
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGenerating structural alerts from toxicology datasets using the local interpretable model-agnostic explanations method†
Cayque Monteiro Castro
Nascimento
,
Paloma Guimarães
Moura
and
Andre Silva
Pimentel
 *
*
Departamento de Química, Pontifícia Universidade Católica do Rio de Janeiro, Rio de Janeiro, RJ 22453-900, Brazil. E-mail: a_pimentel@puc-rio.br
First published on 21st July 2023
Abstract
The local interpretable model-agnostic explanations method was used to interpret a machine learning model of toxicology generated by a neural network multitask classifier method. The model was trained and validated using the Tox21 dataset and tested against the Clintox and Sider datasets, which are datasets of marketed drugs with adverse reactions and drugs approved by the Federal Drug Administration that have failed clinical trials for toxicity reasons. The stability of the explanations is proved here with a reasonable reproducibility of the sampling process, making very similar and trustful explanations. The explanation model was created to produce structural alerts with more than 6 heavy atoms that serve as toxic alerts for researchers in many fields of academics, regulatory agencies and industry such as organic synthesis, pharmaceuticals, toxicology, and so on.
Introduction
Currently, one of the main causes for drug failures in clinical trials is unacceptable toxicity. As a result, computational models become increasingly relevant to pre-laboratory analyses.1,2 Studies already point out the importance of computational toxicology as a tool for the future of environmental health sciences and regulatory decisions in public health.3,4 In fact, this tool combined with machine learning has a range of applications in many areas of science such as pharmacology,5–10 genetics and biochemistry,11–15 and drug discovery for COVID-19.16 However, the model explainability in machine learning is a highly essential issue17–21 because machine learning models are mostly considered as black boxes,17,21–24 indicating an ambitious challenge to the progress of machine learning.With the advancement of data science and big data, the availability of information about chemical structures has increased considerably. Filter structures with undesired physical–chemical properties in virtual libraries can reduce the universe from millions to a few thousand drug candidates.25 In addition, it is highly desirable to have filters for functional groups or fragments commonly considered as inappropriate26 to increasingly develop virtual screenings.27–31 Web servers and expert systems in structural alerts are highly developed in the literature,2,21,22,31–39 but interpretable machine learning models and model explainers are still not widely investigated with the intent to obtain structural alerts and toxic alerts.21
While expert systems in structural alerts can highlight the potential dangers of chemical substructures, automated methods with machine learning techniques and neural networks can be superior in terms of their predictive performance.3,40,41 It is important to note that, despite all the progress made by these tools, the correlation between the interpretation of toxicological data and the mechanism of action of the compound in the human body is still a challenge.5,22,31,32,40
Molecular featurization may effectively extract structural and chemical insights from any given drug using fingerprints42 and molecular descriptors3,22,29,30,32,43,44 with a prediction-based approach applied to large datasets. After doing that and building a model, it is usual to perform the dataset validation.5,24,45,46 This validation delivers a comprehensive view of the model performance using an unknown data.34,47 It is feasible to identify which features are the most striking in a model prediction.21,35,36 To obtain a full comprehension of machine learning models, its interpretation is highly necessary. Although it is difficult to understand why some predictions are incorrect while others are correct, local interpretable model-agnostic explanations (LIME)48 may be used to explain the model in a way that is understandable to humans. It is important to note that SHapley Additive exPlanations (SHAP)48–51 is more commonly used compared with LIME, but it is extremely relevant to apply the latter to understand its interpretation capacity. LIME was proposed in 2016.52 In the LIME proposal, LIME has been applied to image recognition and text classification. Since then, LIME has not been applied to toxicology as well as the method SHAP. Some tabular classification on non-related toxicology subjects has been described, but not published at all. As far as we know, LIME has not been used in toxicology yet, and its application to toxicology has not been scrutinized, especially for structural alerts. The innovation of our study is the application of LIME to generate structural alerts which is very important to the pharmaceutical industry and toxicology research. It is interesting that many developers of model explainability algorithms do not exactly understand why these algorithms make such explanations.53 Currently, there are many methods to enlighten the explanations of a model such as feature importance, local explanations, rule extraction, layer-wise relevance propagation, attention mechanisms, model distillation, and counterfactual explanations. These approaches can be used individually or in combination, depending on the specific requirements and nature of the problem. It is important to note that model explainability is an active research area, and new techniques continue to emerge, offering further possibilities to enhance the interpretability of machine learning models.
Although SHAP is more used, considered more robust, and slower than LIME,21 it is important to explore a simpler and faster method to determine structural alerts with different databases and test its limitations. LIME can approximate any black box machine learning model to a local interpretable model to explain each individual prediction, in addition to being an extremely popular explainer.52–54 However, it is believed that the search for a faster method with lower computational cost can be relevant and add novelty to the area of toxicology, data science and chemoinformatics. SHAP and LIME are reasonable methods to interpret models. In principle, SHAP is better than LIME because the first mathematically guarantees the consistency and accuracy of explanations. However, the model agnostic implementation of SHAP (KernelExplainer) is much slower than that of LIME. For large datasets such as Clintox, Sider, and Tox21, SHAP is computationally more expensive than LIME to use the entire dataset. As we must rely on approximations, this reduces the accuracy of the explanation. For example, SHAP explains the prediction deviation from the expected value calculated using the training dataset. Depending on that, it is faster to calculate the expected value using a specific subset of the training set as compared to the use of an entire training set. With the subset of the training set, this considerably reduces the accuracy of the explanation. Therefore, we did not compare SHAP and LIME because LIME is much faster than SHAP for the purpose of our study. Although LIME has been widely explored recently in interpreting the severity of patients diagnosed with COVID-19,55 the consistency and instability of LIME, which is a target of criticism,56 must be analyzed in more studies for advancement in the subject.
Here, a comprehensive outline is given of the explainability of toxicity models of drugs created using machine learning. The purpose is to explain how these models may in fact be remarkably explainable. This study shows advances in the field because LIME51 is used to interpret toxicology models of drugs using the Tox21 dataset57 of qualitative toxicity measurements on biological targets, including nuclear receptors and stress response pathways. Then, the explained model is applied to the SIDER dataset of marketed drugs with adverse reactions58 and to the Clintox dataset of drugs approved by the Federal Drug Administration (FDA)57 that have failed clinical trials for toxicity reasons. From these interpretations, a list of unwanted substructures, i.e., a structural alert list, is built to filter possible toxic drugs from large libraries, which avoids waste of time and effort doing useless synthesis and research of compounds that may fail in clinical trials for toxicity or undesirable reasons. It is always good to remember that “alerts are just alerts” and should be seen as hypotheses for undesirable mechanisms and never as rules.24
Background
LIME
LIME explains the prediction of any classifier by learning an interpretable model and providing a consistent model agnostic explainer. The method selects a representative set with explanations that provides an intuitive understanding of the model. It slightly adjusts the input and tests the forecast changes. This adjustment should be small so that it is still close to the original local region. LIME has the following properties: (1) it provides an easy and qualitative understanding of the response and the variables of the model; (2) it must be at least locally trustful replicating the model behavior with local fidelity; (3) it must explain any model making no assumptions about the model and being agnostic to the model; and (4) it must explain a representative set providing a comprehensive intuition of the model.21,48,59Under the assumption of an explainer being trustful and interpretable, LIME minimizes the following explanation model equation:
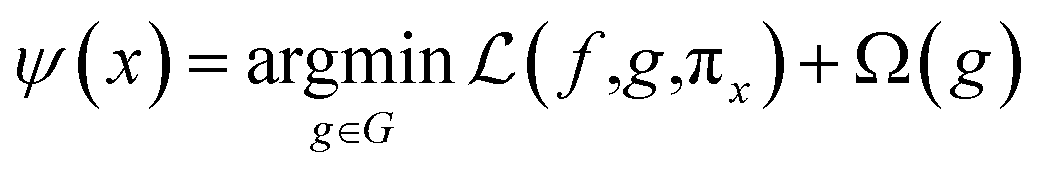
The random uniform sampling for local exploration is performed from x to create a full training data set, i.e., create multiple z from a single row of x, which is weighted by πx to be focused on z data closer to x. The sparse linear explanation assumes that (1) g(z) = wz; (2) the locality-aware loss is a square loss; and (3) the proximity weighing for the samples is:
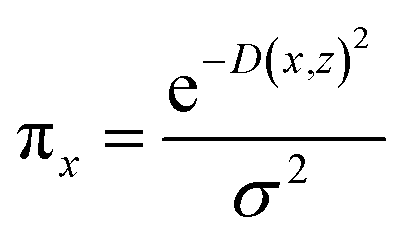
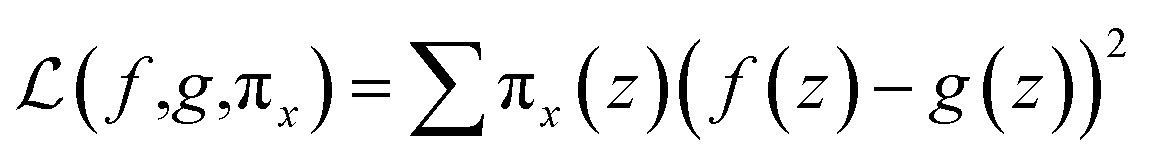
Methodology
Coding and curated data
The code was written using Python (version 3.7) on the Google Colaboratory platform. DeepChem (version 2.7.2.dev),60 Pandas (version 1.5.3)61 and RDKit (version rdkit-pypi-2022.3.5) libraries60,62 were used. Other auxiliary libraries such as Matplotlib (version 3.7.1)63 and Numpy (version 1.21.6)64 were also utilized. Then, the curated Tox21 dataset65 was used to train the machine learning model of qualitative experimental measurements (binary label: 0 for non-toxic and 1 for toxic) of the activity of molecules in 12 biological targets including nuclear receptors and response pathways to stress. In addition, the Tox21 dataset65 was compared with the curated Sider66 and Clintox67 datasets found on MoleculeNet57 to find FDA-approved or marketed drugs that presented adverse reactions in 27 organ system classes or that failed clinical trials for toxicological reasons in the Tox21 dataset.65 These datasets include drug molecules with the corresponding SMILES representations.68Finding the identical molecules in Tox21, Clintox, and Sider datasets
The Tox21 dataset65 was featurized using the extended connectivity fingerprints (ECFPs)69 without splitting the dataset. To obtain a model as general as possible, it was necessary to previously remove from the Tox21 the molecules that are shared with the Clintox and Sider datasets. In this manner, the molecules in the test dataset were not found in the training and validation datasets as mentioned before. These molecules in common were removed using the Tanimoto coefficient70 as a measure of similarity and the Morgan fingerprint71 as a vector of bits. For this, it was necessary to transform the Tox21 dataset into a Pandas data frame.In the first step of the method, a column was created in the data frames dfclintox, dfsider, and dftox21 using the AddMoleculeColumnToFrame function of the RDKit library60,62 in which each value corresponded to the sketch of the molecular structure of the corresponding SMILES representation (ROMol structure).68 Thus, Morgan fingerprints71 were generated as a vector of bits for all molecules of the data frames using the rdFingerprintGenerator.GetFPs class to create a column. As this command works only when there is a structure drawn in the ROMol column, it was therefore necessary to previously remove the lines of all smiles that could not be transformed into ROMol structures.
The droppingIdenticals function was created to calculate the Tanimoto similarity of a given smile representation to compare with all molecules in the Tox21 dataset. All molecules that had similarity equal to 1.0 were eliminated from the Tox21 data frame. Finally, the droppingIdenticals function returned two data frames that contained all molecules in common between the Tox21 dataset and the Clintox and Sider datasets.
Data splitting
The Tox21 dataset was randomly split into training and validation datasets to train and validate the model using the simple hold-out technique. In this sense, it was initially necessary to convert the data frame dftox21 into a Numpy object using the class dc.data.NumpyDataset.from_dataframe(). Then, the Tox21 dataset was split using the random splitter object, separating 80% of the data for training and 20% for validation. It is important to emphasize that the training model is randomly generated for each run. Therefore, the testing results must be a little different from each other even though the test dataset is the same for each run. It was decided to not save the training model to evaluate the results from each run to check the consistency of results. The test dataset was created with the compounds of Clintox that are found in the Tox21 dataset, and it was converted to a dataset object to utilize DeepChem tools.62 The only molecules that were used in the model were the overlapping ones, the ones that Clintox/Sider and Tox21 datasets had in common. As mentioned before, we removed the common molecules from Tox21 and separated them into a different dataframe that we used as a test. The remaining Clintox/Sider molecules are not used because we need the task information for each molecule; without this task information (toxic (1) or non-toxic (0) for each of 12 different toxic effects by specifically designed assays) it is not possible to perform the analysis. Many Clintox/Sider molecules do not have this information, and the procedure proposed here adds this information. Using the multitask classifier function from the DeepChem library,60 12 tasks with 1024 features were classified to create a multitask classification model. The MultiTaskClassifier model is a fully connected deep residual network for multitask classification composed from pre-activation residual blocks.72 The hyperparameter optimization was made using different fingerprints and numbers of epochs.To ensure that there were no improper deviations, the receiver operating characteristics (ROC) curve (AUC) statistical method was used to calculate the accuracy score between the model and the test.73 The ROC curve showed how well the model could distinguish between two binary labels (0 for non-toxic or 1 for toxic). The best model can accurately distinguish the binomial. Thus, to simplify the ROC analysis, the area under the ROC curve is nothing more than a way of summarizing the ROC curve into a single value, aggregating all ROC thresholds, calculating the area under the curve. The ROC curve is generated by changing the threshold between classes; the AUC value is not dependent on the threshold. That is, above this threshold, the algorithm classifies in one class and below in the other class. The higher the AUC, the better. An AUC equal to 0.5 indicates a random prediction result, and AUC equal to 0 indicates a perfectly reversed prediction (the prediction values of all positive samples are below those of negative samples). The interesting thing about the AUC is that the metric is scale invariant, as it works with classification accuracy instead of their absolute values. In addition, it also measures the quality of the model predictions, regardless of the classification threshold.
Explaining the model and generating the structural alerts
With the model trained, the next step was to explain the model using the LIME library (version 0.2.0.1).48,74 The agnostic explainer was created using the LimeTabularExplainer function and the training dataset. To investigate a possible correlation between the 12 biological receptors (tasks) and the fragments that activate a specific characteristic (fingerprints) of a molecule, a code was written to describe some information about the fragments. Worksheets are created to make it possible to understand why a molecule, predicted by the model as toxic, is classified as such. More details about this code are summarized in Fig. 1 and explained in the ESI.†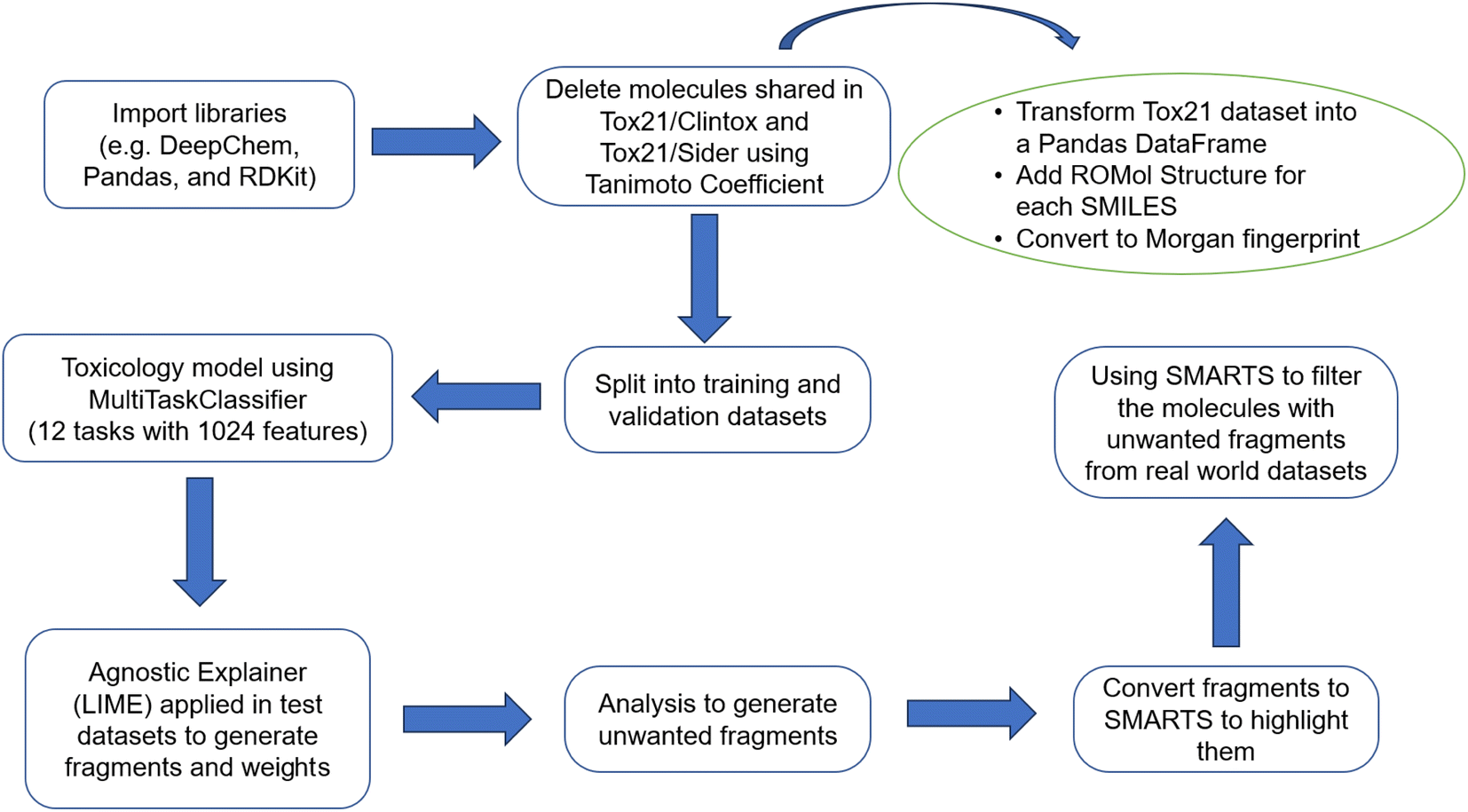 | ||
| Fig. 1 Flowchart of the methodology. | ||
At the end, the classification data of each active molecule for each task is exported. The number of times a fragment is found in molecules that are predicted to be toxic, and how much that fragment contributes to the toxicity of those molecules, are two important factors in understanding how toxicity is predicted. The data is grouped by task. For each of these groups, dictionaries are obtained with the counting frequency of each fragment and the total sum of associated weights for each fragment. Then, these dictionaries are broken up in such a way that the desired data frame is obtained. In addition, the new column with active molecules is created in this same data frame and the index is that of the fragment for a given group. Thus, a list is returned with all molecule ids belonging to the group. Finally, RDKit resources62 are used to highlight the fragment and visualize them in the associated molecule.
Application of the structural alert list in a large dataset
After different structural alerts are found and the likely unwanted substructures are highlighted in molecules, they can be used to remove them with RDKit60,62 to not include into a screening library. These substructures may have unfavorable pharmacokinetic properties due to their toxicity or reactivity, or because they may interfere with certain toxicity assays. Filtering these structural alerts can save time and resources, by supporting to assemble more efficiently screening libraries.All molecules that have been tested against the epidermal growth factor receptor (EGFR) kinase are downloaded by using the ChEMBL web resource client.75 Then, the resource objects for API access are created to get the target data using the UniProt ID P00513.76 The target data is fetched and downloaded from ChEMBL and the result of the query is stored. After checking the entries, the CHEMBL203 entry is selected as the target of interest, which is a single protein of the human EGFR, also named erbB1. The bioactivity data for the target of interest is fetched and only human proteins, bioactivity type IC50, exact measurements, and binding data (assay type ‘B’) are considered. Finally, the query set is downloaded in the form of a Pandas data frame.
The bioactivity data is preprocessed and filtered by converting the datatype of standard value from object to float, deleting entries with missing values, keeping only entries with standard units (nanomolar), deleting duplicate molecules, resetting the data frame index, and renaming the columns. The molecular structures are linked to respective bioactivity ChEMBL IDs. Thus, the compound data are also preprocessed and filtered by removing entries with missing entries, deleting duplicate molecules, and getting molecules with canonical SMILES.68 Then, the values of interest found in bioactivities_df and compounds_df data frames are merged in the data frame output_df based on the ChEMBL IDs of compounds.
The Lipinski rule of five77,78 and PAINS filtering26 were applied to the EGFR dataset (in output_df data frame) for compliancy as already implemented in RDKit.60,62 The number of compounds with and without PAINS was obtained. Then, an external list provided by Brenk et al.25 was used to further filter the EGFR dataset to get the substructure matches and search the filtered data frame for these matches with the unwanted substructures. Then, the structural alert list found here was applied to filter the EGFR dataset. The underlying SMARTS patterns79 in these fragments were used to highlight the substructures within the filtered molecules using RDKit.60,62
Results and discussion
The droppingIdenticals function was used to calculate the Tanimoto similarity70 using Morgan fingerprints71 of each molecule in the data frames dfclintox and dfsider with respect to all molecules in the Tox21 dataset. Thus, the molecules with similarity equal to 1.0 were eliminated from the Tox21 dataset (originally with a total of 7831 molecules) and moved to two data frames (dfdropOutClintox_Tox21 and dfdropOutSider_Tox21) containing the molecules of the Tox21 dataset in common with the Clintox and Sider datasets. From the original Tox21 data frame, the dfdropOutClintox_Tox21 data frame was created with 7240 molecules after removing 591 molecules, and the dfdropOutSider_Tox21 data frame with 6949 molecules after removing 882 molecules.From the multitask classifier function from the DeepChem library,60 12 tasks and 1024 features using the ECFP fingerprint were classified to create a multitask classification model that had the best loss function after 200 to 300 epochs as part of the hyperparameter optimization. The featurizers MACCS,80 MAT,60 PubChem,60 and TokenizerSmiles81 were tested as part of the hyperparameter optimization but all results were like those found by the ECFP featurizer so that only this last one is presented here. Table 1 presents the results regarding the metrics obtained in the model validation and testing. It is noted that the data are reasonably promising because the accuracies of the model validation and testing are in the range of 0.71–0.77 in both Clintox and Sider datasets. This means that the model is considerably accurate and coherent for the two test datasets, not showing significant overfitting or underfitting.
| Dataset | Run | Validation set | Testing set |
|---|---|---|---|
| Clintox | #1 | 0.719 | 0.750 |
| #2 | 0.746 | 0.765 | |
| #3 | 0.735 | 0.761 | |
| Sider | #1 | 0.731 | 0.721 |
| #2 | 0.731 | 0.731 | |
| #3 | 0.730 | 0.727 |
Using the LIME explainer module, it is important to mention that all the values of kernel width tested to explain the trained model did not yield unstable results. It was also verified that the fragments produced in the explanation are mostly similar for the different kernel values tested. Therefore, we decided to use the default value suggested by LIME developers.52 Another important attempt to use only meaningful results in the explanation is the elimination of unimportant fragments. For the sake of conciseness, we decided to eliminate some unimportant fragments with small or negative weights by using the average of all weights. We also attempted to use only the maximum weight or the maximum and minimum weights, but the fragments generated in both attempts were one third of those produced by using the average of weights. Mathematically, the elimination of fragments with negative or small weights seems to be incorrect because we are eliminating fragments in the model that may cause the toxic behavior in a molecule to vanish. So, it makes sense to only look at the maximum and minimum weights mathematically. But chemically speaking, the fragment with minimum weight (non-toxic fragment) does not make the behavior of a fragment with maximum weight (toxic fragment) vanish. At the end, what matters for the behavior in a molecule is the toxic fragment that justifies the use of this cutoff.
Ideally, the molecules to be explained should be only those predicted to be toxic by the model as explained in the Methodology section. However, this is not exactly what is shown in Tables S1 and S2,† which expose a certain number of non-toxic molecules. Despite being considered toxic compounds by the model with a probability of at least 0.8 for a given task, this is not correctly calculated when analyzed individually. This is because the final balance of the sum resulted in negative numbers when considering all the toxicity weight contributions of their respective fragments. For example, Table S1† shows that of the 61 Clintox molecules predicted to be toxic for this task, only 56 are toxic in the NR-AR-LBD task, which gives an accuracy of more than 91% in a separate run. The sum of all weights was less than zero for the other five, even with positive toxic contributions. However, it is noticeable that most molecules that entered the classification were correctly predicted. Thus, the appearance of non-toxic molecules in the classification table is acceptable. Therefore, the model does not have perfect accuracy as observed in the previous metrics (Table 1). Table S2† shows an example of one of the leaderboards produced in the classification of Clintox molecules for task SR-p53. As observed, there are seven molecules classified as toxic (78%) and two molecules as non-toxic (22%) in this separate run. This classification is made using the model results calculated in LIME. It is determined by the balance of toxicity and non-toxicity. It is important to note that the difference between toxicity and non-toxicity for some compounds is subtle. For these molecules, it is not possible to conclude that molecules 190, 216, and 252 are very toxic. It is also important to note that no fragment of any molecule contributed to the toxicity of task NR-PPAR-γ in this separate run. Table 2 presents the number of molecules classified as toxic or non-toxic for each task in Clintox and Sider datasets in three different runs. It also shows that none of the fragments of any molecule contributed to the toxicity of tasks NR-PPAR-γ, SR-MMP and SR-p53 for all runs presented in this study. However, it is important to reaffirm that the model predicts with reasonable accuracy with an acceptable reproducibility as it was repeated a certain number of times (more than shown here).
| Run | #1 | #2 | #3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dataset | Clintox | Sider | Clintox | Sider | Clintox | Sider | ||||||
| Task | Toxic | Non-toxic | Toxic | Non-toxic | Toxic | Non-toxic | Toxic | Non-toxic | Toxic | Non-toxic | Toxic | Non-toxic |
| NR-AR | 67 | 0 | 57 | 0 | 73 | 0 | 62 | 0 | 73 | 0 | 52 | 0 |
| NR-AR-LBD | 55 | 2 | 47 | 1 | 61 | 1 | 46 | 2 | 59 | 2 | 41 | 1 |
| NR-AhR | 5 | 0 | 2 | 1 | 3 | 3 | 4 | 0 | 3 | 1 | 2 | 1 |
| NR-Aromatase | 3 | 1 | 4 | 1 | 4 | 3 | 2 | 1 | 0 | 6 | 2 | 1 |
| NR-ER | 30 | 4 | 30 | 6 | 35 | 4 | 31 | 6 | 34 | 2 | 26 | 6 |
| NR-ER-LBD | 22 | 0 | 21 | 5 | 26 | 1 | 23 | 1 | 23 | 2 | 17 | 3 |
| NR-PPAR-γ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| SR-ARE | 11 | 3 | 9 | 4 | 7 | 10 | 9 | 4 | 9 | 6 | 8 | 4 |
| SR-ATAD5 | 2 | 0 | 2 | 0 | 2 | 1 | 2 | 1 | 2 | 0 | 2 | 0 |
| SR-HSE | 1 | 1 | 1 | 1 | 3 | 0 | 1 | 1 | 3 | 0 | 1 | 0 |
| SR-MMP | 0 | 0 | 0 | 0 | 12 | 7 | 0 | 0 | 0 | 0 | 0 | 0 |
| SR-p53 | 0 | 0 | 0 | 0 | 7 | 3 | 0 | 0 | 0 | 0 | 0 | 0 |
Table S3† shows only the first two lines of one of the data frames generated from the Sider database. These two fragments that contributed to the toxicity in the biological target NR-AR (task 0) are presented according to the model. In this case, despite the CC(C)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O fragment having a lower frequency in the task than the CC(C)(C)C fragment, the total weight of the toxic contribution is higher. In the last column of Table S3,† it is possible to visualize the highlight where such fragments are found in one of the molecules activated by them. For simplicity, the fragment was highlighted only on the first active molecule in the list presented in the previous column (active molecules). Table S3† may lead to a wrong interpretation that fragments that have the largest contribution are those with high frequency in the task. The very small and simple fragments are found in most molecules, so their frequencies are very high in the task. Fig. S1 and S2† present the results after running LIME. They show the small fragments (less than 7 heavy atoms) that influenced the increase in toxicity for the 12 tasks all together in the Clintox and Sider databases, respectively, in terms of counting the frequency of appearance in the analysis. It is interesting to note that most of the fragments found in Fig. S1 and S2† are very simple structures. Because of that, it was decided to pay more attention to the number of heavy atoms of each fragment. It was noticed that the fragments with less than 7 heavy atoms are mostly saturated and unsaturated hydrocarbon fragments, and to a lesser extent, oxygenated and nitrogenated groups. Most importantly, these fragments are also part of the larger and complex fragments with more than 6 heavy atoms that have a low frequency in the task. Thus, it was decided to filter the small fragments from the list, keeping only the fragments with more than 6 heavy atoms to avoid redundancy. The most solid evidence supporting this choice is that the cutoff is in the center of the range. Although this cutoff is easily changed by the user, we tested the cutoffs from 4 to 10. It seems the lower end of the range gives too many fragments, and the higher end gives only a few fragments. So, the middle of the range seems to be the best choice.
O fragment having a lower frequency in the task than the CC(C)(C)C fragment, the total weight of the toxic contribution is higher. In the last column of Table S3,† it is possible to visualize the highlight where such fragments are found in one of the molecules activated by them. For simplicity, the fragment was highlighted only on the first active molecule in the list presented in the previous column (active molecules). Table S3† may lead to a wrong interpretation that fragments that have the largest contribution are those with high frequency in the task. The very small and simple fragments are found in most molecules, so their frequencies are very high in the task. Fig. S1 and S2† present the results after running LIME. They show the small fragments (less than 7 heavy atoms) that influenced the increase in toxicity for the 12 tasks all together in the Clintox and Sider databases, respectively, in terms of counting the frequency of appearance in the analysis. It is interesting to note that most of the fragments found in Fig. S1 and S2† are very simple structures. Because of that, it was decided to pay more attention to the number of heavy atoms of each fragment. It was noticed that the fragments with less than 7 heavy atoms are mostly saturated and unsaturated hydrocarbon fragments, and to a lesser extent, oxygenated and nitrogenated groups. Most importantly, these fragments are also part of the larger and complex fragments with more than 6 heavy atoms that have a low frequency in the task. Thus, it was decided to filter the small fragments from the list, keeping only the fragments with more than 6 heavy atoms to avoid redundancy. The most solid evidence supporting this choice is that the cutoff is in the center of the range. Although this cutoff is easily changed by the user, we tested the cutoffs from 4 to 10. It seems the lower end of the range gives too many fragments, and the higher end gives only a few fragments. So, the middle of the range seems to be the best choice.
Structural alerts provide the basis for grouping compounds into categories that can allow comparisons. In addition, they must be progressively developed to allow the understanding of the mechanism of action of chemical compounds.82 Some scientific evidence can be shown that supports our results. Furans, phenols, nitroaromatics, and thiophenes are found to be toxic alerts in the literature,5 but only some alcohols, nitro compounds and sulfur compounds were found in our study. Li and collaborators83 investigated toxic fragments by frequency analysis and identified some substructures present in highly toxic and moderately toxic compounds. Some of these substructures were found by LIME, such as an alkylfluoride, sulfenic derivate, phosphonic trimester, cyanohydrin and nitrile. Yang and collaborators22 compared small toxic radicals obtained by different techniques (such as machine learning, graphs and expert systems). Many of these alerts were also found by LIME (such as NO2, NN and R–O–X). In addition, this work also shows figures with highlights obtained by different techniques. Most of these substructures were also found by LIME, such as alkyl radicals. Lei et al.84 presented some toxic fragments generated from a database of acute oral toxicity in rats such as alkylfluoride and amines. These fragments are also generated by LIME using the Tox21/Clintox/Sider datasets. Our methodology also found structural alerts with halogenated compounds that are found in studies of endocrine disruption with androgen and estrogen receptors.85 Our study also found nitrogen compounds that are mentioned as structural alerts in the literature.86
It is important to point out that some issues could not be interpreted by LIME. It does not recognize two identical structures with different SMILES representations. For example, CCC[C@H](C)C and CCCC(C)C are the same structures for the NR-ER and NR-ER-LBD tasks in Fig. S1 and S2,† respectively. LIME does not recognize the difference between these two SMILES because these representations are not canonical. Some other similar results appeared during the analysis, but they are not shown here for the sake of simplicity. Sometimes, new molecules are randomly sorted in the splitting of the original dataset that slightly changes the amount that a fragment appears and/or its toxicity contribution. This is probably because the RandomSplitter class was used to train the machine learning model and cross-validate the original database. Because of that, LIME was run several times (three repetitions are shown here, but it was done with many more repetitions for consistency), giving robust results as presented in Fig. 2 and 3. They show 109 and 113 fragments with more than 6 heavy atoms for the Clintox and Sider datasets, respectively. Although LIME interpreted the datasets finding some structural alerts that are present in other webservers or software, it also generated structural alerts that would be difficult for human beings to suggest by expert knowledge.23,48 It is important to mention that this number of heavy atoms is somewhat arbitrary. However, several runs were performed changing the number of heavy atoms from 4 to 8 heavy atoms. It was found that 6 heavy atoms are reasonable because the largest fragment usually has around 13 heavy atoms, e.g., right in the center of the range. Fig. S3 and S4, and S5 and S6† present the results of Runs #2 and #3, respectively, to confirm the reasonable LIME reproducibility. The number of fragments of each run for the Clintox and Sider datasets is shown in Table 3.
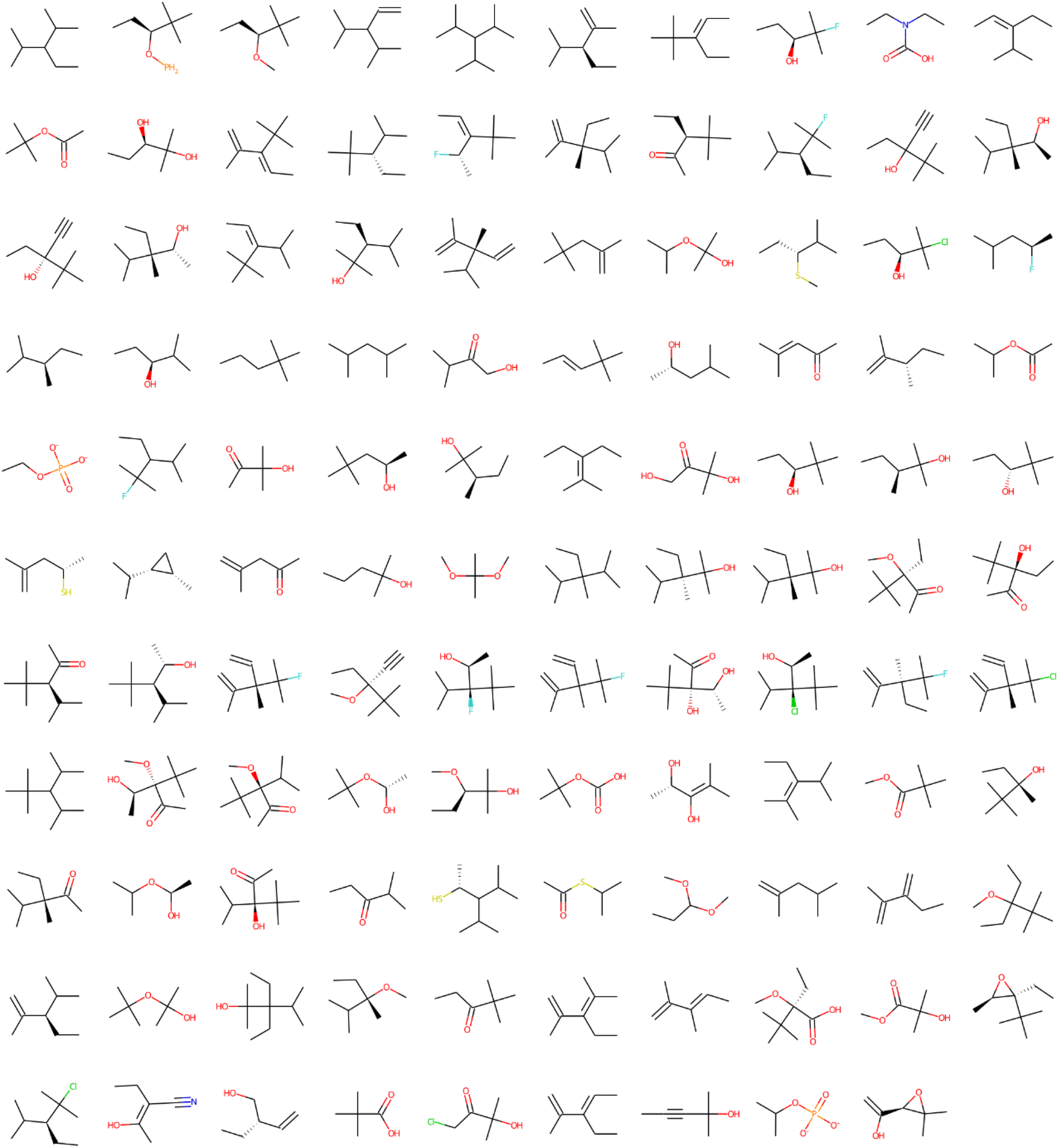 | ||
| Fig. 2 Structural alerts that influenced the increase in toxicity for all the 12 tasks in the Clintox dataset (Run #1). The order does not represent the toxic influence of each fragment. The other two runs are presented in the ESI.† | ||
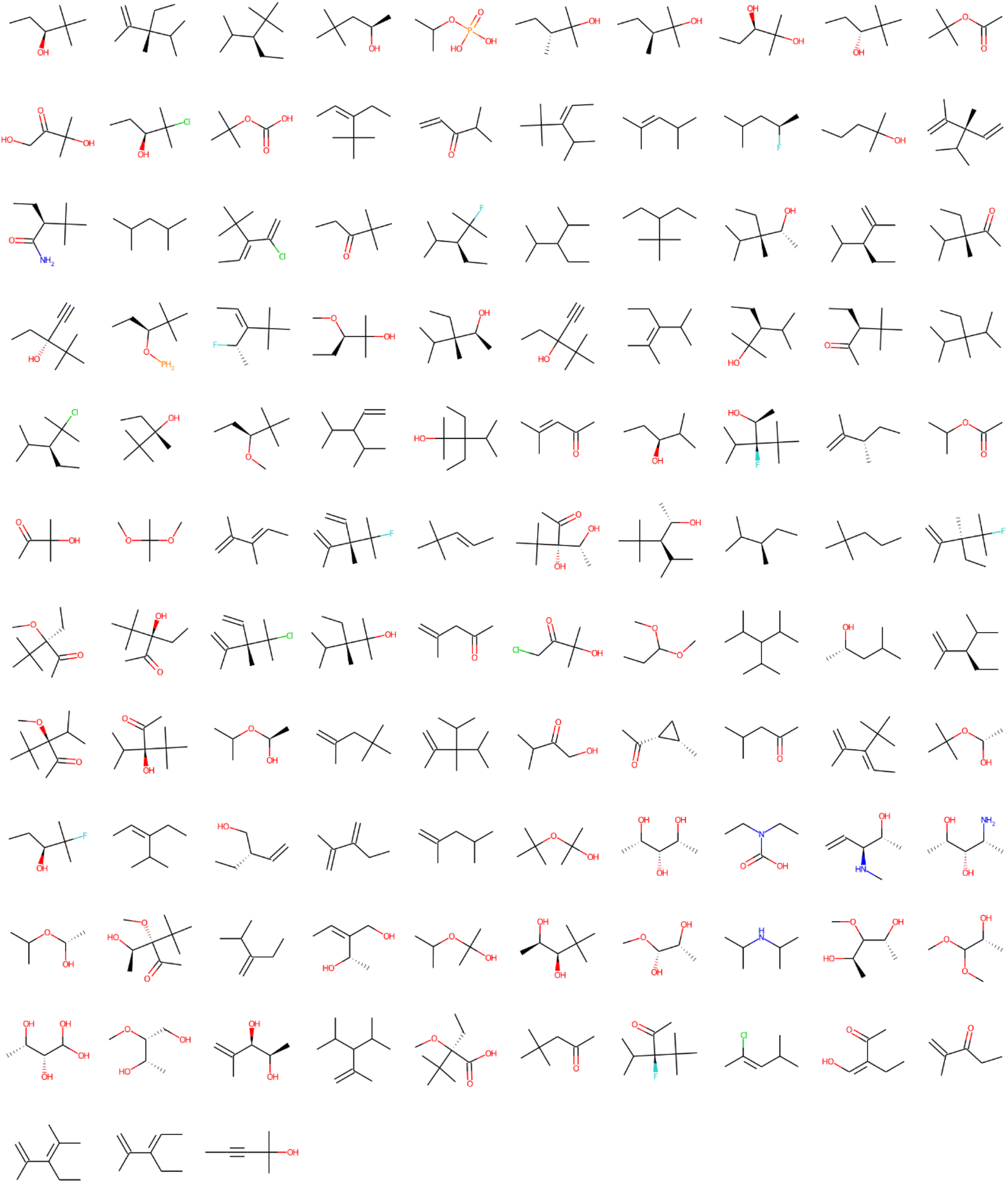 | ||
| Fig. 3 Structural alerts that influenced the increase in toxicity for all the 12 tasks in the Sider dataset (Run #1). The order does not represent the toxic influence of each fragment. The other two runs are shown in the ESI.† | ||
| Dataset | Run | Unwanted fragments | Number of found unwanted substructures | Number of compounds without unwanted substructures |
|---|---|---|---|---|
| Clintox | #1 | 109 | 147 | 4183 |
| #2 | 121 | 163 | 4174 | |
| #3 | 113 | 110 | 4218 | |
| Sider | #1 | 113 | 237 | 4165 |
| #2 | 133 | 311 | 4123 | |
| #3 | 114 | 380 | 4043 |
Table 3 also shows the results of the filtering of likely unwanted fragments obtained using the ECFP fingerprints in the EGFR dataset with compounds that show high binding affinity to EGFR. The dataset has 4266 compounds with RO5 and PAINS compliance, e.g., after Lipinski rules of 5 (RO5) and PAINS filtering were applied to the original EGFR dataset. The number of unwanted substructures found by LIME and the number of compounds without unwanted substructures are presented to confirm the filtering of structural alerts in the EGFR dataset. Fig. 4 and 5 present the structural alerts generated by using LIME. These structural alerts are highlighted in the first active molecule of some tasks for the Clintox and Sider datasets (Run #1 in each dataset). Fig. S7 and S8, and S9 and S10† show the other two runs (Runs #2 and #3) for the Clintox and Sider datasets, respectively, which are presented in the ESI.† As observed, LIME interpreted the datasets and found some structural alerts. It would be difficult for human beings to propose them. However, unique knowledge-based expert web-based platforms or software may be used for suggesting structural alerts for toxic compounds that may cause adverse drug reactions.5,6,48,87,88 It is important to mention that most structural alerts are present in different runs, and some are like each other, showing the LIME robustness.
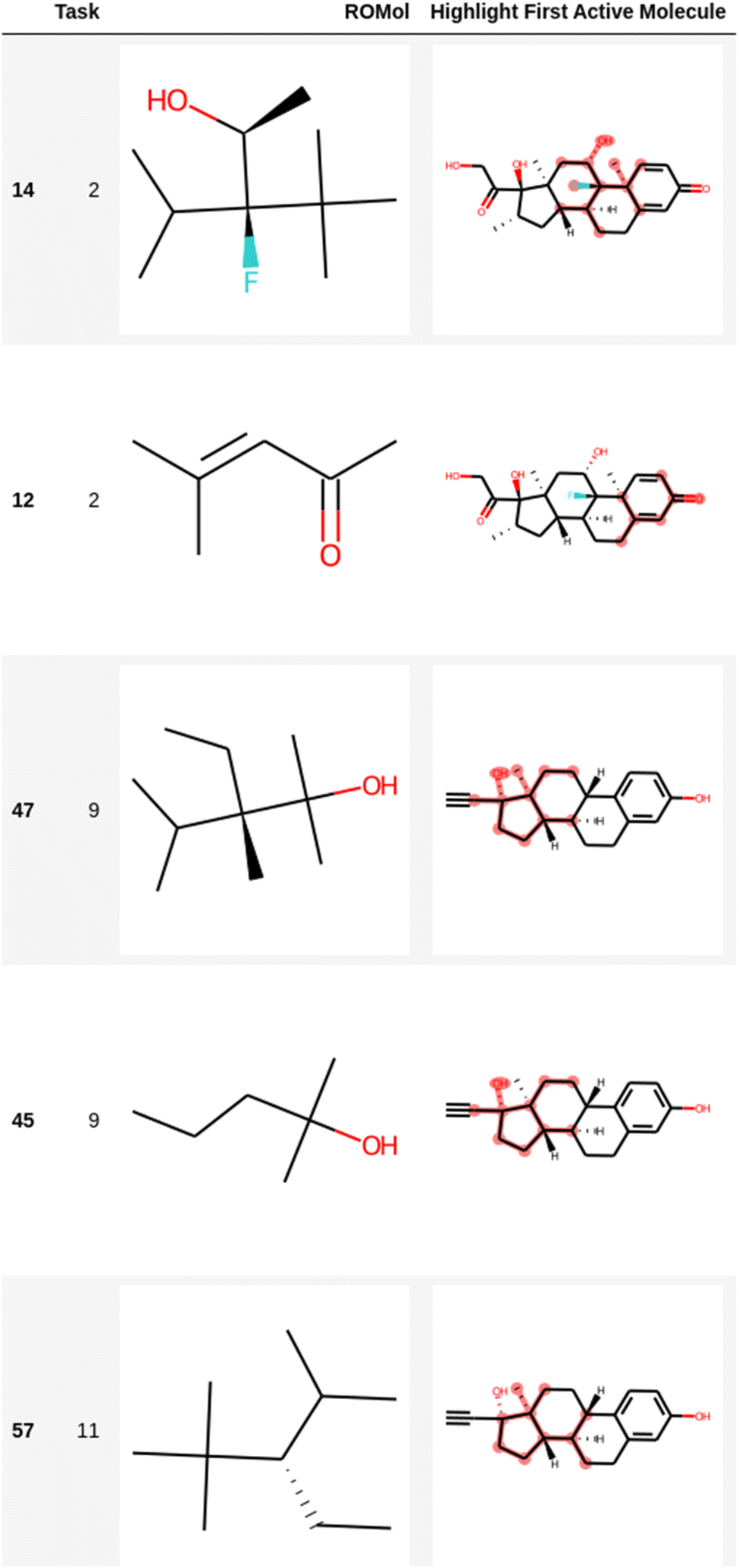 | ||
| Fig. 4 Representation of several fragments highlighted in the first active molecule of some tasks for the Clintox dataset (Run #1). The other two runs are presented in the ESI.† The number in the first column is the index in the data frame. | ||
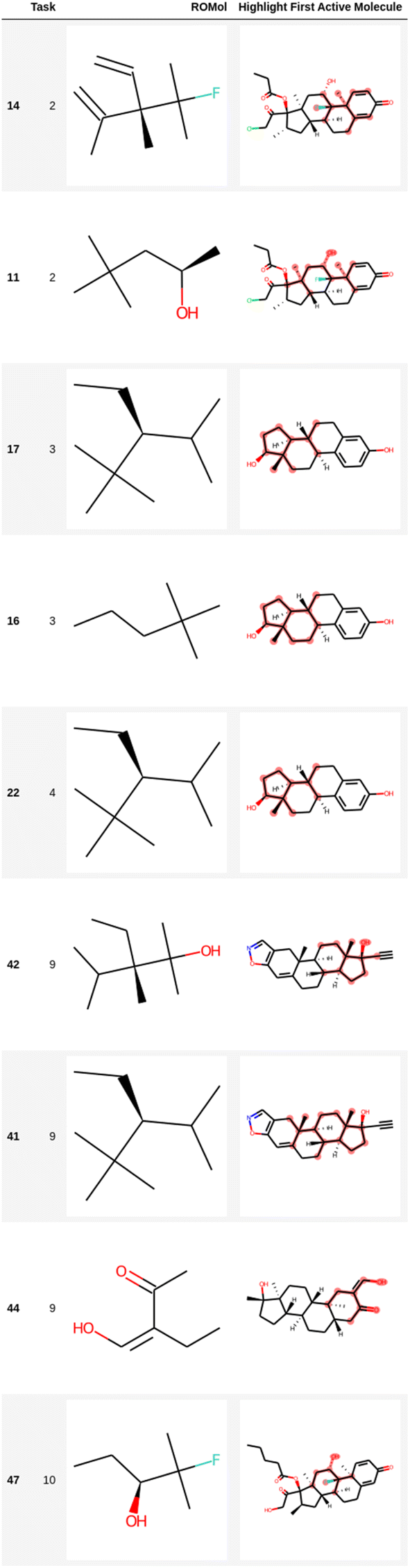 | ||
| Fig. 5 Representation of several fragments highlighted in the first active molecule of some tasks for the Sider dataset (Run #1). The other two runs are shown in the ESI.† The number in the first column is the index in the data frame. | ||
The potential of the structural alerts found here in this study was assessed based on the frequency of fragments interpreted using the toxicological information of thousands of compounds found in Tox21, Clintox, and Sider datasets. From the point of view of statistics, it is necessary to perform the search in a large dataset with hundreds or thousands of compounds to make the frequency of fragments significant. Therefore, the structural alerts found in this investigation were used to filter 50 to 250 chemical compounds in the large dataset with 4266 EGFR inhibitors. Thus, the structural alerts proposed here are suitable to filter chemical compounds as toxic alerts. However, it is important to emphasize that the structural alerts are only toxic alerts to flag potential toxic compounds and serve to help organic synthetic and pharmaceutical researchers on toxicology issues of complex chemical compounds in both academics and industry.
The attachment in the end of the ESI† shows key structural alerts found here using datasets of marketed drugs with adverse drug reactions58 and qualitative datasets of drugs approved by the FDA57 that have failed clinical trials for toxicity reasons. Many identified fragments for toxic alert were similar and sometimes identical within the 12 tasks in both Clintox and Sider datasets, showing the robustness of the method. However, it is important to mention the LIME limitations as follows.
Meanwhile, the fragments CCC[C@H](C)C and CCCC(C)C were interpreted as different substructures. However, they both may be part of the same molecules, and thus it is possible to indicate they are redundant fragments. Unfortunately, the explainable model used here to get structural alerts does not consider or is not able to solve this issue. LIME can generate dozens of substructures that are probably related, but further work needs to be performed to eliminate the redundant substructures. The other methods below also present issues on redundancy.
Another issue in structural alerts is the existence of two substructures in the same molecule. LIME does apply to this issue. There are some methods that also consider the existence of two or more substructures in the same compound.40 The emerging pattern40 is a method widely used, where a molecular pattern is identified as a set of molecular fragments. The jumping emerging pattern89 is a mining algorithm to find the patterns assigning atom pairs as descriptors. Then, the emerging pattern mining method uses the contrast pattern tree algorithm to find toxic features.90 Another issue may probably come from a structural alert generated from a dataset with false positives. The explainable model to find structural alerts may not correctly interpret the effect of existing redundant structural alerts in a set of false positive compounds. Although some methods exist, avoiding false positives in datasets is still challenging. Therefore, the future development of explainable models to find structural alerts should consider the generalization of specific structural alerts to avoid redundancy.
It is important to mention about the stability and consistency of LIME. The canonical SMILES representation used here ideally guarantees exclusive codes to uniquely define molecules.68 Despite this, molecules with different SMILES representations can be the same in practice. This occurs especially in 2-hydroxy-2,3-dimethylpentane, 2,2-dimethyl-3-hydroxypentane and 2-hydroxy-3-ethane-2,3,4-trimethylpentane which appear in the results of Clintox and Sider datasets. It is important to point out that these structural alerts are fragments of molecules. Although they are the same, they can be found in different spatial dispositions, being understood by LIME as different fragments. Fig. 6A shows the repeated structures in the result for the Clintox dataset (Run #1). For Runs #2 and #3, the repeated structures are shown in Fig. S11 and S12 in the ESI,† respectively. For the Sider dataset (Run #1), the repeated structures are shown in Fig. 6B. Runs #2 and #3 have the same repeated structures as compared with Run #1.
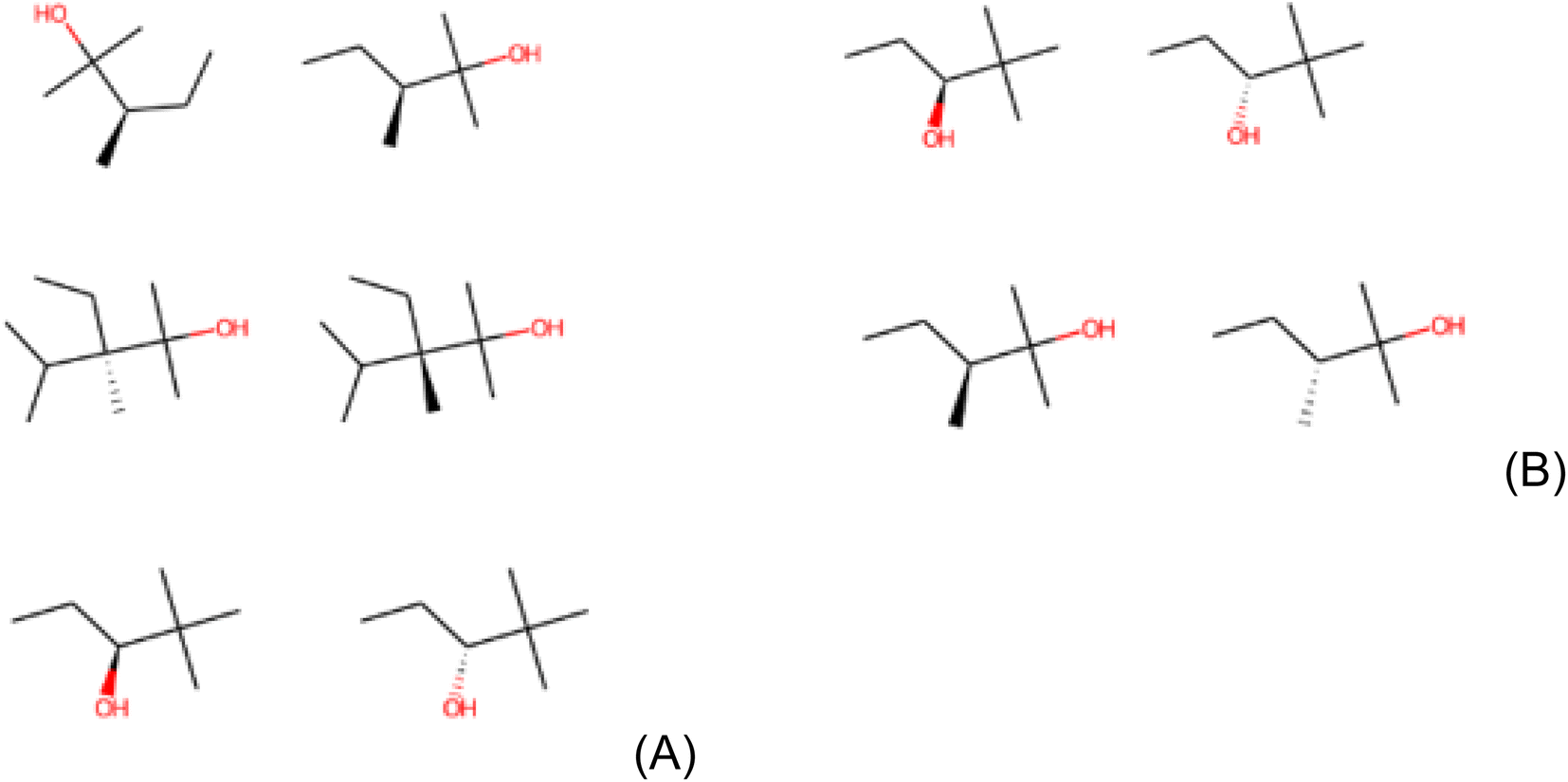 | ||
| Fig. 6 Representation of repeated structures in (A) Clintox and (B) Sider datasets (Run #1). The representations of the other two runs are shown in the ESI.† | ||
From the SMARTS patterns (available in the end of the ESI†), it is possible to verify the similarity between the three runs of Clintox dataset and the three runs of Sider dataset only to analyze the consistency of the LIME model applied in these datasets. Taking the 109 structural alerts generated by the Clintox dataset (Run #1), 98 appear in Run #2 and 89 are repeated in Run #3, corresponding to 90% and 82% similarities, respectively. Furthermore, Runs #2 and #3 have 96 similar structural alerts, corresponding to 79% and 85% of their structures, respectively.
Analyzing the 113 structural alerts generated by Run #1 in the Sider dataset, 96 structures are repeated in Run #2 (85% similarity) and 85 in Run #3 (75% similarity). In addition, Runs #2 and #3 have 95 similar structural alerts, corresponding to 71% and 83% of their structures, respectively. Finally, it is important to analyze all structures generated for the Clintox dataset (Runs #1, #2 and #3), excluding the repeated ones (total of 143 alerts), and for the Sider dataset (Runs #1, #2 and #3), excluding the repeated ones (total of 160 alerts). It is observed that there are only 38 non-repeated structures, showing high similarity and consistency between both results. For better visualization, Fig. S13–S15† present these data discussed here.
The proof of concept
Previously, we trained the model with the Tox21 datasets, and tested the model with the Clintox and Sider datasets, which are datasets of marketed drugs with adverse reactions and drugs approved by the Federal Drug Administration that have failed clinical trials for toxicity reasons. For sure, none of these datasets contain drugs with mutagenicity issues, so the model would not be capable of highlighting aromatic rings, nitrosamines, epoxides, and nitro compounds, for example. We also run the code with the Tox21 dataset with data splitting (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10) to check if this dataset would be capable of highlighting these fragments, but it does not yield any highlight of those. The explanation for this result is that the tasks (NR-AR, NR-AR-LBD, NR-AhR, NR-Aromatase, NR-ER, NR-ER-LBD, NR-PPAR-γ, SR-ARE, SR-ATAD5, SR-HSE, SR-MMP, and SR-p53) are not related to mutagenicity, so the models should not highlight aromatic rings. For our model, benzene is not toxic in any of the 12 tasks in the Tox21 dataset, as expected.
:10:10) to check if this dataset would be capable of highlighting these fragments, but it does not yield any highlight of those. The explanation for this result is that the tasks (NR-AR, NR-AR-LBD, NR-AhR, NR-Aromatase, NR-ER, NR-ER-LBD, NR-PPAR-γ, SR-ARE, SR-ATAD5, SR-HSE, SR-MMP, and SR-p53) are not related to mutagenicity, so the models should not highlight aromatic rings. For our model, benzene is not toxic in any of the 12 tasks in the Tox21 dataset, as expected.
One example of the proof-of-concept experiments will be predicting if a molecule that contains mutagenic fragments such as an aromatic ring, nitro group, nitrosamine group or an epoxide group would be caught by LIME. Different from toxicity tasks, these experiments should have a very clear ground truth for explanations. LIME should highlight these groups and it is a perfect experiment to see if LIME could pick the correct structures in such tasks. To prove this concept, we performed a preliminary LIME analysis using the Bursi mutagenicity dataset. It is a reasonably large data set, containing 4337 molecular structures with the relative Ames test results. We found many mutagenic fragments, including the C–CC group found in the aromatic ring, nitrosamines, (poly)halogenated compounds, thiols, nitro compounds, epoxides, etc. It is important to mention that all these groups are very well recognized as mutagenic. The top-5 mutagenic compounds found in this preliminary experiment are presented in Fig. 7.
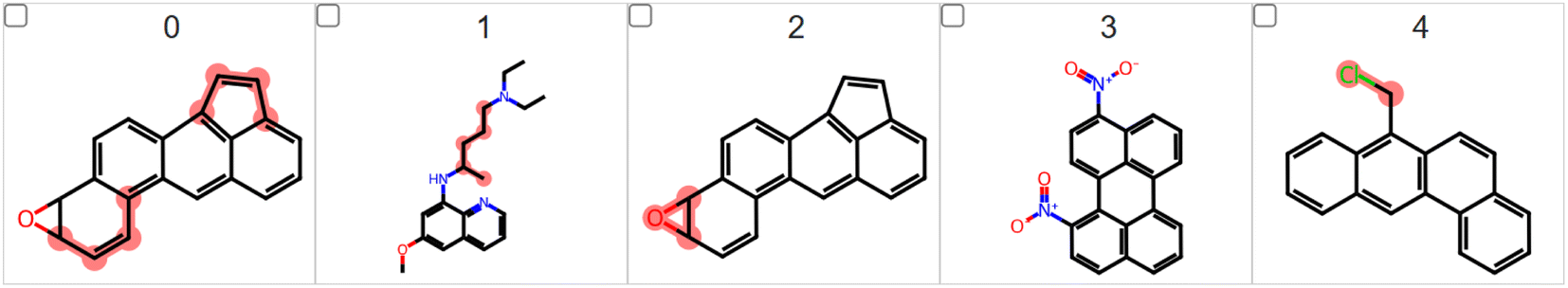 | ||
| Fig. 7 The top-5 mutagenic compounds found in the LIME analysis applied to the Bursi mutagenicity dataset choosing fragments larger than one heavy atom. | ||
Concluding remarks
The explanation model generated by LIME was successfully used to interpret datasets of marketed drugs with adverse reactions and qualitative datasets of drugs approved by the FDA that have failed clinical trials for toxicity reasons. These datasets are diverse and representative, yielding a non-redundant explanation in most toxic compounds. It was found that the local interpretable explanation model may be used to replace the machine learning model; the explanations were contrastive and simple, worked very well for the tabular data found in toxicological datasets, and the AUC measure indicated a good idea of how reliable the interpretable model is. However, as the data points were sampled using a Gaussian distribution, ignoring the feature correlation, LIME produced some redundancies in structural alerts. The generation of structural alerts by LIME might identify redundant and overspecific substructures. Therefore, it is still challenging to automatically detect structural alerts that are more convincing and beneficial to researchers. The instability of the explanations was not a real issue here because of the high reproducibility of the LIME method, making the explanations that come out from the analysis very similar. That means it is possible to trust in the explanations. The structural alerts found here may be used as toxic alerts by organic synthesis and pharmaceutical researchers in academics and industry.Abbreviations
| LIME | Local Interpretable Model-Agnostic Explanations |
| SHAP | SHapley Additive exPlanations |
| FDA | Federal Drug Administration |
| ECFPs | Extended Connectivity FingerPrints |
| EGFR | Epidermal Growth Factor Receptor |
| RO5 | Lipinski Rule of Five |
| PAINS | Pan-Assay Interference compounds |
| NR | Nuclear Receptor |
| SR | Stress Response |
| NR-AR | Androgen Receptor using the MDA cell line |
| NR-AR-LBD | Androgen Receptor Ligand Binding Domain |
| NR-AhR | Aryl hydrocarbon Receptor |
| NR-Aromatase | Aromatase enzyme |
| NR-ER | Estrogen Receptor α using the BG1 cell line |
| NR-ER-LBD | Estrogen Receptor α Ligand Binding Domain |
| NR-PPAR-γ | Peroxisome Proliferator-Activated Receptor γ |
| SR-ARE | Antioxidant Response Element |
| SR-ATAD5 | Luciferase-tagged ATAD5 in human embryonic kidney cells |
| SR-HSE | Heat Shock Response |
| SR-MMP | Mitochondrial membrane potential p53 response |
| SR-p53 | p53 response |
| RO | Receiver Operating Characteristics |
| AUC | Area Under Curve Statistical Method |
Data availability
Code, input data and processing scripts for this paper are available at github https://github.com/andresilvapimentel/SARAlerts (https://doi.org/10.5281/zenodo.7416484). The data analysis scripts of this paper are also available in the interactive notebook Google Colab.Author contributions
A. S. P. managed the project and funding acquisition, and supervised and conceptualized the investigation. All authors carried out the investigation including methodology, data curation, validation, testing, and graphic preparation. P. G. M. wrote most of the python code. A. S. P. and C. M. C. N. wrote, edited, and reviewed the manuscript.Conflicts of interest
No potential conflict of interest was reported by the authors.Acknowledgements
The authors are thankful to CNPq (Grants 573560/2008-0, 465259/2014-6 and 302554/2017-3), CAPES (Finance Code 001), and FAPESP (Grant 2014/50983-3). The authors also acknowledge the FAPERJ NanoHealth Research Network (E-26/010.000983/2019), the FAPERJ Support Program for Thematic Projects in the State of Rio de Janeiro (210.104/2020) and the National Institute of Science and Technology Complex Fluids (INCT-FCx) for funding. ASP was recently awarded the Scientist of Our State by FAPERJ (201.186/2022). He is also thankful for the research productivity fellowship granted by CNPq (310166/2020-9).References
- A. Lysenko, A. Sharma, K. A. Boroevich and T. Tsunoda, An integrative machine learning approach for prediction of toxicity-related drug safety, Life Sci. Alliance, 2018, 1, e201800098, DOI:10.26508/lsa.201800098.
- A. Mahmoud, A Survey of Computational Toxicology Approaches, J. Inf. Sci., 2021, 2, 1–7, DOI:10.21608/kjis.2021.41013.1008.
- J. Hemmerich and G. F. Ecker, Computational toxicology: a tool for all industries, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 2, 424–434, DOI:10.1002/wcms.100.
- I. Rusyn and G. P. Daston, Computational toxicology: Realizing the promise of the toxicity testing in the 21st century, Environ. Health Perspect., 2010, 118, 1047–1050, DOI:10.1289/ehp.1001925.
- N. le Dang, T. B. Hughes, G. P. Miller and S. J. Swamidass, Computational Approach to Structural Alerts: Furans, Phenols, Nitroaromatics, and Thiophenes, Chem. Res. Toxicol., 2017, 30, 1046–1059, DOI:10.1021/acs.chemrestox.6b00336.
- C. Helma, V. Schöning, J. Drewe and P. Boss, A Comparison of Nine Machine Learning Mutagenicity Models and Their Application for Predicting Pyrrolizidine Alkaloids, Front. Pharmacol., 2021, 12, 708050, DOI:10.3389/fphar.2021.708050.
- Y. Yang, Z. Wu, X. Yao, Y. Kang, T. Hou, C.-Y. Hsieh and H. Liu, Exploring Low-Toxicity Chemical Space with Deep Learning for Molecular Generation, J. Chem. Inf. Model., 2022, 62(13), 3191–3199, DOI:10.1021/acs.jcim.2c00671.
- Y. Kim, J. H. Jang, N. Park, N. Y. Jeong, E. Lim, S. Kim, N. K. Choi and D. Yoon, Machine Learning Approach for Active Vaccine Safety Monitoring, J. Korean Med. Sci., 2021, 36, e198, DOI:10.3346/jkms.2021.36.e198.
- C. Limban, D. C. Nuţă, C. Chiriţă, S. Negreş, A. L. Arsene, M. Goumenou, S. P. Karakitsios, A. M. Tsatsakis and D. A. Sarigiannis, The use of structural alerts to avoid the toxicity of pharmaceuticals, Toxicol. Rep., 2018, 5, 943–953, DOI:10.1016/j.toxrep.2018.08.017.
- T. C. de Campos and T. C. L. de Vasconcelos, Aplicação de algoritmos de machine learning na área farmacêutica: uma revisão, Res., Soc. Dev., 2021, 10, e140101522862 CrossRef.
- J. Hemmerich, F. Troger, B. Füzi and G. F. Ecker, Using Machine Learning Methods and Structural Alerts for Prediction of Mitochondrial Toxicity, Mol. Inf., 2020, 39, e2000005, DOI:10.1002/minf.202000005.
- J. Klekota and F. P. Roth, Chemical substructures that enrich for biological activity, Bioinformatics, 2008, 24, 2518–2525, DOI:10.1093/bioinformatics/btn479.
- Y. Wu and G. Wang, Machine learning based toxicity prediction: From chemical structural description to transcriptome analysis, Int. J. Mol. Sci., 2018, 19, 2358–2378, DOI:10.3390/ijms19082358.
- P. Cui, T. Zhong, Z. Wang, T. Wang, H. Zhao, C. Liu and H. Lu, Identification of human circadian genes based on time course gene expression profiles by using a deep learning method, Biochim. Biophys. Acta, Mol. Basis Dis., 2018, 1864, 2274–2283, DOI:10.1016/j.bbadis.2017.12.004.
- T. B. Hughes and S. J. Swamidass, Deep Learning to Predict the Formation of Quinone Species in Drug Metabolism, Chem. Res. Toxicol., 2017, 30, 642–656, DOI:10.1016/j.bbadis.2017.12.004.
- K. Ghosh, S. A. Amin, S. Gayen and T. Jha, Unmasking of crucial structural fragments for coronavirus protease inhibitors and its implications in COVID-19 drug discovery, J. Mol. Struct., 2021, 1237, 130366–130377, DOI:10.1016/j.molstruc.2021.130366.
- Y. Hua, X. Cui, B. Liu, Y. Shi, H. Guo, R. Zhang and X. Li, SApredictor: An Expert System for Screening Chemicals Against Structural Alerts, Front. Chem., 2022, 10, 916614, DOI:10.3389/fchem.2022.916614.
- A. Varnek and I. Baskin, Machine Learning Methods for Property Prediction in Chemoinformatics: Quo Vadis?, J. Chem. Inf. Model., 2012, 52, 1413–1437, DOI:10.1021/ci200409x.
- A. Lavecchia, Machine-learning approaches in drug Discovery: methods and applications, Drug Discovery Today, 2015, 20, 318–331, DOI:10.1016/j.drudis.2014.10.012.
- R. Rodríguez-Pérez, T. Miyao, S. Jasial, M. Vogt and J. Bajorath, Prediction of Compound Profiling Matrices Using Machine Learning, ACS Omega, 2018, 3, 4713–4723, DOI:10.1021/acsomega.8b00462.
- R. Rodríguez-Pérez and J. Bajorath, Interpretation of compound activity predictions from complex machine learning models using local approximations and shapley values, J. Med. Chem., 2020, 63, 8761–8777, DOI:10.1021/acs.jmedchem.9b01101.
- H. Yang, J. Li, Z. Wu, W. Li, G. Liu and Y. Tang, Evaluation of Different Methods for Identification of Structural Alerts Using Chemical Ames Mutagenicity Data Set as a Benchmark, Chem. Res. Toxicol., 2017, 30, 1355–1364, DOI:10.1021/acs.chemrestox.7b00083.
- N. Barr Kumarakulasinghe, T. Blomberg, J. Liu, A. Saraiva Leao and P. Papapetrou, Evaluating local interpretable model-agnostic explanations on clinical machine learning classification models, in Proceedings – IEEE Symposium on Computer-Based Medical Systems, Institute of Electrical and Electronics Engineers Inc., Rochester, 2020, pp. 7–12, DOI:10.1109/CBMS49503.2020.00009.
- V. M. Alves, E. N. Muratov, S. J. Capuzzi, R. Politi, Y. Low, R. C. Braga, A. v. Zakharov, A. Sedykh, E. Mokshyna, S. Farag, C. H. Andrade, V. E. Kuz'Min, D. Fourches and A. Tropsha, Alarms about structural alerts, Green Chem., 2016, 18, 4348–4360, 10.1039/c6gc01492e.
- R. Brenk, A. Schipani, D. James, A. Krasowski, I. H. Gilbert, J. Frearson and P. G. Wyatt, Lessons learnt from assembling screening libraries for drug discovery for neglected diseases, ChemMedChem, 2008, 3, 435–444, DOI:10.1002/cmdc.200700139.
- J. B. Baell and G. A. Holloway, New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays, J. Med. Chem., 2010, 53, 2719–2740, DOI:10.1021/jm901137j.
- T. Biniashvili, E. Schreiber and Y. Kliger, Improving classical substructure-based virtual screening to handle extrapolation challenges, J. Chem. Inf. Model., 2012, 52, 678–685, DOI:10.1021/ci200472s.
- O. Ursu, A. Rayan, A. Goldblum and T. I. Oprea, Understanding drug-likeness, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 760–781, DOI:10.1002/wcms.52.
- C. G. Bologa, O. Ursu and T. I. Oprea, How to prepare a compound collection prior to virtual screening, Methods Mol. Biol., 2019, 1939, 119–138, DOI:10.1007/978-1-4939-9089-4_7.
- T. I. Oprea, Property distribution of drug-related chemical databases, J. Comput.-Aided Mol. Des., 2000, 14, 251–264, DOI:10.1023/a:1008130001697.
- I. Sushko, E. Salmina, V. A. Potemkin, G. Poda and I. V. Tetko, ToxAlerts: A web server of structural alerts for toxic chemicals and compounds with potential adverse reactions, J. Chem. Inf. Model., 2012, 52, 2310–2316, DOI:10.1021/ci300245q.
- A. Lepailleur, G. Poezevara and R. Bureau, Automated detection of structural alerts (chemical fragments) in (eco)toxicology, Comput. Struct. Biotechnol. J., 2013, 5, e201302013, DOI:10.5936/csbj.201302013.
- H. Yang, L. Sun, W. Li, G. Liu and Y. Tang, In Silico Prediction of Chemical Toxicity for Drug Design Using Machine Learning Methods and Structural Alerts, Front. Chem., 2018, 6, 30, DOI:10.3389/fchem.2018.00030.
- C. A. Marchant, Computational toxicology: A tool for all industries, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 424–434, DOI:10.1002/wcms.100.
- R. Guha, On the interpretation and interpretability of quantitative structure-activity relationship models, J. Comput.-Aided Mol. Des., 2008, 22, 857–871, DOI:10.1007/s10822-008-9240-5.
- A. M. Doweyko, QSAR: Dead or alive?, J. Comput.-Aided Mol. Des., 2008, 22, 81–89, DOI:10.1007/s10822-007-9162-7.
- L. H. Bruner, G. J. Carr_F, M. Chamberlains and R. D. Currenq, Validation of Alternative Methods for Toxicity Testing, Environ. Health Perspect., 1998, 106, 477–484, DOI:10.1289/ehp.98106477.
- J. Ridings, R. Gary, C. Earnshawd, C. Eggingtond, M. Ellis, P. Judsonf, J. Langowskig, C. Marchantg, W. Watson, T. Yih and M. Payneh, Computer prediction of possible toxic action from chemical structure: an update on the DEREK system, Toxicon, 1996, 106, 267–279, DOI:10.1016/0300-483x(95)03190-q.
- G. Roberts, G. J. Myatt, W. P. Johnson, K. P. Cross and P. E. Blower, LeadScope : Software for Exploring Large Sets of Screening Data, J. Chem. Inf. Comput. Sci., 2000, 40, 1302–1314, DOI:10.1021/ci0000631.
- H. Yang, C. Lou, W. Li, G. Liu and Y. Tang, Computational Approaches to Identify Structural Alerts and Their Applications in Environmental Toxicology and Drug Discovery, Chem. Res. Toxicol., 2020, 33, 1312–1322, DOI:10.1021/acs.chemrestox.0c00006.
- M. Von Korff and T. Sander, On Exploring Structure Activity Relationships, J. Chem. Inf. Model., 2006, 46, 536–544, DOI:10.1007/978-1-62703-342-8_6.
- L. Xue and J. Bajorath, Molecular Descriptors in Chemoinformatics, Computational Combinatorial Chemistry, and Virtual Screening, Comb. Chem. High Throughput Screening, 2000, 3, 363–367, DOI:10.2174/1386207003331454.
- A. Liu, M. Walter, P. Wright, A. Bartosik, D. Dolciami, A. Elbasir, H. Yang and A. Bender, Prediction and mechanistic analysis of drug-induced liver injury (DILI) based on chemical structure, Biol. Direct, 2021, 16, 6, DOI:10.1186/s13062-020-00285-0.
- J. Dong, D. S. Cao, H. Y. Miao, S. Liu, B. C. Deng, Y. H. Yun, N. N. Wang, A. P. Lu, W. Bin Zeng and A. F. Chen, ChemDes: an integrated web-based platform for molecular descriptor and fingerprint computation, J. Cheminf., 2015, 7, 60, DOI:10.1186/s13321-015-0109-z.
- H. Yang, L. Sun, W. Li, G. Liu and Y. Tang, Identification of Nontoxic Substructures: A New Strategy to Avoid Potential Toxicity Risk, Toxicol. Sci., 2018, 165, 396–407, DOI:10.1093/toxsci/kfy146.
- A. Seal, A. Passi, U. C. Abdul Jaleel, D. J. Wild and O. S. D. D. Consortium, In-silico predictive mutagenicity model generation using supervised learning approaches, J. Cheminf., 2012, 4, 10, DOI:10.1186/1758-2946-4-10.
- I. Shah, J. Liu, R. S. Judson, R. S. Thomas and G. Patlewicz, Systematically evaluating read-across prediction and performance using a local validity approach characterized by chemical structure and bioactivity information, Regul. Toxicol. Pharmacol., 2016, 79, 12–24, DOI:10.1016/j.yrtph.2016.05.008.
- M. R. Zafar and N. Khan, Deterministic Local Interpretable Model-Agnostic Explanations for Stable Explainability, Mach. Learn. Knowl. Extr., 2021, 3, 525–541, DOI:10.3390/make3030027.
- A. Wojtuch, R. Jankowski and S. Podlewska, How can SHAP values help to shape metabolic stability of chemical compounds?, J. Cheminf., 2021, 13, 74, DOI:10.1186/s13321-021-00542-y.
- K. Jaganathan, H. Tayara and K. T. Chong, An Explainable Supervised Machine Learning Model for Predicting Respiratory Toxicity of Chemicals Using Optimal Molecular Descriptors, Pharmaceutics, 2022, 14, 832, DOI:10.3390/pharmaceutics14040832.
- B. Ramsundar, Molecular machine learning with deepchem, PhD thesis, Stanford University, 2018 Search PubMed.
- M. T. Ribeiro, S. Singh and C. Guestrin, arXiv, 2016, preprint, arXiv:1602.04938, DOI:10.48550/arXiv.1602.04938.
- S. M. Lundberg, P. G. Allen and S. I. Lee, arXiv, 2017, preprint, arXiv:1705.07874, DOI:10.48550/arXiv.1705.0787.
- N. B. Kumarakulasinghe, T. Blomberg, J. Liu, A. Saraiva Leao and P. Papapetrou, 33rd International Symposium on Computer-Based Medical Systems (CBMS), IEEE, Rochester, 2020 Search PubMed.
- F. Gabbay, S. Bar-Lev, O. Montano and N. Hadad, A LIME-Based Explainable Machine Learning Model for Predicting the Severity Level of COVID-19 Diagnosed Patients, Appl. Sci., 2021, 11, 10417, DOI:10.3390/app112110417.
- B. Ledel and S. Herbold, arXiv, 2022, preprint, arXiv:2209.07623, DOI:10.48550/arXiv.2209.07623.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. Pande, MoleculeNet: A benchmark for molecular machine learning, Chem. Sci., 2018, 9, 513–530, 10.1039/c7sc02664a.
- H. Altae-Tran, B. Ramsundar, A. S. Pappu and V. Pande, Low Data Drug Discovery with One-shot Learning, ACS Cent. Sci., 2017, 3, 283–293, DOI:10.1021/acscentsci.6b00367.
- M. T. Ribeiro, S. Singh and C. Guestrin, Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Association for Computing Machinery, San Francisco, 2016 Search PubMed.
- B. Ramsundar, P. Eastman, P. Walters, and V. Pande, Deep Learning for the Life Sciences, O'Reilly Media, Inc., Sebastopol, 2014 Search PubMed.
- pandas-dev/pandas: Pandas, DOI:10.5281/zenodo.3509134, (accessed June 2023).
- rdkit/rdkit: 2022_03_5 (Q1 2022) Release, https://zenodo.org/record/6961488#.YxtZBaDMLcc, (accessed June 2023) Search PubMed.
- J. D. Hunter, MATPLOTLIB: A 2D graphics environment, Comput. Sci. Eng., 2007, 9, 90–95, DOI:10.1109/MCSE.2007.55.
- C. R. Harris, K. J. Millman, S. J. van der Walt, R. Gommers, P. Virtanen, D. Cournapeau, E. Wieser, J. Taylor, S. Berg, N. J. Smith, R. Kern, M. Picus, S. Hoyer, M. H. van Kerkwijk, M. Brett, A. Haldane, J. F. del Río, M. Wiebe, P. Peterson, P. Gérard-Marchant, K. Sheppard, T. Reddy, W. Weckesser, H. Abbasi, C. Gohlke and T. E. Oliphant, Array programming with NumPy, Nature, 2020, 585, 357–362, DOI:10.1038/s41586-020-2649-2.
- A. M. Richard, R. Huang, S. Waidyanatha, P. Shinn, B. J. Collins, I. Thillainadarajah, C. M. Grulke, A. J. Williams, R. R. Lougee, R. S. Judson, K. A. Houck, M. Shobair, C. Yang, J. F. Rathman, A. Yasgar, S. C. Fitzpatrick, A. Simeonov, R. S. Thomas, K. M. Crofton, R. S. Paules, J. R. Bucher, C. P. Austin, R. J. Kavlock and R. R. Tice, The Tox21 10K Compound Library: Collaborative Chemistry Advancing Toxicology, Chem. Res. Toxicol., 2021, 34, 189–216, DOI:10.1021/acs.chemrestox.0c00264.
- M. Kuhn, I. Letunic, L. J. Jensen and P. Bork, The SIDER database of drugs and side effects, Nucleic Acids Res., 2016, 44, D1075–D1079, DOI:10.1093/nar/gkv1075.
- A. Tasneem, L. Aberle, H. Ananth, S. Chakraborty, K. Chiswell, B. J. McCourt and R. Pietrobon, The database for aggregate analysis of Clinicaltrials.gov (AACT) and subsequent regrouping by clinical specialty, PLoS One, 2012, 7, e33677, DOI:10.1371/journal.pone.0033677.
- D. Weininger, SMILES, a Chemical Language and Information System: 1: Introduction to Methodology and Encoding Rules, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36, DOI:10.1021/ci00057a005.
- D. Rogers and M. Hahn, Extended-Connectivity Fingerprints, J. Chem. Inf. Model., 2010, 50, 742–754, DOI:10.1021/ci100050t.
- A. Rácz, D. Bajusz and K. Héberger, Life beyond the Tanimoto coefficient: Similarity measures for interaction fingerprints, J. Cheminf., 2018, 10, 1–12, DOI:10.1186/s13321-018-0302-y.
- A. Capecchi, D. Probst and J. L. Reymond, One molecular fingerprint to rule them all: Drugs, biomolecules, and the metabolome, J. Cheminf., 2020, 12, 1–15, DOI:10.1186/s13321-020-00445-4.
- K. He, X. Zhang, S. Ren and J. Sun, arXiv, 2016, preprint, arXiv:1603.05027, DOI:10.48550/arXiv.1603.05027.
- A. P. Bradley, The use of the area under the ROC curve in the evaluation of machine learning algorithms, Pattern Recognit., 1997, 30, 1145–1159, DOI:10.1016/S0031-3203(96)00142-2.
- V. Giorgio, B. Enrico and C. Frederico, arXiv, 2020, preprint, arXiv:2006.05714, DOI:10.48550/arXiv.2006.05714.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani and J. P. Overington, ChEMBL: A large-scale bioactivity database for drug discovery, Nucleic Acids Res., 2012, 40, D1100–D1107, DOI:10.1093/nar/gkr777.
- E. Boutet, D. Lieberherr, M. Tognolli, M. Schneider, P. Bansal, A. J. Bridge, S. Poux, L. Bougueleret and I. Xenarios, UniProtKB/Swiss-Prot, the Manually Annotated Section of the UniProt KnowledgeBase: How to Use the Entry View, Methods Mol. Biol., 2016, 1374, 23–54, DOI:10.1007/978-1-4939-3167-5_2.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 2001, 46, 3–26, DOI:10.1016/s0169-409x(00)00129-0.
- C. A. Lipinski, Lead-and drug-like compounds: the rule-of-five revolution, Drug Discovery Today: Technol., 2004, 1, 337–341, DOI:10.1016/j.ddtec.2004.11.007.
- E. S. R. Ehmki, R. Schmidt, F. Ohm and M. Rarey, Comparing Molecular Patterns Using the Example of SMARTS: Applications and Filter Collection Analysis, J. Chem. Inf. Model., 2019, 59, 2572–2586, DOI:10.1021/acs.jcim.9b00249.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, Reoptimization of MDL keys for use in drug discovery, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280, DOI:10.1021/ci010132r.
- P. Schwaller, D. Probst, A. C. Vaucher, V. H. Nair, D. Kreutter, T. Laino and J. L. Reymond, Mapping the space of chemical reactions using attention-based neural networks, Nat. Mach. Intell., 2021, 3, 144–152, DOI:10.1038/s42256-020-00284-w.
- M. T. D. Cronin, S. J. Enoch, C. L. Mellor, K. R. Przybylak, A. N. Richarz and J. C. Madden, In Silico Prediction of Organ Level Toxicity: Linking Chemistry to Adverse Effects, Toxicol. Res., 2017, 33, 173–182, DOI:10.5487/TR.2017.33.3.173.
- X. Li, L. Chen, F. Cheng, Z. Wu, H. Bian, C. Xu, W. Li, G. Liu, X. Shen and Y. Tang, In Silico Prediction of Chemical Acute Oral Toxicity Using Multi-Classification Methods, J. Chem. Inf. Model., 2014, 54, 1061–1069, DOI:10.1021/ci5000467.
- T. Lei, Y. Li, Y. Song, D. Li, H. Sun and T. Hou, ADMET evaluation in drug discovery: 15. Accurate prediction of rat oral acute toxicity using relevance vector machine and consensus modeling, J. Cheminf., 2016, 8, 6, DOI:10.1186/s13321-016-0117-7.
- Y. Chen, F. Cheng, L. Sun, W. Li, G. Liu and Y. Tang, Computational models to predict endocrine-disrupting chemical binding with androgen or estrogen receptors, Ecotoxicol. Environ. Saf., 2014, 110, 280–287, DOI:10.1016/j.ecoenv.2014.08.026.
- R. D. Snyder, An update on the genotoxicity and carcinogenicity of marketed pharmaceuticals with reference to in silico predictivity, Environ. Mol. Mutagen., 2009, 50, 435–450, DOI:10.1002/em.20485.
- I. Cortes-Ciriano, Bioalerts: A python library for the derivation of structural alerts from bioactivity and toxicity data sets, J. Cheminf., 2016, 8, 13, DOI:10.1186/s13321-016-0125-7.
- A. Morger, M. Mathea, J. H. Achenbach, A. Wolf, R. Buesen, K. J. Schleifer, R. Landsiedel and A. Volkamer, KnowTox: Pipeline and case study for confident prediction of potential toxic effects of compounds in early phases of development, J. Cheminf., 2020, 12, 24, DOI:10.1186/s13321-020-00422-x.
- R. Sherhod, V. J. Gillet, P. N. Judson and J. D. Vessey, Automating knowledge discovery for toxicity prediction using jumping emerging pattern mining, J. Chem. Inf. Model., 2012, 52, 3074–3087, DOI:10.1021/ci300254w.
- R. Sherhod, P. N. Judson, T. Hanser, J. D. Vessey, S. J. Webb and V. J. Gillet, Emerging pattern mining to aid toxicological knowledge discovery, J. Chem. Inf. Model., 2014, 54, 1864–1879, DOI:10.1021/ci5001828.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dd00136e |
| This journal is © The Royal Society of Chemistry 2023 |