
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards a comprehensive data infrastructure for redox-active organic molecules targeting non-aqueous redox flow batteries†
Rebekah
Duke
 ab,
Vinayak
Bhat
ab,
Parker
Sornberger
ab,
Susan A.
Odom
a and
Chad
Risko
*ab
ab,
Vinayak
Bhat
ab,
Parker
Sornberger
ab,
Susan A.
Odom
a and
Chad
Risko
*ab
aDepartment of Chemistry, University of Kentucky, Lexington, Kentucky 40506, USA. E-mail: chad.risko@uky.edu
bCenter for Applied Energy Research, University of Kentucky, Lexington, Kentucky 40511, USA
First published on 4th July 2023
Abstract
The shift of energy production towards renewable, yet at times inconsistent, resources like solar and wind have increased the need for better energy storage solutions. An emerging energy storage technology that is highly scalable and cost-effective is the redox flow battery comprised of redox-active organic materials. Designing optimum materials for redox flow batteries involves balancing key properties such as the redox potential, stability, and solubility of the redox-active molecules. Here, we present the data-enabled discovery and design to transform liquid-based energy storage (D3TaLES) database, a curated data collection of more than 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 redox-active organic molecules that are of potential interest as the redox-active species for redox flow batteries with the aim to offer readily accessible and uniform data for big data metanalyses. D3TaLES raw data and derived properties are organized into a molecule-centric schema, and the database ontology contributes to the establishment of community reporting standards for electrochemical data. Data are readily accessed and analyzed through an easy-to-use web interface. The data infrastructure is coupled with data upload and processing tools that extract, transform, and load relevant data from raw computation or experimental data files, all of which are available to the public via a D3TaLES API. These processing tools along with an embedded high-throughput computational workflow enable community contributions and versatile data sharing and analyses, not only in redox-flow battery research but also in any field that applies redox-active organic molecules.
000 redox-active organic molecules that are of potential interest as the redox-active species for redox flow batteries with the aim to offer readily accessible and uniform data for big data metanalyses. D3TaLES raw data and derived properties are organized into a molecule-centric schema, and the database ontology contributes to the establishment of community reporting standards for electrochemical data. Data are readily accessed and analyzed through an easy-to-use web interface. The data infrastructure is coupled with data upload and processing tools that extract, transform, and load relevant data from raw computation or experimental data files, all of which are available to the public via a D3TaLES API. These processing tools along with an embedded high-throughput computational workflow enable community contributions and versatile data sharing and analyses, not only in redox-flow battery research but also in any field that applies redox-active organic molecules.
Introduction
Increasing use of renewable yet inconsistent energy sources like solar and wind demands better energy storage solutions. An emerging energy storage technology that is highly scalable and cost-effective is the redox flow battery (RFB).1–3 The RFB decouples energy capacity and power by separating the electrochemical reactions from stored electrochemical energy, allowing the battery to store large quantities of energy cheaply and safely.1 The battery consists of two tanks of solvated redox-active molecules—the catholyte in one tank and the anolyte in the other. During charge, the catholyte and anolyte are pumped through a reaction cell where a membrane separates them. The catholyte is comprised of redox-active molecules that are oxidized at a porous electrode, while the anolyte contains redox-active molecules that are reduced at another porous electrode. At discharge, the oxidized catholyte and reduced anolyte are pumped back through the reaction cell, where the reverse reaction occurs, releasing stored electrochemical energy.While current commercially available RFB use vanadium, organic-based RFB show promise as organic molecules can be more widely available and cheaper than mined and/or rare metals.4–6 Additionally, redox-active organic molecules are highly tunable and can be synthesized from sustainable materials.3,7–9 While commercial and other promising RFB materials are comprised of aqueous solvents, nonaqueous solvents afford large potential windows, increasing the battery's voltage and thus its potential energy storage.10 Even so, there is limited research targeting redox-active molecules for nonaqueous solvents in RFB, so-called nonaqueous RFB (NARFB).
Both experimental and computational methods exist for deciphering fundamental material properties for NARFB materials (Fig. 1), and the computational/simulation-based approaches have been vital in identifying candidates for NARFB by simulating redox potential, stability, and reversibility, to name a few.11–13 However, while there have been efforts to identify redox-active molecules suitable for NARFB catholyte and anolyte materials, there remains a lack of fundamental chemical understanding for these systems, especially considerations as to how to appropriately balance critical properties such as redox potential, stability, and solubility.3,9,14,15
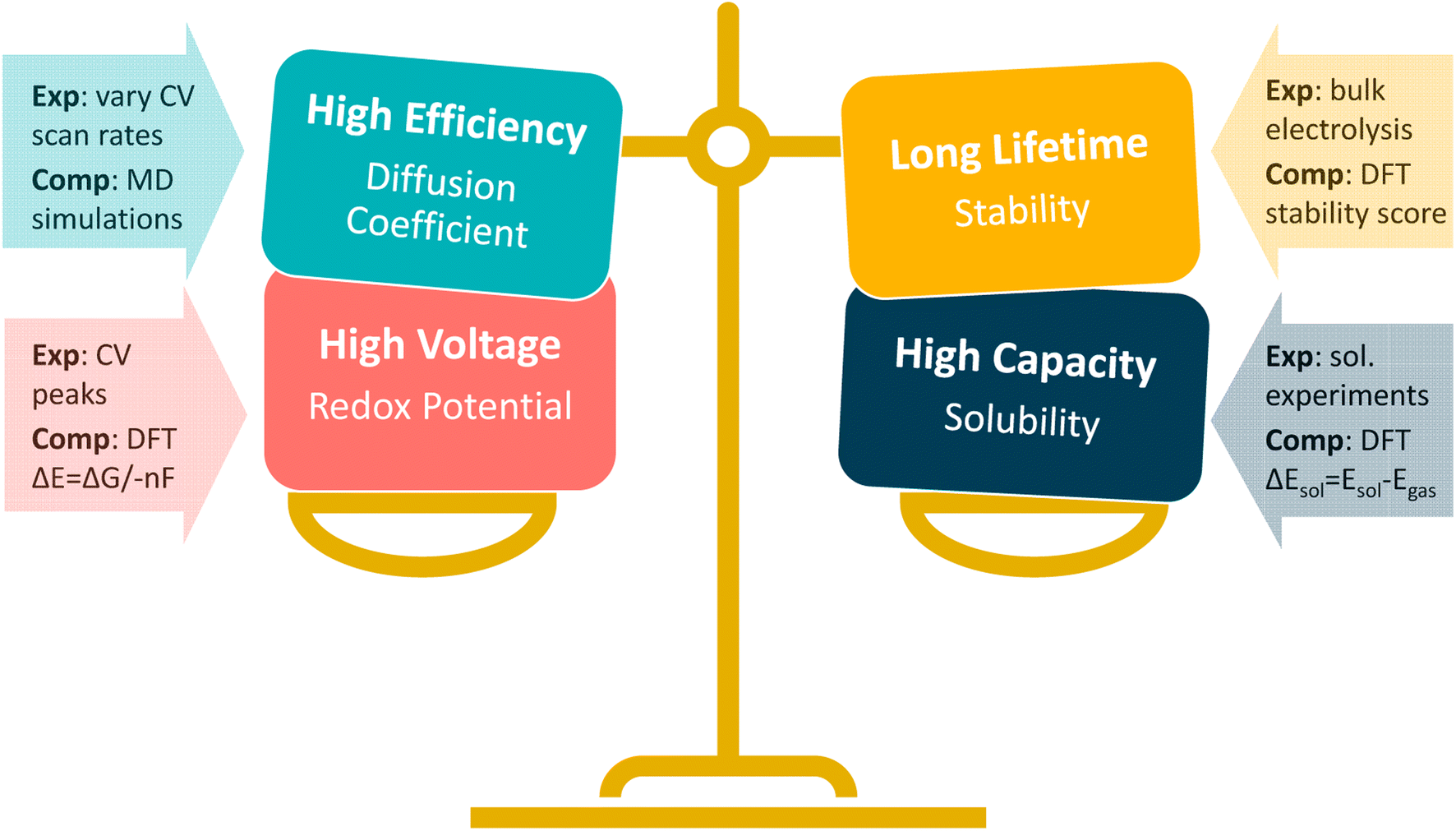 | ||
| Fig. 1 Fundamental redox-active material properties that must be balanced for use as the catholyte or anolyte in an RFB. Each fundamental property can be estimated experimentally via techniques like cyclic voltammetry (CV) or computationally via density functional theory (DFT) and/or molecular dynamics (MD) simulations. | ||
Fortunately, when data are amassed from both computational and experimental sources, big-data analyses can inform structure–property relationships. Previous data-enabled insights have been achieved in similar fields. For example, big data analyses elucidated a “stability cliff” in quinones, a prevalent molecular class in aqueous RFB, encouraging researchers to explore other chemical spaces.16 Efforts are already underway to develop data-driven pipelines and apply big-data analyses for vanadium RFB17,18 and aqueous organic RFB19–21 materials. Some big data approaches have been applied to the search for NARFB materials; for example, data-enabled high-throughput screening of redox-active molecules for NARFB has been demonstrated in a small-scale proof-of-concept study where several theoretically viable molecules for NARFB anolytes were selected from ∼1400 quinoxaline-based systems a with funnel-based screening approach focusing on reduction potential, solvation energy, and structural changes with oxidation.22
Unfortunately, the few studies that examine systems for NARFB are smaller scale and (like those in the field of aqueous RFB) often focus on quinone-based systems alone. Additionally, elucidating structure–property relationships for properties such as solubility in nonaqueous environments for NARFB can be much more challenging than in aqueous environments.23 Thus, in the field of NARFB, the metanalyses necessary to elucidate structure–property relationships are often prohibited by the lack of large-scale, broad, accessible, and uniform data.
Here we present a curated database of redox-active organic molecules as a part of a multidisciplinary, collaborative platform entitled data-enabled discovery and design to transform liquid energy storage (D3TaLES).24 We collect and curate data from various sources, including computational analyses and original experimentation, and we build the infrastructure to accept data submissions from the community. The data infrastructure includes data upload and processing tools that extract, transform, and load (ETL) relevant data from raw computation or experimental data files and organize it into a molecule-centric schema. Here the data are easily accessed and analyzed, and a layered, redundant database structure provides critical opportunities for manual and automated data curation. A high-throughput computational workflow begins populating the database with DFT computational data. Similar data structures involving high-throughput computation for π-conjugated organic molecules exist,25–30 and there also exist databases targeting batteries.19,31,32 But unlike these existing databases, D3TaLES provides a data infrastructure and framework for multiple data types targeting NARFB. The database enables deeper physiochemical understanding and opportunities for meta-analysis. Though the focus presented here is on identifying systems that hold potential for NARFB, the platform has a broad scope and can be used or expanded to search for characteristics of redox-active organic molecules in other fields of application.
D3TaLES database
Design
The schema, or organizational data structure, provides the foundation for the database. A No-SQL schema was chosen because of its flexibility and scalability.33 D3TaLES data is segmented into two databases to accommodate the complexity and breadth of the data collected—one for raw data (backend) and the other for processed data (frontend).The backend database contains data parsed directly from experiment data files. It uses computation- and/or experiment-centric schema where each data instance is a calculation or experiment with associated attributes (Fig. 2). Attributes include calculation/experiment identifier, submission information, and a collection of raw data values, including computational/experimental conditions. For example, the backend database might hold raw data values like the computed energies for a molecule's ground and oxidized or reduced states. The backend database can also hold cyclic voltammetry (CV) data extracted from a potentiostat output file. Thus, the backend schema relates directly to the files that supply data and often incorporates features from existing community schema.34,35 Because D3TaLES contains many types of data, the backend schema has several sub-schemas—one for each data type. The sub-schemas share common fields such as “mol_id”, “submission_info”, and “data”. The backend schemas are broad and accommodate different types of data including but not limited to computations at different levels of theory (e.g., molecular dynamics simulations), experiments with various processing or data collection conditions, and literature-extracted data from various learning models (see ESI Section 1†); efforts are ongoing to add more schema.
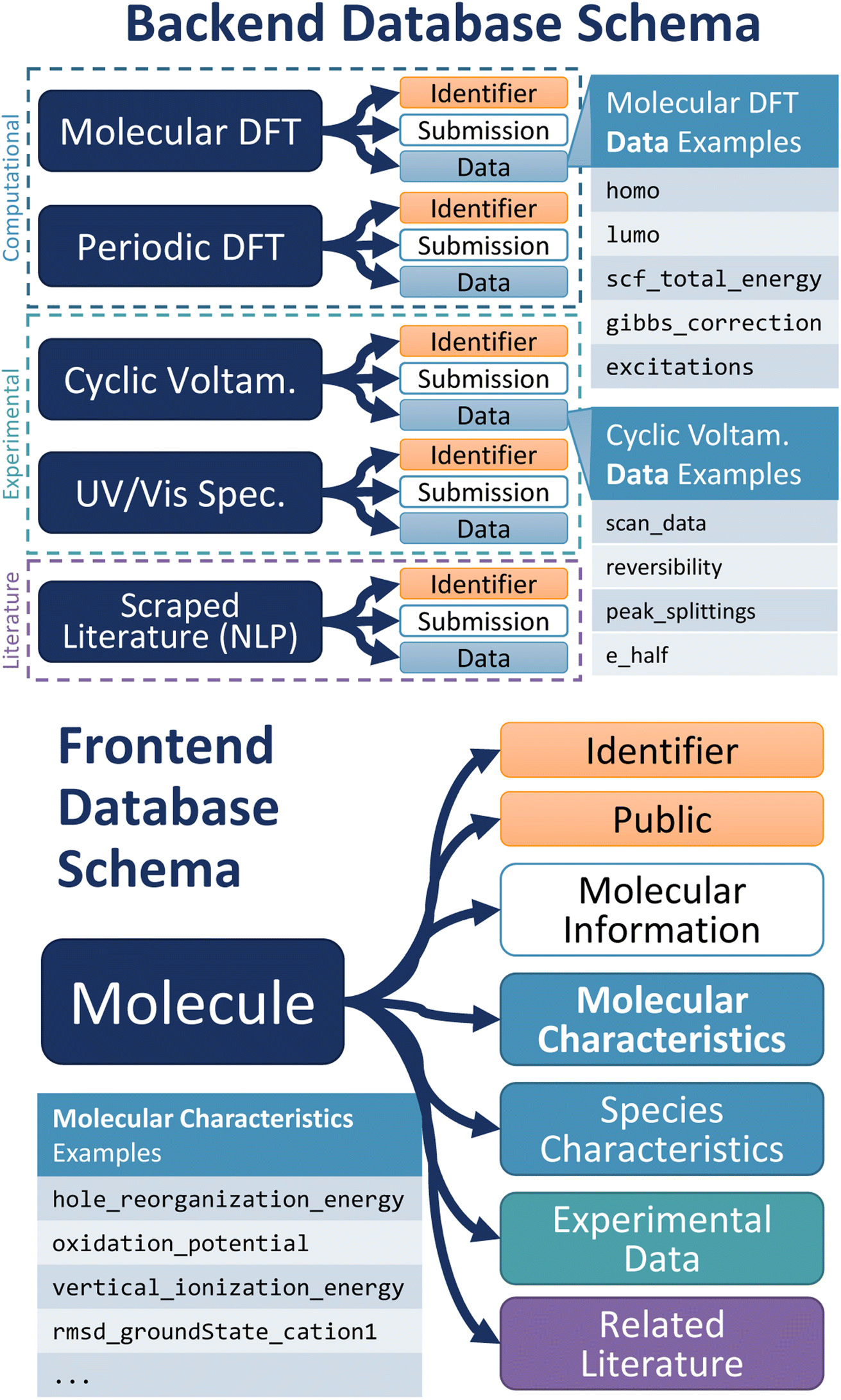 | ||
| Fig. 2 (Top) Depiction of the backend D3TaLES schema and collection types. Note that the figure shows a sampling of the types of collections that exist in the backend database; to view the full D3TaLES database schema, visit the documentation.36 Tables with example “data” attributes are also shown. (Bottom) Schematic showing the first property level for the molecule-centric, frontend D3TaLES schema along with a table showing example attributes in the “molecular characteristics” attribute group. | ||
The frontend database holds data that are more useful for analysis. For example, the frontend database contains ionization potentials calculated from the ground- and oxidized-state energies in the backend database. Likewise, the frontend database contains estimated redox potentials calculated from CV data. The frontend database uses a molecule-centric schema where each data instance is a molecule (Fig. 2). Molecule attributes include a molecule identifier and public/private status, while the remaining attributes are grouped into the following sub-categories: molecule characteristics, species characteristics, raw experiment data (which connects to the backend database), and related literature. Molecular characteristics include properties of the entire molecule (usually involving multiple species), such as oxidation potential or relaxation energies. Species characteristics include properties relating to a single charge species for the molecule, such as ground-state species HOMO or oxidized-state solvation energy. The complete D3TaLES schema is available online.36
Population
A processing workflow populates the database when raw data files and associated metadata are uploaded to the D3TaLES website (Fig. 3). The raw data files are parsed to extract key values. Existing parsing packages (such as Pymatgen,35 RDKit,37 and SciPy38) are integrated with original code to parse raw computational and experimental data files. These processing tools are packaged in the D3TaLES application programming interface (API; discussed in the D3TaLES tools section). The raw data files are then compressed and stored, while extracted key values are inserted into the backend database. At this stage, an administrator inspects the backend data to ensure some degree of fidelity. Upon administrator approval, the backend data is transformed into frontend properties. Users may view the frontend database via interactive molecule viewing webpages on the D3TaLES website.24 More information about the database software can be found in the ESI Section 2.†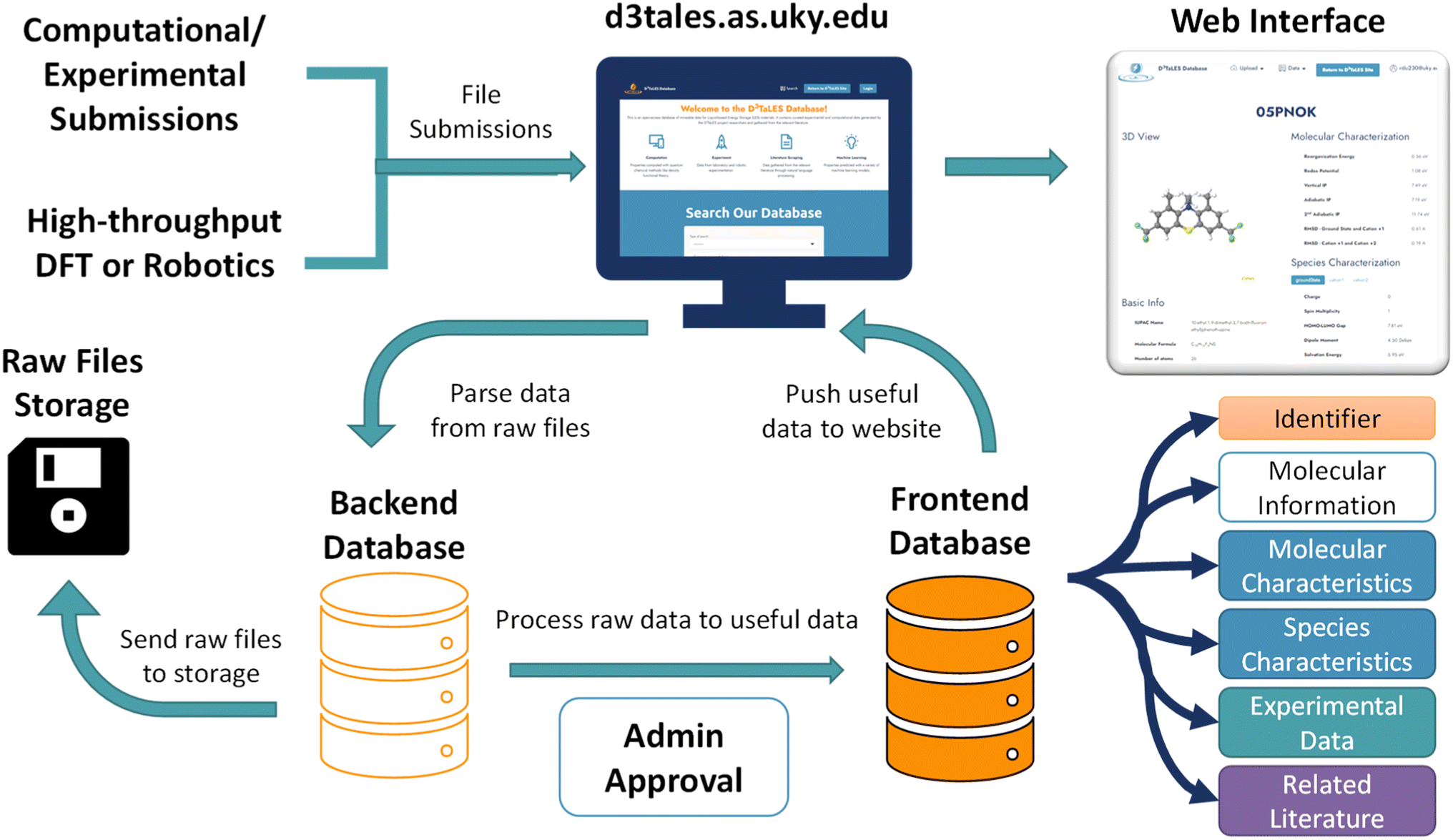 | ||
| Fig. 3 Schematic showing D3TaLES data processing. Data flows from external sources, such as high-throughput computation or robotics, through the D3TaLES website to the backend database. From here, raw data is stored while administrator (admin) approval allows data transformation to the frontend database. Frontend data is displayed through a user interface on the D3TaLES website. | ||
Currently, the D3TaLES database contains primarily computational data generated through a high-throughput molecular computational workflow using density functional theory (DFT) carried out at the (IP-tuned) LC-ωHPBE/Def2SVP level of theory via the Gaussian16 (rev A.03) software suite.39–42 The data produced in this workflow cover several fundamental properties of redox-active molecules, including oxidation and reduction potentials, stability, and solubility; see ESI Section 4† for more details.
Data composition
Molecules in the D3TaLES database are collected from those appearing in the NARFB literature,23,43,44 scraped from the Cambridge Structural Database (CSD)45 and ZINC46 datasets (Fig. S4 and S5†), and combinatorically generated from fragments of molecules commonly used in NARFB; see ESI Section 3† for more details. While these datasets contain inherent biases (e.g., CSD molecules are crystallizable, ZINC molecules are already commercially available, combinatorically generated molecules conform to current conceptions in the field about what structures will work in NARFB, etc.), this collection provides an initial dataset of small organic molecules covering a relatively diverse chemical space. The scraped data number over 600000 molecules, along with a few dozen experimental molecules from collaborators and a few hundred auto-generated molecules from common motifs used in NARFB (Fig. 4). The following criteria were then used to filter this extensive molecular dataset: A molecule must have at least one aromatic ring, contain no rings with more than six atoms and no rings with less than five atoms, contain no rings with more than three heteroatoms, and not exist already in the OCELOT26 database (a database of large organic molecules and their corresponding crystal properties targeting organic semiconductors developed by our lab). This narrowed the dataset to approximately 115000 molecules. Finally, the dataset was narrowed further because of limited computational resources. To ensure diversity of the chemical space, the 33000 filtered ZINC molecules most different from the rest of the dataset (CSD, generate, and NARFB literature molecules) were chosen. The similarity was determined with the RDKit Tanimoto fingerprint method.37,47 The final chemical space consists of 43168 molecules, where approximately 3500 are proprietary and 39500 are public. Of these structures, 31583 have a complete oxidation profile.
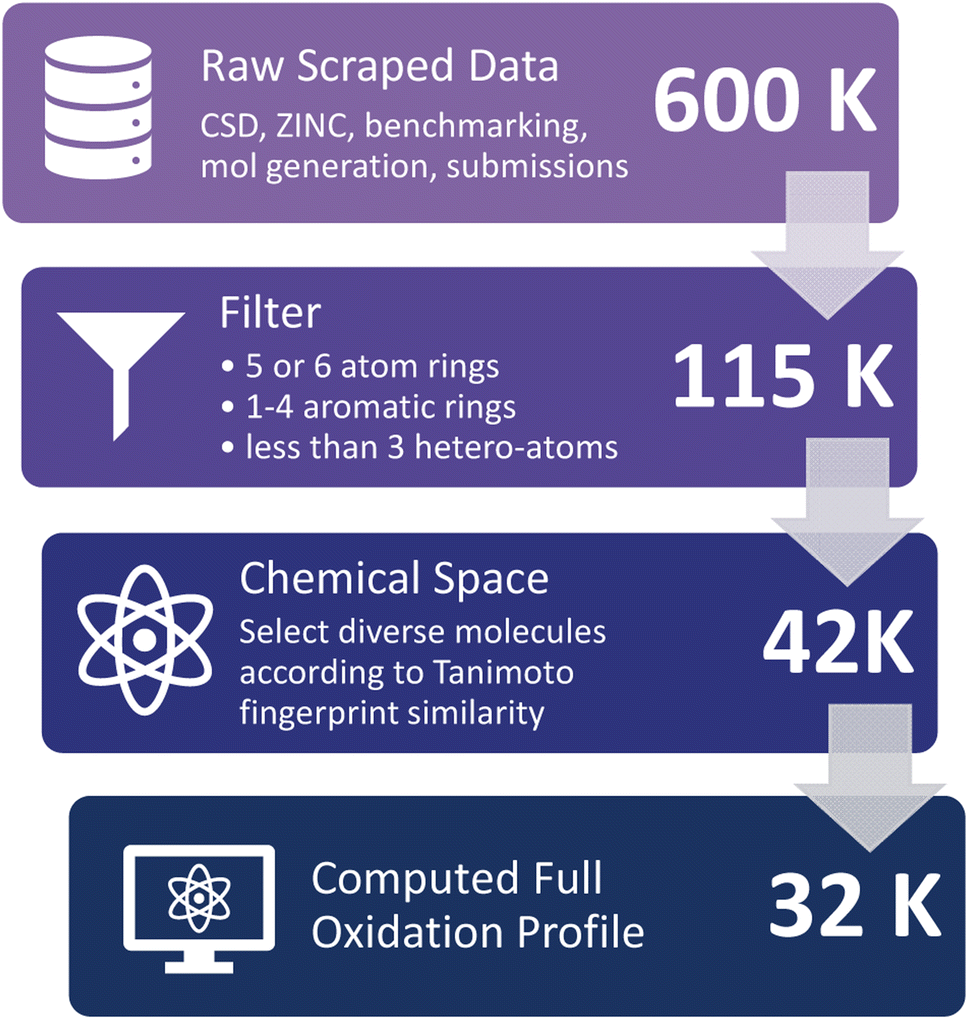 | ||
| Fig. 4 Molecule screening process for the D3TaLES database. | ||
The 43168 unique structures in the D3TaLES database have a mean molecular weight of 329 g mol−1 (Fig. 5A). All properties generated for the oxidation profile are listed in the D3TaLES database documentation,36 but notable properties include oxidation potential, relaxation energies, vertical and adiabatic ionization potentials, solvation energies, and a radical-cation stability score developed by Sowndarya et al.48Fig. 5B shows a UMAP49 chemical space plot of the calculated oxidation potentials where groupings of higher and lower potentials are viable. The plot includes 10-ethylphenothiazine (EPT) and (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO), two widely-reported molecules of interest for organic RFB.7Fig. 5C shows the database structures plotted by oxidation potential and the radical-cation stability score.48 The marginal histogram depicts a normally distributed radical stability score (RSS), with the highest stability scores observed for larger molecules. In contrast, there exists little correlation between size and oxidation potential, though most oxidation potentials are concentrated just above zero eV (relative to the standard hydrogen electrode, SHE). The database is now being populated with reduction profiles for many of the structures. These profiles contain the reduction analog for each of the oxidation profile properties. Currently, the database contains over 28000 reduction profiles.
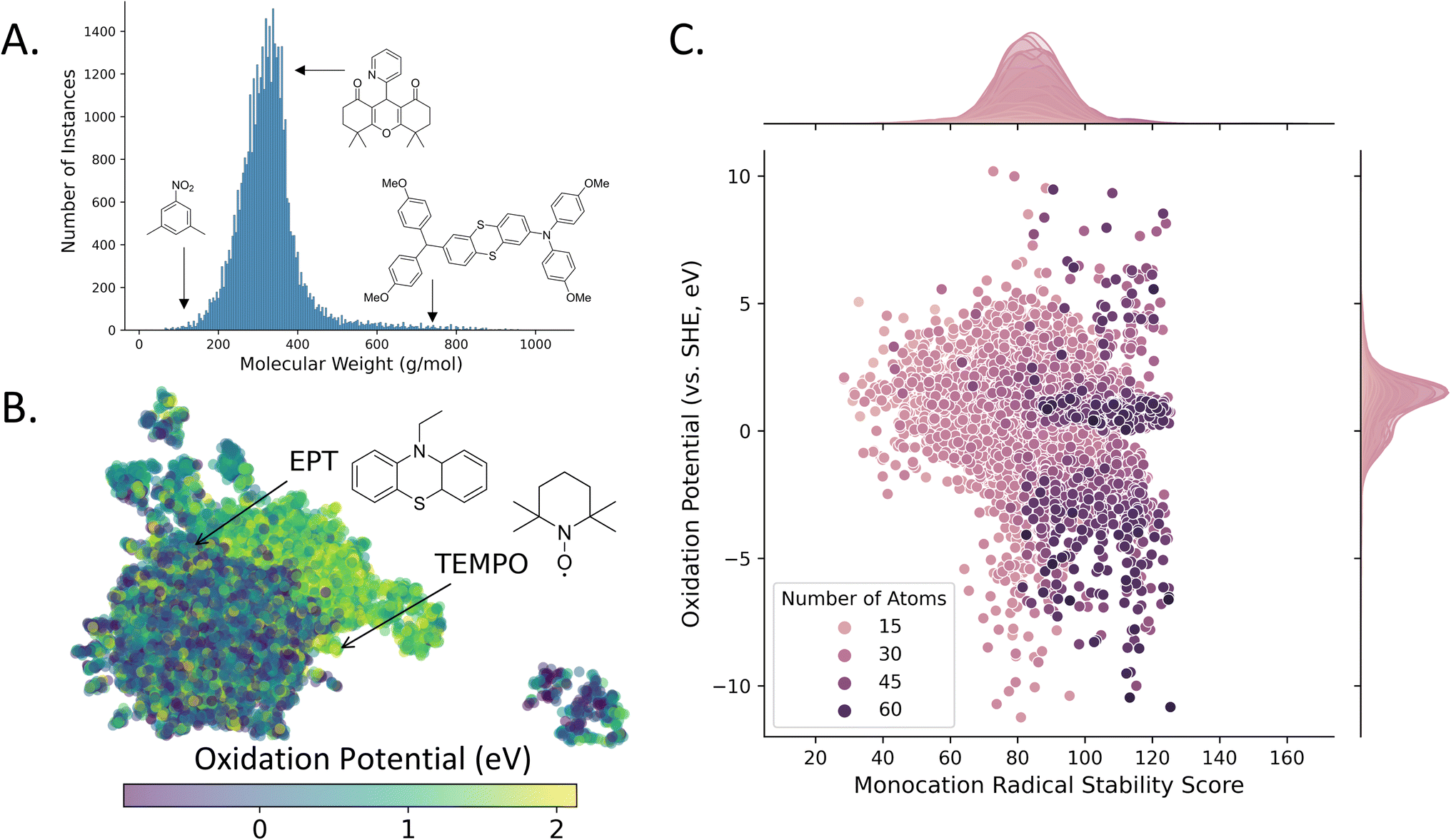 | ||
| Fig. 5 The D3TaLES frontend database contains over 43000 molecules. (A) Histogram showing molecular weight distribution for the D3TaLES database. (B) The computed values for oxidation potential (a molecular characteristic) are mapped onto a two-dimensional chemical space with ChemPlot50 and UMAP49 dimension reduction. (C) Scatter plot with marginal histograms showing D3TaLES molecules plotted by calculated oxidation potential (versus the standard hydrogen electrode, SHE) and radical stability score, colored by number of atoms.48 | ||
D3TaLES tools
The D3TaLES database is coupled with several data interaction and management tools including the D3TaLES website24 and the D3TaLES API.51 The D3TaLES website is integral for many of the processes described above. Website features include file upload systems, backend data viewing and approval, database search functions, and molecule viewing pages. All user data submissions and administrator approval of the processed data occur through the website. Users may search the database by molecule name or structure. All data for a given molecule can be viewed on the molecule property viewing page (Fig. 6). Alternatively, for those wishing to access large quantities of data through code, the D3TaLES REST API allows data access through HTML according to REST (representational state transfer) standards.52 Finally, the site contains links to the D3TaLES database documentation,36 D3TaLES API documentation,51 and the D3TaLES calculators interactive Python notebooks.53 | ||
| Fig. 6 (Top) D3TaLES molecule viewing page.54 (Bottom) The organizational structure of the D3TaLES API. Full documentation for the D3TaLES API is available.51 | ||
Several tools for moving, processing, and transforming data accompany the D3TaLES database. These tools are compiled in the D3TaLES API.51 The D3TaLES API includes three modules: Processors for data processing, D3database for database access, and Calculators for property calculations (Fig. 6). The Processors module contains various parsing classes for extracting useful data from instrument-produced computational and experimental data files. Among other database access functions, the D3database module contains a class for accessing the D3TaLES database via Python through the REST API. This module also contains functions for gathering and plotting D3TaLES properties as one- and two-dimensional histograms. Finally, the Calculators module, perhaps the most useful module for the general community, allows users to calculate useful computational and experimental properties from nested data. All calculators contain unit conversion features. Useful molecular DFT calculators include redox potential, radical buried volume,55 and radical spin density,56 while useful CV calculators include diffusion constant using the Randles–Ševčík equation and charge-transfer rate. The D3TaLES API documentation51 explains basic usage for these calculators, and we also provide interactive Python notebooks that use the calculators to perform calculations without the need for the user to know Python coding.53 For more information about the D3TaLES API, see ESI Section 6.†
D3TaLES database utility
To demonstrate the D3TaLES database utility in identifying candidates for redox flow batteries, we used the compiled computational data to perform a proof-of-concept funnel pipeline (Fig. 7).57–60 The funnel pipeline iteratively narrows the D3TaLES chemical space through a series of tests to identify candidates for a NARFB catholyte material. The tests are ordered from least to most computationally intensive. The first test (∼1 ms) selects molecules with less than 30 atoms. Redox-active systems with fewer atoms per charge event increase the atom economy,61 and thus the capacity for a RFB. Subsequently, the second test (∼1 s) filters out molecules that would be difficult to synthesize by selecting systems with a synthetic accessibility score below 4.1.56,62 The next two tests filter by stability and solvation energy, respectively, relative to the properties of a known candidate for NARFB: N-(2-(2-methoxyethoxy)ethyl)phenothiazine (MEEPT).7,63 MEEPT is known to be soluble, especially in its ground state, and it shows stable cycling of one oxidation event. The third test (∼21 core hours) filters out molecules with an RSS greater than MEEPT's score of 81, while the fourth test (∼21 core hours) identifies molecules with solvation energy lower than MEEPT's −0.19 eV. The final test (∼43 core hours) finds molecules with an oxidation potential of 1.96 V or higher, as higher oxidation potentials are most desirable for catholyte materials. (To view structures from the funnel pipeline and for more information about the core-hour estimations, see ESI Section 5.†) The funnel pipeline down-selects the 43168 D3TaLES structures to 364 potential systems for NARFB. While all calculations were performed for all molecules used here, this approach could be employed to explore a large chemical space without performing all resource-intensive calculations for all systems. Additionally, the existing D3TaLES data can be used to train machine learning (ML) models that quickly estimate resource-intensive properties such as oxidation potential; these models could be added as an upper level of the funnel pipeline.64
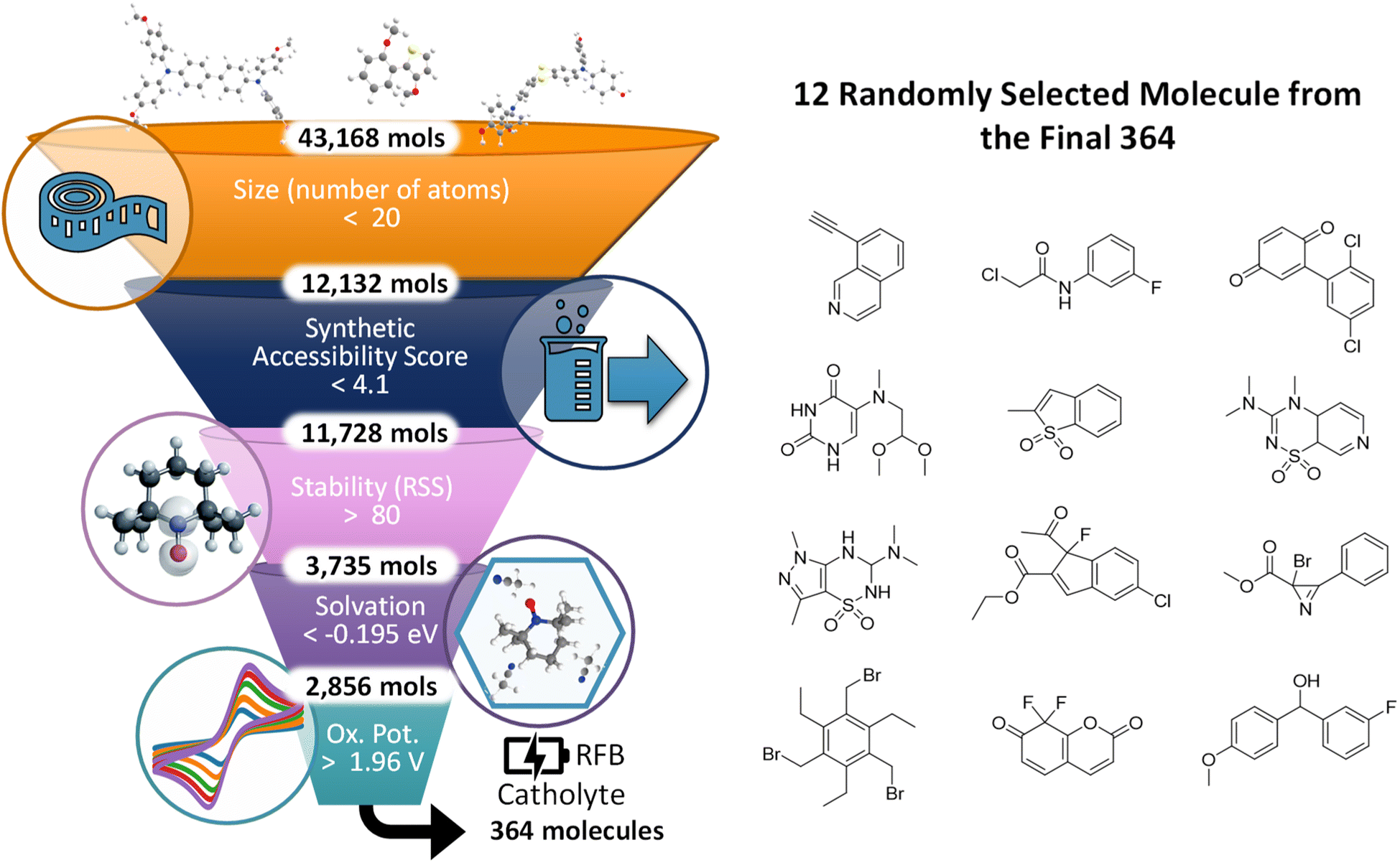 | ||
| Fig. 7 (Left) Schematic demonstrating the proof-of-concept funnel pipeline using D3TaLES computational data. The five tests narrow the chemical space by number of atoms, synthetic accessibility score, radical stability score (RSS), solvation energy, and oxidation potential, respectively. (Right) Twelve randomly selected structures from the final 364 structures that emerged from the funnel pipeline. | ||
Conclusion
We demonstrate a comprehensive data infrastructure for redox-active small molecules for use in NARFBs. For the over 43000 molecules currently in the D3TaLES database, a high-throughput computational workflow has determined over 31000 oxidation profiles and other properties of interest to date. While the database currently consists almost exclusively of DFT computational data, the schema and processing infrastructure exist for incorporating experimental and literature-reported data. Future work will focus on exploiting the data processing tools and data storage infrastructure to continue populating the D3TaLES database, especially in areas outside of molecular DFT, such as periodic DFT, molecular dynamics simulations, and cyclic voltammetry and UV-Vis spectroscopy experiments.
We demonstrate the utility of the D3TaLES infrastructure by screening the over 43000 molecules in the database for NARFB application. This preliminary screening predicts 364 candidates with characteristics superior to the current standard MEEPT. We note that a thorough analysis is warranted to confirm these predictions. The D3TaLES database and data infrastructure will enable integrated meta-analytical and machine-learning-based evaluation in the NARFB field, with the aim to expedite materials discovery and pave the way for predictive models for properties such as redox potentials and radical cation stability. The uniform and accessible D3TaLES data will enable machine learning and robotic experimentation towards better exploring relevant chemical space for application-suitable redox molecules.
Data availability
The data presented here are accessible via the D3TaLES website (https://d3tales.as.uky.edu/), and the public portion of the dataset (∼39500 molecules) can be downloaded at https://d3tales.as.uky.edu/datasets. The D3TaLES website also includes documentation for the database structure and more information about the data composition (https://d3tales.as.uky.edu/docs/). The processing tools associated with the D3TaLES API exist in an open-access Python package documented at https://d3tales.github.io/d3tales_api/. The Fireworks-based65 code used for the high-throughput quantum chemical calculations is available publicly at https://github.com/D3TaLES/d3tales_fw. Additional details and information can be found in the accompanying ESI.†
Author contributions
R. D.: conceptualization, data curation, formal analysis, investigation, methodology, software, validation, visualization, writing – original draft, writing – review & editing. V. B.: conceptualization, software, supervision, writing – review & editing. P. S.: data curation, writing – review & editing. S. A. O.: conceptualization, funding acquisition. C. R.: conceptualization, supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by the National Science Foundation (NSF) under Cooperative Agreement Number 2019574. Computational resources were provided through an NSF Extreme Science and Engineering Discovery Environment (XSEDE) Resource Allocation Award (CHE200119) and Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) DISCOVER Allocation Award (PHY220121). We further acknowledge the University of Kentucky (UK) Center for Computational Sciences and Information Technology Services Research Computing for their fantastic support and collaboration, and use of the Lipscomb Compute Cluster and associated research computing resources. Finally, we wholeheartedly thank the entire D3TaLES team for their insights into the development of this data architecture.References
- J. Luo, B. Hu, M. Hu, Y. Zhao and T. L. Liu, Status and Prospects of Organic Redox Flow Batteries toward Sustainable Energy Storage, ACS Energy Lett., 2019, 4, 2220–2240, DOI:10.1021/acsenergylett.9b01332.
- F. Pan and Q. Wang, Redox Species of Redox Flow Batteries: A Review, Molecules, 2015, 20, 20499–20517, DOI:10.3390/molecules201119711.
- M. Li, S. A. Odom, A. R. Pancoast, L. A. Robertson, T. P. Vaid, G. Agarwal, H. A. Doan, Y. Wang, T. M. Suduwella and S. R. Bheemireddy, et al., Experimental Protocols for Studying Organic Non-aqueous Redox Flow Batteries, ACS Energy Lett., 2021, 3932–3943, DOI:10.1021/acsenergylett.1c01675.
- R. A. Scott, B. Hu, J. Luo, C. DeBruler, M. Hu, W. Wu and T. L. Liu, Redox-Active Inorganic Materials for Redox Flow Batteries, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2019, pp. 1–25, DOI:10.1002/9781119951438.eibc2679.
- V. Viswanathan, A. Crawford, D. Stephenson, S. Kim, W. Wang, B. Li, G. Coffey, E. Thomsen, G. Graff and P. Balducci, et al., Cost and performance model for redox flow batteries, J. Power Sources, 2014, 247, 1040–1051, DOI:10.1016/j.jpowsour.2012.12.023.
- W. Wang, Q. Luo, B. Li, X. Wei, L. Li and Z. Yang, Recent Progress in Redox Flow Battery Research and Development, Adv. Funct. Mater., 2013, 23, 970–986, DOI:10.1002/adfm.201200694.
- M. Li, Z. Rhodes, J. R. Cabrera-Pardo and S. D. Minteer, Recent advancements in rational design of non-aqueous organic redox flow batteries, Sustainable Energy Fuels, 2020, 4, 4370–4389, 10.1039/d0se00800a.
- X. Fang, Z. Li, Y. Zhao, D. Yue, L. Zhang and X. Wei, Multielectron Organic Redoxmers for Energy-Dense Redox Flow Batteries, ACS Mater. Lett., 2022, 277–306, DOI:10.1021/acsmaterialslett.1c00668.
- X. Wei, W. Pan, W. Duan, A. Hollas, Z. Yang, B. Li, Z. Nie, J. Liu, D. Reed and W. Wang, et al., Materials and Systems for Organic Redox Flow Batteries: Status and Challenges, ACS Energy Lett., 2017, 2, 2187–2204, DOI:10.1021/acsenergylett.7b00650.
- S.-H. Shin, S.-H. Yun and S.-H. Moon, A review of current developments in non-aqueous redox flow batteries: characterization of their membranes for design perspective, RSC Adv., 2013, 3, 9095, 10.1039/c3ra00115f.
- E. C. Montoto, Y. Cao, K. Hernández-Burgos, C. S. Sevov, M. N. Braten, B. A. Helms, J. S. Moore and J. Rodríguez-López, Effect of the Backbone Tether on the Electrochemical Properties of Soluble Cyclopropenium Redox-Active Polymers, Macromolecules, 2018, 51, 3539–3546, DOI:10.1021/acs.macromol.8b00574.
- Y. Yan, S. G. Robinson, M. S. Sigman and M. S. Sanford, Mechanism-Based Design of a High-Potential Catholyte Enables a 3.2 V All-Organic Nonaqueous Redox Flow Battery, J. Am. Chem. Soc., 2019, 141, 15301–15306, DOI:10.1021/jacs.9b07345.
- Y. Yan, T. P. Vaid and M. S. Sanford, Bis(diisopropylamino)cyclopropenium-arene Cations as High Oxidation Potential and High Stability Catholytes for Non-aqueous Redox Flow Batteries, J. Am. Chem. Soc., 2020, 142, 17564–17571, DOI:10.1021/jacs.0c07464.
- M.-A. Goulet, L. Tong, D. A. Pollack, D. P. Tabor, S. A. Odom, A. Aspuru-Guzik, E. E. Kwan, R. G. Gordon and M. J. Aziz, Extending the Lifetime of Organic Flow Batteries via Redox State Management, J. Am. Chem. Soc., 2019, 141, 8014–8019, DOI:10.1021/jacs.8b13295.
- F. Zhong, M. Yang, M. Ding and C. Jia, Organic Electroactive Molecule-Based Electrolytes for Redox Flow Batteries: Status and Challenges of Molecular Design, Front. Chem., 2020, 8, 451, DOI:10.3389/fchem.2020.00451.
- D. P. Tabor, R. Gómez-Bombarelli, L. Tong, R. G. Gordon, M. J. Aziz and A. Aspuru-Guzik, Mapping the frontiers of quinone stability in aqueous media: implications for organic aqueous redox flow batteries, J. Mater. Chem. A, 2019, 7, 12833–12841, 10.1039/c9ta03219c.
- Z. Cheng, K. M. Tenny, A. Pizzolato, A. Forner-Cuenca, V. Verda, Y.-M. Chiang, F. R. Brushett and R. Behrou, Data-driven electrode parameter identification for vanadium redox flow batteries through experimental and numerical methods, Appl. Energy, 2020, 279, 115530, DOI:10.1016/j.apenergy.2020.115530.
- R. Li, B. Xiong, S. Zhang, X. Zhang, Y. Li, H. Iu and T. Fernando, A novel U-Net based data-driven vanadium redox flow battery modelling approach, Electrochim. Acta, 2023, 444, 141998, DOI:10.1016/j.electacta.2023.141998.
- E. Sorkun, Q. Zhang, A. Khetan, M. C. Sorkun and S. Er, RedDB, a Computational Database of Electroactive Molecules for Aqueous Redox Flow Batteries, American Chemical Society (ACS), 2021 Search PubMed.
- P. Gao, A. Andersen, J. Sepulveda, G. U. Panapitiya, A. Hollas, E. G. Saldanha, V. Murugesan and W. Wang, SOMAS: a platform for data-driven material discovery in redox flow battery development, Sci. Data, 2022, 9, 740, DOI:10.1038/s41597-022-01814-4.
- Q. Zhang, A. Khetan, E. Sorkun, F. Niu, A. Loss, I. Pucher and S. Er, Data-driven discovery of small electroactive molecules for energy storage in aqueous redox flow batteries, Energy Storage Mater., 2022, 47, 167–177, DOI:10.1016/j.ensm.2022.02.013.
- L. Cheng, R. S. Assary, X. Qu, A. Jain, S. P. Ong, N. N. Rajput, K. Persson and L. A. Curtiss, Accelerating Electrolyte Discovery for Energy Storage with High-Throughput Screening, J. Phys. Chem. Lett., 2015, 6, 283–291, DOI:10.1021/jz502319n.
- A. S. Perera, T. M. Suduwella, N. H. Attanayake, R. K. Jha, W. L. Eubanks, I. A. Shkrob, C. Risko, A. P. Kaur and S. A. Odom, Large variability and complexity of isothermal solubility for a series of redox-active phenothiazines, Mater. Adv., 2022, 3, 8705–8715, 10.1039/d2ma00598k.
- D3TaLES, https://d3tales.as.uky.edu/ Search PubMed.
- S. Gallarati, P. Van Gerwen, R. Laplaza, S. Vela, A. Fabrizio and C. Corminboeuf, OSCAR: an extensive repository of chemically and functionally diverse organocatalysts, Chem. Sci., 2022, 13, 13782–13794, 10.1039/d2sc04251g.
- Q. Ai, V. Bhat, S. M. Ryno, K. Jarolimek, P. Sornberger, A. Smith, M. M. Haley, J. E. Anthony and C. Risko, OCELOT: An infrastructure for data-driven research to discover and design crystalline organic semiconductors, J. Chem. Phys., 2021, 154, 174705, DOI:10.1063/5.0048714.
- S. Curtarolo, W. Setyawan, G. L. W. Hart, M. Jahnatek, R. V. Chepulskii, R. H. Taylor, S. Wang, J. Xue, K. Yang and O. Levy, et al., AFLOW: An automatic framework for high-throughput materials discovery, Comput. Mater. Sci., 2012, 58, 218–226, DOI:10.1016/j.commatsci.2012.02.005.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner and G. Ceder, et al., Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Mater., 2013, 1, 011002, DOI:10.1063/1.4812323.
- J. Hachmann, R. Olivares-Amaya, S. Atahan-Evrenk, C. Amador-Bedolla, R. S. Sánchez-Carrera, A. Gold-Parker, L. Vogt, A. M. Brockway and A. Aspuru-Guzik, The Harvard Clean Energy Project: Large-Scale Computational Screening and Design of Organic Photovoltaics on the World Community Grid, J. Phys. Chem. Lett., 2011, 2, 2241–2251, DOI:10.1021/jz200866s.
- Ö. H. Omar, T. Nematiaram, A. Troisi and D. Padula, Organic materials repurposing, a data set for theoretical predictions of new applications for existing compounds, Sci. Data, 2022, 9, 54, DOI:10.1038/s41597-022-01142-7.
- S. Huang and J. M. Cole, A database of battery materials auto-generated using ChemDataExtractor, Sci. Data, 2020, 7, 260, DOI:10.1038/s41597-020-00602-2.
- L. Ward, S. Babinec, E. J. Dufek, D. A. Howey, V. Viswanathan, M. Aykol, D. A. C. Beck, B. Blaiszik, B.-R. Chen and G. Crabtree, et al., Principles of the Battery Data Genome, Joule, 2022, 6, 2253–2271, DOI:10.1016/j.joule.2022.08.008.
- R. Duke, V. Bhat and C. Risko, Data storage architectures to accelerate chemical discovery: data accessibility for individual laboratories and the community, Chem. Sci., 2022, 13, 13646–13656, 10.1039/d2sc05142g.
- O. Andriuc, M. Siron, J. H. Montoya, M. Horton and K. A. Persson, Automated Adsorption Workflow for Semiconductor Surfaces and the Application to Zinc Telluride, J. Chem. Inf. Model., 2021, 61, 3908–3916, DOI:10.1021/acs.jcim.1c00340.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis, Comput. Mater. Sci., 2013, 68, 314–319, DOI:10.1016/j.commatsci.2012.10.028.
- D3TaLES Database Documentation, https://d3tales.as.uky.edu/docs/ Search PubMed.
- G. Landrum, RDKit, 2010 Search PubMed.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser and J. Bright, et al., SciPy 1.0: fundamental algorithms for scientific computing in Python, Nat. Methods, 2020, 17, 261–272, DOI:10.1038/s41592-019-0686-2.
- A. F. Izmaylov, G. E. Scuseria and M. J. Frisch, Efficient evaluation of short-range Hartree-Fock exchange in large molecules and periodic systems, J. Chem. Phys., 2006, 125, 104103, DOI:10.1063/1.2347713.
- Gaussian 16 Rev. A.03, Wallingford, CT, 2016, accessed Search PubMed.
- T. M. Henderson, A. F. Izmaylov, G. Scalmani and G. E. Scuseria, Can short-range hybrids describe long-range-dependent properties?, J. Chem. Phys., 2009, 131, 044108, DOI:10.1063/1.3185673.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297, 10.1039/b508541a.
- A. Preet Kaur, B. J. Neyhouse, I. A. Shkrob, Y. Wang, N. Harsha Attanayake, R. Kant Jha, Q. Wu, L. Zhang, R. H. Ewoldt and F. R. Brushett, et al., Concentration-dependent Cycling of Phenothiazine-based Electrolytes in Nonaqueous Redox Flow Cells, Chem.–Asian J., 2023, 18, e202201171, DOI:10.1002/asia.202201171.
- M. D. Casselman, A. P. Kaur, K. A. Narayana, C. F. Elliott, C. Risko and S. A. Odom, The fate of phenothiazine-based redox shuttles in lithium-ion batteries, Phys. Chem. Chem. Phys., 2015, 17, 6905–6912, 10.1039/c5cp00199d.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179, DOI:10.1107/s2052520616003954.
- T. Sterling and J. J. Irwin, ZINC 15 – Ligand Discovery for Everyone, J. Chem. Inf. Model., 2015, 55, 2324–2337, DOI:10.1021/acs.jcim.5b00559.
- D. Rogers and M. Hahn, Extended-Connectivity Fingerprints, J. Chem. Inf. Model., 2010, 50, 742–754, DOI:10.1021/ci100050t.
- S. V. S. Sowndarya, P. C. St. John and R. S. Paton, A quantitative metric for organic radical stability and persistence using thermodynamic and kinetic features, Chem. Sci., 2021, 12, 13158–13166, 10.1039/d1sc02770k.
- L. McInnes, J. Healy and J. Melville, UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction, 2020 Search PubMed.
- M. Cihan Sorkun, D. Mullaj, J. M. V. A. Koelman and S. Er, ChemPlot, a Python Library for Chemical Space Visualization, Chem.: Methods, 2022, 2, e202200005, DOI:10.1002/cmtd.202200005.
- D3TaLES API Docs, https://d3tales.github.io/d3tales_api/ Search PubMed.
- R. T. Fielding and R. N. Taylor, Principled design of the modern Web architecture, ACM Trans. Internet Technol., 2002, 2, 115–150, DOI:10.1145/514183.514185.
- D3TaLES Google Collaboratory Calculators, https://d3tales.as.uky.edu/tools/calculators Search PubMed.
- https://d3tales.as.uky.edu/database/06TNKR/ .
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, SambVca: A Web Application for the Calculation of the Buried Volume of N-Heterocyclic Carbene Ligands, Eur. J. Inorg. Chem., 2009, 2009, 1759–1766, DOI:10.1002/ejic.200801160.
- S. V. Shree Sowndarya, J. N. Law, C. E. Tripp, D. Duplyakin, E. Skordilis, D. Biagioni, R. S. Paton and P. C. St. John, Multi-objective goal-directed optimization of de novo stable organic radicals for aqueous redox flow batteries, Nat. Mach. Intell., 2022, 4(8), 720–730, DOI:10.1038/s42256-022-00506-3.
- J. Peng, D. Schwalbe-Koda, K. Akkiraju, T. Xie, L. Giordano, Y. Yu, C. J. Eom, J. R. Lunger, D. J. Zheng and R. R. Rao, et al., Human- and machine-centred designs of molecules and materials for sustainability and decarbonization, Nat. Rev. Mater., 2022, 7(12), 991–1009, DOI:10.1038/s41578-022-00466-5.
- Ö. H. Omar, M. Del Cueto, T. Nematiaram and A. Troisi, High-throughput virtual screening for organic electronics: a comparative study of alternative strategies, J. Mater. Chem. C, 2021, 9, 13557–13583, 10.1039/d1tc03256a.
- C. Kunkel, J. T. Margraf, K. Chen, H. Oberhofer and K. Reuter, Active discovery of organic semiconductors, Nat. Commun., 2021, 12(2422) DOI:10.1038/s41467-021-22611-4.
- E. O. Pyzer-Knapp, C. Suh, R. Gómez-Bombarelli, J. Aguilera-Iparraguirre and A. Aspuru-Guzik, What Is High-Throughput Virtual Screening? A Perspective from Organic Materials Discovery, Annu. Rev. Mater. Res., 2015, 45, 195–216, DOI:10.1146/annurev-matsci-070214-020823.
- J. A. Kowalski, M. D. Casselman, A. P. Kaur, J. D. Milshtein, C. F. Elliott, S. Modekrutti, N. H. Attanayake, N. Zhang, S. R. Parkin and C. Risko, et al., A stable two-electron-donating phenothiazine for application in nonaqueous redox flow batteries, J. Mater. Chem. A, 2017, 5, 24371–24379, 10.1039/c7ta05883g.
- P. Ertl and A. Schuffenhauer, Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions, J. Cheminf., 2009, 1, 8, DOI:10.1186/1758-2946-1-8.
- J. D. Milshtein, A. P. Kaur, M. D. Casselman, J. A. Kowalski, S. Modekrutti, P. L. Zhang, N. Harsha Attanayake, C. F. Elliott, S. R. Parkin and C. Risko, et al., High current density, long duration cycling of soluble organic active species for non-aqueous redox flow batteries, Energy Environ. Sci., 2016, 9, 3531–3543, 10.1039/c6ee02027e.
- R. Gómez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D.-G. Ha and T. Wu, et al., Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach, Nat. Mater., 2016, 15, 1120–1127, DOI:10.1038/nmat4717.
- A. Jain, S. P. Ong, W. Chen, B. Medasani, X. Qu, M. Kocher, M. Brafman, G. Petretto, G. M. Rignanese and G. Hautier, et al., FireWorks: a dynamic workflow system designed for high-throughput applications, Concurr. Comput. Pract. Exp., 2015, 27, 5037–5059, DOI:10.1002/cpe.3505.
Footnote |
| † Electronic supplementary information (ESI) available: Document containing details about molecular generation, computational methods, funnel workflow details, more information about the D3TaLES API, etc. See DOI: https://doi.org/10.1039/d3dd00081h |
| This journal is © The Royal Society of Chemistry 2023 |