---|---|
| 1.00 | 19.73% | 16.09% |
| ≥0.99 | 80.57% | 75.90% |
| ≥0.90 | 89.03% | 85.96% |
| ≥0.50 | 97.01% | 95.94% |
| ≤0.10 | 0.53% | 0.77% |
2.3 Molecular electronic property optimisation
In our previous publications, we have discussed the objective of the optimisation of molecular electronic properties like frontier molecular orbital energies.21 The energy of the last occupied level (HOMO) or the first empty level (LUMO) is an essential characteristic for the design of components in organic electronics for example. Thus, the optimisation of the HOMO energy is linked to the design of electron donor molecules when the LUMO is associated with acceptors. In a real application, it will rather be envisaged to optimise these levels in an energy range dictated by the other constituents of the device (other molecules and electrode potentials). The interest here is to maximise the energy of the HOMO and minimise that of the LUMO in order to approach the frontier of stable chemistry. A very good donor or a very good acceptor would probably be quite unstable and so this problem pushes the evolutionary algorithm to produce unrealistic molecules. Thus, our last objective is to study the impact of connectivity and cycle filters on the HOMO energy maximisation and LUMO energy minimisation problems.All the compounds listed by EvoMol (Evo10) with a number of heavy atoms lower than or equal to 8 and respecting the connectivity filters were optimised in DFT (see computational details). Out of 38013 molecules, only 569 generate an error in molecular mechanics. This may be due to the non-existence of the HF bond in the force field, to the fact that the molecule is not a singlet (O2), but for many other cases, it is due to the tension between the rings. Indeed, only 25 of those 569 molecules pass the GCF filter based on ChEMBL, and 164 pass the GCF filter based on ChEMBL and ZINC. In addition, 73 molecules have failed or diverged geometric DFT optimisation and 1335 molecules have a different topology after optimisation. To compare the molecular topologies, we have used the first layer of the InchI. It is a more robust method than using SMILES due to the conjugated unsaturated bonds that correspond to different SMILES for equivalent topologies. Of those 1335 molecules, only 115 and 148 pass, respectively, the GCF filters of ChEMBL or ChEMBL and ZINC. Thus, thanks to the connectivity and cyclic feature filtering it is possible to define chemical spaces where less than 1% of the generated molecules could not be evaluated in DFT. This is a much lower proportion than that observed during the BOINC computational campaign associated with the generation of diversity without filters, where the proportion of failures was rather 66%.29
In Fig. 11, the structural formulae of the five molecules, passing filter 1, with the highest possible HOMO energies are plotted as a function of the number of heavy CNOF atoms ranging from 1 to 8. This figure is identical when we use filter 2. It can be seen that nitrogen is the best donor group and therefore the amino derivatives dominate the list. Anti-aromatic compounds such as cyclobutadiene are also found. In fact, a previous optimisation resulted in a top score for tetraaminocyclobutadiene.42 This configuration is excluded by the filters, and the best solution with 8 heavy atoms becomes the derivative with four alcohol functions. The application of the cyclic filters eliminates only one compound in this figure, which is the double square fused with the two amine functions. We will see that the impact is much greater in the case of the LUMO.
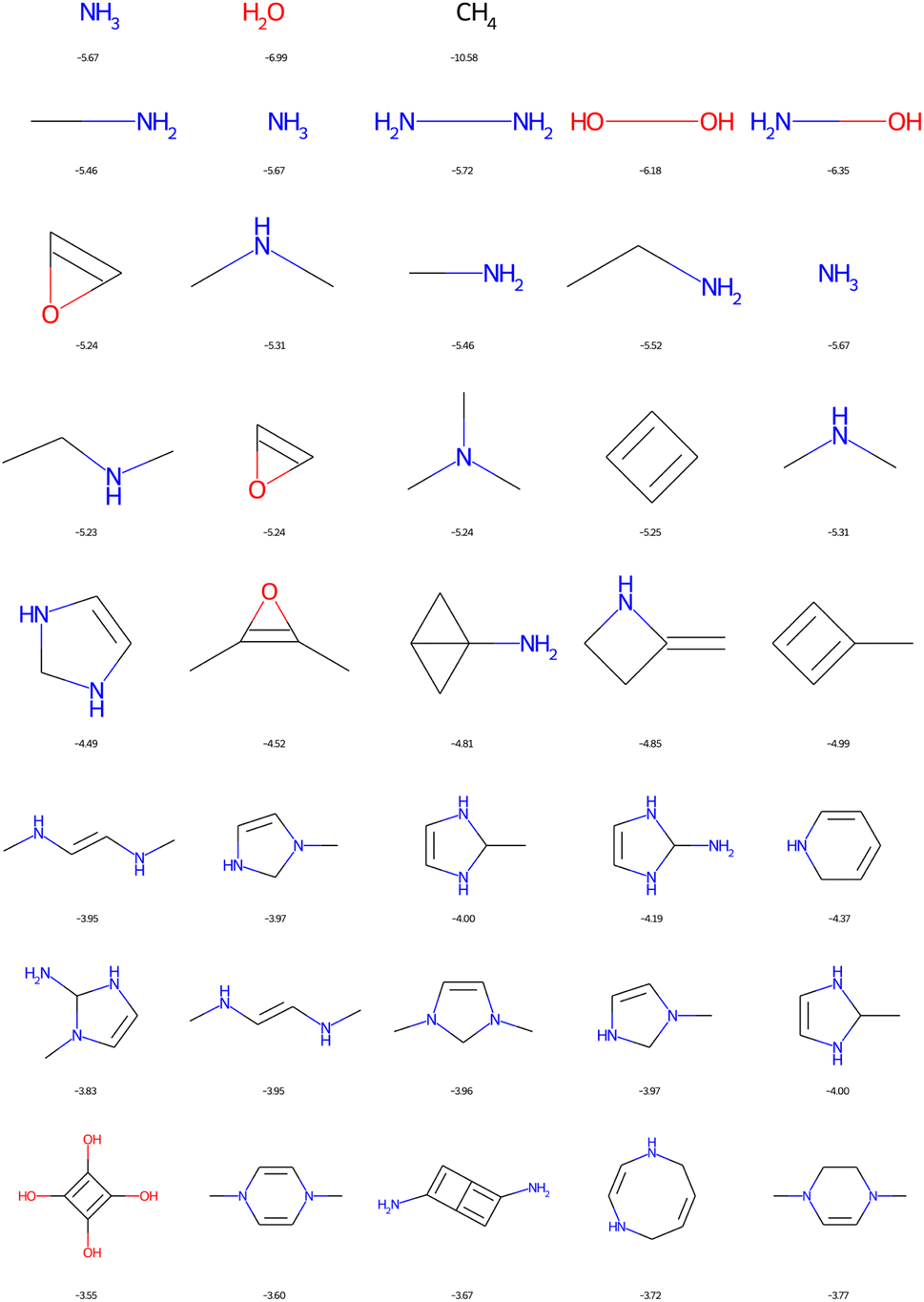 | ||
| Fig. 11 Top 5 enumerated molecules with the highest HOMO energy (in eV), respecting filter 1, depending on the CNOF HAC (from 1 up to 8 down). | ||
The dominant chemical functions in molecules with the lowest LUMO are the carbonyl, nitrile and nitric acid derivatives, see Fig. 12. It is important here to recall that EvoMol does not currently handle cases with formal charges, which unfortunately excludes nitro compounds. From HAC 4 onwards, particularly unstable unsaturated rings appear, such as cyclobutyne, or unsaturated derivatives of prisman. The optimisation of the LUMO energy is thus biased by the lack of information on the ring size in ECFPs. SAscores, CLscores, and RAscores were calculated for the molecules in Fig. 12. It can be noted that none of these three scores is able to quickly rule out the set of molecules with constrained topology. The SAscore does not penalise small rings. The CLscore presents values lower than 3.3 for known and realistic molecules. The RAscore which could have been used as a discriminator in principle would eliminate only one compound (having an RAscore of 0.10). The RAscore allows the same constrained molecules as our ECFP filtering even if it is based on ECFP6. Therefore, the contribution of the filtering by the cyclic features appears to be crucial. After filtering by GCF1, all these fused rings disappear, leaving only realistic molecules, see Fig. 13. The smallest LUMO molecule of HAC 6 to 8 would be mesoxalonitrile. This is followed by tetrazine, pyrimidine, and carbonyl derivatives.
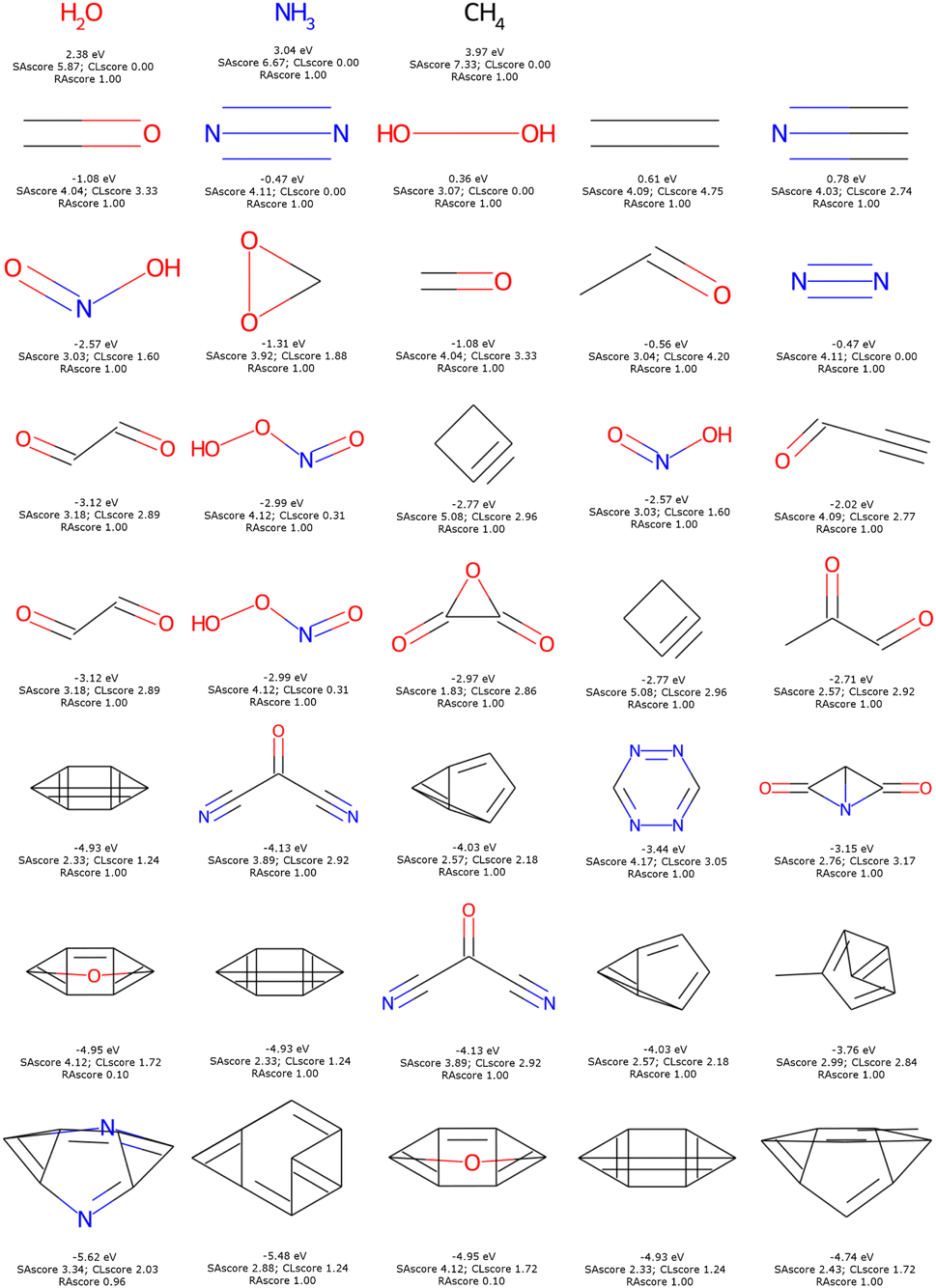 | ||
| Fig. 12 Top 5 enumerated molecules with the lowest LUMO energy (in eV), respecting filter 1, depending on the CNOF HAC (from 1 up to 8 down) with their corresponding energy, SAscore, CLscore, and RAscore. | ||
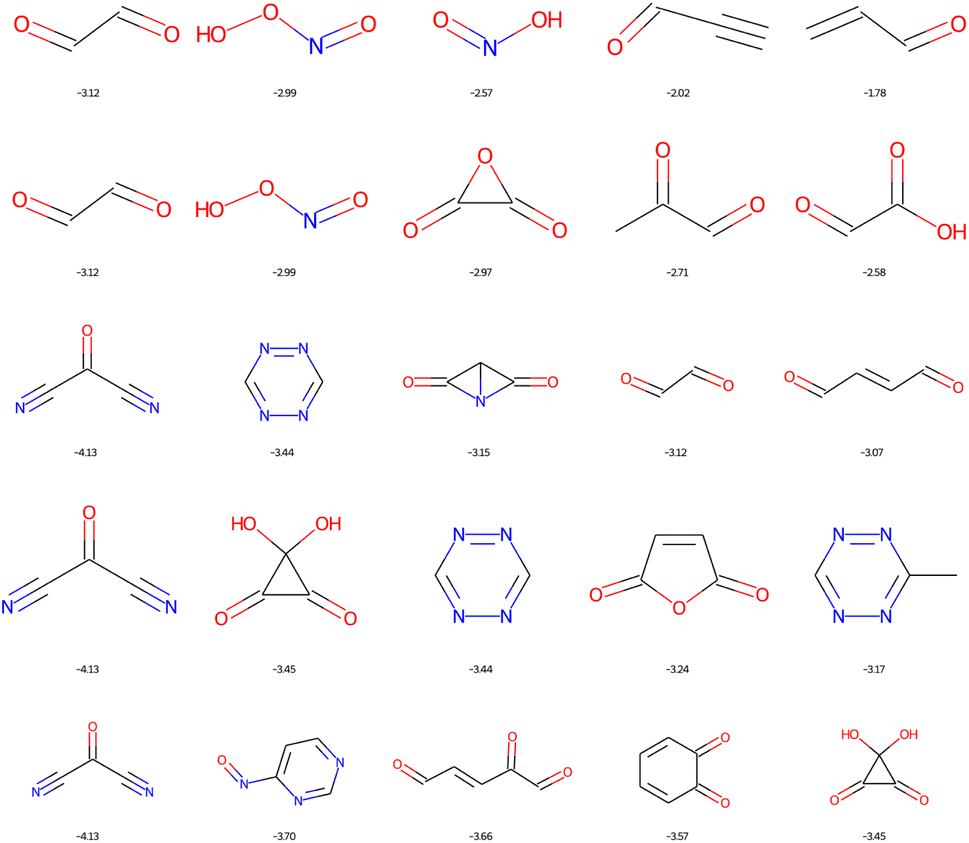 | ||
| Fig. 13 Top 5 enumerated molecules with the lowest LUMO energy (in eV), respecting ECFP filter 1 and the GCF1, depending on the CNOF HAC (from 4 up to 8 down). | ||
3 Conclusions
When we consider several types of heavy atoms, such as the simple set of C, N, O, F, and S, a phenomenal amount of combinations are then possible while respecting the valence rules. Many of these combinations contain very exotic associations. It is fairly accepted that scores can be used to select more realistic molecules and some of these scores are based on connectivity descriptors such as ECFP4. We present in this work new descriptors, called generic cyclic features (GCF), to complete the local information of the ECFP4 with the cyclic substructures of a molecule. For both ECFP4 and GCF, we have chosen to limit the possible combinations to those that exist in two datasets based on real molecules, ChEMBL and ZINC. These two full datasets encompass 1156416 connectivity features (based on ECFP4). And on a filtered subset (see the Methodology section) composed of 288000000 molecules of the ChEMBL and ZINC, 15431 cyclic features were extracted. With these whitelists, it is possible to evaluate the amount of features of a molecule belonging to ChEMBL, or ChEMBL and ZINC. It is not a matter of assessing whether on average a molecule resembles the most common in these databases, but whether each piece that constitutes it exists, even rarely. This is how we have defined a realistic molecule.
We have implemented this approach in our evolutionary generator EvoMol, but we believe that all generation methods can benefit from these whitelists. The chemical space of realistic molecules is much smaller than the set of possible combinations. It still contains an estimated amount of more than 1022 molecules with at least 40 heavy atoms. We were able to enumerate starting from methane more than 676000 molecules having up to 10 heavy atoms of the set C, N, O, F, and S, and only having connectivities and cyclic features known in ChEMBL and ZINC. We were able to navigate through the chemical space of realistic molecules and indeed rediscover all molecules passing these same filters from the reference datasets which are here ChEMBL, ZINC, QM9, PC9, GDB11, and GDBChEMBL. It cannot be said that all synthesizable molecules are represented in this set. This is a common limit of whitelist-based filtering. The list needs to be updated if features that exist in reality happen to have been overlooked. However, our work is based on a very large sample of the known chemical space.
It is especially the visualisation of the proposed solutions after filtering that convinces us of the relevance of this approach. The comparison between free optimisation and optimisation under connectivity constraints shows the obvious impact on the chemistry of the heteroelements. However, the filter based on connectivity features still allows for the generation of nested small cycles with unsaturations that are probably very unstable. A filtering of the cyclic features further limits the proposed solutions to visually very interesting molecules. Optimisations of electronic properties of molecules (HOMO and LUMO energies) confirm the impact and interest of these two filters.
This realistic chemical space is a major breakthrough for de novo generation of optimised molecules in domains where prior knowledge is too limited for robust deep learning models or restricted to too few examples and therefore requires an exploratory approach. In such cases, an evolutionary generator optimisation can be a good approach now that it can be restricted to realistic solutions.
Data availability
The code for the molecular generator EvoMol can be found at https://github.com/jules-leguy/EvoMol. The version of the code employed for this study is version 1.4.1. The code for the general cyclic feature scoring program GCF can be found at https://github.com/BenoitDamota/gcf. Data as JSON dictionaries for this paper are available as a collection in figshare at https://doi.org/10.6084/m9.figshare.c.6041117.Author contributions
Methodology, investigation, software, and writing (all). Conceptualization, data curation, visualization, and supervision (TC and BDM).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Angers, and the French Ministry of Education and Research (JL PhD grant). The calculation resources were provided by the LERIA and MOLTECH-Anjou laboratories. The authors would therefore like to thank Jean-Mathieu Chantrein for his technical help. TC and BDM would especially like to thank the BOINC community involved in our project, called QuChemPedIA@home (https://quchempedia.univ-angers.fr/athome/). While no results published here came from the community, it is the large failure ratio observed in the DFT calculations during the collaborative computing effort that paved the ideas of this article.Notes and references
- M. Olivecrona, T. Blaschke, O. Engkvist and H. Chen, J. Cheminf., 2017, 9, 48 Search PubMed
.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Science, 2018, 361, 360–365 CrossRef CAS PubMed
- H. Chen, O. Engkvist, Y. Wang, M. Olivecrona and T. Blaschke, Drug Discovery Today, 2018, 23, 1241–1250 CrossRef PubMed
- D. C. Elton, Z. Boukouvalas, M. D. Fuge and P. W. Chung, Mol. Syst. Des. Eng., 2019, 4, 828–849 RSC
- K. Terayama, M. Sumita, R. Tamura and K. Tsuda, Acc. Chem. Res., 2021, 54, 1334–1346 CrossRef CAS PubMed
- W. Ma, Z. Liu, Z. A. Kudyshev, A. Boltasseva, W. Cai and Y. Liu, Nat. Photonics, 2021, 15, 77–90 CrossRef CAS
- T. Sousa, J. Correia, V. Pereira and M. Rocha, J. Chem. Inf. Model., 2021, 61, 5343–5361 CrossRef CAS PubMed
- C. Bilodeau, W. Jin, T. Jaakkola, R. Barzilay and K. F. Jensen, WIREs Comput. Mol. Sci., 2022, 12(5), e1608 Search PubMed
- P. Schneider, W. P. Walters, A. T. Plowright, N. Sieroka, J. Listgarten, R. A. Goodnow, J. Fisher, J. M. Jansen, J. S. Duca, T. S. Rush, M. Zentgraf, J. E. Hill, E. Krutoholow, M. Kohler, J. Blaney, K. Funatsu, C. Luebkemann and G. Schneider, Nat. Rev. Drug Discovery, 2020, 19, 353–364 CrossRef CAS PubMed
- C. W. Coley, N. S. Eyke and K. F. Jensen, Angew. Chem., Int. Ed., 2020, 59, 23414–23436 CrossRef CAS PubMed
- H. S. Kwak, Y. An, D. J. Giesen, T. F. Hughes, C. T. Brown, K. Leswing, H. Abroshan and M. D. Halls, Front. Chem., 2022, 9, 800370 CrossRef PubMed
- N. C. Forero-Martinez, K.-H. Lin, K. Kremer and D. Andrienko, Adv. Sci., 2022, 9, 2200825 CrossRef CAS PubMed
- P. Ertl and A. Schuffenhauer, J. Cheminf., 2009, 1, 8 Search PubMed
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, J. Chem. Inf. Model., 2018, 58, 252–261 CrossRef CAS PubMed
- W. Gao and C. W. Coley, J. Chem. Inf. Model., 2020, 60, 5714–5723 CrossRef CAS PubMed
- P. Polishchuk, J. Chem. Inf. Model., 2020, 60, 6074–6080 CrossRef CAS PubMed
- A. Thakkar, V. Chadimová, E. J. Bjerrum, O. Engkvist and J.-L. Reymond, Chem. Sci., 2021, 12, 3339–3349 RSC
- J. D. Harris, M. J. Moran and I. Aprahamian, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9414–9422 CrossRef CAS PubMed
- J. H. Jensen, Chem. Sci., 2019, 10, 3567–3572 RSC
- P. Polishchuk, J. Cheminf., 2020, 12, 28 Search PubMed
- J. Leguy, T. Cauchy, M. Glavatskikh, B. Duval and B. Da Mota, J. Cheminf., 2020, 12, 55 CAS
- S. Bühlmann and J.-L. Reymond, Front. Chem., 2020, 8, 46 CrossRef PubMed
- M. Voršilák, M. Kolář, I. Čmelo and D. Svozil, J. Cheminf., 2020, 12, 35 Search PubMed
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez, P. Mutowo, F. Atkinson, L. J. Bellis, E. Cibrián-Uhalte, M. Davies, N. Dedman, A. Karlsson, M. P. Magariños, J. P. Overington, G. Papadatos, I. Smit and A. R. Leach, Nucleic Acids Res., 2017, 45, D945–D954 CrossRef CAS PubMed
-
ChEMBL Database, https://www.ebi.ac.uk/chembl/ Search PubMed
- J. J. Irwin, K. G. Tang, J. Young, C. Dandarchuluun, B. R. Wong, M. Khurelbaatar, Y. S. Moroz, J. Mayfield and R. A. Sayle, J. Chem. Inf. Model., 2020, 60, 6065–6073 CrossRef CAS PubMed
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Sci. Data, 2014, 1, 140022 CrossRef CAS PubMed
- M. Glavatskikh, J. Leguy, G. Hunault, T. Cauchy and B. Da Mota, J. Cheminf., 2019, 11, 69 Search PubMed
- J. Leguy, M. Glavatskikh, T. Cauchy and B. Da Mota, J. Cheminf., 2021, 13, 76 Search PubMed
- J.-L. Reymond, L. Ruddigkeit, L. Blum and R. v. Deursen, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 717–733 CAS
- M. Nakata and T. Shimazaki, J. Chem. Inf. Model., 2017, 57, 1300–1308 CrossRef CAS PubMed
-
P. Walters, silly_walks, 2022, https://github.com/PatWalters/silly_walks, original-date: 2020-12-26T17:25:07Z Search PubMed
-
RDKit: Open-source cheminformatics, https://www.rdkit.org Search PubMed
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed
-
A. A. Hagberg, D. A. Schult and P. J. Swart, Proceedings of the 7th Python in Science Conference, Pasadena, CA USA, 2008, pp. 11–15 Search PubMed
- A. H. Lipkus, Q. Yuan, K. A. Lucas, S. A. Funk, W. F. Bartelt, R. J. Schenck and A. J. Trippe, J. Org. Chem., 2008, 73, 4443–4451 CrossRef CAS PubMed
- N. M. OLBoyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed
-
RDKit: Open-source cheminformatics, https://www.rdkit.org Search PubMed
- P. Tosco, N. Stiefl and G. Landrum, J. Cheminf., 2014, 6, 37 Search PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09 Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS
-
J. Leguy, B. Duval, B. D. Mota and T. Cauchy, 2021 IEEE 33rd International Conference on Tools with Artificial Intelligence (ICTAI), 2021, pp. 780–785 Search PubMed
| This journal is © The Royal Society of Chemistry 2023 |