
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving molecular machine learning through adaptive subsampling with active learning†
Yujing
Wen
,
Zhixiong
Li
,
Yan
Xiang
 and
Daniel
Reker
*
and
Daniel
Reker
*
Department of Biomedical Engineering, Duke University, Durham, North Carolina 27705, USA. E-mail: daniel.reker@duke.edu
First published on 23rd June 2023
Abstract
Data subsampling is an established machine learning pre-processing technique to reduce bias in datasets. However, subsampling can lead to the removal of crucial information from the data and thereby decrease performance. Multiple different subsampling strategies have been proposed, and benchmarking is necessary to identify the best strategy for a specific machine learning task. Instead, we propose to use active machine learning as an autonomous and adaptive data subsampling strategy. We show that active learning-based subsampling leads to better performance of a random forest model trained on Morgan circular fingerprints on all four established binary classification tasks when compared to both training models on the complete training data and 16 state-of-the-art subsampling strategies. Active subsampling can achieve an increase in performance of up to 139% compared to training on the full dataset. We also find that active learning is robust to errors in the data, highlighting the utility of this approach for low-quality datasets. Taken together, we here describe a new, adaptive machine learning pre-processing approach and provide novel insights into the behavior and robustness of active machine learning for molecular sciences.
I. Introduction
Machine learning algorithms are increasingly deployed to predict the properties of small molecules to hasten and de-risk the discovery and development of new drug candidates and materials.1,2 Although advances in computing power and algorithms have improved machine learning capabilities, the quality and characteristics of the available training data remain major determinants of machine learning performance.3–6 Therefore, careful dataset curation continues to be a key step in model development to ensure high-quality models and reliable predictions.5,6One of the classic challenges for machine learning is biases in the training dataset, such as class imbalance.7–9 A significant difference in the number of datapoints with specific labels biases the model towards the majority class, leading to poor performance of the machine learning model on the minority class.10 This is a critical challenge for molecular machine learning since many of the utilized datasets are highly imbalanced and the minority class is commonly the more important category for many molecular classification tasks. For example, hit rates in high-throughput screens can be as low as 0.01%,11 meaning that large molecular screening datasets can contain up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold more inactive than active compounds and only a few hits that could be developed into life-saving therapeutics. Naïve training of machine learning methods on such imbalanced datasets results in models that predominantly predict molecules as “inactive”, which can create models that appear to be highly predictive (with accuracy values >99%) but do not actually facilitate drug discovery tasks given a poor ability to predict the desired “active” compound class. In molecular machine learning, additional biases, such as limited scaffold diversity and skewed distribution of protein targets, can adversely affect predictive performance.12–15 Therefore, extensive parameter optimizations or dataset curation are performed to enable the development of more powerful predictive models.10
000-fold more inactive than active compounds and only a few hits that could be developed into life-saving therapeutics. Naïve training of machine learning methods on such imbalanced datasets results in models that predominantly predict molecules as “inactive”, which can create models that appear to be highly predictive (with accuracy values >99%) but do not actually facilitate drug discovery tasks given a poor ability to predict the desired “active” compound class. In molecular machine learning, additional biases, such as limited scaffold diversity and skewed distribution of protein targets, can adversely affect predictive performance.12–15 Therefore, extensive parameter optimizations or dataset curation are performed to enable the development of more powerful predictive models.10
One of the state-of-the-art approaches for data curation is sampling of the training data, i.e., algorithmic selection of training data to reduce class imbalances. The community mainly distinguishes two types of sampling methods, oversampling and subsampling (also called “undersampling”). In oversampling, datapoints are duplicated from the minority class or new synthetic datapoints are created to increase the number of minority class samples. In subsampling, the number of majority samples and other biases are reduced to mitigate imbalances in the training data. While subsampling is often the method of choice since it does not artificially create new or duplicated samples, subsampling can lead to a loss of information as datapoints are removed from the training data. Therefore, hundreds of different subsampling strategies have been conceived and implemented to minimize the amount of information loss.10 The performance of different subsampling methods depends on the underlying dataset and model, requiring extensive benchmarking to identify the best-performing strategy per machine learning task.
Active machine learning has become an increasingly popular strategy in machine learning model development, in which a model can request additional data to further improve the performance of the employed machine learning models.16 Although active learning was originally developed for prospective applications to guide the acquisition of novel data, the retrospective application of active learning on datasets with known labels has become an established strategy to benchmark different active learning strategies.16 Most recently, studies have shown that active machine learning can also be applied retrospectively as a subsampling method to maintain high predictive performance of machine learning models while reducing the amount of necessary training data by up to 10-fold.14,17–20
With the increasing pervasiveness of machine learning methods and the growing amount of available, complex, and biased datasets, there is an unmet need for novel automated data curation strategies that can further boost the predictive performance of machine learning models without the need for manual benchmarking and intervention of data sampling strategies.10 We hypothesize that active machine learning-based subsampling can serve as an autonomous and adaptive data curation strategy to significantly improve machine learning model performance compared to training on the complete dataset or using other state-of-the-art data sampling strategies.21 The primary objective of the here presented study is to quantify the performance of machine learning models trained on data subsampled using active learning compared to models trained on the full datasets. Furthermore, we contextualize the performance of our novel approach to 16 state-of-the-art data sampling approaches. Additionally, we investigate the reasons for model improvements and study the robustness of our approach when introducing errors into our datasets. We expect our developed pipeline to be generalizable to other machine learning and optimization tasks to boost performance based on automated data curation and to provide further insights into the performance and behavior of active machine learning.
II. Methods and materials
Datasets
All four single-task binary classification datasets from the MoleculeNet AI benchmarking repository9 were accessed via DeepChem.22 These datasets are “BBBP” (molecules annotated for their ability to cross the blood–brain barrier, accessed 1/11/22), “BACE” (molecules annotated for their ability to inhibit human β-secretase 1, accessed 12/29/21), “ClinTox” (molecules annotated according to whether they exhibited toxicity in clinical trials, accessed 1/11/22) and “HIV” (molecules annotated for their ability to inhibit HIV replication, accessed 10/24/21). Molecules were described as Morgan fingerprints with radius 2 and 1024 bits using RDKit.23 Additionally, we extracted the “Breast Cancer” dataset24,25 from scikit-learn26 (accessed 2/15/22) as a non-molecular, established machine learning benchmarking dataset.Active machine learning
For all datasets, we employed scikit-learn's (version 0.24.2) Random Forest (RF) classifier with default parameters (100 trees, Gini impurity).26 At the beginning of the active learning process, the dataset is split into an active learning set and a validation set
and a validation set 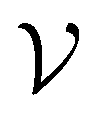 . We carried out a 50:50 scaffold split (as implemented in DeepChem22) for the molecular datasets and a 50:50 stratified split for the Breast Cancer dataset since this dataset does not contain molecular structures and can therefore not be split based on scaffold groups.
. We carried out a 50:50 scaffold split (as implemented in DeepChem22) for the molecular datasets and a 50:50 stratified split for the Breast Cancer dataset since this dataset does not contain molecular structures and can therefore not be split based on scaffold groups.
We then followed established protocols for active learning (see pseudocode in Algorithm 1).27 Briefly, we randomly selected one positive datapoint  and one negative datapoint
and one negative datapoint 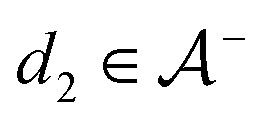 , where
, where 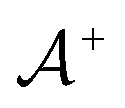 is the positive data and
is the positive data and 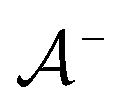 is the negative data in
is the negative data in 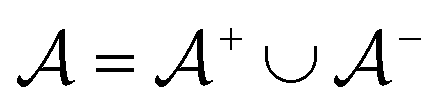 . The iterative active learning is then started with the training set
. The iterative active learning is then started with the training set 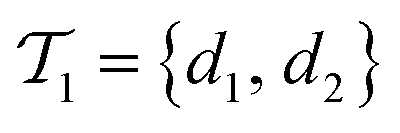 and the pool set
and the pool set 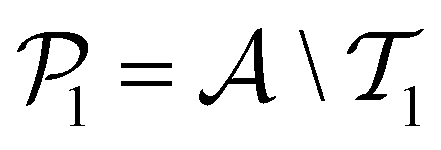 . In each iteration
. In each iteration 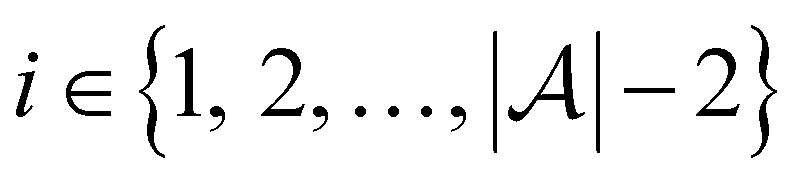 , a RF classifier is trained using the training set
, a RF classifier is trained using the training set 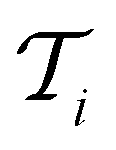 , and the resulting RF model is then used to predict the class of every datapoint in the pool set
, and the resulting RF model is then used to predict the class of every datapoint in the pool set 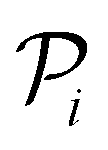 and to quantify the predictive uncertainty of every prediction as the disagreement among the decision trees of the RF model (ensemble-based uncertainty, variance of the output of “predict_proba” function in sklearn)
and to quantify the predictive uncertainty of every prediction as the disagreement among the decision trees of the RF model (ensemble-based uncertainty, variance of the output of “predict_proba” function in sklearn)

| Algorithm 1 Pseudocode of active learning-based subsampling for single active learning run. For statistical assessment, the algorithm is repeated 20 times and the maxIter is instead defined using the maximum average performance across all runs |
|---|

|
The datapoint dk with the highest uncertainty (maximum disagreement among the trees) is selected
| k = argmaxi(ui) |
The selected datapoint dk is then removed from the pool set
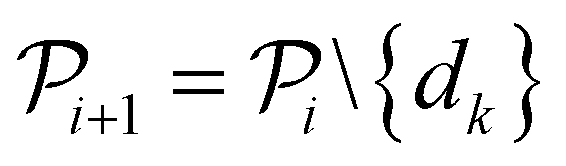
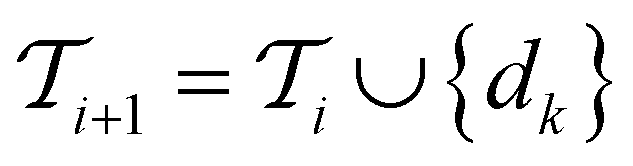
Active learning terminates after 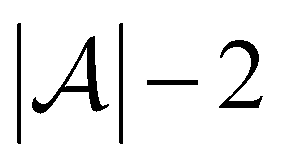 iterations, at which point all data has been added to the training dataset
iterations, at which point all data has been added to the training dataset
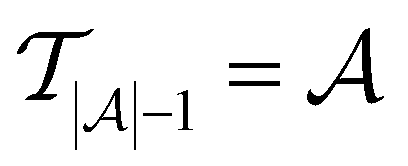
such that there is no further data to select. The performance of active learning is evaluated by making predictions on the validation set
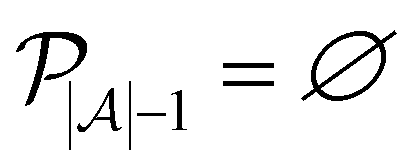
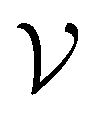 at each iteration after training the RF model on training data
at each iteration after training the RF model on training data 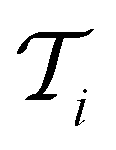 . Performance is quantified using four metrics: Mathews Correlation Coefficient (MCC), F1 score, accuracy, and balanced accuracy.
. Performance is quantified using four metrics: Mathews Correlation Coefficient (MCC), F1 score, accuracy, and balanced accuracy.
The active learning pipeline is repeated 20 times using different initial training sets  by randomly selecting different initial datapoints (d1, d2) from
by randomly selecting different initial datapoints (d1, d2) from 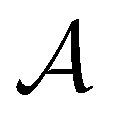 . We use the Kolmogorov–Smirnov test to assess the normality of the performance distribution. The results of the 20 runs are averaged, and the iteration nmax with the highest average MCC on
. We use the Kolmogorov–Smirnov test to assess the normality of the performance distribution. The results of the 20 runs are averaged, and the iteration nmax with the highest average MCC on 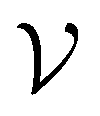 across all 20 active learning repeats is defined as “maxIter”. The “maxIter models” refers to the RF models trained at maxIter using
across all 20 active learning repeats is defined as “maxIter”. The “maxIter models” refers to the RF models trained at maxIter using 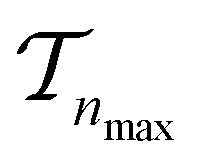 in every of the 20 active learning trajectories. We compare the performance of these “maxIter models” to the “full models”, which refers to the RF models trained at the end of the active learning trajectory, i.e., the full active learning set
in every of the 20 active learning trajectories. We compare the performance of these “maxIter models” to the “full models”, which refers to the RF models trained at the end of the active learning trajectory, i.e., the full active learning set 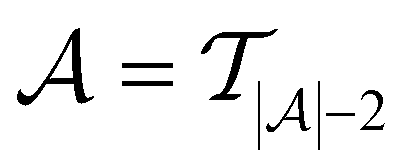 is the training data and a RF model is trained 20 times on this full training dataset.
is the training data and a RF model is trained 20 times on this full training dataset.
For further comparison and contextualizing of the learning process (i.e., the changes of the model performance as measured through the MCC on 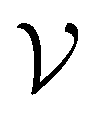 ), we also implemented a “random selection” process in which datapoints are selected from the pool set
), we also implemented a “random selection” process in which datapoints are selected from the pool set 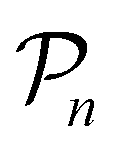 randomly without considering the predictive uncertainty. For both active learning and random selection, we track the predictive performance on
randomly without considering the predictive uncertainty. For both active learning and random selection, we track the predictive performance on 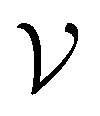 and the number of positive datapoints in the training data
and the number of positive datapoints in the training data 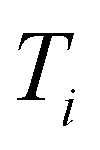 (“positive selection ratio”) at every iteration i of the active learning subsampling process.
(“positive selection ratio”) at every iteration i of the active learning subsampling process.
Error introduction
To assess the robustness of models to errors in the data, we incorporated pre-specified ratios of corrupted labels in our learning data. To this end, we randomly selected a subset of data from the active learning set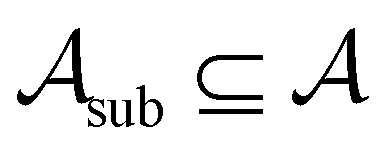 without replacement and flipped the class labels for all datapoints in
without replacement and flipped the class labels for all datapoints in 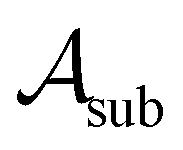 (i.e., the annotation of “active” compounds was switched to “inactive” and vice versa). This was done before the first datapoints d1, d2 are selected to be added to
(i.e., the annotation of “active” compounds was switched to “inactive” and vice versa). This was done before the first datapoints d1, d2 are selected to be added to 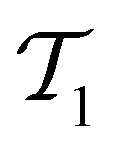 , meaning that the complete active learning run (including the initial training data for the first model) was potentially affected by this erroneous data. We repeated this experiment using different ratios of corrupted data, from 0% (none of the data is affected) to 50% (half of the learning data has been inverted, and all machine learning models collapse because all information has been deleted) in increments of 10%.
, meaning that the complete active learning run (including the initial training data for the first model) was potentially affected by this erroneous data. We repeated this experiment using different ratios of corrupted data, from 0% (none of the data is affected) to 50% (half of the learning data has been inverted, and all machine learning models collapse because all information has been deleted) in increments of 10%.
Other subsampling strategies
Sixteen established subsampling algorithms are used in this study to contextualize our performance. “Balanced” uses random supervised subsampling to create a training data subset with an equal number of instances belonging to either of the two binary class labels. The “diverse” sampling strategy uses the MaxMin selection algorithm (RDKit) based on the Tanimoto similarity of molecules to select a diverse training set. We also implemented “balanced-diverse” and “diverse-balanced”, which apply both the “balanced” and the “diverse” strategies in sequence to sample training data that is both diverse and balanced, emphasizing diversity or class balance depending on which sampling method was selected first. Additionally, we used all 12 established sampling strategies from the imblearn Python library (version 0.8)28 with default parameters. Imblearn is a Python library that provides various imbalanced learning techniques to address the issue of imbalanced datasets. Some of the implemented undersampling methods in imblearn include AllKNN, which applies the k-NN algorithm to every sample to remove majority class samples, and CondensedNearestNeighbour, which uses 1-NN to reduce majority class samples while retaining all minority class samples. Another approach, EditedNearestNeighbours, removes majority class samples based on the k-NN algorithm's classification errors. InstanceHardnessThreshold removes samples that are misclassified by a classifier with high hardness scores. NearMiss selects majority class samples based on distance to minority class samples, while NeighbourhoodCleaningRule removes noisy samples by applying k-NN to every sample. OneSidedSelection uses TomekLinks to remove noisy samples, and RandomUnderSampler randomly removes samples from the majority class. RandomOverSampler duplicates samples from the minority class. SMOTEN as an extension of SMOTE that works with categorical data creates synthetic examples of the minority class. We note that we excluded SMOTE and its extensions except for SMOTEN since it does not intrinsically support categorical or binary features as used in this study.III. Results and discussion
Active learning subsampling outperforms training on full dataset
We downloaded all single task classification datasets from MoleculeNet9 using DeepChem22 and described them using Morgan fingerprints (http://RDKit.org).23 The datasets are “BBBP”, “BACE”, “ClinTox”, and “HIV”. “BBBP” contains 2039 molecular structures annotated for whether they can cross the blood–brain-barrier. “BACE” is a dataset of 1513 molecules annotated for their ability to inhibit human beta-secretase 1. A total of 1478 molecules are annotated in “ClinTox” for whether they caused toxicity in clinical trials. “HIV” is the largest dataset in our benchmark and contains 41127 molecules annotated for their ability to inhibit HIV replication. Importantly, our datasets thereby cover a range of different sizes (from 1478 for ClinTox up to 41127 for HIV), different class imbalances (e.g., 76% positive for BBBP, balanced for BACE, 4% positive HIV), include both in vitro and in vivo readouts, and are of different modelling complexity based on previously published benchmarking results.9 We split our data into train and test sets of equal size based on molecular scaffolds using DeepChem.22 Training random forest models with default parameters on the training data and evaluating their performance on the test data gave the expected performance according to the published benchmark values.9
We then investigated how active learning performed on these datasets by providing two random data points from the training data for model initialization and then letting the active learning algorithm select additional data from the training data iteratively based on uncertainty sampling until the complete training data had been selected. Corroborating previous studies,14,18 active learning yielded good predictive performance based on only a fraction of the data for all the five datasets used in this study (Fig. 1A).
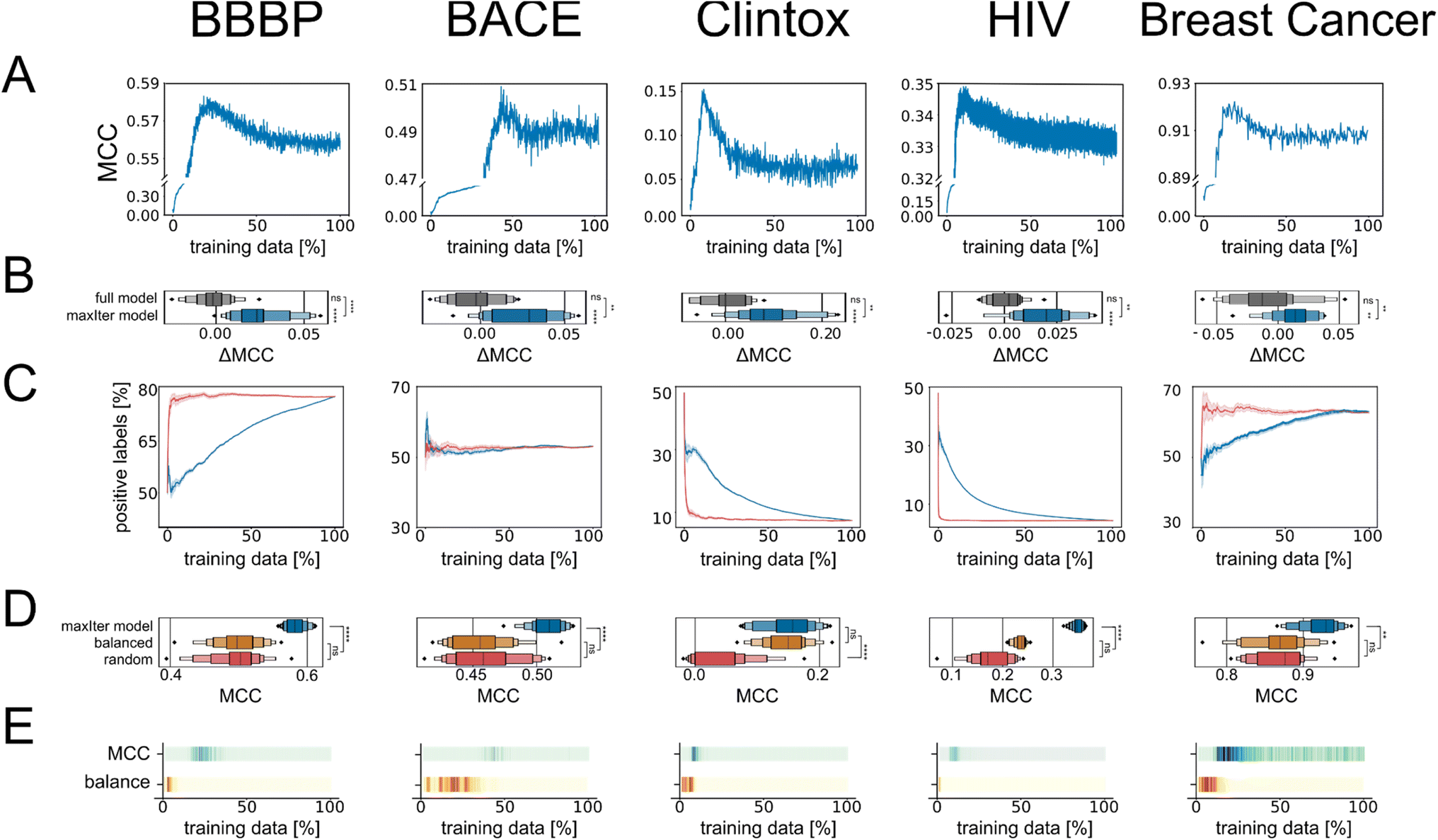 | ||
| Fig. 1 (A) Active learning curves track the performance of models on the validation set. (B) Boxplots of the distribution of the MCC improvements (ΔMCC) of the “maxIter models” using the active learning subsample of the data and the “full models” trained on the complete training data, compared to the average MCC of the “full models”. (C) Relationship between the fraction of datapoints labeled as “positive” and the percentage of data selected by active learning (orange curve) or selected by random sampling (gray curve). Note that the random sampling will be initialized in the same way as the active learning algorithm, with one positive and one negative example – therefore all curves start at 50%. (D) Boxplots show the distribution of the absolute MCC values on the validation split for models trained on the active learning-based subsample of data (“maxIter models”, blue), for models trained on randomly selected subsamples of data (red), and for models trained on data that was randomly subsampled into balanced training data based on class labels (“balanced control models”, orange). (E) Colorbar showing the p values for Student's t-tests comparing the MCC values at every active learning iteration with the average MCC values of “maxIter models” (top) and for comparing the positive selection ratio at every active learning iteration with the most balanced positive selection ratio achieved during active learning (bottom). Darker color indicates higher p value and therefore values more similar to the maximum performance and the maximum dataset balance, respectively. | ||
In contrast to previous studies,14,18,29,30 we observed a “turning point” at which a global maximum performance is reached and after which the performance of the model decreases when more data is added. This was consistent using different evaluation metrics (Fig. S1†), indicating that this effect was not simply an artifact observed for a single performance metric. Previous active learning implementations had shown that actively trained models rapidly converge to a maximum predictive performance value and then stagnate.14,18,29,30 The main differences between our work and previous studies are: (1) previous studies commonly terminate active learning before adding the full learning data,14,17–20i.e., they may have stopped active learning before identifying a “turning point”; and (2) we used scaffold-based splitting to quantify the model's ability to generalize across different chemotypes, and previous studies have tracked training data performance or used random train-test splitting,14,17–20 which means that the training set and the test set were drawn from the same molecular distribution. When using random stratified splitting instead of scaffold splitting, we observed a less pronounced benefit of active learning-base subsampling (Fig. S2†), suggesting that the generalizability of the actively trained model is at least in-part responsible for the observed “turning point”. This implied that long active learning runs on the same, constrained training set improve or maintain performance but might lead to overfitting and decreasing generalizability of the model.
To determine whether the improvement in performance for models trained at the “turning point” compared to performance for models trained on the complete dataset was statistically significant, we repeated our active learning runs 20 times with different initial training datapoints. The mean MCC value of the 20 models trained at the “maxIter” iteration (i.e., trained on a subset of training data of size nmax which corresponds to the point where the average MCC value was highest for the 20 models trained on data selected by each active learning run) was significantly higher than the mean MCC value of the models trained on the full dataset for all our benchmarks (p < 5 × 10−7, n = 20, paired t-test). We defined the absolute improvement in performance for every active learning run compared to the average performance of models trained on the full dataset as ΔMCC and found that the mean improvements were significantly larger than zero (Fig. 1B). The improvements in performance ranged from as little as 0.5% to as much as 139% of the original performance value depending on the dataset and performance metric used (Table S1†). Overall, this indicated that active learning could identify a subset of data that leads to reproducibly stronger performance compared to training the machine learning models on the full training data – positioning active learning as a useful subsampling approach.
Balancing data is a major feature but not the sole reason for active learning performance
We then investigated the reason for the improved performance of the models trained on the active learning-selected data subsets. One of the known properties of active learning is its ability to sample training data in a more balanced manner compared to random sampling.14,21,27 Rebalancing imbalanced data is a well-known strategy in machine learning to improve performance.10 We tracked the ratio of positive datapoints to negative datapoints (positive selection ratio) across the complete active learning campaign and noticed that active learning sampled data in a more balanced manner than random sampling on our data (Fig. 1C and Table S2†).However, we also noticed that one of our benchmark datasets (“BACE”) was more balanced than the other datasets and showed similar improvements through active learning-based subsampling (Fig. 1B). Additionally, as a control experiment, we trained RF models on balanced training data that we created by randomly sampling balanced subsets of equal size as the active learning-based subsampling strategies. The RF models trained on these “balanced” datasets did not outperform the RF model trained on the complete training data (p > 0.05, Fig. 1D), except for the Clintox dataset, possibly due to its small size and extreme class label imbalance. Additionally, when directly comparing the performance of our models at different learning iterations with the positive selection ratio throughout the active learning process, we noticed that the peak dataset balance and the peak performance did not coincide (Fig. 1E). Based on these three observations, we concluded that rebalancing the class imbalance is a benefit of active learning but is insufficient to explain the full extent of the improved performance.
Active learning selects a core set of datapoints independent of the starting point
We next wondered whether the set of molecules selected by active learning would be consistent across different active learning runs, even if the active learning campaign was started on distinct initial training data. To this end, we extracted the datapoints that were selected in every active learning run and found that the number of times a specific molecule was selected differed widely. We discovered that some of the available molecules for training were never selected by active learning even when repeating the process 20 times, indicating that they do not encode useful information for the machine learning algorithm. Conversely, out of the molecules that were selected at least once by active learning, between approximately 5% to 20% of them were selected during every active learning run (Table S3†). To quantify the difference in selection frequency for different molecules across active learning runs, we calculated the Gini index per active learning task and found a large discrepancy in selection frequency, which indicates that the selection of datapoints does not follow a uniform distribution but instead favors certain molecules (Fig. 2A). This indicated that the adaptive active learning process was able to sample a core set of datapoints that result in the overall improved performance.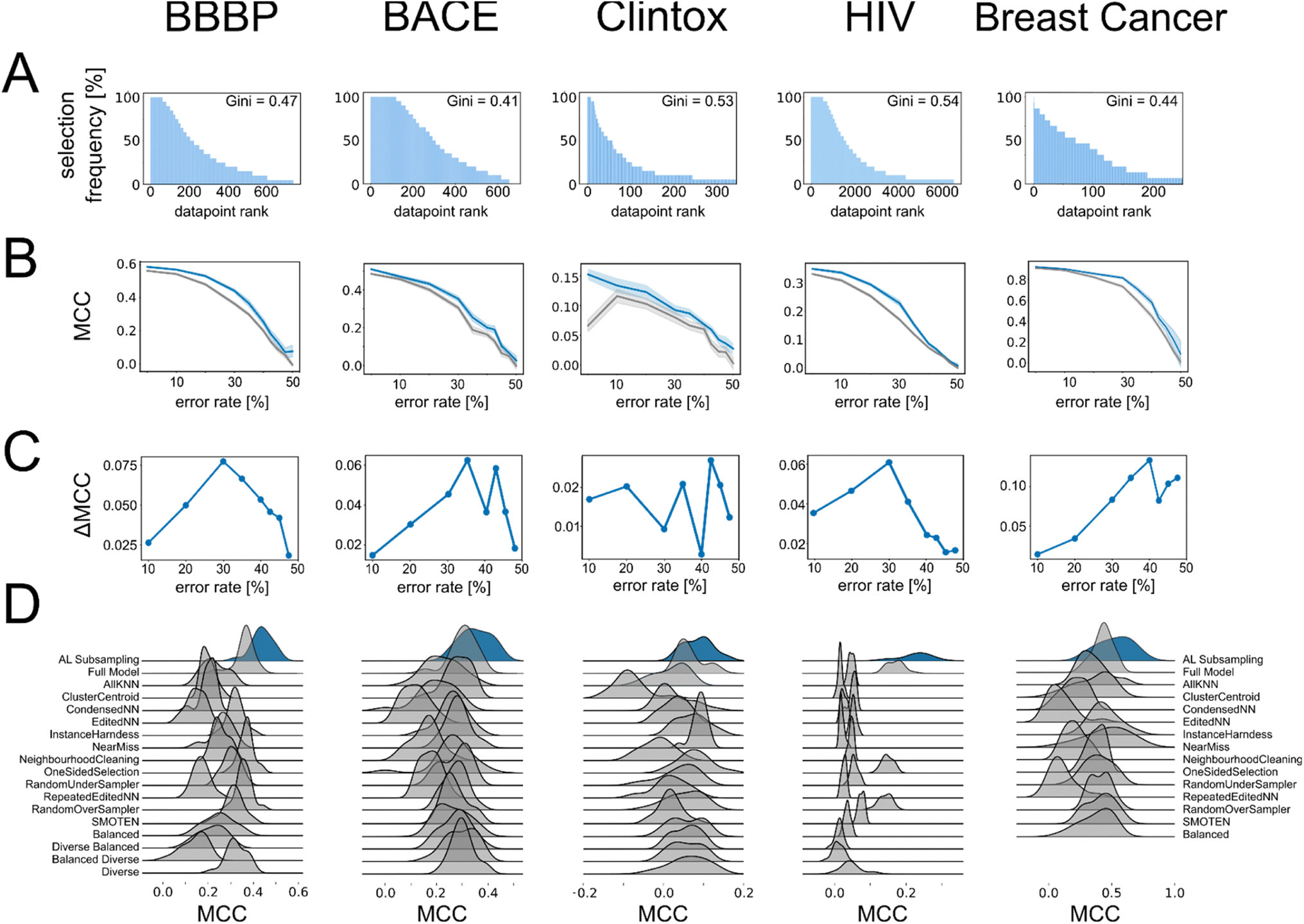 | ||
| Fig. 2 (A) Bar chart shows the number of times a specific molecule was selected during the 20 repeated active learning runs. Every bar corresponds to a specific molecule and molecules are sorted according to occurrence (from largest to smallest number of occurrences). X axis is limited to show only molecules that occur at least once for easier readability. (B) Relationship between the average MCC values and rate of error manually introduced in the training data for the “maxIter models” (blue) and the “full models” (gray). Shading corresponds to one standard deviation. (C) Relationship between error rate and improvement of the “maxIter models” compared to “full models” trained on the same data measured as difference in MCC values. (D) Ridgeplot showing the performance of the active learning subsampling approach and other state-of-the-art sampling approaches on our datasets with error rates selected based on the maximum benefit of active learning compared to the full model (cf. panel D). Note that the molecular diversity measures could not be implemented for the “breast cancer” dataset and were therefore omitted. | ||
Active learning is robust to low-quality data
The ability of active learning to sample a more balanced dataset in terms of class imbalance could imply that it can also circumvent other imbalances and biases in the data. For example, in molecular modeling, we and others have shown that active learning can sample the space of ligand–protein interactions while circumventing underlying biases to specific protein families and chemotypes.14 Active learning is also known to avoid redundant information in the training data.16 We wondered whether this means that active learning should also be robust to potential errors in the training data. To analyze the behavior of active learning-based subsampling on low-quality data, we performed active learning on label-corrupted datasets, i.e., we introduced errors in the data by flipping the class labels of randomly selected subsets of data in the active learning set .
.
As expected, the performance of the machine learning models trained on these corrupted datasets decreased rapidly (gray lines in Fig. 2B): on the external validation set, which maintained its correct class labels, predictive performance decreased with increasing amount of error in the training data. When 50% of the training labels were flipped, all information in the training data was lost and all models collapsed to random guessing, as expected. We then evaluated the behavior of our active learning-subsampling strategy and found that models trained on subsets of the corrupted datasets selected by active learning maintained higher predictive performance (blue lines in Fig. 2B) compared to training models on the complete, corrupted datasets. This was noteworthy because the active learning process was unaware which datapoints were erroneous and could have been misled by the incorrect data to sample irrelevant data subspaces. The improvement in performance for active learning-based subsampling compared to full model training increased with increasing error rate, suggesting that the benefits of active learning subsampling can increase when the quality of the dataset decreases. When the error rate was very high (>30% of the training data corrupted), the benefit of active learning started to diminish (Fig. 2C).
To further contextualize the robustness of the active learning-based subsampling on erroneous data, we compared the predictive performance of active learning against a range of other state-of-the-art subsampling methods trained on this corrupted data, including but not limited to “balanced sampling”, “diverse sampling”, and SMOTEN. None of these established data processing methods outperformed active learning-based subsampling on these corrupted datasets (Fig. 2D), indicating that adaptive subsampling through active learning provides a robust and competitive approach to autonomously curate datasets and improve predictive machine learning performance. For additional context, we also compared the performance of active learning-based subsampling against all the other state-of-the-art subsampling methods without error introduction. Although active learning-based subsampling does not outcompete every other method on all datasets, it was the only method that performed best in more than one dataset (BBBP and BACE) and also showed the highest median performance across all datasets (Table S4†). This shows that, although active learning-based subsampling appears particularly attractive for erroneous data, it also presents a competitive approach for other types of data.
IV. Conclusions
In spite of advances in complex model architectures, data quality remains a key determinant of machine learning performance and data curation continues to be a key step in model development. We implemented an automated data curation pipeline based on active machine learning that can improve performance of a random forest model using binary Morgan fingerprints for a range of different machine learning applications. Our analysis shows that active learning can identify data subsets that lead to improved performance compared to training on the complete data. This effect was consistent across all our datasets, indicating that active learning as a subsampling technique could be useful for molecular datasets of various sizes, class imbalances, and describing different types of properties. It appears the benefits of subsampling are most pronounced when introducing error to the datasets, indicating that this technique could be particularly useful for data with incorrect annotations, for example including artifactual readouts from high-throughput screens. We have made the code of this work available and hope that broad deployment will not only aid other researchers in their data curation workflows but also help to further characterize the most beneficial use cases for this novel sampling technique. Although improvements were modest for some of the datasets, all observed improvements were statistically significant and often in-line with magnitudes of advances that are reported for improved predictive architectures, thus highlighting the potential for data curation instead of model optimization to improve predictive performance of molecular machine learning models. Instead of manually benchmarking data subsampling strategies for this purpose, our pipeline relies on active learning to autonomously determine the best data to subsample and is never outperformed by other state-of-the-art sampling strategies. Admittingly, running a full retrospective active learning campaign is computationally more expensive than many other currently implemented sampling approaches, but we expect this additional computation time to be offset by not having to benchmark multiple different sampling approaches. In the future, it will be important to test whether other machine learning models beyond random forest could be used for sampling and whether a data sample extracted by one machine learning model might be transferable to another machine learning model.Additionally, our pipeline further characterizes important properties of active machine learning. We show that decreasing data quality affects active machine learning workflows less than classic machine learning, indicating an underexplored capability of active learning to train robust models on poor quality data and using noisy experimental “oracles”. Most commonly, active learning is considered the method of choice when the “oracle” is expensive or slow, but we show that active learning can also provide benefits when the “oracle” is inaccurate. In line with previous studies, we observed that active learning can extract a more balanced training dataset compared to random sampling, but we note that class balance alone was insufficient to replicate the improved performance through active learning, suggesting that additional factors might be involved. Furthermore, a drop in model performance for late active learning iterations indicates that further improvements are potentially limited by the constrained selection in a small pool of data. It is reasonable to assume that a more unrestricted sampling could provide more sustained benefits. This has important implications for the application of active learning to constrained and small (chemical) spaces and must be considered in future active learning implementations and during “stopping criterion” development. Overall, we expect the implementation of active learning-based subsampling as an autonomous data curation pipeline to become a powerful tool to boost machine learning performance while reducing data storage needs and costs, thus making molecular machine learning more reproducible, accessible, and powerful.
Data availability
The datasets used in this study are available through the freely available DeepChem and Scikit-Learn libraries. The code used in this study is available on GitHub at https://github.com/RekerLab/Active-Subsampling. Additional dependencies for the code are the Python libraries numpy, scipy, Scikit-Learn, numpy, DeepChem, and matplotlib – all of which are freely available.Author contributions
Y. W. and D. R. conceived the study. Y. W., Z. L., Y. X., and D. R. designed experiments, implemented code, and analyzed data. Y. W., Y. X., and D. R. wrote the manuscript. All authors read and approved the final manuscript.Conflicts of interest
D. R. acts as a consultant to the pharmaceutical and biotechnology industry.Acknowledgements
We are grateful to the Duke Science & Technology Initiative for funding. Y. W. and Z. L. acknowledge support from a Duke Master's Student Research Fellowship. All computations were run on the Duke Compute Cluster. We are grateful to Khalimat Murtazaliev for technical support.References
- N. Brown, P. Ertl, R. Lewis, T. Luksch, D. Reker and N. Schneider, Artificial Intelligence in Chemistry and Drug Design, J. Comput.-Aided Mol. Des., 2020, 34(7), 709–715, DOI:10.1007/s10822-020-00317-x.
- E. Smalley, AI-Powered Drug Discovery Captures Pharma Interest, Nat. Biotechnol., 2017, 35(7), 604–605, DOI:10.1038/nbt0717-604.
- P. M. Pflüger and F. Glorius, Molecular Machine Learning: The Future of Synthetic Chemistry?, Angew. Chem., Int. Ed., 2020, 59(43), 18860–18865, DOI:10.1002/anie.202008366.
- J. Vamathevan, D. Clark, P. Czodrowski, I. Dunham, E. Ferran, G. Lee, B. Li, A. Madabhushi, P. Shah, M. Spitzer and S. Zhao, Applications of Machine Learning in Drug Discovery and Development, Nat. Rev. Drug Discovery, 2019, 18(6), 463–477, DOI:10.1038/s41573-019-0024-5.
- A. Nandy, C. Duan and H. J. Kulik, Audacity of Huge: Overcoming Challenges of Data Scarcity and Data Quality for Machine Learning in Computational Materials Discovery, Curr. Opin. Chem. Eng., 2022, 36, 100778, DOI:10.1016/j.coche.2021.100778.
- V. Gudivada, A. Apon and J. Ding, Data Quality Considerations for Big Data and Machine Learning: Going beyond Data Cleaning and Transformations, International Journal on Advances in Software, 2017, 10(1), 1–20 Search PubMed.
- P. S. Kutchukian, N. Y. Vasilyeva, J. Xu, M. K. Lindvall, M. P. Dillon, M. Glick, J. D. Coley and N. Brooijmans, Inside the Mind of a Medicinal Chemist: The Role of Human Bias in Compound Prioritization during Drug Discovery, PLoS One, 2012, 7(11), e48476, DOI:10.1371/journal.pone.0048476.
- X. Jia, A. Lynch, Y. Huang, M. Danielson, I. Lang'at, A. Milder, A. E. Ruby, H. Wang, S. A. Friedler, A. J. Norquist and J. Schrier, Anthropogenic Biases in Chemical Reaction Data Hinder Exploratory Inorganic Synthesis, Nature, 2019, 573(7773), 251–255, DOI:10.1038/s41586-019-1540-5.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. Pande, MoleculeNet: A Benchmark for Molecular Machine Learning, Chem. Sci., 2018, 9(2), 513–530, 10.1039/C7SC02664A.
- H. He and E. A. Garcia, Learning from Imbalanced Data, IEEE Transactions on Knowledge and Data Engineering, 2009, 21(9), 1263–1284, DOI:10.1109/TKDE.2008.239.
- T. Zhu, S. Cao, P.-C. Su, R. Patel, D. Shah, H. B. Chokshi, R. Szukala, M. E. Johnson and K. E. Hevener, Hit Identification and Optimization in Virtual Screening: Practical Recommendations Based on a Critical Literature Analysis, J. Med. Chem., 2013, 56(17), 6560–6572, DOI:10.1021/jm301916b.
- L. Hakes, J. W. Pinney, D. L. Robertson and S. C. Lovell, Protein-Protein Interaction Networks and Biology—What's the Connection?, Nat. Biotechnol., 2008, 26(1), 69–72, DOI:10.1038/nbt0108-69.
- J. Mestres, E. Gregori-Puigjané, S. Valverde and R. V. Solé, Data Completeness—The Achilles Heel of Drug-Target Networks, Nat. Biotechnol., 2008, 26(9), 983–984, DOI:10.1038/nbt0908-983.
- D. Reker, P. Schneider, G. Schneider and J. Brown, Active Learning for Computational Chemogenomics, Future Med. Chem., 2017, 9(4), 381–402, DOI:10.4155/fmc-2016-0197.
- M. Glavatskikh, J. Leguy, G. Hunault, T. Cauchy and B. Da Mota, Dataset's Chemical Diversity Limits the Generalizability of Machine Learning Predictions, J. Cheminf., 2019, 11(1), 69, DOI:10.1186/s13321-019-0391-2.
- D. Reker, Chapter 14: Active Learning for Drug Discovery and Automated Data Curation, in Artificial Intelligence in Drug Discovery, 2020, pp. 301–326, 10.1039/9781788016841-00301.
- J. S. Smith, B. Nebgen, N. Lubbers, O. Isayev and A. E. Roitberg, Less Is More: Sampling Chemical Space with Active Learning, J. Chem. Phys., 2018, 148(24), 241733, DOI:10.1063/1.5023802.
- T. Lang, F. Flachsenberg, U. von Luxburg and M. Rarey, Feasibility of Active Machine Learning for Multiclass Compound Classification, J. Chem. Inf. Model., 2016, 56(1), 12–20, DOI:10.1021/acs.jcim.5b00332.
- C. Rakers, D. Reker and J. B. Brown, Small Random Forest Models for Effective Chemogenomic Active Learning, Journal of Computer Aided Chemistry, 2017, 18, 124–142, DOI:10.2751/jcac.18.124.
- B. Li and S. Rangarajan, Designing Compact Training Sets for Data-Driven Molecular Property Prediction through Optimal Exploitation and Exploration, Mol. Syst. Des. Eng., 2019, 4(5), 1048–1057, 10.1039/C9ME00078J.
- S. Ertekin, J. Huang, L. Bottou and L. Giles, Learning on the Border: Active Learning in Imbalanced Data Classification, in Proceedings of the sixteenth ACM conference on conference on information and knowledge management, CIKM '07, Association for Computing Machinery, New York, NY, USA, 2007, pp. 127–136, DOI:10.1145/1321440.1321461.
- B. Ramsundar, P. Eastman, P. Walters and V. Pande, Deep Learning for the Life Sciences: Applying Deep Learning to Genomics, Microscopy, Drug Discovery, and More, O'Reilly Media, Inc., 2019 Search PubMed.
- G. Landrum, RDKit: Open-Source Cheminformatics, 2006 Search PubMed.
- W. N. Street, W. H. Wolberg and O. L. Mangasarian, Nuclear Feature Extraction for Breast Tumor Diagnosis, in Biomedical Image Processing and Biomedical Visualization, SPIE, 1993, vol. 1905, pp. 861–870, DOI:10.1117/12.148698.
- O. L. Mangasarian, W. N. Street and W. H. Wolberg, Breast Cancer Diagnosis and Prognosis via Linear Programming, Oper. Res., 1995, 43(4), 570–577, DOI:10.1287/opre.43.4.570.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, Scikit-Learn: Machine Learning in Python, Journal of Machine Learning Research, 2011, 12, 2825–2830 Search PubMed.
- D. Reker and J. B. Brown, Selection of Informative Examples in Chemogenomic Datasets, in Computational Chemogenomics, ed. J. B. Brown, Methods in Molecular Biology, Springer, New York, NY, 2018, pp. 369–410, DOI:10.1007/978-1-4939-8639-2_13.
- G. Lemaître, F. Nogueira and C. K. Aridas, Imbalanced-Learn: A Python Toolbox to Tackle the Curse of Imbalanced Datasets in Machine Learning, Journal of Machine Learning Research, 2017, 18(1), 559–563 Search PubMed.
- Y. Fujiwara, Y. Yamashita, T. Osoda, M. Asogawa, C. Fukushima, M. Asao, H. Shimadzu, K. Nakao and R. Shimizu, Virtual Screening System for Finding Structurally Diverse Hits by Active Learning, J. Chem. Inf. Model., 2008, 48(4), 930–940, DOI:10.1021/ci700085q.
- M. K. Warmuth, J. Liao, G. Rätsch, M. Mathieson, S. Putta and C. Lemmen, Active Learning with Support Vector Machines in the Drug Discovery Process, J. Chem. Inf. Comput. Sci., 2003, 43(2), 667–673, DOI:10.1021/ci025620t.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dd00037k |
| This journal is © The Royal Society of Chemistry 2023 |