
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benchmarking protein structure predictors to assist machine learning-guided peptide discovery†
Victor Daniel
Aldas-Bulos‡
a and
Fabien
Plisson
 *ab
*ab
aCentre for Research and Advanced Studies of the National Polytechnic Institute (CINVESTAV-IPN), Advanced Genomic Unit, National Laboratory of Genomics for Biodiversity (LANGEBIO), 36824 Irapuato, Guanajuato State, Mexico. E-mail: fabien.plisson@cinvestav.mx
bCentre for Research and Advanced Studies of the National Polytechnic Institute (CINVESTAV-IPN), Irapuato Unit, Department of Biotechnology and Biochemistry, 36824 Irapuato, Guanajuato State, Mexico
First published on 2nd June 2023
Abstract
Machine learning models provide an informed and efficient strategy to create novel peptide and protein sequences with the desired profiles. Nevertheless, they are primarily trained on sequences where the tridimensional structures of peptides and proteins are often overlooked. We need a fast and reliable approach to estimate the structural diversity of medium-large training sets before building models. This study benchmarked four protein structure prediction methods (Jpred4, PEP2D, PSIPRED, AlphaFold2) using 261 curated and experimentally known structures from the PDBe database. We applied our best predictor to map the structural landscape of GRAMPA, the giant and vastly uncharted repository of 5980 antimicrobial peptides. The dataset was predominantly made of loose helices (65.1%), followed by random coils (17.8%), and β-stranded and mixed structures accounted for the rest.
1. Introduction
Advances in computer sciences, accessible high-performing machines and the proliferation of large public databases have accelerated the development of computational models for peptide design and protein engineering. The advent of artificial intelligence (AI) in the biological sciences has led to the creation of machine learning (ML) models capable of predicting and generating new peptides and proteins with the desired characteristics. Predictive models are trained on small-to-large datasets to learn the relationships between the biological sequences and their respective functional measurements (e.g., thermostability, bacterial growth inhibition, protein binding affinity). Generative models can learn meaningful representations (e.g., a conserved cysteine framework, a catalytic site) to create new peptide/protein sequences that resemble the native counterparts. The interplay between predictive and generative AI models provide an informed and efficient sequence design by predicting the outcomes of different peptide/protein sequences. Many comprehensive reviews have referenced the successful applications of ML-guided sequence design to antimicrobial peptides (AMPs),1–4 protein binders,5 antigen-specific monoclonal antibodies,6–9 protein families10–15 and enzymes.16–20 The field of ML-guided peptide/protein sequence design is snowballing; the reader is encouraged to consult these two GitHub repositories21,22 to stay informed.Machine learning models primarily predict or generate novel peptides and proteins from sequential representation, lacking structural information. To minimise the impact that structural factors might have upon biological prediction or sequence generation, researchers have voluntarily selected sequences based on structural or evolutionary constraints, so they presumably adopt the same tridimensional structure(s). In recent years, computational peptide designers have capitalised on neural network architectures to predict or generate novel α-helical AMPs,23–26 α-helical non-hemolytic AMPs27 or α-helical non-hemolytic anticancer peptides.28,29 Likewise, Batra and co-workers trained their models on peptides susceptible to form β-sheets to develop self-assembling materials.30 In machine learning-guided directed evolution,17 researchers have used directed evolution to assemble a set of homologous sequences, then devised robust ML strategies to engineer the next batch of proteins (e.g., channelrhodopsins,31 fluorescent proteins32) or enzymes (e.g., glycosyltransferase superfamily 1 (ref. 33)). Alternatively, several upcoming “structure-first” ML strategies tackle the sequence–structure–function problem upside down; by designing sequences that would fold into a pre-determined backbone structure derived from native topologies or generated de novo.34–37 These approaches are often regrouped under the terms inverse protein folding, structure-based protein design or fixed-backbone protein design.
Experimentally solving the structure of a peptide or a protein by techniques like X-ray crystallography or nuclear magnetic resonance is time-consuming and costly, making it challenging to have known structures for the vast number of available sequences.38 This issue is even more pronounced in peptides, as their short length and high flexibility make it difficult to obtain stable structures experimentally. As a result, various computational methods have been developed to estimate peptide/protein structures, from the propensities of specific amino acids to form secondary structures39,40 to the predictions of secondary structures38,41 and tertiary structures.42 Basic methods are inaccurate but easy to use, while advanced methods have high accuracy but require significant computational resources. We need a fast and reliable approach to estimate the structural diversity of medium-large training datasets used prior building ML models. Secondary structure predictors offer a moderate cost and good performance, and are often used as a preliminary step before predicting the tridimensional structure. They are particularly handy for analysing the structural landscape of medium-large peptide/protein databases.
In the present study, we used GRAMPA, the giant repository of AMPs, counting 6760 unique sequences.43 Despite the abundance of sequences, the structural information of most AMPs remains unclear as only a tiny fraction (2.5%) have a resolved structure.44 Not many databases provide information on the number of structures they contain, but in the case of APD,45 the majority (60.41%) of its sequences have no known structure, 15.20% are peptides with identified disulfide bonds but lack a tridimensional structure, while 14.38% are α-helical peptides, and just 2.59% have β-sheet structures. This distribution is likely similar across other databases, indicating a significant gap in our understanding of AMP structures and the greater prevalence of α-helices. Here, we benchmarked four protein structure prediction methods – Jpred4,46 PEP2D,47 PSIPRED,48 AlphaFold2 (ref. 49) – using 261 curated and experimentally known structures from the PDBe database. We applied our best structure predictor to map the structural landscape of GRAMPA, the giant yet vastly uncharted repository of antimicrobial peptides.
2. Materials and methods
2.1. GRAMPA dataset
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 345 experimental minimum inhibitory concentration (MIC) values for 766 different bacteria, the most represented being E. coli (n = 9150), S. aureus (n = 8954), and P. aeruginosa (n = 4966), expressed in μM. MIC is the standard measure of antimicrobial activity and refers to the lowest drug concentration capable of inhibiting bacterial growth. In the present study, we only used the MIC values to filter GRAMPA into subsets. The GRAMPA repository and detailed information are available at: https://github.com/zswitten/Antimicrobial-Peptides.
345 experimental minimum inhibitory concentration (MIC) values for 766 different bacteria, the most represented being E. coli (n = 9150), S. aureus (n = 8954), and P. aeruginosa (n = 4966), expressed in μM. MIC is the standard measure of antimicrobial activity and refers to the lowest drug concentration capable of inhibiting bacterial growth. In the present study, we only used the MIC values to filter GRAMPA into subsets. The GRAMPA repository and detailed information are available at: https://github.com/zswitten/Antimicrobial-Peptides.
2.2. Benchmarking dataset
2.3. Protein secondary structure prediction (PSSP) methods
Two-hundred sixty one GRAMPA sequences with related experimental structure were used to test the performance of three secondary structure prediction tools: Jpred4, PEP2D and PSIPRED. The results are shown in ESI Table S1.† Jpred4 uses the JNet 2.3.1 algorithm based on neural networks for the prediction of secondary structure, solvent accessibility and supercoiled helices of proteins. It can be used in single sequences, sequence batches or multiple alignments and is available as a web server at: https://www.compbio.dundee.ac.uk/jpred/. It also has a REST API to easily automate the predictions, we use this method in this work. PEP2D is a tool developed in 2019 for the prediction of peptide secondary structure, it uses a random forest type multiclass classification algorithm and was built with a database balanced by ordinary folding type with sequences between 5 and 50 residues of length47 (http://crdd.osdd.net/raghava/pep2d/). Finally, we used PSIPRED version 2.0, which is based on neural networks to predict the ordinary states of protein secondary structure,48 included in the PHYRE2 (ref. 57) protein secondary structure prediction (http://www.sbg.bio.ic.ac.uk/%7Ephyre2/html/page.cgi?id=index). Although version 4.0 of PSIPRED is available as a web server at: http://bioinf.cs.ucl.ac.uk/psipred, it only allows secondary structure prediction of one sequence at a time, so it was unfeasible to use it with our dataset.2.4. Protein tertiary structure prediction method
We predicted the tridimensional structures of 261 AMP sequences using ColabFold,58 an easy-to-use interface using the AlphaFold2 (ref. 49) technology within the Google Colab environment. Their batch mode allows for the simultaneous protein structure prediction of medium-large datasets. In order to compare AlphaFold2 results with the aforementioned PSSP tools, we assigned secondary structure ranges (H, E, C) to our best-predicted structures using STRIDE.592.5. Comparing H, E, C distributions
After the secondary structure prediction of the GRAMPA sequences with experimentally resolved structure, we calculated the Jensen–Shannon distance between the secondary structure probability distributions of each prediction method and the experimental reference. The Jensen–Shannon distance is calculated from the square root of the Jensen–Shannon divergence and makes it possible to measure the similarity between two probability distributions,60 it is based on the Kullback–Leibler divergence, with the advantage of being symmetric. The Jensen–Shannon distance is defined as: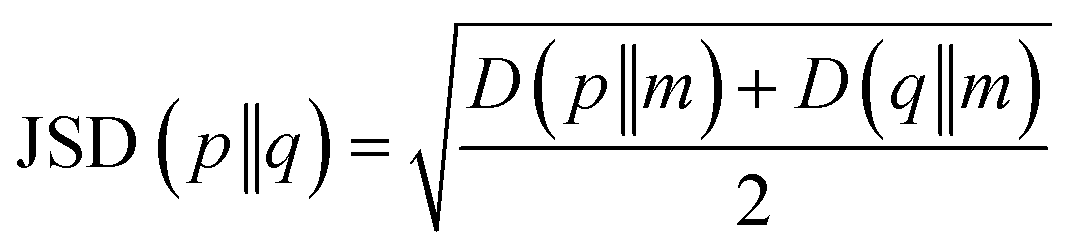 | (1) |
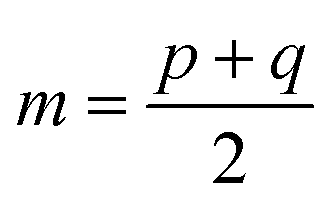 and D is the Kullback–Leibler divergence.
and D is the Kullback–Leibler divergence.
2.6. GRAMPA structural prediction
We calculated the secondary structure of the GRAMPA sequences with PEP2D, filtering out sequences with unusual amino acids and keeping only those with a length of 50 residues or less due to program limitations. Subsequently, we obtained the density of sequences by secondary structure to identify the largest structure represented, we repeated the procedure with three different data subsets for the most represented microorganisms in GRAMPA, and we evaluated the distribution of antimicrobial activity for each type of secondary structure. We then divided the original dataset into subclassifications based on their percentage composition of secondary structure into seven groups: helical and coil, mostly helical, helical and stranded, mostly stranded (β-sheet), stranded and coil, and mixed structures and mostly coil.2.7. Graphics
We displayed the predictions of the three states of secondary structure (H, E, C) using a ternary plot with R version 4.1.2 (2021-11-01)61 in R Studio62 with libraries ggtern and ggplot. The reported H/E/C values must be non-null to make sure that all data points are considered for density estimation.3. Results and discussion
3.1. Building the benchmarking dataset
345 experimental minimum inhibitory concentration (MIC) values for 766 different bacteria, the most represented being E. coli (n = 9150), S. aureus (n = 8954), and P. aeruginosa (n = 4966). After filtering for sequences with undetermined or non-canonical amino acids, we obtained 6169 unique sequences and 45498 associated MIC values for 738 unique bacteria. The sequences against E. coli, S. aureus and P. aeruginosa will be used later in the study to compare the predicted structural landscapes across the three bacterial strains. We obtained 66805 associations/pairs of a GRAMPA sequence with a PDBe structure. However, these results only comprised 2143 GRAMPA sequences related to 6087 unique identifiers. Many of these structures were related to more than one GRAMPA sequence and that, of the total number of sequences, only 34.7% had a related experimental structure (Fig. 1A). After reviewing the most prevalent biomolecules in our results (Fig. 1B), we observed large proteins that were only distantly related to the GRAMPA sequences through a short sequence motif with a significant degree of sequence identity between the two. Therefore, they did not truly represent the structures of the peptides present in GRAMPA. For example, we identified 36 GRAMPA sequences related to 364 PDBe identifiers belonging only to our most represented α-subunit of haemoglobin (Fig. 1C), leading to 10788 associations or pairs.
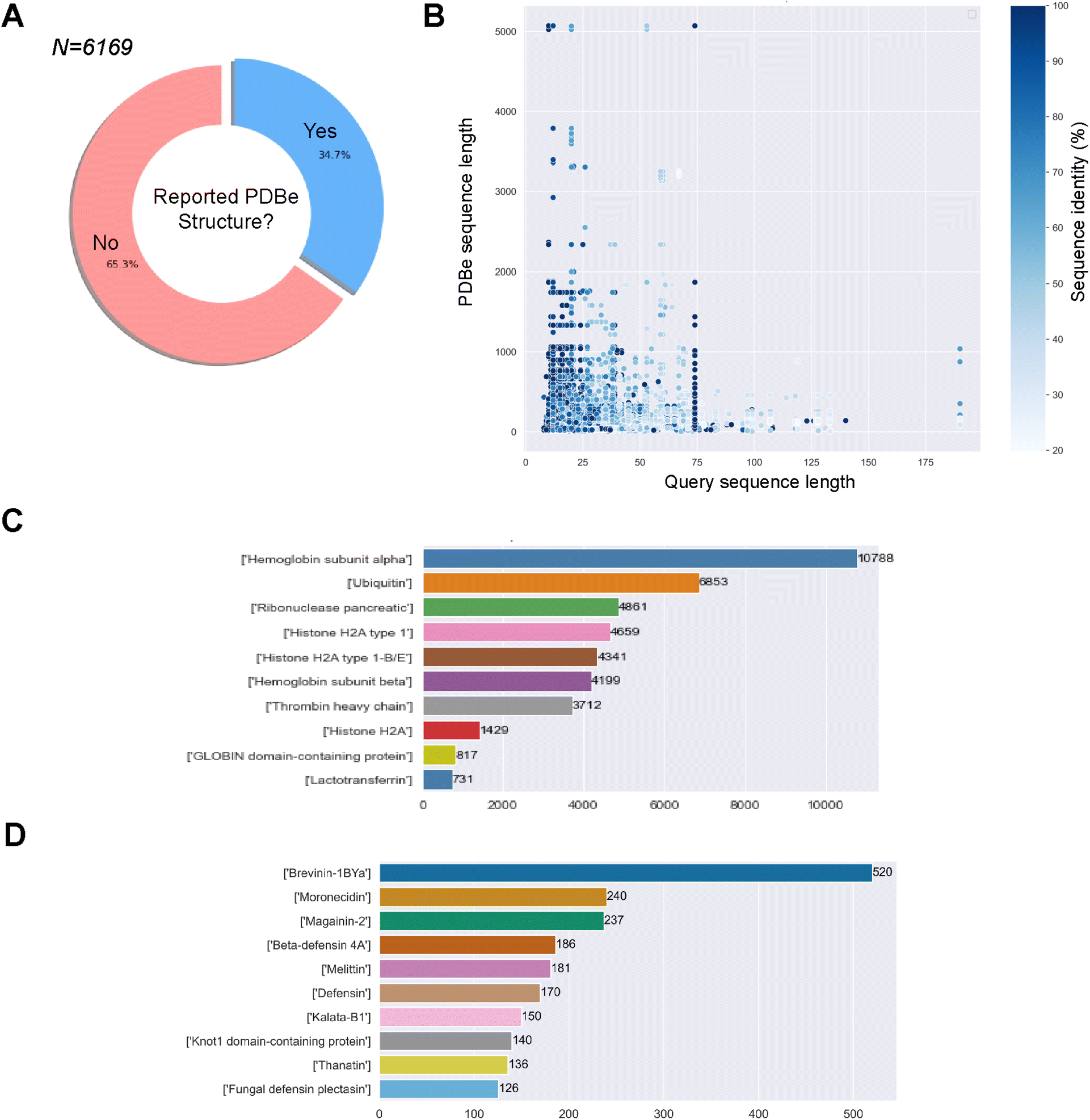 | ||
| Fig. 1 Obtaining our benchmarking dataset of GRAMPA sequences and related PDBe structures. (A) Percentages of GRAMPA sequences with or without related experimental structures, (B) comparing length and identity between GRAMPA sequences and matching PDBe sequences, (C) peptides or proteins with the largest number of related GRAMPA sequences, and (D) most represented AMP families after filtering the repository. | ||
266 pairs of GRAMPA sequences and PDBe structures, with 1475 GRAMPA sequences linked to 1905 PDBe structures that belong to 1015 PDBe sequences. We then calculated the Smith–Waterman distance between the GRAMPA and PDBe sequences, retaining only those with a minimum similarity of 70%. Our dataset consists of 3158 pairs, with 787 unique GRAMPA sequences and 723 PDBe structures. Finally, we removed sequences longer than 50 residues (due to the limits of secondary structure prediction tools), reducing the number to 2811 pairs, with 737 unique GRAMPA sequences, 435 PDBe structures and 320 PDBe sequences. We confirmed the usefulness of our filters by re-assessing the most prevalent biomolecules in our results, AMP families (Fig. 1D).
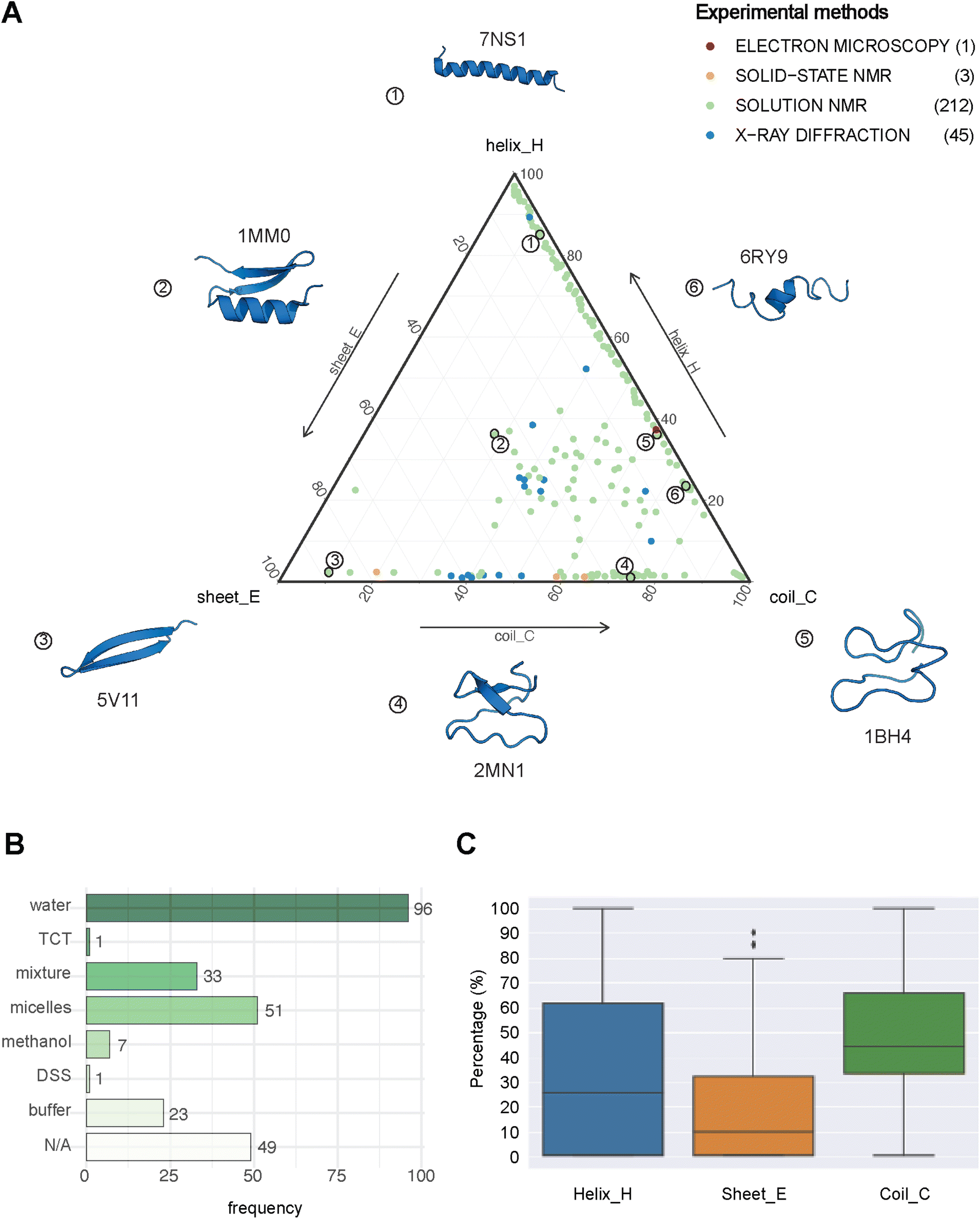 | ||
| Fig. 2 The structural landscape of our benchmarking dataset. (A) Ternary plot illustrating the secondary structure compositions for the 261 GRAMPA-related PDBe structures based on their experimental methods. The following six examples serve as structural markers; (1) pepG1 (PDB ID: 7NS1), (2) termicin (1MM0), (3) synthetic arenicin-3 analogue (5V11), (4) kalata B1[W23WW] (2MN1), (5) circulin A (1BH4) and (6) pleurocidin-like peptide 1a-1 (6RY9). (B) Solvents used to solve the 212 NMR solution structures (TCT: tri-N-acetylchitotriose, DSS: sodium 4,4-dimethyl-4-silapentane-1-sulfonate). (C) General distribution of each secondary structure (H, E, C) across our experimental references. | ||
3.2. Evaluating the performances of four protein predictors
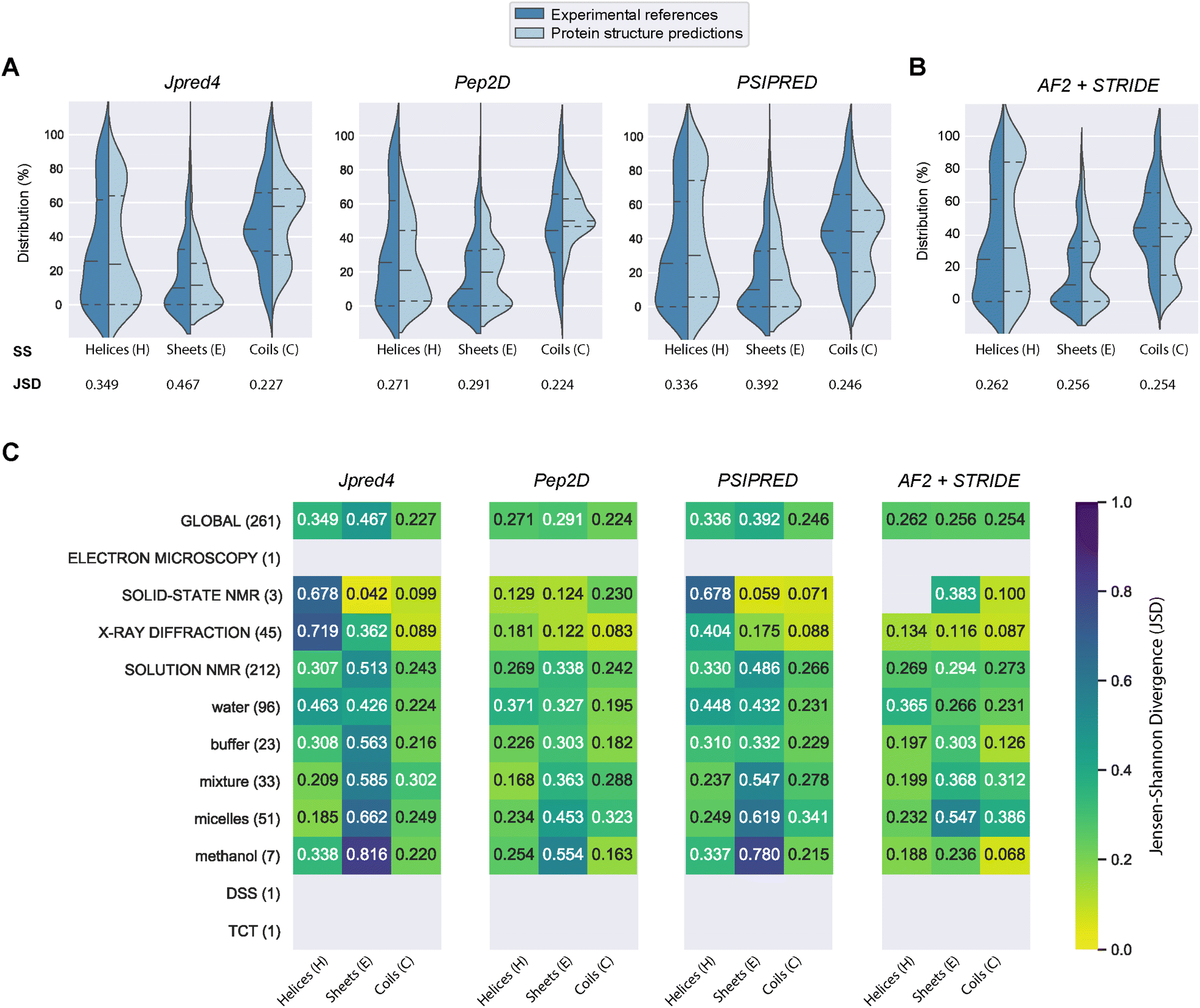 | ||
| Fig. 3 Comparing four structural prediction methods. (A) Violin plots showing the distributions of the three secondary structure states – helix (H), strand (E), and coil (C) – across the three PSSP methods Jpred4, PEP2D, PSIPRED for our benchmarking dataset. (B) Violin plots showing the performances of AlphaFold2 with STRIDE. Jensen–Shannon divergences (JSD) indicate similarities between the predicted and experimental distributions, a low value is synonymous with high similarity. Horizontal dash lines indicate the median and quartiles lines. (C) JSD values across different experiment methods and nuclear magnetic resonance (NMR) solvent conditions. | ||
PEP2D predictions remained the closest to the experimental distributions (lowest JSD values) across the three secondary structure states, the experimental methods, and the NMR solvent conditions, as illustrated in Fig. 3C. PEP2D underwent training with consideration given to the imbalance of secondary structures.47 Indeed, its training dataset was heavily populated with coiled motifs, leading to a skewed outcome towards this particular secondary structure. To address this issue, the authors employed a balancing technique, where the weight each secondary structure holds in the prediction was adjusted based on the ratio between the most abundant secondary structure and the one that needed to be balanced. This could account for its good performance in predicting the three secondary structure states. In addition, PEP2D was solely trained using peptide sequences unlike Jpred4 and PSIPRED. As a result, we anticipated that PEP2D would perform better when tested with sequences of similar length distributions that it was trained on. Finally, Fig. S2† displayed the distribution of PEP2D secondary structure predictions (cyan) compared to the ground truth (blue). The six structures mentioned above also depicted the differences between PEP2D predictions and experimental references. The lines indicated the error between the three-state secondary structures (H, E, C) measured between PEP2D predictions and corresponding experimental references. For example, the small lines for (1) pepG1 (7NS1), (2) termicin (1MM0), and (4) kalata B1[W23WW] (2MN1) suggested minor errors. In contrast, the lines for (3) arenicin-3 analogue (5V11), (5) circulin A (1BH4) and (6) pleurocidin-like peptide 1a-1 (6RY9) were wider suggesting more noticeable prediction errors. However, these errors remained local as the predictions and the experimental structures were predominantly in the same structural “regions”.
:methanol conditions, whereas the highest scores were observed with experiments in buffers, micelles and water.
In order to compare our newly predicted AF2 structures to the protein secondary structure predictions, we submitted each selected AF2 structure to the STRIDE webserver59 and measured the 3-state secondary structures (H, E, C) of each structure. The selected 261 AF2 predictions, their corresponding STRIDE assessments were summarised in Table S2.† In Fig. 3B and S5,† we depicted the AF2+STRIDE predictions (cyan) against experimental distributions (blue) across the three secondary structure states (H, E, C). The results indicated that AF2+STRIDE outperformed PEP2D with its predictions closer to the experimental distributions for the helical and stranded states (respective JSD values of 0.262 and 0.256). These observations remain true with PDBe structures solved using X-ray crystallography and certain NMR solvents (i.e., methanol, buffers), as depicted in Fig. 3C. In contrast, all three PSSP methods accounted for better predictions of the coiled state than AF2+STRIDE (C: JSD = 0.254), particularly for PDBe structures solved in aqueous NMR solutions like water, micelles or mixtures, see Fig. 3C. Our results corroborate the recent findings by McDonald and co-workers regarding the performance of AlphaFold2 in predicting 588 peptide structures between 10 and 40 amino acids.63 The authors also reported that AF2 predicted helical (H) and stranded/β-sheets structures with high accuracy, but the program failed with segments presenting low pLDDT scores, often associated with coils (C). Our approach combining AlphaFold2 and STRIDE was more time-consuming and computer-intensive than PSSP methods. Despite these shortcomings, the coupled method represents an excellent alternative to PEP2D, particularly for peptide sequences with 50 or more residues.
3.3. Mapping the structural landscape of GRAMPA
Considering the rapid implementation of PEP2D to estimate secondary structures of our benchmarking dataset, we chose to apply this tool to the entire GRAMPA repository. After eliminating sequences with unusual amino acids and length greater than 50 residues, we submitted 5980 GRAMPA sequences to PEP2D for secondary structure prediction.The results are shown in Fig. 4, where each dot represents the PEP2D prediction of a GRAMPA sequence across the 3-state secondary structures (H, E, C). We also depicted the density of structures present in GRAMPA using a colour gradient (green “low” – red “high”). Our analysis showed that the peptide dataset is heavily populated with sequences that are likely forming helices and sequences with a fully extended structure (coils). The abundance of helical structures may be due to evolutionary forces that have preferred them, but it may also stem from certain studies that purposely skew their predictive and generative models towards these AMP structures.23,26 We noticed that specific dots followed each other forming straight lines. They corresponded to PEP2D predictions that presented similar or identical values for two of the 3-state secondary structures (H, E, C); they were not homologous sequences.
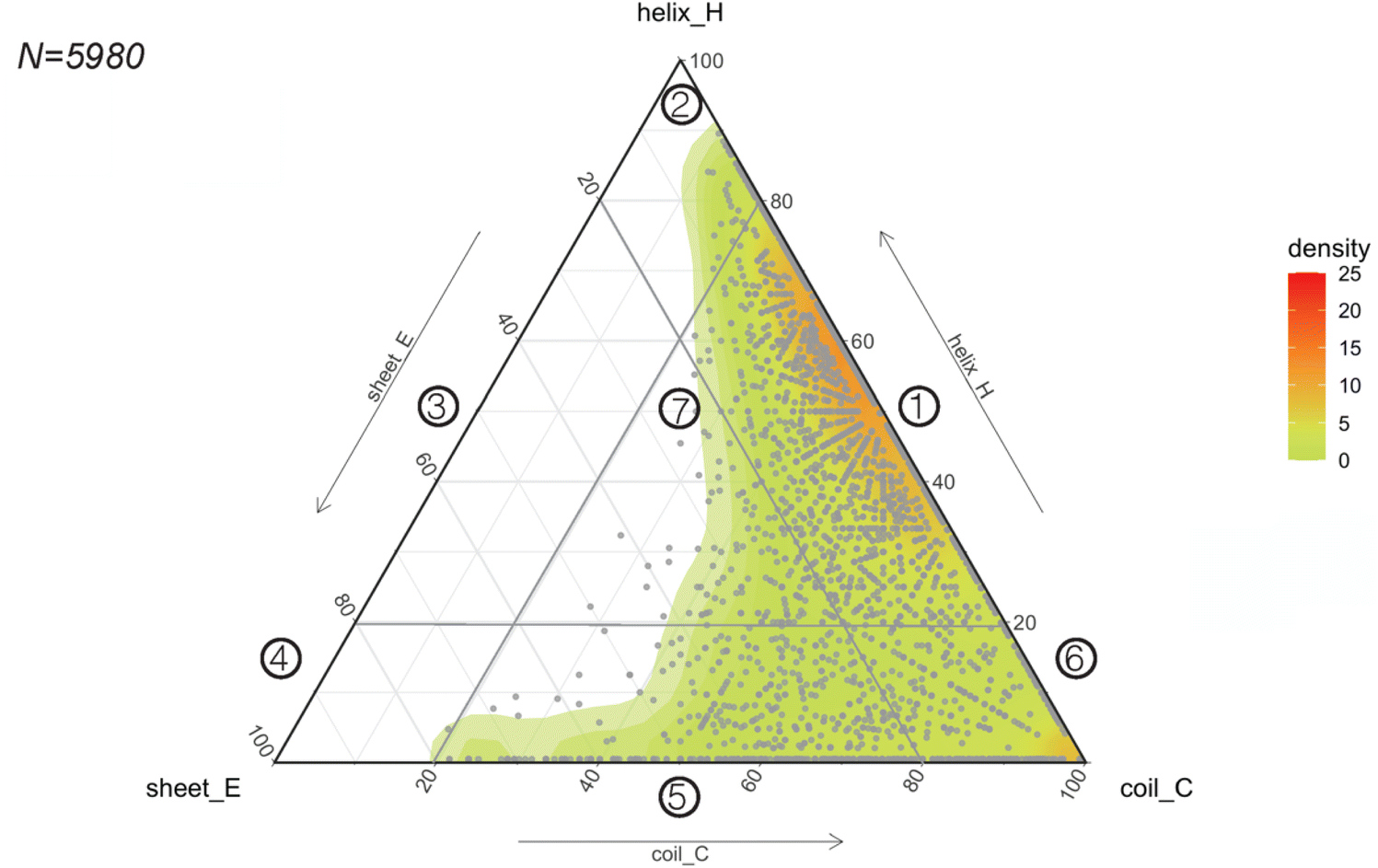 | ||
| Fig. 4 Predicting the structural space of GRAMPA. Ternary plot (points and density map) showing the PEP2D secondary structure predictions. | ||
To ease readership of GRAMPA structures based on their secondary structure composition, we created a classification system using a graphical representation of the percentage of structural composition. The segmentation was done by dividing the ternary plot into three triangles and assigning a structural class to each intersection of their edges (Fig. 4). The resulting seven classes were: helical and coiled structures (1), mostly helical structures (2), helical and stranded/β-sheet structures (3), mostly β-sheet structures (4), stranded/β-sheet and coiled structures (5), mixed structures (7), and mostly coiled structures (7). Here, we report the largest structural prediction of AMPs with 5980 sequences. Our segmented analysis further confirmed that the GRAMPA repository is dominated by helical and coiled structures (1), with three-four times more sequences than the second most represented classification, mostly coiled structures (6), i.e. 65.1% versus 17.8% (Table 1). In contrast, structural classes (2)–(4) were disregarded due to the few structures they contained (<2%). Finally, stranded and coiled structures (5) and mixed structures (7) represent about 15% of the entire dataset.
| Structural class | Benchmarking dataset (N = 261) | GRAMPA (N = 5980) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PDBe | % | PEP2D | % | AF2+STRIDE | % | PEP2D | % | ||
| 1 | Helices and coils | 90 | 34.5 | 106 | 40.6 | 46 | 17.6 | 3892 | 65.1 |
| 2 | Mostly helices | 36 | 13.8 | 3 | 1.1 | 72 | 27.6 | 88 | 1.5 |
| 3 | Helices and strands | 1 | 0.4 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| 4 | Mostly strands (β-sheets) | 4 | 1.5 | 0 | 0.0 | 0 | 0.0 | 1 | 0.0 |
| 5 | Strands and coils | 63 | 24.1 | 113 | 43.3 | 89 | 34.1 | 754 | 12.6 |
| 6 | Mostly coiled structures | 27 | 10.3 | 15 | 5.7 | 4 | 0.0 | 1063 | 17.8 |
| 7 | Mixed structures | 40 | 15.3 | 24 | 9.2 | 50 | 19.2 | 182 | 3.0 |
Before our study, Kozic and co-workers conducted the large-scale Rosetta ab initio modelling of 184 AMPs containing between 20 and 120 residues.64 The authors measured PSIPRED secondary structure predictions and clustered all 184 peptides into one of 4 structural classes; all-α, all-β, αβ and coil. About half were predicted to fold into α-helices, supporting our general observation that helical structures dominate fold spaces of the benchmarking dataset and GRAMPA (structural classes 1 and 2, Table 1). In addition, stranded/β-sheets structures represented about 15% of their dataset in agreement with GRAMPA (structural classes 4 and 5). These percentages were higher in the benchmarking dataset for PDBe structures and PEP2D predictions. Unlike our study, the authors indicated few coiled structures – i.e., 0.5% vs. 17.6% (structural class 6) – and many αβ structures – i.e., 33.7% vs. 3.1% (structural classes 3 and 7). The differences in the number of peptide sequences, ranges in sequence length, PSSP predictions and disulfide-rich structures between the three datasets might explain these variations in percentages (Table 1). In Fig. 3A, PSIPRED was less successful than PEP2D in predicting the 3-state secondary structures (H, E, C) for the 261 AMPs.
In 2021, Morita and co-workers implemented PSIPRED predictions to protein sequences from multiple species.65 The authors found that proteins from eukaryotic origins were rich in helices and coils, whereas bacteria and archaea abound in stranded proteins. Some of the public AMP databases that form GRAMPA indicate their taxon diversity. For example, 73% of APD3 include peptides from animals.45
3.4. Mapping the structural landscapes of GRAMPA subsets
The release of GRAMPA43 in 2019 has led several research groups to develop generative ML models capable of designing broad-spectrum AMPs25,44 and strain-specific AMPs.26,27,66–69 Most studies described their AI-generated AMPs to share similar physicochemical properties (i.e., hydrophobicity, hydrophobic moment, global charge) and similar amino acid composition (i.e., moderate-high fractions in alanine, valine, glycine, lysine, arginine) to their training sets. In addition, some would predict the newly generated peptides to fold into α-helices using helical wheel representation and circular dichroism,25,27 protein structure predictors,26,66,69 or molecular dynamics simulations.68 The training sets often consisted of sequences with proteinogenic residues (except cysteine), positively charged, between 10 and 52 residues in length, and potentially amidated on their C-terminus. Their secondary or tertiary structures were often ignored but assumed to be helical.Thus, we explored the structural composition of GRAMPA sequences inhibiting the three bacterial strains E. coli, S. aureus or P. aeruginosa (FASTA sequences, see Data availability). After eliminating sequences with unusual amino acids and lengths greater than 50 residues, we displayed PEP2D secondary structure predictions of the three GRAMPA subsets in Fig. 5 – top-left: E. coli N = 4,567, top-right: S. aureus N = 4,146, and bottom-right: P. aeruginosa N = 2519. In addition, we reported the secondary structures for 3367 GRAMPA sequences (Fig. 5, bottom-left); the subset is quasi-identical to the 3280 training sequences used to build PepVAE,26 showing antimicrobial activity against E. coli. The results, summarised in Table 2, alluded that the first three subsets would mimic the fold landscape of GRAMPA; where most of the sequences (i.e., 67–69.5%) may fold into α-helices (1) and another 16.4–17.5% would predict as mostly coiled structures (6). The structural classes (2)–(4) were merely observed (<2%). Stranded and coiled structures (5) and mixed structures (7) represented 12.4–13.9% of the subsets.
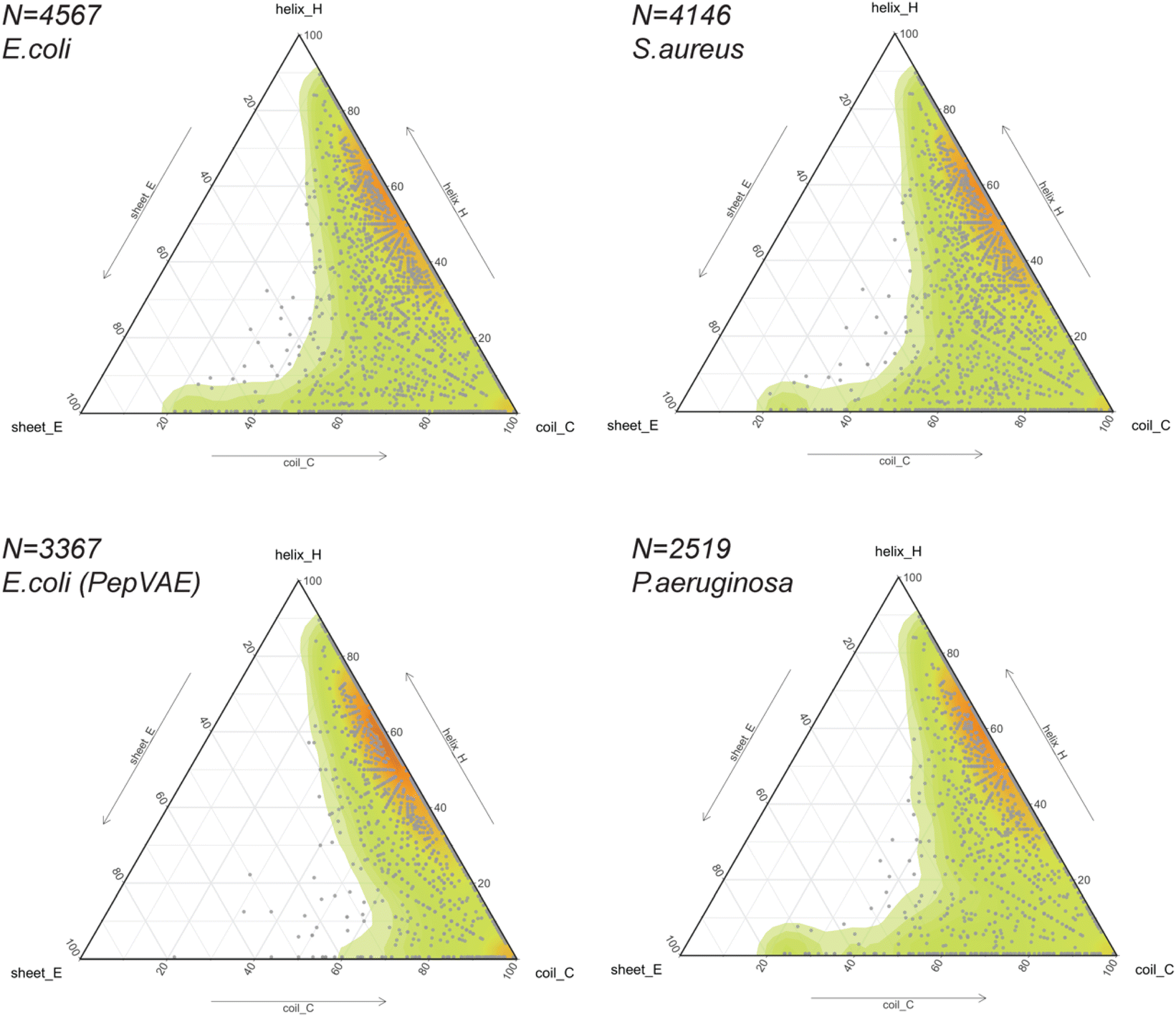 | ||
| Fig. 5 Predicting the structural spaces of GRAMPA subsets. Ternary plots (points and density map) showing the PEP2D secondary structure predictions applied to 4 subsets against E.coli (top-left and bottom-left), S. aureus (top right) and P. aeruginosa (bottom-right). | ||
| Structural class | E. coli | S. aureus | P. aeruginosa | E.coli (PepVAE) | |||||
|---|---|---|---|---|---|---|---|---|---|
| N = 4567 | % | N = 4146 | % | N = 2519 | % | N = 3367 | % | ||
| 1 | Helices and coils | 3059 | 67.0 | 2805 | 67.6 | 1751 | 69.5 | 2578 | 76.6 |
| 2 | Mostly helices | 71 | 1.6 | 66 | 1.6 | 42 | 1.7 | 56 | 1.7 |
| 3 | Helices and strands | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| 4 | Mostly strands (β-sheets) | 0 | 0.0 | 1 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| 5 | Strands and coils | 525 | 11.5 | 466 | 11.2 | 260 | 10.3 | 97 | 2.9 |
| 6 | Mostly coiled structures | 800 | 17.5 | 694 | 16.7 | 413 | 16.4 | 610 | 18.1 |
| 7 | Mixed structures | 112 | 2.4 | 114 | 2.7 | 53 | 2.1 | 26 | 0.8 |
Our structural analysis of the PepVAE-like subset (Fig. 5, bottom-left) predominantly showed three-four times more helical and coiled structures (1) than mostly coiled structures (6). The structural classes (2)–(4) were quasi-inexistent. Removing cysteine-rich sequences to the original E. coli subset has drastically reduced PEP2D predictions across stranded and coiled structures (5) and mixed structures (7), i.e., <4% (Table 2). These observations coincided with Dean and co-workers’ observations, where their generated AMPs against E. coli were likely folding into α-helices (Group B, Fig. 3 and 5A).26 Likewise, the authors reported that the generated AMPs against S. aureus or P. aeruginosa would mostly be α-helical structures. We can therefore assume that removing cysteine-rich sequences from the relevant subsets would enrich their training sets with folds from structural classes (1), (2) and (6). Notably, their strain-specific predictive models towards the three bacterial strains were prone to fewer errors between predicted and experimental MICs with the α-helical subset (Group B) than the one with more diverse structures (Fig. 5B). These results suggested a bias towards α-helical structures from model training and models that might not generalise well over other structural classes. It further highlights the importance of estimating the structures of medium-large training datasets before building predictive or generative ML models.
4. Conclusions
The present study searched for a fast and reliable approach to estimate the structural diversity of medium-large training datasets for general fold discovery. We considered three protein secondary structure predictors (PSSP) Jpred4, PEP2D, PSIPRED and the 3D structure predictor AlphaFold2 (batch mode) in combination with STRIDE for secondary structure annotation. We benchmarked the four PSP methods comparing 261 curated and experimentally known PDBe structures with their predicted 3-state secondary structures (H, E, C). PEP2D predictions were the closest to the experimental distributions across the three states among the PSSP methods. Our results also revealed that the AlphaFold2+STRIDE approach provided more accurate predictions of helical and stranded/β-sheet structures, but PSSP methods performed better for coiled structures. The protein secondary structure predictor PEP2D is fast, and its results were comparable to those of AlphaFold2+STRIDE to estimate the structural landscape of sequential datasets with less than 50 residues. The coupled method represents an excellent alternative to PEP2D, particularly for peptide or protein sequences with 50 or more residues.Considering the rapid implementation of PEP2D, we explored the structural landscape of GRAMPA, the giant yet vastly uncharted repository of antimicrobial peptides (AMPs). Our analysis showed that most 5980 peptide sequences would adopt helical structures (65.1%), random coils (17.8%), and β-stranded and mixed structures accounted for the rest. We observed similar structural compositions across three strain-specific GRAMPA subsets against E. coli, S. aureus or P. aeruginosa. Removing cysteine-rich sequences further enriches the subset with helical and coiled structures. Finally, we introduced a new classification system for peptide structures based on their secondary structure composition, which provided a convenient way to visualize and compare the diversity of AMP folds. The abundance of helical structures may be due to evolutionary forces that have preferred them, but it may also stem from specific studies that skew their sequence-based predictive and generative models towards this structural class. Early peptide/protein structure prediction of medium-large training datasets becomes crucial prior to building predictive or generative ML models.
Data availability
Fig. S1 and Tables S1, S2 are available in ESI.† Data and scripts used to reproduce the computational experiments: (1) extracting information from Protein Data Bank in Europe (PDBe) database, (2) annotating secondary structure annotation of AlphaFold2-predicted structures with STRIDE, (3) benchmarking secondary structure states (H/E/C) for different protein structure predictors (AlphaFold2, PEP2D, Jpred, PHYRE2) and (4) plotting peptide/protein structural landscape(s) are available at: https://github.com/DanielAldas/Benchmark-PSP.Author contributions
F. P. conceptualised the investigation. Both authors (V. D. A.-B. and F. P.) carried out the investigation including methodology, data curation, bioinformatic analysis and figures. Both authors wrote, edited and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to the Mexican research council Consejo Nacional de Ciencia y Tecnología (CONACYT), grant number A1-S-32579. V. D. A.-B. was the recipient of a national CONACYT postgraduate scholarship (No 772091). F. P. was supported by a Cátedras CONACYT fellowship – 2017–2022. We thank Dr David Armstrong from European Molecular Biology Laboratory (EMBL), European Bioinformatics Institute (EBI), Hinxton, United Kingdom for providing access to the original PDBe tutorials.Notes and references
- C. D. Fjell, J. A. Hiss, R. E. W. Hancock and G. Schneider, Designing antimicrobial peptides: form follows function, Nat. Rev. Drug Discovery, 2012, 11, 37–51 CrossRef CAS PubMed.
- M. H. Cardoso, R. Q. Orozco, S. B. Rezende, G. Rodrigues, K. G. N. Oshiro, E. S. Cândido and O. L. Franco, Computer-Aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates?, Front. Microbiol., 2020, 10, 1–15 CrossRef PubMed.
- M. C. R. Melo, J. R. M. A. Maasch and C. de la Fuente-Nunez, Accelerating antibiotic discovery through artificial intelligence, Commun. Biol., 2021, 4, 1–13 CrossRef PubMed.
- F. Wan, D. Kontogiorgos-Heintz and C. de la Fuente-Nunez, Deep generative models for peptide design, Digital Discovery, 2022, 1, 195–208 RSC.
- N. Bennett, B. Coventry, I. Goreshnik, B. Huang, A. Allen, D. Vafeados, Y. P. Peng, J. Dauparas, M. Baek, L. Stewart, F. DiMaio, S. D. Munck, S. N. Savvides and D. Baker, Improving de novo Protein Binder Design with Deep Learning, Nat. Commun., 2023, 14(2625), 1–9 Search PubMed.
- J. Graves, J. Byerly, E. Priego, N. Makkapati, S. V. Parish, B. Medellin and M. Berrondo, A Review of Deep Learning Methods for Antibodies, Antibodies, 2020, 9, 12 CrossRef CAS PubMed.
- M. Pertseva, B. Gao, D. Neumeier, A. Yermanos and S. T. Reddy, Applications of Machine and Deep Learning in Adaptive Immunity, Annu. Rev. Chem. Biomol. Eng., 2021, 12, 39–62 CrossRef CAS PubMed.
- R. Akbar, P. A. Robert, C. R. Weber, M. Widrich, R. Frank, M. Pavlović, L. Scheffer, M. Chernigovskaya, I. Snapkov, A. Slabodkin, B. B. Mehta, E. Miho, F. Lund-Johansen, J. T. Andersen, S. Hochreiter, I. Hobæk Haff, G. Klambauer, G. K. Sandve and V. Greiff, In silico proof of principle of machine learning-based antibody design at unconstrained scale, mAbs, 2022, 14, 2031482 CrossRef PubMed.
- J. Kim, M. McFee, Q. Fang, O. Abdin and P. M. Kim, Computational and artificial intelligence-based methods for antibody development, Trends Pharmacol. Sci., 2023, 44, 175–189 CrossRef CAS PubMed.
- D. Ofer, N. Brandes and M. Linial, The language of proteins: NLP, machine learning & protein sequences, Comput. Struct. Biotechnol. J., 2021, 19, 1750–1758 CrossRef CAS PubMed.
- T. Bepler and B. Berger, Learning the protein language: evolution, structure, and function, Cell Syst., 2021, 12, 654–669 CrossRef CAS PubMed.
- B. L. Hie and K. K. Yang, Adaptive machine learning for protein engineering, Curr. Opin. Struct. Biol., 2022, 72, 145–152 CrossRef CAS PubMed.
- S. Unsal, H. Atas, M. Albayrak, K. Turhan, A. C. Acar and T. Doğan, Learning functional properties of proteins with language models, Nature Machine Intelligence, 2022, 4, 227–245 CrossRef.
- B. E. Clifton, D. Kozome and P. Laurino, Efficient Exploration of Sequence Space by Sequence-Guided Protein Engineering and Design, Biochemistry, 2023, 62, 210–220 CrossRef CAS PubMed.
- A. Madani, B. Krause, E. R. Greene, S. Subramanian, B. P. Mohr, J. M. Holton, J. L. Olmos, C. Xiong, Z. Z. Sun, R. Socher, J. S. Fraser and N. Naik, Large language models generate functional protein sequences across diverse families, Nat. Biotechnol., 2023, 1–8 Search PubMed.
- P. A. Romero, A. Krause and F. H. Arnold, Navigating the protein fitness landscape with Gaussian processes, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E193–E201 CrossRef CAS PubMed.
- K. K. Yang, Z. Wu and F. H. Arnold, Machine-learning-guided directed evolution for protein engineering, Nat. Methods, 2019, 16, 687–694 CrossRef CAS PubMed.
- S. Mazurenko, Z. Prokop and J. Damborsky, Machine Learning in Enzyme Engineering, ACS Catal., 2020, 10, 1210–1223 CrossRef CAS.
- Z. Wu, K. E. Johnston, F. H. Arnold and K. K. Yang, Protein sequence design with deep generative models, Curr. Opin. Chem. Biol., 2021, 65, 18–27 CrossRef CAS PubMed.
- W. D. Jang, G. B. Kim, Y. Kim and S. Y. Lee, Applications of artificial intelligence to enzyme and pathway design for metabolic engineering, Curr. Opin. Biotechnol., 2022, 73, 101–107 CrossRef CAS PubMed.
- K. K. Yang, https://github.com/yangkky/Machine-learning-for-proteins.
- S. P. Zhang, https://github.com/Peldom/papers_for_protein_design_using_DL.
- A. T. Müller, J. A. Hiss and G. Schneider, Recurrent Neural Network Model for Constructive Peptide Design, J. Chem. Inf. Model., 2018, 58, 472–479 CrossRef PubMed.
- D. Nagarajan, T. Nagarajan, N. Roy, O. Kulkarni, S. Ravichandran, M. Mishra, D. Chakravortty and N. Chandra, Computational antimicrobial peptide design and evaluation against multidrug-resistant clinical isolates of bacteria, J. Biol. Chem., 2018, 293, 3492–3509 CrossRef CAS PubMed.
- A. Tucs, D. P. Tran, A. Yumoto, Y. Ito, T. Uzawa and K. Tsuda, Generating Ampicillin-Level Antimicrobial Peptides with Activity-Aware Generative Adversarial Networks, ACS Omega, 2020, 5, 22847–22851 CrossRef CAS PubMed.
- S. N. Dean, J. A. E. Alvarez, D. Zabetakis, S. A. Walper and A. P. Malanoski, PepVAE: Variational Autoencoder Framework for Antimicrobial Peptide Generation and Activity Prediction, Front. Microbiol., 2021, 12, 725727 CrossRef PubMed.
- A. Capecchi, X. Cai, H. Personne, T. Köhler, C. van Delden and J.-L. Reymond, Machine learning designs non-hemolytic antimicrobial peptides, Chem. Sci., 2021, 12, 9221–9232 RSC.
- F. Grisoni, C. S. Neuhaus, G. Gabernet, A. T. Müller, J. A. Hiss and G. Schneider, Designing Anticancer Peptides by Constructive Machine Learning, ChemMedChem, 2018, 13, 1300–1302 CrossRef CAS PubMed.
- E. Zakharova, M. Orsi, A. Capecchi and J.-L. Reymond, Machine Learning Guided Discovery of Non-Hemolytic Membrane Disruptive Anticancer Peptides, ChemMedChem, 2022, 17, e202200291 CrossRef CAS PubMed.
- R. Batra, T. D. Loeffler, H. Chan, S. Srinivasan, H. Cui, I. V. Korendovych, V. Nanda, L. C. Palmer, L. A. Solomon, H. C. Fry and S. K. R. S. Sankaranarayanan, Machine learning overcomes human bias in the discovery of self-assembling peptides, Nat. Chem., 2022, 14, 1427–1435 CrossRef CAS PubMed.
- C. N. Bedbrook, K. K. Yang, A. J. Rice, V. Gradinaru and F. H. Arnold, Machine learning to design integral membrane channelrhodopsins for efficient eukaryotic expression and plasma membrane localization, PLoS Comput. Biol., 2017, 13, e1005786 CrossRef PubMed.
- Y. Saito, M. Oikawa, H. Nakazawa, T. Niide, T. Kameda, K. Tsuda and M. Umetsu, Machine-Learning-Guided Mutagenesis for Directed Evolution of Fluorescent Proteins, ACS Synth. Biol., 2018, 7, 2014–2022 CrossRef CAS PubMed.
- M. Yang, C. Fehl, K. V. Lees, E.-K. Lim, W. A. Offen, G. J. Davies, D. J. Bowles, M. G. Davidson, S. J. Roberts and B. G. Davis, Functional and informatics analysis enables glycosyltransferase activity prediction, Nat. Chem. Biol., 2018, 14, 1109–1117 CrossRef CAS PubMed.
- S. Ovchinnikov and P.-S. Huang, Structure-based protein design with deep learning, Curr. Opin. Chem. Biol., 2021, 65, 136–144 CrossRef CAS PubMed.
- X. Pan and T. Kortemme, Recent advances in de novo protein design: principles, methods, and applications, J. Biol. Chem., 2021, 296, 100558 CrossRef CAS PubMed.
- N. Ferruz, M. Heinzinger, M. Akdel, A. Goncearenco, L. Naef and C. Dallago, From sequence to function through structure: deep learning for protein design, Comput. Struct. Biotechnol. J., 2023, 21, 238–250 CrossRef CAS PubMed.
- A. H.-W. Yeh, C. Norn, Y. Kipnis, D. Tischer, S. J. Pellock, D. Evans, P. Ma, G. R. Lee, J. Z. Zhang, I. Anishchenko, B. Coventry, L. Cao, J. Dauparas, S. Halabiya, M. DeWitt, L. Carter, K. N. Houk and D. Baker, De novo design of luciferases using deep learning, Nature, 2023, 614, 774–780 CrossRef CAS PubMed.
- T. Smolarczyk, I. Roterman-Konieczna and K. Stapor, Protein Secondary Structure Prediction: A Review of Progress and Directions, Curr. Bioinf., 2020, 15, 90–107 CrossRef CAS.
- A.-M. Fernandez-Escamilla, F. Rousseau, J. Schymkowitz and L. Serrano, Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins, Nat. Biotechnol., 2004, 22, 1302–1306 CrossRef CAS PubMed.
- M. Levitt, Conformational preferences of amino acids in globular proteins, Biochemistry, 1978, 17, 4277–4285 CrossRef CAS PubMed.
- Q. Jiang, X. Jin, S.-J. Lee and S. Yao, Protein secondary structure prediction: a survey of the state of the art, J. Mol. Graphics Modell., 2017, 76, 379–402 CrossRef CAS PubMed.
- B. Kuhlman and P. Bradley, Advances in protein structure prediction and design, Nat. Rev. Mol. Cell Biol., 2019, 20, 681–697 CrossRef CAS PubMed.
- J. Witten and Z. Witten, Deep learning regression model for antimicrobial peptide design, bioRxiv, 2019, 692681 Search PubMed.
- C. M. Van Oort, J. B. Ferrell, J. M. Remington, S. Wshah and J. Li, AMPGAN v2: Machine Learning-Guided Design of Antimicrobial Peptides, J. Chem. Inf. Model., 2021, 61, 2198–2207 CrossRef CAS PubMed.
- G. Wang, X. Li and Z. Wang, APD3: the antimicrobial peptide database as a tool for research and education, Nucleic Acids Res., 2016, 44, D1087–D1093 CrossRef CAS PubMed.
- A. Drozdetskiy, C. Cole, J. Procter and G. J. Barton, JPred4: a protein secondary structure prediction server, Nucleic Acids Res., 2015, 43, W389–W394 CrossRef CAS PubMed.
- H. Singh, S. Singh and G. P. S. Raghava, Peptide Secondary Structure Prediction using Evolutionary Information, bioRxiv, 2019, 558791, DOI:10.1101/558791.
- D. W. A. Buchan and D. T. Jones, The PSIPRED Protein Analysis Workbench: 20 Years on, Nucleic Acids Res., 2019, 47, W402–W407 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- M. Novković, J. Simunić, V. Bojović, A. Tossi and D. Juretić, DADP: the database of anuran defense peptides, Bioinformatics, 2012, 28, 1406–1407 CrossRef PubMed.
- M. Pirtskhalava, A. Gabrielian, P. Cruz, H. L. Griggs, R. B. Squires, D. E. Hurt, M. Grigolava, M. Chubinidze, G. Gogoladze, B. Vishnepolsky, V. Alekseev, A. Rosenthal and M. Tartakovsky, DBAASP v.2: an enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides, Nucleic Acids Res., 2016, 44, D1104–D1112 CrossRef CAS PubMed.
- X. Kang, F. Dong, C. Shi, S. Liu, J. Sun, J. Chen, H. Li, H. Xu, X. Lao and H. Zheng, DRAMP 2.0, an updated data repository of antimicrobial peptides, Sci. Data, 2019, 6, 148 CrossRef PubMed.
- S. P. Piotto, L. Sessa, S. Concilio and P. Iannelli, YADAMP: yet another database of antimicrobial peptides, Int. J. Antimicrob. Agents, 2012, 39, 346–351 CrossRef CAS PubMed.
- M. Steinegger and J. Söding, MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets, Nat. Biotechnol., 2017, 35, 1026–1028 CrossRef CAS PubMed.
- D. R. Armstrong, J. M. Berrisford, M. J. Conroy, A. Gutmanas, S. Anyango, P. Choudhary, A. R. Clark, J. M. Dana, M. Deshpande, R. Dunlop, P. Gane, R. Gáborová, D. Gupta, P. Haslam, J. Koča, L. Mak, S. Mir, A. Mukhopadhyay, N. Nadzirin, S. Nair, T. Paysan-Lafosse, L. Pravda, D. Sehnal, O. Salih, O. Smart, J. Tolchard, M. Varadi, R. Svobodova-Vařeková, H. Zaki, G. J. Kleywegt and S. Velankar, PDBe: improved findability of macromolecular structure data in the PDB, Nucleic Acids Res., 2020, 48, D335–D343 CAS.
- T. F. Smith, M. S. Waterman and C. Burks, The statistical distribution of nucleic acid similarities, Nucleic Acids Res., 1985, 13, 645–656 CrossRef CAS PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, The Phyre2 web portal for protein modeling, prediction and analysis, Nat. Protoc., 2015, 10, 845–858 CrossRef CAS PubMed.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, ColabFold: making protein folding accessible to all, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- M. Heinig and D. Frishman, STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins, Nucleic Acids Res., 2004, 32, W500–W502 CrossRef CAS PubMed.
- J. Lin, Divergence measures based on the Shannon entropy, IEEE Trans. Inf. Theory, 1991, 37, 145–151 CrossRef.
- R Core Team, R: a language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, 2020 Search PubMed.
- RStudio Team, RStudio: Integrated Development for R, RStudio, PBC, Boston, MA, 2020, http://www.rstudio.com/ Search PubMed.
- E. F. McDonald, T. Jones, L. Plate, J. Meiler and A. Gulsevin, Benchmarking AlphaFold2 on peptide structure prediction, Structure, 2023, 31, 111–119 CrossRef CAS PubMed.
- M. Kozic, S. J. Fox, J. M. Thomas, C. S. Verma and D. J. Rigden, Large scale ab initio modeling of structurally uncharacterized antimicrobial peptides reveals known and novel folds, Proteins: Struct., Funct., Bioinf., 2018, 86, 548–565 CrossRef CAS PubMed.
- R. Morita, Y. Shigeta and R. Harada, Comprehensive predictions of secondary structures for comparative analysis in different species, J. Struct. Biol., 2021, 213, 107735 CrossRef CAS PubMed.
- S. N. Dean and S. A. Walper, Variational Autoencoder for Generation of Antimicrobial Peptides, ACS Omega, 2020, 5, 20746–20754 CrossRef CAS PubMed.
- K. Boone, C. Wisdom, K. Camarda, P. Spencer and C. Tamerler, Combining genetic algorithm with machine learning strategies for designing potent antimicrobial peptides, BMC Bioinf., 2021, 22, 239 CrossRef PubMed.
- P. Das, T. Sercu, K. Wadhawan, I. Padhi, S. Gehrmann, F. Cipcigan, V. Chenthamarakshan, H. Strobelt, C. dos Santos, P.-Y. Chen, Y. Y. Yang, J. P. K. Tan, J. Hedrick, J. Crain and A. Mojsilovic, Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations, Nat. Biomed. Eng., 2021, 5, 613–623 CrossRef CAS PubMed.
- C. Wang, S. Garlick and M. Zloh, Deep Learning for Novel Antimicrobial Peptide Design, Biomolecules, 2021, 11, 471 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dd00045a |
| ‡ Current address: Stowers Institute for Medical Research, 1000 E 50th Street, Kansas City, Missouri, USA. |
| This journal is © The Royal Society of Chemistry 2023 |