
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA high-throughput workflow for the synthesis of CdSe nanocrystals using a sonochemical materials acceleration platform†
Maria
Politi
 a,
Fabio
Baum
a,
Kiran
Vaddi
a,
Edwin
Antonio
a,
Joshua
Vasquez
b,
Brittany P.
Bishop
a,
Nadya
Peek
b,
Vincent C.
Holmberg
a and
Lilo D.
Pozzo
*a
a,
Fabio
Baum
a,
Kiran
Vaddi
a,
Edwin
Antonio
a,
Joshua
Vasquez
b,
Brittany P.
Bishop
a,
Nadya
Peek
b,
Vincent C.
Holmberg
a and
Lilo D.
Pozzo
*a
aDepartment of Chemical Engineering, University of Washington, Seattle, WA, USA. E-mail: dpozzo@uw.edu
bDepartment of Human Centered Design & Engineering, University of Washington, Seattle, WA, USA
First published on 23rd June 2023
Abstract
We present a complete open-hardware and software materials acceleration platform (MAP) for sonochemical synthesis of nanocrystals using a versatile tool-changing platform (Jubilee) configured for automated ultrasound application, a liquid-handling robot (Opentrons OT2) and a well-plate spectrometer. An automated high-throughput protocol was developed demonstrating the synthesis of CdSe nanocrystals using sonochemistry and different combinations of sample conditions, including precursor and ligand compositions and concentrations. Cavitation caused by ultrasound fields causes local and transient increases in temperature and pressure sufficient to drive the decomposition of organometallic precursors to drive the chemical reaction leading to nanocrystal formation. A total of 625 unique sample conditions were prepared and analyzed in triplicate with an individual sample volume of as little as 0.5 mL, which drastically reduced chemical waste and experimental times. The rapid onset of cavitation and quick dissipation of energy result in fast nucleation with little nanocrystal growth leading to the formation of small nanocrystals or magic-size clusters (MSCs) depending on composition. Using the effective mass approximation, the calculated QD diameters obtained under all our experimental conditions ranged between 1.3 and 2.1 nm, which was also validated with small angle X-ray scattering (SAXS). Polydispersity, QD shape and optical properties largely varied depending on the concentration of ligands present in solution. Statistical analysis of the spectroscopic data corroborates the qualitative relationships observed from the optical characterization of the samples with the model-agnostic SHAP analysis. The complete workflow relies on relatively low-cost and open-source systems. Automation and the reduced volumes also allow for cost-efficient experimentation, increasing the accessibility of this MAP. The high-throughput capabilities of the automated sonication platform, the extensible nature of the Jubilee system, and the modular nature of the protocol, make the workflow adaptable to a variety of future studies, including other nanocrystal design spaces, emulsification processes, and nanoparticle re-dispersion or exfoliation.
1 Introduction
Quantum dots (QDs) are materials with great scientific and technological interest, with applications in solar cells,1 light-emitting diodes (LEDs),2 biofluorescence tagging,3 sensors,4 and recently in neuromorphic hardware.5 They consist of semiconductor nanoparticles with sizes below the exciton Bohr radius. Their remarkable optical and electronic properties arise from the quantum confinement effect, which is strongly dependent on the QD size. Among QDs, there is a special class of nanocrystals known as magic-sized clusters (MSCs). These are formed under thermodynamic local minimum conditions that favor the growth of species with a precise number of atoms (the magic size) instead of continuous growth towards a large particle. Typically, CdSe MSCs show diameters below 2 nm. Their optical characterization presents narrow and sharp absorption peaks and broad photoluminescence peaks. These features limit the application of MSCs in devices that require color purity, but enable them to be used as white-light emitters.6 The optical properties of QDs and their subgroup, MSCs, are also strongly dependent on their surface functionalization. In this context, one possible method to improve surface passivation is the use of ligands through colloidal synthetic routes.7The most widely used technique for the synthesis of QDs and MSCs is the hot injection method.8 Its protocol includes precursor solution preparation under an inert atmosphere, followed by the injection step, which starts the decomposition of the organometallic species, leading to nucleation and growth processes. This synthesis usually involves the use of ligands with long carbon chains to stabilize the nanoparticle surface. This method presents great versatility and has been used to obtain metallic,9 chalcogenide,10 and perovskite nanocrystals11 with various shapes and sizes. The advantage of this method is the relationship between QD size and reaction time, which can provide great size control. Additionally, the fast nucleation events generated in the injection step allow the formation of monodisperse particles.8 However, this synthesis route requires high temperatures for precursor decomposition and the use of a Schlenk line to ensure an inert atmosphere. Under these conditions, only a single set of experimental parameters can be tested per batch, leading to lengthy procedures for exploratory or property optimization tasks. Furthermore, this method is prone to batch-to-batch variability, as well as observational and random errors, leading to inconsistent outcomes under the same set of conditions.
Sonochemical synthesis is an interesting alternative technique for the fabrication of nanostructured materials. It was used to obtain noble metal nanostructures,12–17 metal oxides,18,19 metal sulfides,20–24 and quantum dots.25–27 This technique provides thermal energy to the reaction medium through acoustic cavitation, the formation, growth, and implosive collapse of bubbles in a liquid due to a perturbation in the form of pressure waves. As ultrasonic waves travel through the solution, they generate cavitation bubbles, which increase in size and decrease in internal pressure. Finally, the bubbles reach a point of instability, culminating in their collapse. This process generates localized transient regions of extreme conditions in the solution, with temperatures around 5000 K and pressures of 500 atmospheres, with cooling rates as high as 109 °C s−1.28–30 Sonochemistry exploits these conditions to decompose the precursors and promote chemical reactions, leading to target compound formation, followed by its nucleation and growth into a particle. Among the advantages of sonochemical synthesis, its single pot synthetic procedure facilitates the scalability and reproducibility of the experimental design. Furthermore, sonochemistry does not necessarily require inert conditions and the synthesis can be carried out at room temperature, significantly simplifying its implementation.
Conventional nanomaterials synthesis schemes can be labor- and time-intensive, which significantly impedes the pace of new materials discovery and their applications. In this context, materials acceleration platforms (MAPs) have emerged as integrated ecosystems incorporating automation, artificial intelligence (AI), databases, scheduling and orchestration along with human creativity and intuition to speed up processes to discover, optimize, and use new materials.31 An ultimate objective of MAPs is to achieve autonomous experimentation through the combination of artificial intelligence with high-throughput robotic systems without requiring human intervention. High-throughput experimentation involves the use of automated and streamlined protocols for synthesis and characterization of a large number of samples in a short time interval. Moreover, the use of robotic platforms also promises to eliminate batch variability due to human errors, allowing systematic errors to be tracked and quantified, leading to more reproducible procedures. The main advantage of these platforms is the possibility of investigating broader experimental spaces for both synthesis and parameter optimization. Robotic platforms have been successfully applied to synthesize organic compounds of pharmaceutical interest,32 colloidal nanocrystals,33–36 and complex liquid mixtures,37 among others. However, most MAPs still present some limitations, such as ‘rigid’ designs (i.e., only useful for specific reactions or applications) and the high upfront cost of implementation in the laboratory. This limits incentives to integrate such platforms, and most wet-lab researchers still rely on low-throughput, time intensive workflows.38 To contrast this limitation and decrease the price barrier for a more wide-spread use of automation systems, open-source hardware (OSH) principles have made their use more accessible and more easily implemented for a variety of applications.39,40 These include solutions for high-throughput IR bolometry,41 the adaptation of 3D printers and platforms,42,43 platforms for modular automated workflows,38 pipetting robot platforms and autosamplers,44,45 and imaging,46 to name a few. OSH not only allows for free access to the system's design source, but also gives researchers the freedom to study, modify, distribute, and reproduce any component of the setup.47
Considering its characteristics, sonochemical synthesis is a perfect match to combine high-throughput experimentation with low-cost fabrication techniques. Herein, we present a new workflow, using an affordable liquid handling robot and an open-hardware sonication station. The OT-2 liquid handling robot, from Opentrons, is an affordable, open-source tool capable of preparing hundreds of precursor solutions to be synthesized and allows for flexible and reproducible protocols. The sonication station is based on a flexible tool-changing motion platform, called Jubilee, which is integrated with a sonication horn to allow for an array of automated sonochemical synthesis experiments. To demonstrate the implementation of these automation platforms, we explored how the combination of different experimental parameters affects the sonochemical synthesis of CdSe QDs and MSCs. CdSe is one of the most well-studied QD systems, with established relationships between optical properties, particle size, and surface passivation. Therefore, with simple characterization techniques, such as UV-Vis extinction spectroscopy and photoluminescence spectroscopy we could access information about particle shape, particle size, size distribution, particle concentration, surface passivation, and band gap energy to name a few.
In this context, herein we thoroughly investigate how the concentration of ligands (oleic acid and oleylamine) and precursors (cadmium acetate and elemental selenium) influences the properties of the resulting CdSe QDs or MSCs. In our design of experiments (DOE), we explored 5 different conditions or ‘levels’ for these four parameters using a full factorial design (54), resulting in 625 unique synthetic conditions. The experiments were repeated in triplicate to ensure repeatability of the workflow and to assess the reliability of the results obtained from each campaign. This number of syntheses would be infeasible or extremely costly, both in terms of time and resources, to perform using traditional low-throughput synthesis routes such as solvothermal methods. Furthermore, the automated workflow allows researchers to carry out experiments with sample volumes as small as 0.5 mL per condition tested, significantly reducing the cost and the environmental impact of the experimental process. The UV-Vis extinction and photoluminescence spectra were obtained using microplate readers, which allowed fast characterization of up to 96 samples at a time. Design of experiments, data analysis, and visualization routines were created with Python scripts, which facilitated the handling of the large volume of data resulting from the experimental campaigns. Fig. 1 shows a schematic of the workflow.
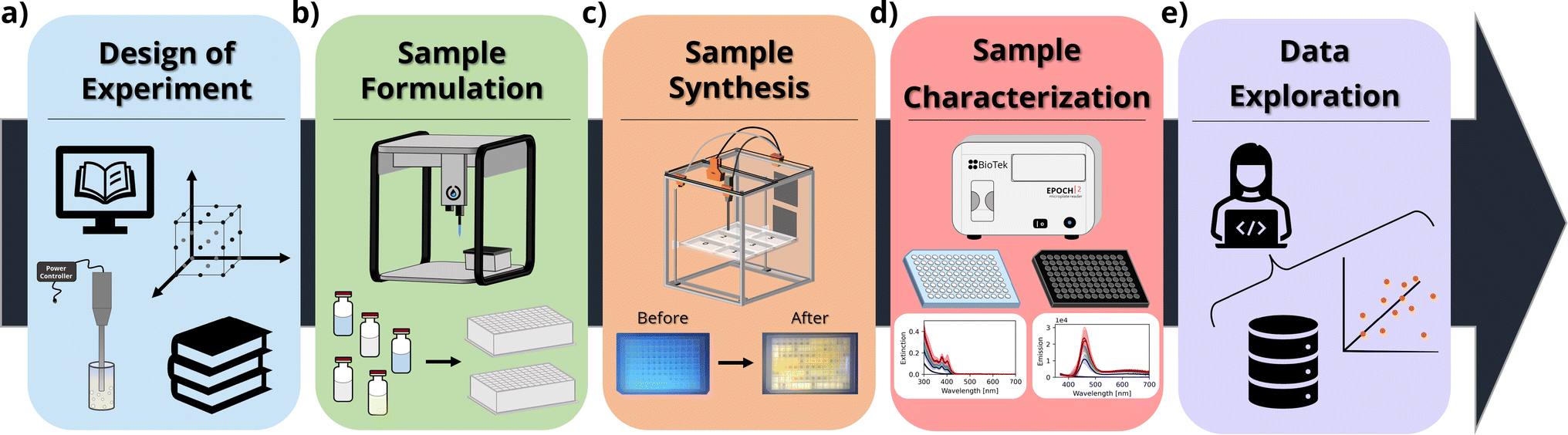 | ||
| Fig. 1 Example workflow schematics for the high-throughput sonochemical synthesis of CdSe nanocrystals. (a) The design of experiments is based on prior design knowledge from the literature and previous experimental campaigns. (b) The sample formulation is completely automated using a liquid handling robot and 96-well well plates. (c) The sample synthesis is conducted using an open-hardware platform for high-throughput sonication protocols. The samples are processed in the same 96-well plates they were formulated in. (d) After processing a plate reader is used to measure the samples' optical properties. (e) The data are then visualized and analyzed using Python scripts and open-source software. | ||
2 Experimental synthesis and characterization
2.1 Materials
An Opentrons OT-2 liquid handling robot was used to mix the precursors for all the samples, as well as for performing all dilution procedures for measurements. The robot was equipped with a single-channel Opentrons P300 GEN2 pipette (20–300 μL) and a single-channel P20 GEN2 pipette (1–20 μL). The pipette tips were stored on an Opentrons 300 μL tip rack and Opentrons 20 μL tip rack, respectively. To interface with the robotic platform, the Opentrons Python API was used to run commands through Jupyter Notebooks. In-house developed open-source software, OT2-DOE, was used to define the design space and communicate with the OT-2 Liquid Handling Robot.49 The calibration files for the labware used in the experimental campaign are also available on the software's GitHub repository.
A second robotic platform was implemented for the sonochemical synthesis of the samples. The platform, called a sonication station, is a configurable and open-source laboratory automation platform for sonochemical applications, based on the Jubilee motion control system. The original Jubilee design presents a modular tool-changing motion platform that accommodates user-created tools and beds.50,51 The sonication station was assembled with a QSonica Q125 sonicator, equipped with a 5/64 (2 mm) sonication probe (#4423). The platform allows for automated single-point sonochemical processing of samples, for a maximum of six plates loaded on the platform's deck at a time. The sonication station was interfaced using a Python script and the machine's API. The sonication station design and control libraries are open-source and available at https://github.com/machineagency/sonication_station.
3 Procedures
3.1 Stock solution preparation
The stock solutions were prepared under inert conditions in a glovebox due to air and water reactivity of trioctylphosphine (TOP). TOP was chosen as the solvent for the metal precursors due to the higher solubility of both anhydrous cadmium acetate and elemental selenium in this medium. For the chalcogenide precursor solution (0.5 M), 1.729 g of cadmium acetate anhydrous and 15 mL of TOP were added into a scintillation flask, heated to 125 °C and stirred until complete dissolution of the salt, usually around 4 h. For the selenium precursor solution (1 M), 1.201 g of selenium pellets and 15 mL of TOP were added into a scintillation flask and stirred at room temperature until complete dissolution, usually 10 h. Both scintillation flasks were sealed and stored for use in the OT-2 robotic platform. All other components, namely oleic acid, oleylamine, and 1-octadecene, were used in their pure form and did not require any processing before use.3.2 Automated sample formulation
The OT-2 Liquid Handling Robot was programmed to formulate different solutions containing a given amount of each of the four components under investigation in square deep-well 96 well plates. Octadecene was added to the solution to achieve a total sample volume of 0.5 mL. Each component was tested at five different concentrations (Table 1), resulting in 54 = 625 possible unique conditions. The synthesis was performed in batches of 96 samples in well plates, processed sequentially. The full list of the 625 different conditions can be found in the ESI† and in the GitHub repository for this study.| Parameter | Concentration (M) | ||||
|---|---|---|---|---|---|
| Cadmium precursor | 0 | 0.025 | 0.05 | 0.075 | 0.1 |
| Selenium precursor | 0 | 0.025 | 0.05 | 0.075 | 0.1 |
| Oleic acid | 0 | 0.125 | 0.25 | 0.375 | 0.5 |
| Oleylamine | 0 | 0.125 | 0.25 | 0.375 | 0.5 |
3.3 Sonochemical synthesis
After the solutions were prepared by the OT-2 robotic liquid handling platform, the 96 well plate was transferred to the sonication station. This station automatically sonicated each well in the well plate for 5 minutes, with a duty cycle of 0.5, a pulse interval of 15 s (7.5 s on/off), and a total time per sample of 10 minutes, using 50% amplitude from the 100 W source (20 kHz). Between the sonication of each sample, the probe underwent a cleaning cycle using four hexanol baths and two ethanol baths, each at maximum sonication power for 30 s, and was finally allowed to dry in air for 1 minute.3.4 Characterization
UV-Vis extinction spectroscopy was performed on a Biotek Epoch 2, using clear 96 well plates loaded with the as-synthesized CdSe solutions, diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 fold with 1-octadecene. The measurement was performed from 300 to 700 nm, with a step of 1 nm. The measurements were performed 24 hours after the synthetic procedure was complete.
:10 fold with 1-octadecene. The measurement was performed from 300 to 700 nm, with a step of 1 nm. The measurements were performed 24 hours after the synthetic procedure was complete.
Photoluminescence spectroscopy was performed on a Biotek Synergy H1, using black 96 well plates, loaded with the crude CdSe solutions diluted 1:100 fold with 1-octadecene. The measurement was performed with an excitation wavelength of 325 nm, from 300 to 700 nm, with a reading step of 1 nm. Once again, the measurements were performed 24 hours after the synthetic procedure was complete.
A subset of the experimental sample space was also reproduced and measured using a Xenocs Xeuss 3.0 (Grenoble, France) SAXS instrument. The samples were loaded into 1.5 mm diameter boron-rich capillaries (Charles Supper, 15BG) and measured at three different detector distances: 50 mm (WAXS), 370 mm (MAXS) and 900 mm (SAXS) to cover a broad q-range. The measurement was obtained using a copper source and the combined configurations allow the investigation of a scattering vector q ranging from 5 × 10−3 to approximately 1 Å−1. In this regime it is possible to obtain information about the particle size, polydispersity, and aggregation state after the data are adjusted by subtracting the scattering contribution from both the solvent and empty capillary. Absolute scaling was not possible due to small variations in the thickness of the sealed capillaries. Samples were run ‘as-synthesized’, as well as diluted 4-fold in n-decane to reduce interparticle correlations and to probe the dilute-limit form factor P(q) that is associated with intrinsic particle shape. The data reduction and processing were completed using the XSACT software, while the data fitting was completed using the open-source McSAS software.52,53
4 Results and discussion
The main appeal of using high-throughput platforms for materials synthesis is the speed at which large experimental spaces can be investigated. In fact, using the high-throughput synthesis and characterization workflow, all 625 unique samples composing the design space were completed (from formulation to data analysis) in less than two weeks. To ensure that both the workflow and the experimental conditions were reproducible, the campaign was repeated two more times, for a total of 1875 samples processed. For each replicate, new precursor solutions and fresh stocks were used in order to track any possible batch-to-batch variability between syntheses. The samples were prepared in a batch fashion, using commercially available 96-well plates to sonochemically synthesize the CdSe QDs. The use of 96-well ANSI/SLAS standards is convenient, as it is widely commercialized and also requires no further customization of sample holders for characterization techniques in, for example, commercial microplate readers. Since insonated wells share walls with neighboring wells, it is important to characterize any bulk temperature changes that could affect the synthesis outcomes. An IR camera was used to monitor bulk temperature changes during sonication as shown in Fig. S2.† The maximum bulk temperature of samples being processed changes from 22 °C to 35 °C after 10 minutes of processing; these correspond to 5 minutes of active sonication at a 50 percent duty cycle. The neighboring wells also present a slight increase in temperature, up to 28 °C due to conduction. While a bulk temperature variation of up to 12 °C is measurable, this is very small compared to the transient extreme conditions caused by the implosive acoustic cavitation events (5000 K), which is what drives the sonochemical reaction.29 The total time for the synthetic procedure was approximately 30 hours per plate (96 samples). This short experimental time was possible as most of the longer processing steps (e.g., the sonication of the samples) were fully automated and required no human intervention. On the other hand, even though the conventional hot-injection synthesis protocol is a more established synthesis route, it would have been prohibitive to implement for this type of experimental study, and the entire campaign run in triplicate would have taken well more than a year to complete. The sonochemical material acceleration platform poses a great advantage compared to the traditional hot-injection synthesis route when comparing the volumes of materials required to complete the experimental campaign. Table 2 summarizes the total volume of each stock solution required to formulate all 1875 samples, at a total volume of 0.5 mL per sample. These values include excess stock, approximately 15% of the required amount for each component, to allow the use of the OT-2 pipetting robot. Values for the thermochemical route would be at least one order of magnitude higher, as its lowest working volume usually amounts to approximately 5 mL. Consequently, high-throughput platforms are generally more cost effective and environmentally friendly (i.e., significantly less waste is produced) for the same amount of information extraction. Finally, the sonochemical synthesis of nanocrystals could also vary depending on the reactor size. Synthetic outcomes in sonochemistry are known to be affected by the energy (W) delivered per volume. It is possible, however, to adjust the total energy delivered by increasing the ultrasound power, the probe size, and the total processing time, to achieve similar conditions to those in vessels of different sizes. In the current study, sonication conditions are selected based on the specific sample volume of our experiments, 0.5 mL. The ultrasound probe was set at 50 percent of maximum power, which is 100 W. This can be scaled up or down based on a changing sample volume, or to probe for the effect of power density in future studies and applications.| Component | Volume (mL) |
|---|---|
| Cadmium acetate/TOP [0.5 M] | 108.0 |
| Selenium/TOP [1.0 M] | 53.8 |
| Oleic acid | 84.5 |
| Oleylamine | 88 |
| 1-Octadecene | 742.4 |
4.1 Effect of ligand concentration and sample composition on CdSe size and optical properties
To determine the effect of the synthetic conditions on the optical properties of the nanocrystals, we measured their absorption and emission spectra. Given the number of dimensions of the experimental space and the large amount of data generated, the data visualization task became a challenge. To address this challenge, the spectra were shown in two different groups: one based on the precursor concentration, and the other based on the ligand concentration. Even though it only allows one to analyze a subset of conditions at a time, it still enables the extraction of general trends in the synthesis from UV-vis extinction and photoluminescence emission spectra. Fig. 2 shows the UV-Vis absorption spectra for the samples synthesized with different Cd(OAc)2 and elemental selenium precursor concentrations, at 0 M and 0.25 M for oleic acid and oleylamine, respectively. As expected, when either cadmium or selenium precursors were absent, no optical signature from quantum dots was observed (e.g., the left-most subplot in Fig. 2). On the other hand, when both precursors are present, for this set of ligand conditions, the absorption intensity tends to increase without variation in the peak number or peak position. This indicates that, at our experimental range, cadmium or selenium concentrations and ratio did not have an influence on the particle size, but only on the number of particles formed. This trend is in line with previous findings by Jiang et al.54 Next, to obtain structural insight into the formed nanocrystals, the effective mass approximation equation, based on the first peak position in the absorption spectra, was used to estimate the particle size.55 Furthermore, the size distribution of the particles was qualitatively estimated from the broadness of the absorption peaks, where broad peaks or ‘shoulders features’ indicate polydispersed particles.56 The calculated QD diameters obtained for all our experimental conditions ranged between 1.3 and 2.1 nm. Finally, looking at the shaded areas indicating the variation across the three repeats of the experimental procedure, we noticed good repeatability of the syntheses and little to no variation in the peak position. This confirmed the fact that the tested experimental conditions are repeatable, which increases the validity of the results obtained. Small differences were only seen for the peak intensity, which could indicate some variability in the concentration of the QDs formed. These variations in concentration are likely due to mixing of the sample right before aliquot sampling for dilution preceding the spectroscopic characterization. However, these were mainly noticed in those samples that did not contain any stabilizing agent. This was expected as ligands not only play an important role in making the particles colloidally stable, but they can also be used for tuning the nucleation and growth of these nanocrystals.57 However, these fluctuations are still within reason (most of them are below 5%) and did not affect the repeatability of the experimental outcomes. Nevertheless, the use of this automated MAP for nanocrystal synthesis provides a significant reduction in random and human errors which are more prominent in traditional, single batch synthesis routes such as the solvothermal or hot-injection method. This once again confirms the great advantage of using an automated workflow for nanocrystal synthesis. This level of repeatability in such a large design space would not have been possible to achieve following the traditional thermochemical route.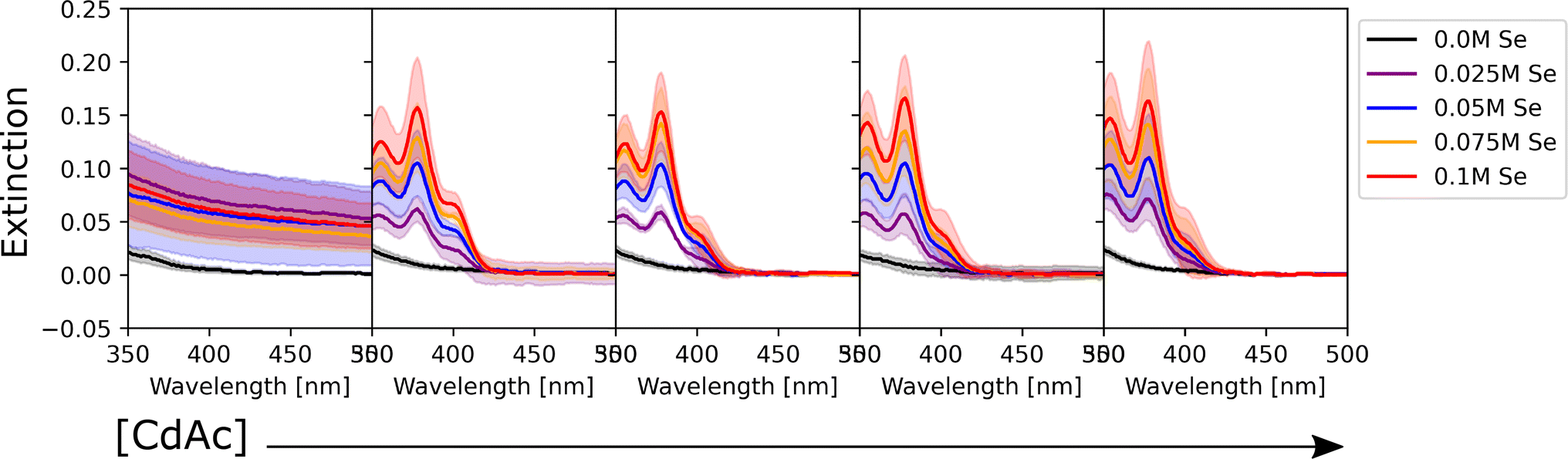 | ||
| Fig. 2 UV-Vis absorption spectra of the CdSe QDs synthesized under varying precursor conditions and with an oleic acid and oleylamine concentration of 0 M and 0.25 M, respectively. The color of the spectra corresponds to different selenium conditions (see legend), whereas the concentration of cadmium acetate increases moving from left to right (0 M, 0.025 M, 0.05 M, 0.075 M, and 0.1 M, respectively). Since the data were collected in triplicate, the shaded area of the corresponding color shows the variation in response across the different batches tested. | ||
As mentioned previously, the data were also grouped with respect to the metal precursor conditions. Fig. 3 presents the UV-Vis spectra of the samples with the highest precursor concentrations (0.1 M for both cadmium acetate and elemental selenium). This representation allowed us to identify two main trends in the data. The former presents broad peaks or ‘shoulders’ at longer wavelengths (450 nm) when oleylamine is absent or its concentration is exceeded by that of oleic acid. This is related to the formation of regular QDs. The latter showed sharp, narrow peaks at specific positions at shorter wavelengths (<450 nm) indicating the formation of magic sized clusters, with absorption first peak positions centered at wavelengths of 380, 405, and 417 nm. MSCs presenting such optical features were reported in the literature for 380,58 405,59 and 417 nm,58,60 and we will refer to these structures as CdSe(380nm), CdSe(405nm), and CdSe(417nm). Magic-sized QDs with similar characteristics were previously reported,58–61 obtained through the traditional hot-injection synthesis. This confirmed the ability of our platform to achieve nanocrystals of similar characteristics as those produced by the traditional synthetic route. In Fig. 3, it is noticed that, as the concentration of oleylamine increases (left to right), the characteristics of magic size cluster formation are more predominant. This was only true if the amount of oleylamine was larger than that of oleic acid. In this case, primarily narrower and sharper absorption peaks were present, while as the fraction of oleic acid increased, the peaks became broader. The broadening of the absorption peak in QDs can be related to the widening of the size distribution. This behavior was previously reported in typical hot-injection syntheses of QDs.56 At the concentrations studied in this paper, oleylamine was reported to act to stabilize the nuclei by reducing the solubility of the monomer (CdSe molecular units) in the solution and promoting nucleation of smaller nuclei.62 This led to the formation of smaller QDs with increased monodispersity or even magic-sized QDs.63 This was reinforced by the observation of the UV-Vis spectra of the samples prior to the sonication step (Fig. S4†). The samples containing higher amounts of oleylamine presented absorption spectra characteristic of magic-sized quantum dots even before undergoing sonochemical processing. On the other hand, increasing the concentration of oleic acid was reported to increase the solubility of the monomer in the solution, de-stabilize the nuclei, and generate less nucleation.56 As expected, from Fig. 3 and S13,† it could also be noticed that generally the samples that did not contain any ligands, had a higher variation in intensity, compared to the surface stabilized samples. Although processing differences are inevitable, knowledge obtained from sonochemical synthesis can still be transferred to other synthesis technique. For example, the effects of variations in coordinating ligands on the morphological properties of nanocrystals are broadly consistent between sonochemical and thermochemical methods. However, with respect to the achievable maximum particle size and distribution, there are significant differences. In fact, due to the transient nature of temperature and pressure conditions during cavitation, sonochemistry favors nucleation over growth. This facilitates the formation of magic-sized clusters and smaller particles, but it makes it more challenging to grow larger ones. Nonetheless, it is often possible to obtain larger particles by increasing the sonication time. A study by Kristl et al. on the same types of nanocrystals reported particle sizes between 6 and 10 nm, after 1 h of insonation.64 Other approaches to increase the size of nanocrystals are to modify the structure and properties of the coordinating ligands or the precursors.
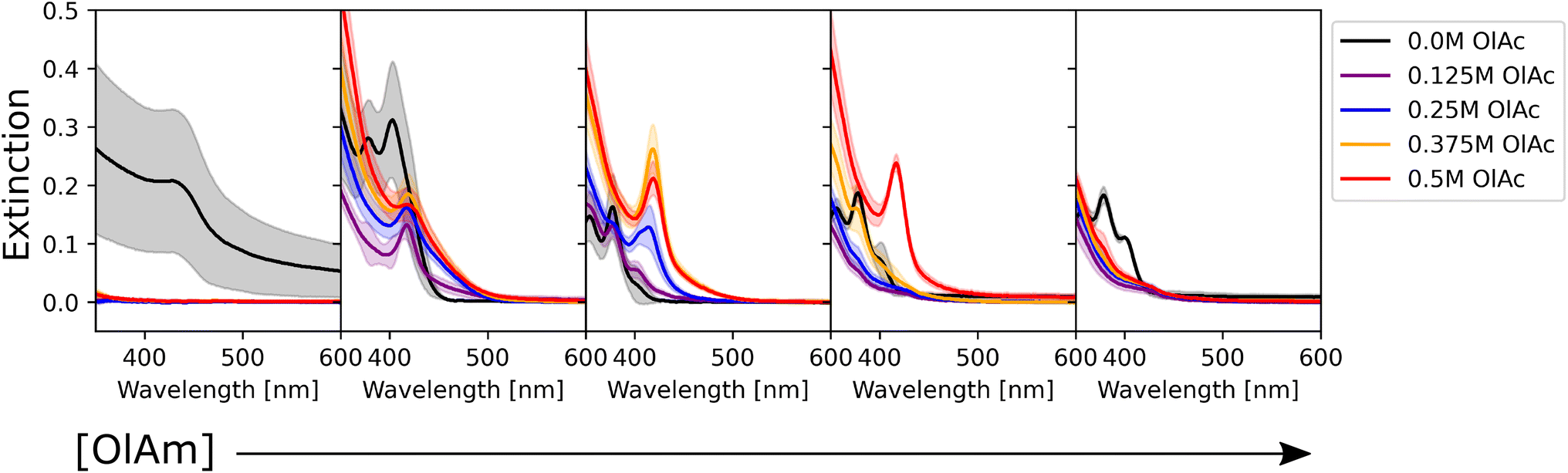 | ||
| Fig. 3 UV-Vis absorption spectra of the CdSe QDs synthesized at the highest precursor concentration (0.1 M for both components) with varying ligand concentrations. The color of the spectra corresponds to different oleic acid conditions (see the legend), whereas the concentration of oleylamine increases moving from left to right (0 M, 0.125 M, 0.25 M, 0.375 M, and 0.5 M, respectively). Since the data were collected in triplicate, the shaded area of the corresponding color shows the variation in response across the different batches tested. | ||
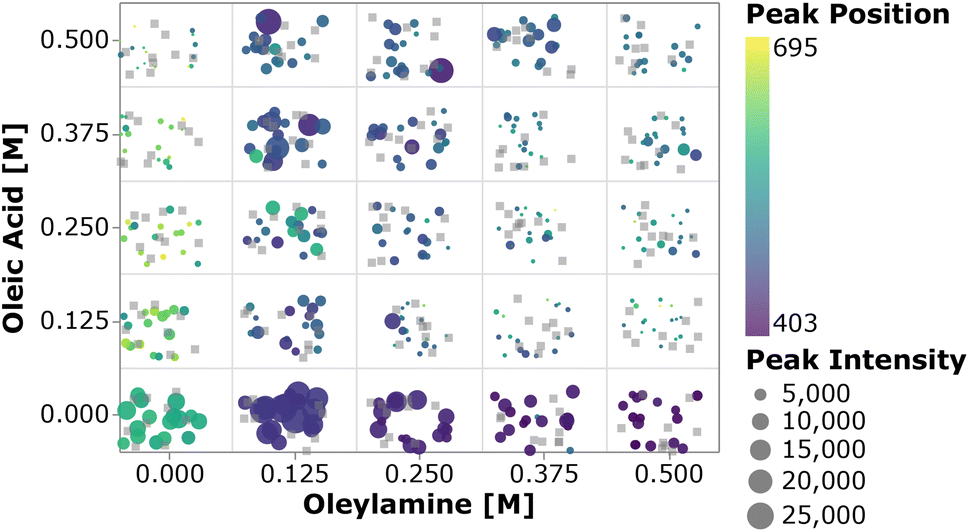
To perform a high-level evaluation of the experimental effects on the synthesis, one needs to consider the entire set of data. However, given the high dimensionality of the experimental space and the large amount of data generated, it becomes impossible to visualize all the spectra at once. Therefore we summarized the most important quantifiable parameters from the absorption spectra using a scatter plot. In Fig. 4, the color of each point represents the first peak position, while the point size represents the average absorption intensity at 300, 320 and 340 nm, which is used as a proxy for the number of particles.56,65 To prevent overlap of the data points and ease the visualization, jitter noise was added to the plot. Therefore, small spatial variations between points which fall into the same quadrant do not carry meaningful information. Each box inside the plot is related to the combination of precursor and ligand concentrations indicated on the axis. When samples are grouped based on the precursor concentration, one cannot see a clear trend in the absorption peak position distribution. The absence of trends indicates that the absorption peak position, related to the QD size, is not greatly affected by the precursor concentration. Additionally, this visual representation confirms that no nanocrystals were formed when cadmium or selenium precursors were absent. In general, for the entire design space the data showed that increasing the concentration of the metal precursors only favored the formation of more particles of the same size, not the growth of the existing ones.66 On the other hand, when grouped based on the ligand concentration, all the samples followed the trends previously observed. This can be discerned by the fact that each quadrant is dominated by a single color for its markers. By drawing a diagonal line from the origin to the top right, the plot can be split into two main regions, based on the ligand amount and proportion. The lower right diagonal region of the plot, in which samples contained higher concentrations of oleylamine and lower amounts of oleic acid, presented peaks below 415 nm. This value corresponds to a particle size of approximately 1.7 nm. Additionally, samples in this region presented lower absorption intensity peaks when compared to those under the remaining experimental conditions. This was mainly related to the fact that alkylamines are reported to bind more strongly to the Cd2+ ion than carboxylic acids, slowing down the reaction to form CdSe monomer units. This led to a smaller number of nanocrystals being formed and consequently lower absorption intensities.63 In summary, increasing the amount of oleylamine or the OLAm/OAc ratio led to a lower concentration of smaller, monodispersed QDs, while the opposite yielded larger concentrations of bigger polydispersed QDs. This was confirmed by looking at the upper left diagonal region in the plot. Here, higher peak positions and generally higher intensities (i.e., larger marker sizes) were present, as well as peaks at wavelengths above 415 nm. It was also interesting to notice that, for the case in which only oleic acid was present in the sample, no absorption feature was observed. It is possible that the cadmium salt contained in these samples formed a stable intermediate cadmium oleate species, therefore inhibiting the nucleation of CdSe nanocrystals. This would explain why these carboxylic acid-terminated ligand containing samples showed no or an extremely small optical response.
 | ||
| Fig. 4 Scatter plot of the average intensity at 300, 320, and 340 nm and the first peak position against the precursor concentration (top) and ligand concentration (bottom). Since each combination of conditions yielded 25 samples, noise was added to the data for plotting purposes. The grey squares indicate that no extinction response was observed for those samples. | ||
We also studied the impact of the synthetic parameters on the photoluminescence. Fig. 5 shows the photoluminescence spectra of the CdSe QDs. The strong signal below 400 nm present in some of the samples is related to scattering of the excitation beam, possibly due to unreacted ligands in the solution. By analyzing the spectra, three main groups of samples were found: (i) samples with no emission or weak emission overwhelmed by the excitation beam scattering; (ii) samples with broad emission peaks between 600 and 700 nm; and (iii) samples with one sharp peak at around 450 nm, with a small or absent secondary peak at higher wavelengths. The optical behavior of the first group of samples was related to the absence of QD formation or the formation of QDs with poor surface passivation, leading to non-radiative recombination. This behaviour was more pronounced at higher concentrations of oleic acid and oleylamine. The spectral characteristics of the second group were related to surface traps,67 which leads to recombination at a lower energy (higher wavelength) than the typical band gap of CdSe QDs. This behavior was prominent when lower concentrations of oleic acid were employed or in TOP-only syntheses (i.e., no ligands were added to the sample formulation). The samples with no ligands showed a similar behavior to those containing only oleic acid, with the surface trap recombination playing an important role in the light emission from the particles. Additionally, increasing the oleic acid concentration generally caused a decrease in the intensity of emission. These results might suggest that in this experimental setup, the presence of oleic acid was deleterious to surface passivation, leading to surface trap-based luminescence or surface trap related non-radiative recombination. A particular characteristic of our experimental setup was the cadmium precursor solution, consisting of cadmium acetate dissolved in trioctylphosphine instead of the commonly used cadmium oleate. This was again chosen as it allowed production of stock solutions with higher concentrations of precursors, which in turn allowed the overall sample volume to be limited to 500 μL. Therefore, it is possible that the addition of oleic acid in the samples containing both ligands might not form cadmium oleate, which is known to play an important role in achieving surface passivation. Instead, this fatty acid would most likely bind as an uncharged type L ligand to the surface of CdSe nanocrystals, decreasing its capacity to passivate the QD's surface. Finally, the last identified trend was present in samples containing only oleylamine or oleylamine in higher amounts than oleic acid. These sample conditions showed a sharp, intense, and well-defined peak, typical of band-edge luminescence. In these samples, the surface-trap luminescence played a small role compared to the main peak. However, as the concentration of oleylamine increased, the intensity of the peak started to decrease (black curves in Fig. 5).
 | ||
| Fig. 5 Photoluminescence spectra of the CdSe QDs synthesized at a higher precursor concentration (0.1 M for both components) with varying ligand concentrations. The color of the spectra corresponds to different oleic acid conditions (see the legend), whereas the concentration of oleylamine increases moving from left to right (0 M, 0.125 M, 0.25 M, 0.375 M, and 0.5 M, respectively). Since the data were collected in triplicate, the shaded area of the corresponding color shows the variation in response across the different batches tested. | ||
Alkylamines are known as excellent surface passivators, and previous reports on syntheses of quantum dots reported their capacity to increase photoluminescence quantum yield.68 In fact, they can coordinate with dangling bonds on the particle surface to mitigate surface trap states, enhancing the photoluminescence intensity. While phosphines, such as TOP, will passivate selenium dangling bonds, alkylamines will coordinate with cadmium ones on the particle surface.68 Finally, we note that there was little variation across the triplicate runs performed, once again confirming the reproducibility of the workflow under a wide range of conditions. Fig. 6 shows how the combination of oleylamine and oleic acid affected the photoluminescence intensity from band edge recombination for the entire collected dataset. It can be noticed that photoluminescence was stronger in the absence of oleic acid and at low concentrations of oleylamine. Most of the remaining samples showed either no feature in their spectra, caused by scattering of unreacted ligands or low intensity peaks at wavelengths above 600 nm, confirming the trends observed from the photoluminescence spectra.
 | ||
| Fig. 6 Scatter plot of emission intensity and the peak position versus the ligand concentration. Since each combination of conditions yielded 25 samples, noise was added to the data for plotting purposes. The grey squares indicate that no emission response was observed for those samples. | ||
Finally, a smaller subset of the samples was repeated and characterized using small-angle X-ray scattering, a non-destructive technique that allows for accessing structural and morphological information in materials across time scales which can range from angstroms to micrometers. Table S1† shows the sample compositions tested. Since the particle size had a higher dependence on the ligand concentration and composition, the samples were all tested at the highest metal precursor concentration, 0.1 M for both cadmium acetate and selenium. Once again, these samples were measured ‘as-synthesized’ and were not purified prior to characterization. SAXS was conducted before and after sonication of the samples to gain more insights into the changes that occur during the processing step. In SAXS measurements, the intensity data are generally plotted as a function of the scattering vector ‘q’, which is related to the wavelength of the X-ray beam and the scattering angle.69 This quantity can be inversely related to the characteristic dimensions of the sample, so smaller particles will present features at larger ‘q’ values; the opposite is observed for larger particles. Fig. 7 shows SAXS profiles for three samples having different ligand compositions. The panel on the left had no ligands in solutions and presents signs of particle aggregation. In fact, the sample profile post sonication (dark blue markers) shows several upturn features at different ‘q’ values and the slope of the SAXS curve keeps increasing towards lower ‘q’ values without reaching a plateau. This feature is not present, or not as prominent as in this sample, in the remainder of the samples tested using this technique, see Fig. 7 and S22–S28.† This behaviour is in line with the UV-Vis spectroscopy data, which showed larger batch-to-batch variation and the formation of large particles for samples synthesized without addition of extra stabilizing ligands.
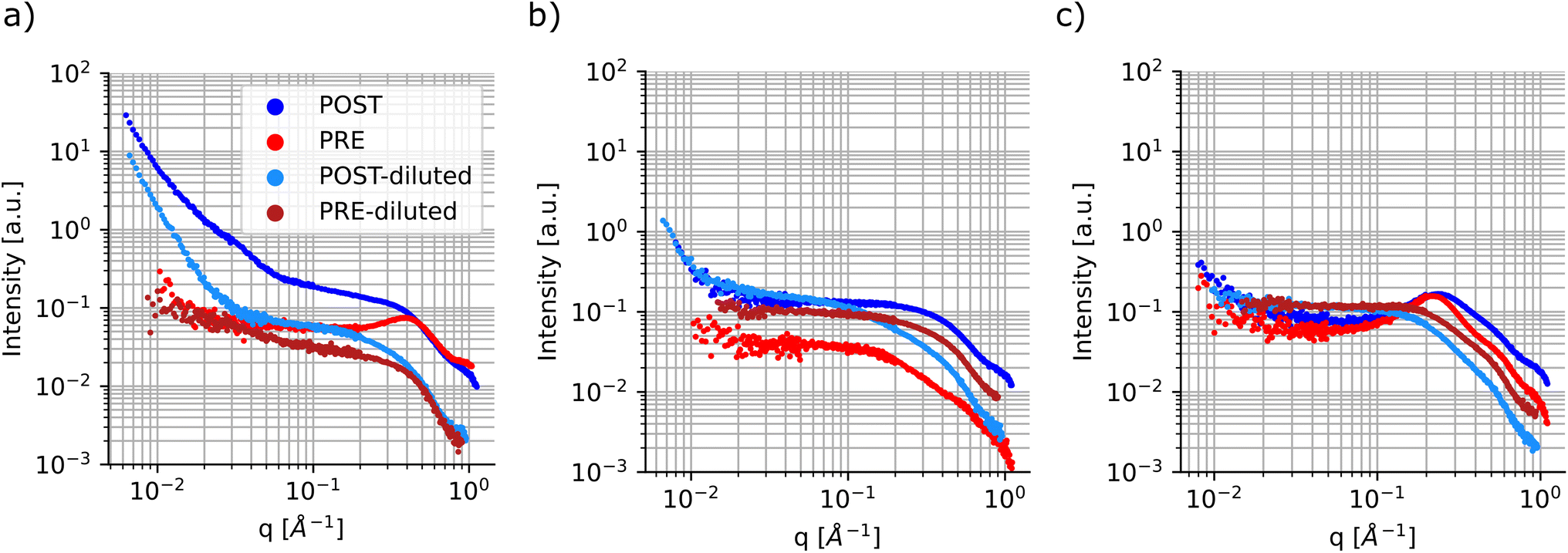 | ||
| Fig. 7 SAXS profiles of CdSe QDs sonochemically synthesized. SAXS profiles were collected for samples before and after sonochemical processing. Furthermore, to gain better insight into the particle size distribution, the samples were also diluted 4-fold in n-decane, see the legend. (a) SAXS profile for CdSe samples containing no ligands and metal precursors at 0.1 M each. (b) SAXS profile for CdSe samples containing only oleylamine at 0.125 M and metal precursors at 0.1 M each. (c) SAXS profile for CdSe samples containing oleylamine at 0.375 M, oleic acid at 0.5 M and metal precursors at 0.1 M each. | ||
Another feature that can be noticed in the SAXS profile is the presence of a structure factor, which appears as a local peak in the intensity curve. This attribute is related to spatial interactions between particles. In this case, as expected, this behavior is more pronounced in the samples prior to sonication and in those samples containing higher concentrations of ligands, especially oleic acid (Fig. 7c). Additionally, this can also be observed in samples post-sonication, again predominantly in those containing larger amounts of coordinating ligands (Fig. S23, S24, S26, and S29†). This dependency on the spatial interactions among samples is minimized or removed by diluting the samples prior to SAXS characterization.
The diluted samples were characterized and fit using a polydisperse sphere model to extract the particle size distributions through the McSAS software. This is an open-source Monte-Carlo regression package that allows for the determination of model parameter distributions, such as the particle size distribution, which allows the polydispersity of the samples to be more quantitatively assessed.52 It is evident that the samples prior to sonication show coordination of the precursors. This can be confirmed by the fact that all samples tested show scattering contributions at ‘high-q’ values in the same range, between 4.5 Å−1 and 6 Å−1 regardless of sample composition (see Fig. 7 and S22–S28†). It is noticeable that some of the samples show small scattering contributions at very high ‘q’ values over 7 Å−1. This feature is caused by slight variations in the diameter of the capillaries that were used to hold the samples during the SAXS measurements, so it was ignored during the fitting protocol. When fitting the data, it was possible to assign a target χ2 parameter, which specifies how well the model function needs to match the experimental data. A low χ2 indicates a good fit, while higher values imply larger uncertainties. A significant difference was noticed when fitting the pre- and post-sonication data, as the software was able to converge when target χ2 values were set between 1.5 and 5 for the former set of samples, while the processed samples were all able to obtain a good fit at target χ2 of 1.25 or below. Once again, McSAS provides a histogram with particle size distribution that is useful to assess differences in synthesis outcomes. The three panels in Fig. 8 show the UV-Vis spectra, SAXS profiles and respective fit outcomes for diluted samples, as well as the size distribution for samples with no ligands (a), 0.125 M oleylamine and no oleic acid (b), and 0.125 M oleylamine and 0.25 M oleic acid (c), respectively. The rest of the data can be found in the ESI (Fig. S29–S35†).
 | ||
| Fig. 8 Summary of results for the characterization of samples CdSe 1 (no ligands) (a), CdSe 2 (oleylamine 0.125 M) (b), and CdSe 3 (0.25 M oleic acid and 0.125 M oleylamine) (c). All samples had 0.1 M for both CdAc and Se precursors. In all the plots displayed, the red colored dataset represents the sample in its pre-sonicated state, whereas the blue dataset depicts the sample in its post-sonication state. (Left) Uv-Vis spectra of the samples diluted in octadecene (1:10). (Center) Reduced SAXS profiles. Both curves represent the samples diluted 25% in n-decane for better data resolution. (Right) A histogram of particle sizes obtained from fitting the SAXS data (center) using the McSAS software. | ||
All samples show at least two populations of particles in their profiles, with the first centering at around 0.35 nm in radius. Again, looking at the distribution of sizes obtained from the pre-sonicated samples, it is clear that this population can be attributed to precursor coordination in solution prior to nanoparticle formation. The second and larger size population can be attributed to the formation of quantum dots and MSCs. Based on the radius found at the peak position of the histograms obtained from the fitting, the final particle size was approximated. For samples displaying three clusters of particle size, the values of the latter two groups were extracted and presented as a range, see Fig. S33–S35.†Table 3 summarizes and compares the findings of particle sizes based on both spectroscopic and SAXS techniques.
| Sample info | UV-Vis | SAXS | |||
|---|---|---|---|---|---|
| Name | Oleic acid | Oleylamine | Peak position | D sphere | D sphere |
| CdSe 1 | 0 | 0 | 432 | 1.82 | 1.16 |
| CdSe 2 | 0 | 0.125 | 379 | 1.36 | 1.3 |
| CdSe 3 | 0.25 | 0.125 | 416 | 1.69 | 1.3 |
| CdSe 4 | 0.5 | 0.125 | 409 | 1.63 | 1.2–1.7 |
| CdSe 5 | 0 | 0.25 | 379 | 1.36 | 1.4 |
| CdSe 6 | 0.25 | 0.25 | 413 | 1.67 | 1.3–2.1 |
| CdSe 7 | 0.5 | 0.25 | 411 | 1.65 | 1.1–2.1 |
| CdSe 8 | 0 | 0.375 | 360 | 1.18 | 1.1–2.0 |
| CdSe 9 | 0.5 | 0.375 | 414 | 1.67 | 1.2–2.1 |
| CdSe 10 | 0 | 0.5 | 363 | 1.21 | 1.4–2.6 |
The data generally show good agreement for the particle size. While perfect matching of sizes was not expected, the data seem to show similar trends in the obtained size. For example the samples containing only oleylamine (CdSe 2, 5, 8, and 10) show the formation of smaller particles with diameters below 1.5 nm, in agreement with the trends observed using the effective mass approximation based on the peak position of the sample's UV-Vis spectrum. It is notable that the sample containing no ligand was fit to a restricted ‘q-range’ as the fitting protocol was not able to converge when smaller ‘q’ values were included. In this region the sample showed signs of aggregation and therefore the particle size presented in Table 3 is not fully representative of the actual particle sizes found in the samples. In this instance, agreement with the UV-Vis data was not expected based on the fitting estimation. Finally, samples that contain higher amounts of ligands, especially oleic acid, tend to present a higher variation in particle sizes. This could also be attributed to possible intermediate particles being formed during synthesis, therefore leading to higher polydispersity. This trend is in line with the findings observed from the UV-Vis data for the entirety of the design space investigated previously.
4.2 Data analysis: impacts of design variables on spectral characteristics
Once again, the challenges posed by the high dimensionality of the experimental space and the large amount of data generated by the experimental campaign can affect not only the visualization of the results but also their holistic interpretation. To address this obstacle, we implemented a model-based approach to better understand the dependencies of the design variables (i.e., concentrations of precursors and ligands) on the optical properties of the samples. The first step involves the selection of a model to predict the feature of interest. This can then be inspected to understand several parameter trends. However, the model can be selected from a variety of classes, from the simple linear model to a more complex, non-linear one. In the first type of model, the value to be predicted (e.g., the absorption peak position of the UV-Vis spectrum) is assumed to be a weighted combination of design variables. Given the linearity of the model, the coefficients associated with each design variable can be used to interpret its relative ‘impact’ or ‘importance’ on the predicted feature. However, these models are very seldom able to capture the complexity of phenomena such as the synthesis of nanocrystals studied in this work. Another approach is to use a class of non-linear models, more commonly associated with the machine learning and statistics communities to improve the predictive accuracy and use a separate explanation model to interpret the data. In fact, given the large amount of data that the experimental workflow generated, it is possible to fit complex and highly accurate models such as a gradient boosting algorithm such as xgboost.70,71 To make the outputs of a framework more interpretable, typical explanation models use a proxy set of simplified inputs (e.g., a binary variable indicating whether a given input is present or missing) and map them to the original inputs. In this work, we make use of SHAP analysis, a strategy based on shapely values from coalition game theory via additive feature attributions.72 At a higher level, the explanation model used in SHAP attributes feature importance based on the change (positive, zero, or negative) in the output of the predictive model relative to its average (over the training data), when conditioned on a particular feature value. The feature attribution values explain how to get from the predictive model average to the actual predicted value of any given input. This is performed by averaging over all possible combinations of the input ordering sequence. One of the advantages of using SHAP is that it is locally accurate, and it can match the model prediction to the explanation prediction for each input. Next, it has missingness, so if a feature has a value of zero or is missing data, it will have no impact on the model outcome. Finally, SHAP has the property of consistency, meaning that for two different predictive models, the impact of the inputs should follow that of the predictive models. This means that if the marginal contribution of a feature changes (i.e., decreases), its shapely value should change in the same manner (i.e., a smaller shapely value).72In this study, we defined the predictive model inputs as the concentration of the precursors and ligands, as well as their respective indicator variables (i.e., a binary variable indicating whether a species is present in the sample or not). Several models were fit for predicting manually annotated features from the spectral data and indicator variables were used to interpret the presence of a peak. Table 4 shows the accuracy of an xgboost model (trained using the Python version in Chen et al.73) for an 80–20 train-test split.
| Feature to be predicted | Test accuracy |
|---|---|
| UV-vis peak indicator (UV_ind) | 0.135 |
| UV-vis peak position (UV_PP) | 0.433 |
| Photoluminescence peak indicator (PL_ind) | 0.013 |
| Photoluminescence peak position (PL_PP) | 0.583 |
| Photoluminescence peak intensity (PL_PI) | 0.781 |
All training data features were standardized to have a mean of zero and a standard deviation of 1. Thanks to this feature transformation, each value of the feature that is modeled represents a change from the mean (i.e. a peak shift or intensity shift from the mean), instead of the absolute values. Furthermore, to avoid data leakage, the test data were only transformed using the corresponding training data before computing the test loss. The design variable values were also transformed similarly to the features but used a quantile transformation instead where the concentrations are binned into 5 groups, corresponding to the tested conditions. All the data transformations were performed using the preprocessing module in scikit-learn74 and we use matplotlib75 for plotting routines. A total of five features were modeled, namely the absorption peak position and peak presence, as well as the photoluminescence peak presence, position, and intensity. Fig. 9 shows the average absolute SHAP values of the different design variables for a given predictive model using a bar chart. The design variables on the y-axis are annotated and ordered (top to bottom) based on their ‘global’ importance in the predictive task. However, we note that each panel in Fig. 9 corresponds to a particular predictive task where we used the exact computation of shapely values since the number of input variables is small (8) and the results would not be sensitive to a random seed used. As expected, the formation of a UV-Vis absorption peak is mainly dependent on the presence of the metal precursors, CdAc and Se, but not on their concentration. This confirms what was previously noted from the spectroscopic characterization. Again, changes to the UV-Vis peak position, which can be related to the size of the QDs or MSCs formed, are mainly driven by the concentration and presence of the coordinating ligands, oleylamine and oleic acid. However, there is no dependence on the concentration of the metal precursors. Looking at the photoluminescence features, the presence of a peak is mainly observed in the presence of CdAc and Se, but the actual concentration is not a significant factor in predicting whether a sample will have photoluminescence emission or not. Again, changes to the photoluminescence peak position (d) and peak intensity (e) are mainly influenced by the presence of oleic acid and the concentration of oleylamine. It is important to consider the fact that the samples were measured ‘as synthesized’ and not cleaned prior to the spectroscopic characterization. Therefore, only a few of the samples presented a feature in their emission spectra. However, the trends learned from SHAP do match the spectroscopic characterization.
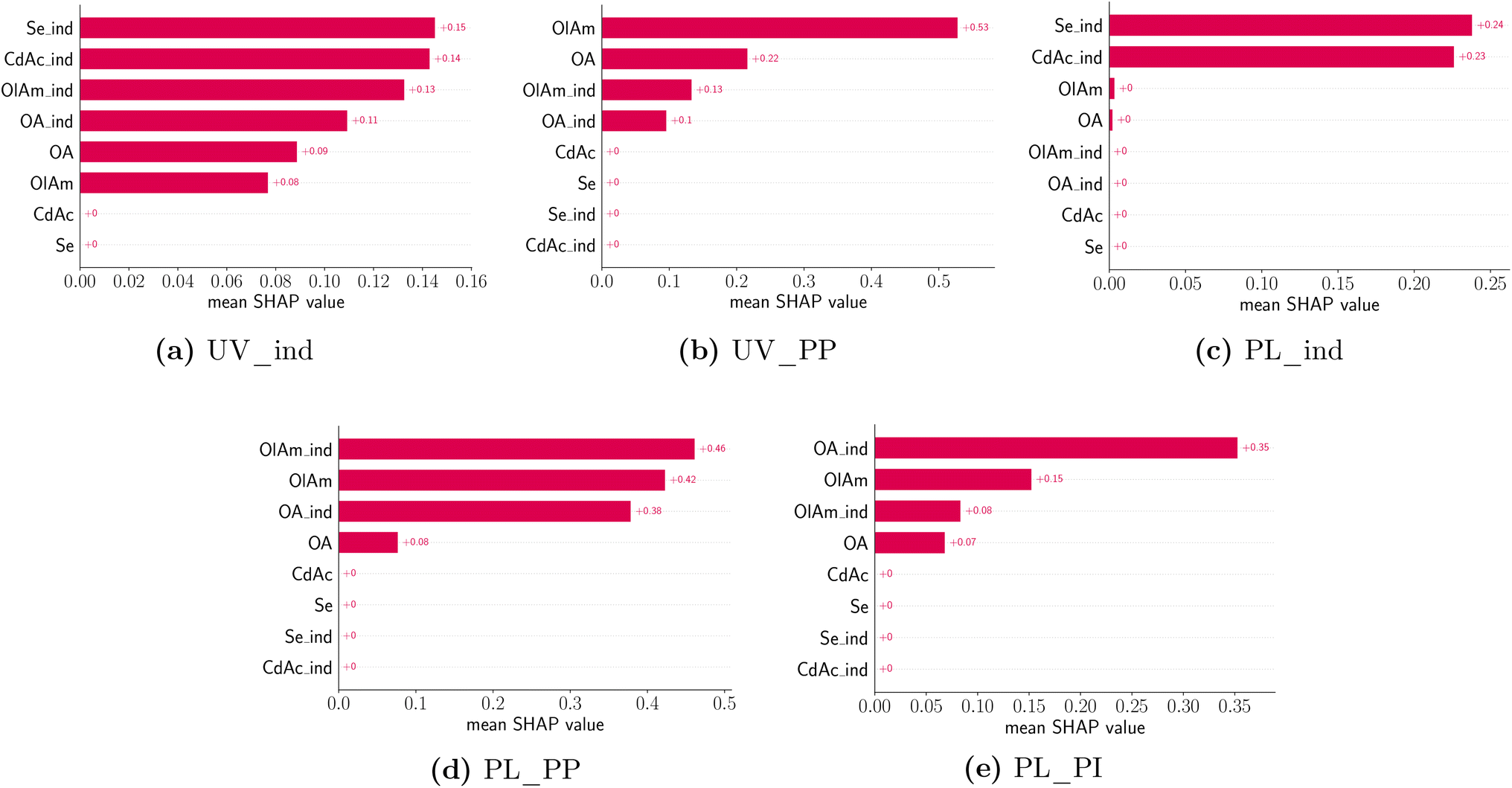 | ||
| Fig. 9 Average absolute SHAP values of different design variables for a given predictive model using a bar chart. Each panel corresponds to a particular predictive task annotated in the figure caption. The design variables on the y-axis are annotated and ordered based on their ‘global’ importance in the predictive task. Design variables are denoted as CdAc for cadmium acetate, Se for selenium, OlAm for oleylamine, and OA for oleic acid. The features are denoted using UV for the UV-Vis extinction spectrum and FL for the photoluminescence spectrum. PP corresponds to the peak position and PI corresponds to peak intensity. In both design variables and features, the suffix _ind corresponds to the respective indicator variable. | ||
While the averages shown in Fig. 9 summarize insights into the major contributions of different design variables to different features of interest, it is also useful to understand how the prediction of a particular feature changes between different samples and the nature of influence (i.e., linear vs. non-linear or if it has any ‘interaction’ effects with other variables). Individual SHAP values are visualized using a ‘bee swarm’ plot shown in Fig. 10. Here the distribution of SHAP values is shown using a scatter plot, one per each design variable in the predictive model. Once again, each panel in Fig. 10 corresponds to a model predicting a feature annotated using the title beneath it. For any given design variable in the model, we plot the SHAP value for all samples on the x-axis, and the value of the design variable is assigned a color code shown on the color bar: high (red) to low (blue) for a continuous variable and 1 (red) and 0 (blue) for an indicator variable. The distribution along the x-axis represents how the design variable's importance changes between different samples studied, while a vertical distribution shows no change, and it is used for data interpretation purposes. Once again, the design variables are ordered on the y-axis using their ‘global’ importance. By analyzing each panel in Fig. 10 and looking at the scatter plot distribution, the following conclusions could be drawn. First, the dependence of both UV-Vis and photoluminescence peak appearance in the presence of both cadmium acetate and elemental selenium is confirmed. This was expected as it is unlikely that we expect a peak corresponding to CdSe quantum dot formation without any traces of either metal precursor. Next, the UV-Vis peak shift is strongly correlated to the specific concentration of oleylamine and oleic acid and there appears to be a non-linear effect. However, it is noteworthy that the model is only trained (and tested) on samples that present an absorption peak since the presence of a peak is already modeled using the indicator function shown in panel (a). Nonetheless, the dependence on the ligand concentration is confirmed to have opposite trends. For higher concentrations of oleylamine, the absorption peak moves towards smaller wavelengths, while the opposite happens for higher concentrations of oleic acid. Finally, the photoluminescence peak shift also shows a competing influence between the presence of oleylamine and oleic acid where a red shift is more likely in the presence of oleic acid and the absence of oleylamine and vice versa. The position and intensity of features in the emission spectra show no dependence on the metal precursor concentration, but mainly on the presence and concentration of ligands. Additionally, the presence of ligands shows a stronger correlation with the modeled features, compared to their respective concentrations.
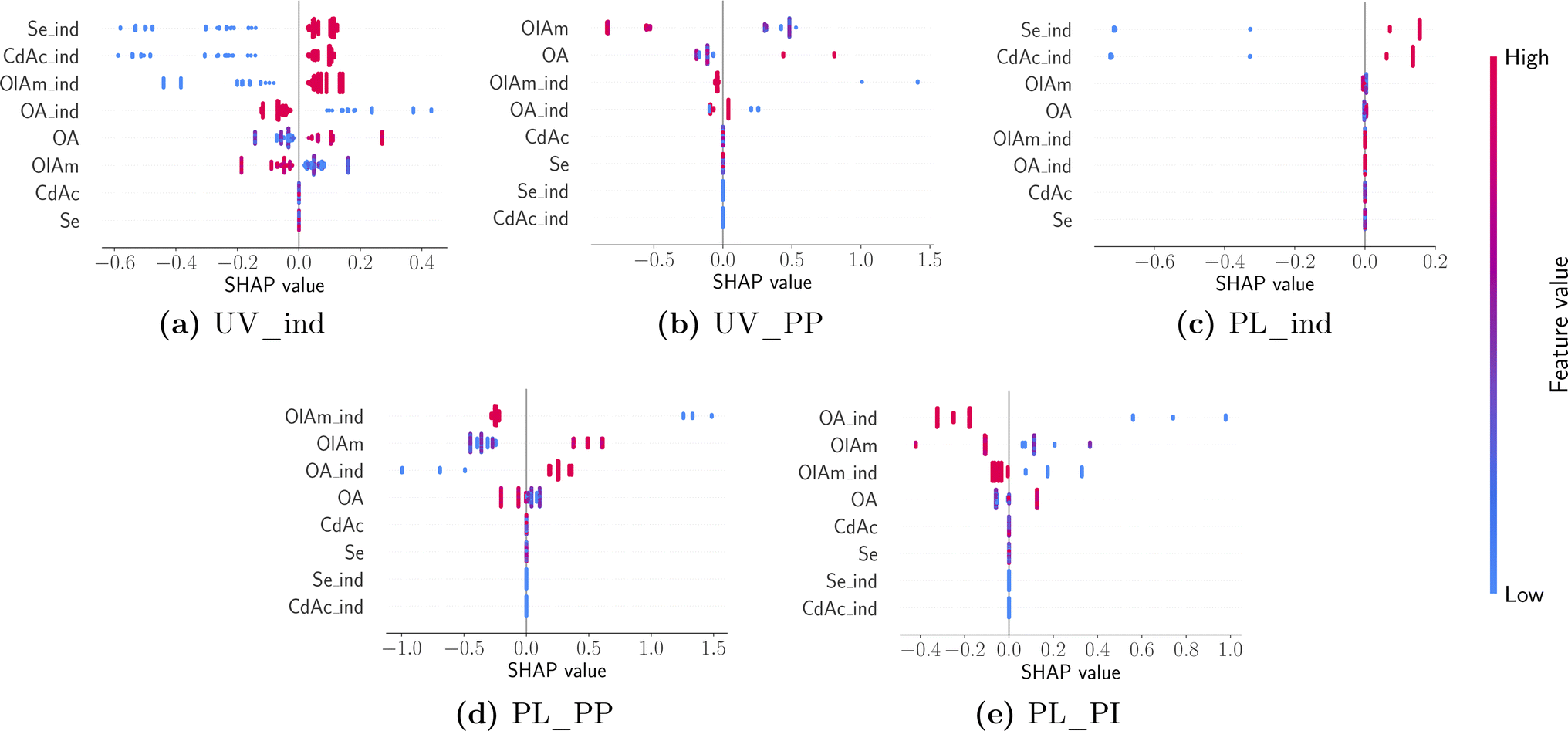 | ||
| Fig. 10 A ‘bee swarm’ plot describing the sample distribution of influence of design variables with panels arranged and annotated in the same manner as in Fig. 9. | ||
5 Conclusion
Here, we demonstrated a workflow for a material acceleration platform based on a sonochemical synthesis to enable the investigation of large experimental designs, which is extremely costly and time consuming by traditional synthetic methods. This platform provides a flexible, open-hardware, and affordable environment that can be implemented for a diverse set of chemical systems. Furthermore, given the speed and simplicity of the experimental workflow, it makes it possible to close the loop and more easily implement AI-driven strategies for achieving a certain spectral target or optimizing materials properties. These high-throughput experimental solutions can allow for a drastic increase in available data on these nanocrystal systems and aid in the discovery of new materials with desired chemical and physical properties. We employed our platform to study the formation of CdSe quantum dots by the sonochemical synthesis. We found that the range of concentrations of selenium and cadmium precursors in this study only affected the amount of particles generated, but did not play an important role in the absorption peak position, related to particle size. On the other hand, the concentration of the ligands is the most important factor in controlling the particle size. The role of the ligands is related to the binding energy of the cadmium ion, which ultimately controls the nucleation rate of the synthesis. Regarding the photoluminescence emission, a similar trend was perceived. However, we found that while the presence of oleylamine favors an increase in photoluminescence intensity, the addition of oleic acid has the opposite effect. This effect is related to the different capabilities of surface passivation between the two ligands. Finally, we implemented a model-agnostic analysis strategy not only to confirm the trends observed in the scatter plot representation of the spectra, but also to provide a quantitative breakdown of which experimental parameter contributed most to the changes in the properties analyzed. The strategy of coupling the high-throughput workflow to a data-driven approach for the interpretation of the results provides a holistic view of the design space investigated. In fact, given the limitation of the traditional representation of the data (i.e., spectra) for this four-dimensional experimental space, the identification of quantitative fingerprints from the data was crucial to the generalization and understanding of how the chosen experimental conditions affected the growth and optical properties of CdSe nanocrystals.Data availability
All of the data and notebooks needed to reproduce the figures in the manuscript can be found at https://doi.org/10.5281/zenodo.7709445. We make the data and code required to generate the SHAP analysis results shown in this study available on GitHub at https://github.com/pozzo-research-group/papers/tree/main/qdots.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This project was supported by the National Science Foundation through NSF-CBET grant no. 1917340, the Data Intensive Research Enabling Clean Technology (DIRECT) National Science Foundation (NSF) National Research Traineeship (DGE-1633216), and the UW Molecular Engineering Materials Center, a Materials Research Science and Engineering Center (Grant No. DMR-1719797). The authors acknowledge the use of facilities and instrumentation supported by the U.S. National Science Foundation through the Major Research Instrumentation (MRI) program (DMR-2116265), as well as by the University of Washington, Department of Chemical Engineering. Part of this work was conducted with instrumentation provided by the Joint Center for Deployment and Research in Earth Abundant Materials (JCDREAM). Finally, the project is based in part upon work supported by the state of Washington through the University of Washington Clean Energy Institute and the UW eScience Institute.Notes and references
- G. H. Carey, A. L. Abdelhady, Z. Ning, S. M. Thon, O. M. Bakr and E. H. Sargent, Chem. Rev., 2015, 115, 12732–12763 CrossRef CAS PubMed.
- X. Dai, Z. Zhang, Y. Jin, Y. Niu, H. Cao, X. Liang, L. Chen, J. Wang and X. Peng, Nature, 2014, 515, 96–99 CrossRef CAS PubMed.
- M. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef CAS PubMed.
- H. Sakaue, A. Aikawa and Y. Iijima, Sens. Actuators, B, 2010, 150, 569–573 CrossRef CAS.
- Z. Lv, Y. Wang, J. Chen, J. Wang, Y. Zhou and S. T. Han, Chem. Rev., 2020, 120, 3941–4006 CrossRef CAS PubMed.
- S. Sadeghi, S. K. Abkenar, C. W. Ow-Yang and S. Nizamoglu, Sci. Rep., 2019, 9, 1–9 CrossRef CAS PubMed.
- K. Kumari, U. Kumar, S. N. Sharma, S. Chand, R. Kakkar, V. D. Vankar and V. Kumar, J. Phys. D: Appl. Phys., 2008, 41, 235409 CrossRef.
- C. D. M. Donegá, P. Liljeroth and D. Vanmaekelbergh, Small, 2005, 1, 1152–1162 CrossRef PubMed.
- Z. Liu, M. Shamsuzzoha, E. T. Ada, W. M. Reichert and D. E. Nikles, J. Power Sources, 2007, 164, 472–480 CrossRef CAS.
- D. Pan, L. An, Z. Sun, W. Hou, Y. Yang, Z. Yang and Y. Lu, J. Am. Chem. Soc., 2008, 130, 5620–5621 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- A. Nemamcha, J. L. Rehspringer and D. Khatmi, J. Phys. Chem. B, 2006, 110, 383–387 CrossRef CAS PubMed.
- K. Okitsu, M. Ashokkumar and F. Grieser, J. Phys. Chem. B, 2005, 109, 20673–20675 CrossRef CAS PubMed.
- H. Xu and K. S. Suslick, ACS Nano, 2010, 4, 3209–3214 CrossRef CAS PubMed.
- Y. Mizukoshi, E. Takagi, H. Okuno, R. Oshima, Y. Maeda and Y. Nagata, Ultrason. Sonochem., 2001, 8, 1–6 CrossRef CAS PubMed.
- C.-H. Su, P.-L. Wu and C.-S. Yeh, J. Phys. Chem. B, 2003, 107, 14240–14243 CrossRef CAS.
- M. Kristl and M. Drofenik, Inorg. Chem. Commun., 2003, 12, 1419–1422 CrossRef.
- S. H. Jung, E. Oh, K. H. Lee, Y. Yang, C. G. Park, W. Park and S. H. Jeong, Cryst. Growth Des., 2008, 8, 265–269 CrossRef CAS.
- L. Yin, Y. Wang, G. Pang, Y. Koltypin and A. Gedanken, J. Colloid Interface Sci., 2002, 246, 78–84 CrossRef CAS PubMed.
- H. Wang, Y. N. Lu, J. J. Zhu and H. Y. Chen, Inorg. Chem., 2003, 42, 6404–6411 CrossRef CAS PubMed.
- G. S. Wu, X. Y. Yuan, T. Xie, G. C. Xu, L. D. Zhang and Y. L. Zhuang, Mater. Lett., 2004, 58, 794–797 CrossRef CAS.
- G. Wang, G. Li, C. Liang and L. Zhang, Chem. Lett., 2001, 30, 344–345 CrossRef.
- Z. A. Peng and X. Peng, J. Am. Chem. Soc., 2001, 123, 183–184 CrossRef CAS PubMed.
- M. Kristl and M. Drofenik, Ultrason. Sonochem., 2008, 15, 695–699 CrossRef CAS PubMed.
- R. A. Hobson, P. Muivaney and F. Grieser, J. Chem. Soc., Chem. Commun., 1994, 823–824 RSC.
- R. Kastilani, B. P. Bishop, V. C. Holmberg and L. D. Pozzo, Langmuir, 2019, 35, 16583–16592 CrossRef CAS PubMed.
- M. J. Murcia, D. L. Shaw, H. Woodruff, C. A. Naumann, B. A. Young and E. C. Long, Chem. Mater., 2006, 18, 2219–2225 CrossRef CAS.
- K. S. Suslick, Sci. Am., 1989, 80–86 CrossRef CAS.
- K. S. Suslick, Science, 1990, 247, 1439–1446 CrossRef CAS PubMed.
- C.-L. Yu, J. C. Yu, H.-B. He and W.-Q. Zhou, Rare Met., 2016, 35, 211–222 CrossRef CAS.
- M. M. Flores-Leonar, L. M. Mejía-Mendoza, A. Aguilar-Granda, B. Sanchez-Lengeling, H. Tribukait, C. Amador-Bedolla and A. Aspuru-Guzik, Curr. Opin. Green Sustainable Chem., 2020, 25, 100370 CrossRef.
- C. W. Coley, D. A. Thomas, J. A. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, Science, 2019, 365, 1–9 CrossRef PubMed.
- J. Li, J. Li, R. Liu, Y. Tu, Y. Li, J. Cheng, T. He and X. Zhu, Nat. Commun., 2020, 11, 1–10 CrossRef CAS PubMed.
- E. M. Chan, C. Xu, A. W. Mao, G. Han, J. S. Owen, B. E. Cohen and D. J. Milliron, Nano Lett., 2010, 10, 1874–1885 CrossRef CAS PubMed.
- A. Vikram, K. Brudnak, A. Zahid, M. Shim and P. J. A. Kenis, Nanoscale, 2021, 13, 17028 RSC.
- R. W. Epps, K. C. Felton, C. W. Coley and M. Abolhasani, Lab Chip, 2017, 17, 4040–4047 RSC.
- P. A. Beaucage and T. B. Martin, Chem. Mater., 2023, 846–852 CrossRef CAS.
- S. Eggert, P. Mieszczanek, C. Meinert and D. W. Hutmacher, HardwareX, 2020, 8, e00152 CrossRef PubMed.
- A. Powell, Media Cult. Soc., 2012, 34, 691–708 CrossRef.
- N. Blow, Nat. Methods, 2008, 5, 109–112 CrossRef CAS PubMed.
- J. Rodriguez, M. Politi, S. Scheiwiller, S. Bonageri, S. Adler, D. Beck and L. D. Pozzo, J. Open Hardw., 2021, 5, 1–13 CrossRef.
- R. Jones, P. Haufe, E. Sells, P. Iravani, V. Olliver, C. Palmer and A. Bowyer, Robotica, 2011, 29, 177–191 CrossRef.
- C. Zhang, B. Wijnen and J. M. Pearce, SLAS Technol., 2016, 21, 517–525 CrossRef CAS PubMed.
- M. C. Carvalho and R. H. Murray, HardwareX, 2018, 3, 10–38 CrossRef.
- R. Keesey, R. LeSuer and J. Schrier, HardwareX, 2022, 12, e00319 CrossRef PubMed.
- A. Bhom, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2019, 75, 531–536 CrossRef PubMed.
- Definition (English) - Open Source Hardware Association, https://www.oshwa.org/definition/.
- pozzo-research group, Automation-Hardware: Pozzo Group OT2 Hardware, https://github.com/pozzo-research-group/Automation-Hardware, 2021.
- E. Antonio and M. Politi, pozzo-research-group/OT2-DOE, 2022, https://github.com/pozzo-research-group/OT2-DOE Search PubMed.
- J. Vasquez, H. Twigg-Smith, J. T. O’leary and N. Peek, CHI 20: Proceedings of the 2020 CHI Conference on Human Factors in Computing Systems, 2020, vol. 1–13 Search PubMed.
- J. Vasquez and N. Peek, machineagency/sonication_station, 2020, https://github.com/machineagency/sonication_station Search PubMed.
- I. Bressler, B. R. Pauw and A. F. Thünemann, J. Appl. Crystallogr., 2015, 48, 962–969 CrossRef CAS PubMed.
- I. Breßler, B. R. Pauw and A. Thünemann, McSAS: A tool for extracting form-free size distributions of small-angle scattering (SAS) patterns using a Monte-Carlo method, 2022, https://github.com/BAMresearch/McSAS Search PubMed.
- Y. Jiang, W.-S. Ojo, B. Mahler, X. Xu, B. Abe and B. Dubertret, ACS Omega, 2018, 3, 6199–6205 CrossRef CAS PubMed.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- S. Abe, R. K. Capek, B. D. Geyter and Z. Hens, ACS Nano, 2013, 7, 943–949 CrossRef CAS PubMed.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak, A. G. Kanaras and U. K. bj, Chem. Rev., 2019, 4819–4880 CrossRef CAS PubMed.
- S. M. Harrell, J. R. McBride and S. J. Rosenthal, Chem. Mater., 2013, 25, 1199–1210 CrossRef CAS.
- Q. Dai, D. Li, J. Chang, Y. Song, S. Kan, H. Chen, B. Zou, W. Xu, S. Xu, B. Liu and G. Zou, Nanotechnology, 2007, 18, 405603 CrossRef.
- D. Zhu, J. Hui, N. Rowell, Y. Liu, Q. Y. Chen, T. Steegemans, H. Fan, M. Zhang and K. Yu, J. Phys. Chem. Lett., 2018, 9, 2818–2824 CrossRef CAS PubMed.
- A. B. Pun, A. S. Mule, J. T. Held and D. J. Norris, Nano Lett., 2021, 21, 7651–7658 CrossRef CAS PubMed.
- X. Huang, V. K. Parashar and M. A. Gijs, Langmuir, 2018, 34, 6070–6076 CrossRef CAS PubMed.
- R. García-Rodríguez and H. Liu, J. Am. Chem. Soc., 2014, 136, 1968–1975 CrossRef PubMed.
- M. Kristl, I. Ban, A. Danč, V. Danč and M. Drofenik, Ultrason. Sonochem., 2010, 17, 916–922 CrossRef CAS PubMed.
- R. K. Čapek, I. Moreels, K. Lambert, D. D. Muynck, Q. Zhao, A. V. Tomme, F. Vanhaecke and Z. Hens, J. Phys. Chem. C, 2010, 114, 6371–6376 CrossRef.
- M. S. Bootharaju, W. Baek, S. Lee, H. Chang, J. Kim and T. Hyeon, Small, 2021, 17, 1–16 CrossRef PubMed.
- N. A. Hill and K. B. Whaley, J. Chem. Phys., 1994, 100, 2831 CrossRef CAS.
- W. Kim, S. J. Lim, S. Jung and S. K. Shin, J. Phys. Chem. C, 2010, 114, 1539–1546 CrossRef CAS.
- T. Li, A. J. Senesi and B. Lee, Chem. Rev., 2016, 116, 11128–11180 CrossRef CAS PubMed.
- J. H. Friedman, Ann. Math. Stat., 2001, 1189–1232 Search PubMed.
- J. Friedman, T. Hastie and R. Tibshirani, Ann. Math. Stat., 2000, 28, 337–407 Search PubMed.
- S. M. Lundberg and S.-I. Lee, Adv. Neural Inf. Process. Syst., 2017, 30, 1–10 Search PubMed.
- T. Chen and C. Guestrin, Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 2016, pp. 785–794 Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss and V. Dubourg, et al. , J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- J. D. Hunter, Comput. Sci. Eng., 2007, 9, 90–95 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dd00033h |
| This journal is © The Royal Society of Chemistry 2023 |