
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
B(3,4,5-F3H2C6)3 Lewis acid-catalysed C3-allylation of indoles†
Nusaybah
Alotaibi
 a,
Rasool
Babaahmadi
a,
Milan
Pramanik
a,
Tanja
Kaehler
a,
Ayan
Dasgupta
ab,
Emma
Richards
a,
Alireza
Ariafard
c,
Thomas
Wirth
d and
Rebecca L.
Melen
*a
a,
Rasool
Babaahmadi
a,
Milan
Pramanik
a,
Tanja
Kaehler
a,
Ayan
Dasgupta
ab,
Emma
Richards
a,
Alireza
Ariafard
c,
Thomas
Wirth
d and
Rebecca L.
Melen
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Translational Research Hub, Maindy Road, Cathays, Cardiff, CF24 4HQ, Cymru/Wales, UK. E-mail: MelenR@cardiff.ac.uk
bChemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK
cSchool of Natural Sciences (Chemistry), University of Tasmania, Private Bag 75, Hobart, Tasmania, 7001 Australia
dSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, Cymru/Wales, UK
First published on 11th April 2023
Abstract
Herein we report the B(3,4,5-F3H2C6)3-catalysed C3-allylation of indoles using allylic esters. 25 examples of C3-allylated products are presented in up to 97% yield. The mechanism for the reaction was explored using detailed Density Functional Theory (DFT) studies.
Indole is one of the most important nitrogen-containing heteroaromatic compounds not only as an essential building block for biologically active natural products and pharmaceuticals, but also for pigments and materials science.1 Therefore, catalytic functionalisation of electron-rich indoles has been explored extensively,2 especially at the nucleophilic C3-position through electrophilic substitution.3 Typically metal catalysts are employed,2 but the major disadvantages associated with these classical metal-catalyzed C3 functionalisation reactions are the post-reaction contamination, use of precious and toxic metals, high temperature, prolonged reaction time, unwanted by-products and several side selectivity issues. In this regard, the implementation of a metal-free catalyst as a sustainable reaction strategy is highly desired in the chemical community. Recently, the use of main group catalysts such as fluorinated triaryl boranes (e.g. B(C6F5)3) has gained popularity due to their potentially lower toxicity and high selectivities (Fig. 1).4 For example, Melen, Morrill, and Pulis et al. used catalytic amounts of B(C6F5)3 for the direct C3 alkylation of indoles with amine-based alkylating reagents.5 Under similar reaction conditions, Melen et al. reported the introduction of alkyl groups with diazoesters via C–H insertion.6 The stereoselective C-glycosylation of indoles with B(C6F5)3 and glycosyl trichloroacetimidates has also been achieved by Mandal et al.7 The synthesis of bis(indolyl)alkanes by a B(C6F5)3-catalysed Markovnikov addition of indoles to aryl alkynes as well as the B(C6F5)3-catalysed dehydrogenative C–C bond formation of indoles with N-tosylhydrazones have also been reported.8,9 An interesting new approach by Tang et al. was the use of B(C6F5)3 as a single-electron oxidant.10 Here, an electron donor–acceptor complex is formed between B(C6F5)3 and the indole which promotes a photoinduced single electron transfer event resulting in an oxidative C–S cross-coupling reaction of indoles with thiophenols.10
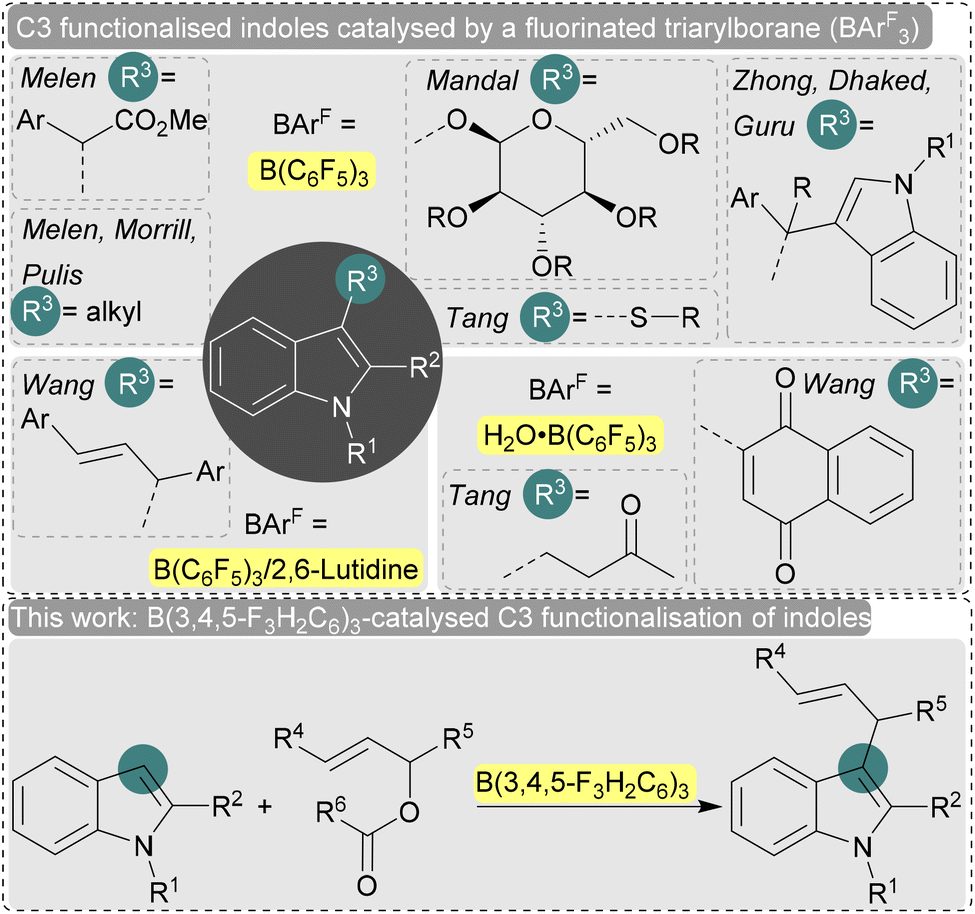 | ||
| Fig. 1 Previous and current work. | ||
The strong Brønsted acidity of the H2O·B(C6F5)3 adduct is well known,11 and has been used by Tang et al. to perform dehydrative Friedel–Crafts alkylations catalytically,12 and for the C–C coupling of 1,4-naphthoquinones with indoles in water as reported by Wang et al.13,14 The same group and others reported the allylation of indole in water catalysed by the B(C6F5)3/2,6-lutidine frustrated Lewis pair.15,16 The introduction of an allyl group yields highly functionable synthetic intermediates that are important for further manipulations.17 However, the previous report with B(C6F5)3/2,6-lutidine used high temperatures and no scope of the allyl reagent was explored.15 We anticipate that a further investigation into this reaction to establish a mild catalytic reaction protocol might be of interest.
Herein we report the high yielding B(3,4,5-F3H2C6)3 Lewis acid-catalysed C3-allylation of indoles with allylic esters including a full DFT supported mechanistic investigation. We started our investigation by searching for the optimal reaction conditions for the C3-allylation of 1-methyl indole 2a (1 equiv.) with allylic ester 1a (1 equiv.) using a Lewis acid catalyst to give allylation product 3a (Scheme 1).
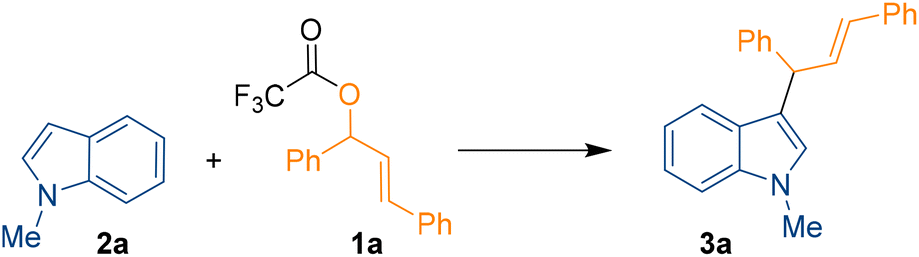 | ||
| Scheme 1 C3-allylation of indoles catalysed by B(3,4,5-F3H2C6)3. | ||
A trifluoroacetic ester was used as the coupling partner since CF3COO− is a good leaving group. Following extensive optimisation studies (see ESI, Table S1†), we found that the optimum conditions for the reaction were 15 mol% B(3,4,5-F3H2C6)3 catalyst loading in chloroform for 24 h at 60 °C. Under these conditions, 3a was formed in 97% yield. Other Lewis acids such as B(C6F5)3, B(2,4,6-F3H2C6)3, BF3·OEt2, BPh3, and Brønsted acid trifluoroacetic acid (TFA) were screened as different catalysts but none of these produced higher yields than B(3,4,5-F3H2C6)3. Again, no product formation was observed in absence of the catalyst. In all reactions, MgSO4 was added to ensure no residual water was helping to form H2O·B(C6F5)3 or H2O·B(3,4,5-F3H2C6)3 and the reaction was not catalysed by a Brønsted acid. Following on from a previous publication from Wang et al.,15 we also attempted the reaction of the alcohol derivative of 1a with 2a using 15 mol% B(C6F5)3 as a catalyst in the presence of MgSO4, which yielded 3a in just 40%, confirming our choice for the ester 1a. Other variables like catalyst loading, solvents, concentration, and stoichiometry of 1a and 2a were studied with B(3,4,5-F3H2C6)3 but did not help to improve the reactivity.
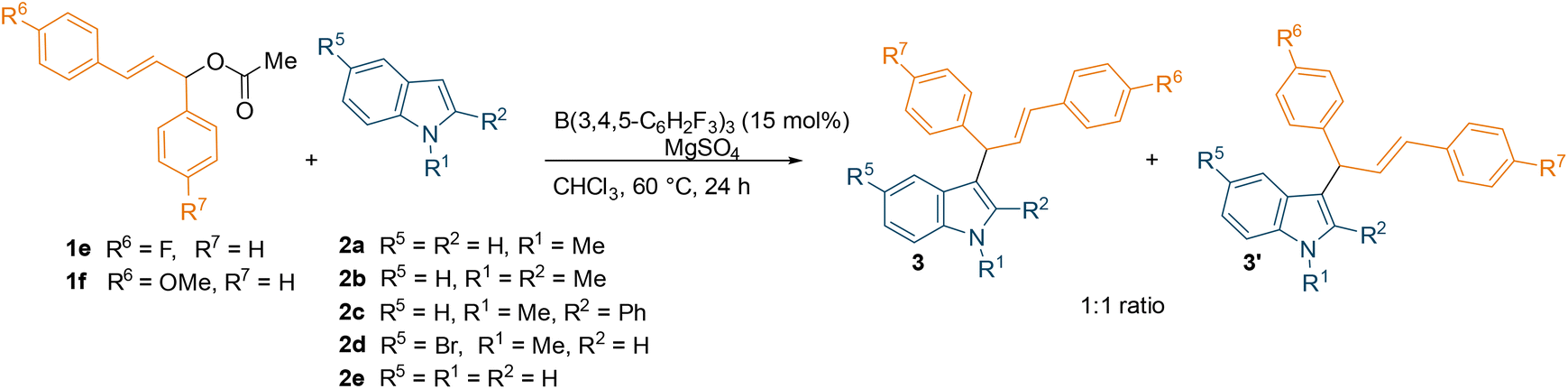
With the optimised conditions in hand, we explored the substrate scope using different commercially available indoles with diphenyl allylic ester 1a (Scheme 2). The 5-bromo-1-methylindole afforded the desired allylated product 3d in near-quantitative yield (93%). On the other hand, relatively lower yields (65% and 50%) were observed with more sterically hindered 1,2-dimethylindole and 1-methyl-2-phenylindole, (2b and 2c respectively). Successful introduction of an allyl moiety to 1-methylindole derivatives was also achieved using allylic esters with symmetrical aryl rings bearing electron-donating group (OMe) or electron-withdrawing group (F) in para-position (1c and 1b, Scheme 2). Regardless of the electronic effect of the substituents on 1, the corresponding allylated products (3f–3i, 3k–3n) were obtained in good to high yields (up to 91%). Interestingly, the unprotected indole also afforded the desired products (3e, 3j and 3o) in moderate yields (up to 57%). The relatively lower yields compared to the allylated 1-methylindole could be attributed to the coordination of the unprotected indole with the borane catalyst, forming a Lewis acid–base adduct. When using unsymmetrical esters 1e or 1f (Table 1), a mixture of two inseparable regio-isomers was formed (3p–y, 3p′–y′, see ESI†) in good to high combined yields (up to 79%) possibly supporting the formation of an allylic carbocation. Despite the different electronic effect of the substituents on the aryl rings of the ester, the ratio of the two isomers determined by NMR spectroscopy was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in all cases. However, we have tried the reaction with C3 methylated indole which did not respond under the standard reaction conditions to give the desired C2-allylation product.
:1 in all cases. However, we have tried the reaction with C3 methylated indole which did not respond under the standard reaction conditions to give the desired C2-allylation product.
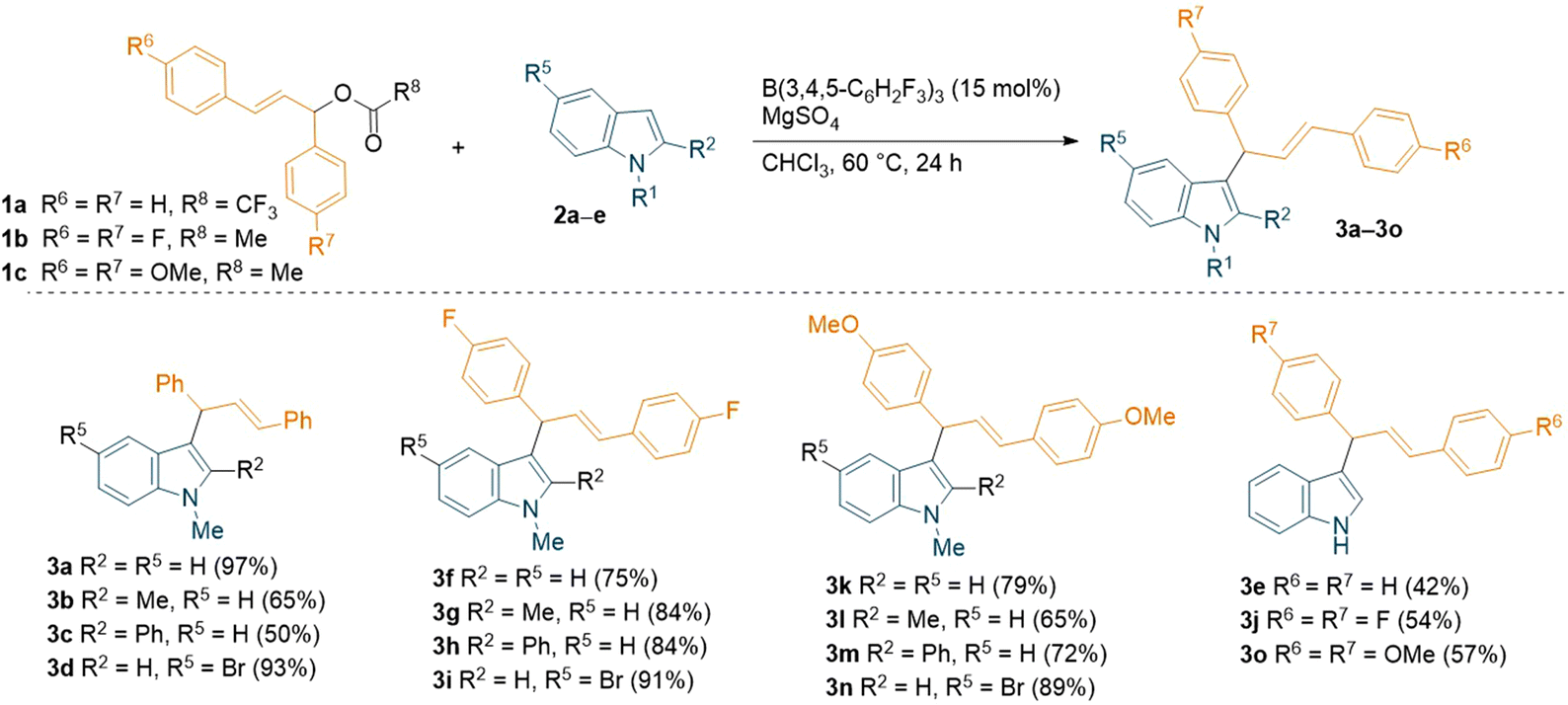 | ||
| Scheme 2 B(3,4,5-F3H2C6)3-catalyzed C3-allylation of indoles using symmetrical diaryl allylic esters. All reactions were carried out on a 0.1 mmol scale; yields reported are isolated. | ||
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Ester | Indole | Combined yield (%) | Products | Entry | Ester | Indole | Combined yield (%) | Products |
| 1 | 1e | 2a | 79 | 3p, 3p′ | 6 | 1f | 2a | 51 | 3u, 3u′ |
| 2 | 1e | 2b | 72 | 3q, 3q′ | 7 | 1f | 2b | 65 | 3v, 3v′ |
| 3 | 1e | 2c | 69 | 3r, 3r′ | 8 | 1f | 2c | 65 | 3w, 3w′ |
| 4 | 1e | 2d | 76 | 3s, 3s′ | 9 | 1f | 2d | 79 | 3x, 3x′ |
| 5 | 1e | 2e | 61 | 3t, 3t′ | 10 | 1f | 2e | 68 | 3y, 3y′ |
To understand the mechanism of the C3-allylation of 1-methyl indole with allylic ester 1a catalysed by B(3,4,5-F3H2C6)3, we performed density functional theory (DFT) calculations in chloroform at the SMD/M06-2X/def2-TZVP//SMD/M06-2X/6-31G(d) level of theory. According to the calculations, the reaction commences with the coordination of the borane catalyst to the carbonyl functionality of allylic ester 1a to form adduct 1a·B(C6F3H2)3. This weakens the C–O bond, allowing it to be cleaved via transition structure TS2 to generate carbocation I and tetrahedral boron anion II. The indole then nucleophilically attacks the ensuing carbocation to form a new C–C bond. This attack is possible via two transition structures, TS3 and TS3′. The transition structure TS3 has a lower energy than TS3′ by 7.4 kcal mol−1, supporting the notion that the C3 position of indole is more nucleophilic than the C2 position.18 Following the completion of the nucleophilic attack through TS3, the iminium ion III is formed. III has a highly acidic proton and is therefore easily deprotonated by tetrahedral boron anion II to produce product 3a and adduct IV. The TFA component of adduct IV is a weakly coordinating species, so it easily leaves the system and regenerates the borane catalyst in an exergonic fashion (Scheme 3 and Fig. 2, top). According to our calculations, the rate-determining step of the catalytic reaction is the nucleophilic attack of the indole on the in situ generated carbocation Ivia transition structure TS3 with a relative free energy of 21.5 kcal mol−1. It is worth noting that dispersive interactions contribute significantly to the stability of this transition structure. This claim is verified by performing single-point calculations at the SMD/B3LYP/def2-TZVP and SMD/B3LYP-D3/def2-TZVP levels of theory on the structures optimised by SMD/M06-2X/6-31G(d).
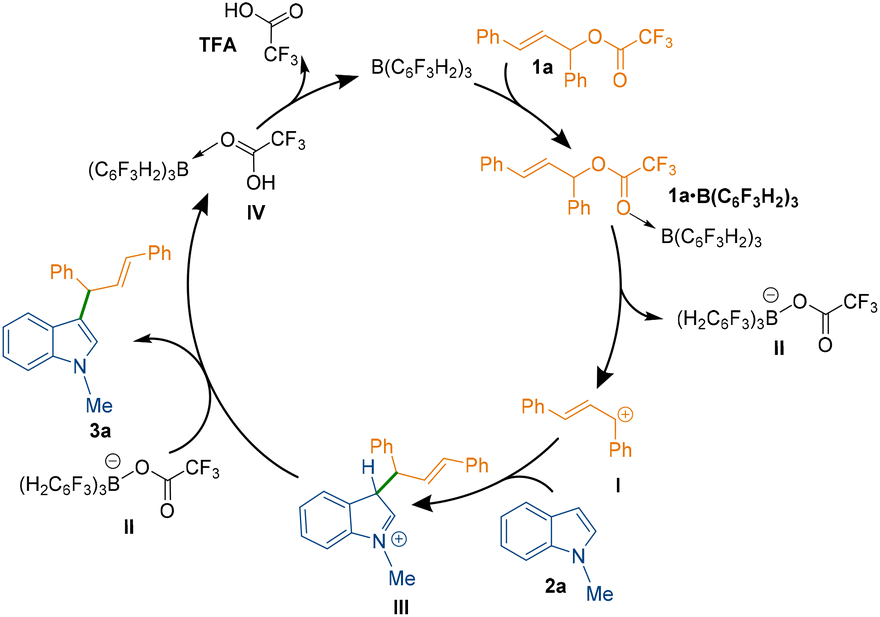 | ||
| Scheme 3 Proposed reaction mechanism for the formation of 3a. | ||
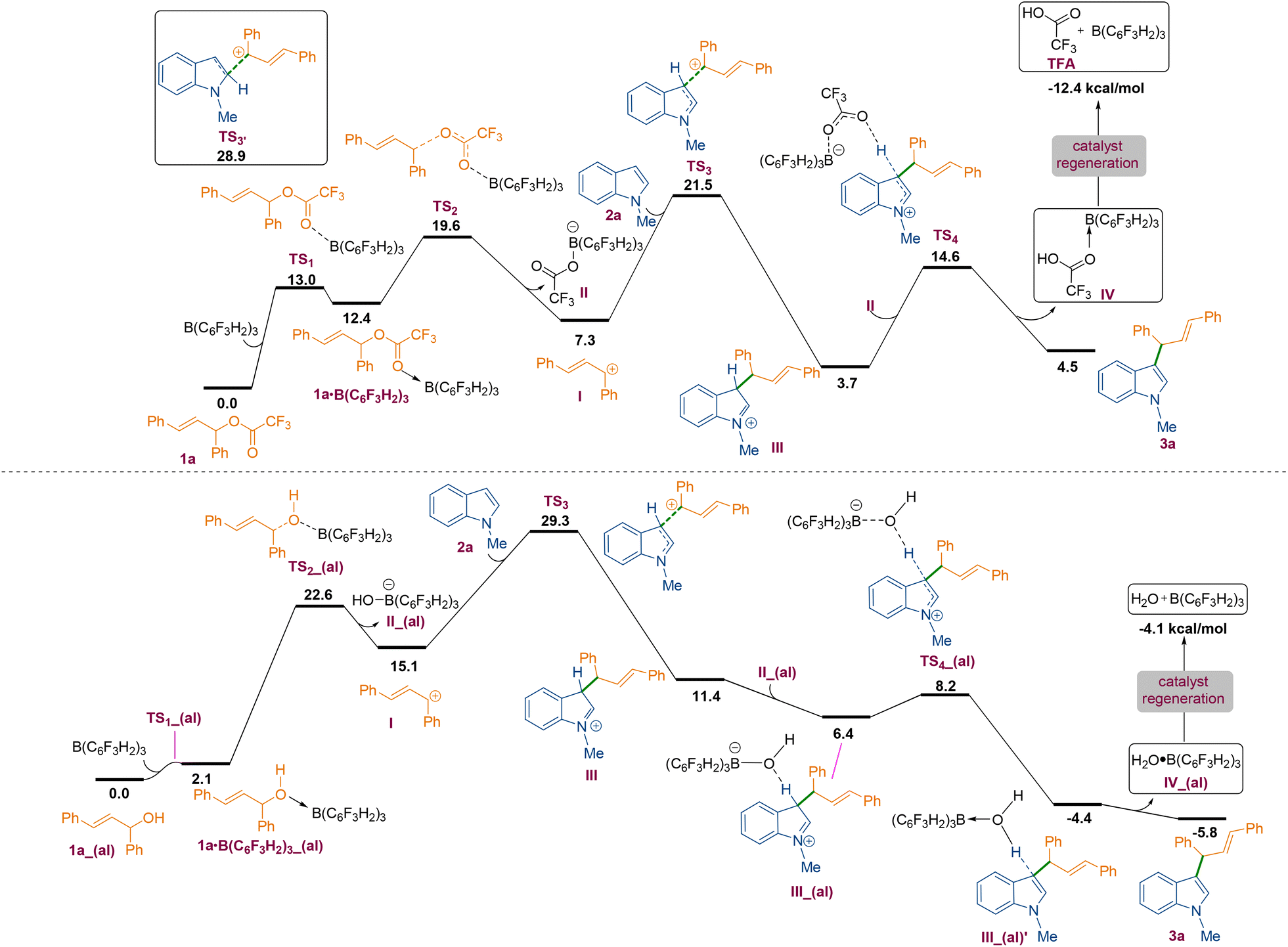 | ||
| Fig. 2 DFT calculated reaction pathways for the formation of 3a compound from the reaction of C3-allylation of 1-methyl indole with allylic ester 1a (top) and alcohol derivative 1a_(al) (bottom) in CHCl3 using SMD/M06-2X/def2-TZVP//SMD/M06-2X/6-31G(d) level of theory. The relative free energies are given in kcal mol−1. | ||
Using B3LYP calculations, the overall activation barrier to the nucleophilic addition of the indole to the carbocation is calculated to be 33.8 kcal mol−1, whereas it is reduced to 15.8 kcal mol−1 when dispersive interactions are considered using B3LYP-D3 calculations. The NCI analysis depicted in Fig. 3 confirms the presence of non-covalent interactions (NCIs) in transition structure TS3, which originate primarily from π–π stacking interactions (Fig. 3). We then turned our attention to determining computationally why the replacement of ester derivative 1a with alcohol derivative 1a_(al) impedes the C3-allylation of 1-methyl indole. Fig. 2 (bottom) depicts the free energy profile for the C3-allylation of 1-methyl indole with allylic alcohol 1a_(al). As depicted, this replacement causes the activation barrier for the allylation reaction to increase to 29.3 kcal mol−1. This increase in activation energy is because the key carbocation I in this instance has much higher energy (15.1 kcal mol−1 for the allylation reaction with alcohol substrate 1a_(al)versus 7.3 kcal mol−1 for the allylation reaction with ester substrate 1). Our calculation indicated that, although the borane catalyst binds more strongly to the alcohol substrate than to the ether substrate, the alcohol substrate is less reactive towards the formation of the key carbocation. Thus, the nature of the leaving group on the allylic substrate largely dictates the ease of such reactions.
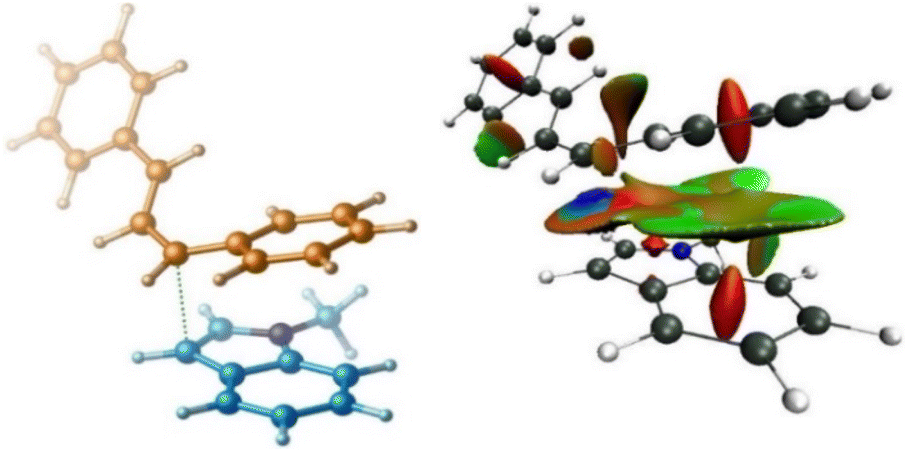 | ||
| Fig. 3 The non-covalent interaction (NCI) analysis of transition structure TS3. | ||
In conclusion, we have demonstrated the B(3,4,5-F3H2C6)3 Lewis acid catalysed C3-allylation of indoles using allylic ester substrates giving 25 examples of allylated products in moderate to excellent yields. The mechanism for the reaction was fully explored by DFT studies which demonstrate that an ester substrate is crucial for the reaction to proceed. Other reactions using the B(3,4,5-F3H2C6)3 Lewis acid are ongoing in our group.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
R.B. would like to thank the Royal Society for a Newton International Fellowship (NIF\R1\211330). A.D., E.R., and R.L.M. thank the Leverhulme Trust (RPG-2020-016)., and M.P.T.K. and R.L.M. thank the EPSRC (EP/R026912/1) for funding. R.B. and A.A. also thank the Australian National Computational Infrastructure Digital Research Services, IT Services at the University of Tasmania for the generous allocation of computing time. N.A. also acknowledges support from the Saudi Ministry of Education and the King Faisal University, Saudi Arabia. Information about the data that underpins the results presented in this article can be found in the Cardiff University data catalogue at http://doi.org/10.17035/d.2023.0252049034.References
- (a) R. J. Sundberg, The Chemistry of Indoles, Academic Press, New York, 1970 Search PubMed; (b) R. K. Brown, Indoles Part I, Wiley-Interscience, New York, 1972 Search PubMed.
- (a) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef CAS PubMed; (b) R. Dalpozzo, Chem. Soc. Rev., 2015, 44, 742 RSC; (c) K. Urbina, D. Tresp, K. Sipps and M. Szostak, Adv. Synth. Catal., 2021, 363, 2723 CrossRef CAS; (d) S. Pradhan, P. B. De, T. A. Shah and T. Punniyamurthy, Chem. – Asian J., 2020, 15, 4184 CrossRef CAS PubMed.
- S. Lakhdar, M. Westermaier, F. Terrier, R. Goumont, T. Boubaker, A. R. Ofial and H. Mayr, J. Org. Chem., 2006, 71, 8088 CrossRef PubMed.
- For reviews see: (a) J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706 RSC; (b) S. Basak, L. Winfrey, B. A. Kustiana, R. L. Melen, L. C. Morrill and A. P. Pulis, Chem. Soc. Rev., 2021, 50, 3720 RSC. For selected examples see: (c) G. Kumar, S. Roy and I. Chatterjee, Org. Biomol. Chem., 2021, 19, 1230 RSC; (d) T. Hackel and N. A. McGrath, Molecules, 2019, 24, 432 CrossRef PubMed; (e) V. Nori, F. Pesciaioli, A. Sinibaldi, G. Giorgianni and A. Carlone, Catalysts, 2022, 12, 5 CrossRef CAS.
- S. Basak, A. Alvarez-Montoya, L. Winfrey, R. L. Melen, L. C. Morrill and A. P. Pulis, ACS Catal., 2020, 10, 4835 CrossRef CAS PubMed.
- A. Dasgupta, R. Babaahmadi, B. Slater, B. F. Yates, A. Ariafard and R. L. Melen, Chem, 2020, 6, 2364 CAS.
- A. Dubey, A. Tiwari and P. K. Mandal, J. Org. Chem., 2021, 86, 8516 CrossRef CAS PubMed.
- F. Ling, L. Xiao, L. Fang, C. Feng, Z. Xie, Y. Lv and W. Zhong, Org. Biomol. Chem., 2018, 16, 9274 RSC.
- Dipika, Y. B. Sharma, S. Pant, D. K. Dhaked and M. M. Guru, Org. Chem. Front., 2022, 9, 3428 RSC.
- W. Yuan, J. Huang, X. Xu, L. Wang and X.-Y. Tang, Org. Lett., 2021, 23, 7139 CrossRef CAS PubMed.
- (a) A. Di Saverio, F. Focante, I. Camurati, L. Resconi, T. Beringhelli, G. D'Alfonso, D. Donghi, D. Maggioni, P. Mercandelli and A. Sironi, Inorg. Chem., 2005, 44, 5030 CrossRef CAS PubMed; (b) A. F. Schneider, Y. Chen and M. A. Brook, Dalton Trans., 2019, 48, 13599 RSC.
- H. H. San, J. Huang, S. L. Aye and X.-Y. Tang, Adv. Synth. Catal., 2021, 363, 2386 CrossRef CAS.
- Y. Dong, H. Zhang, J. Yang, S. He, Z.-C. Shi, X.-M. Zhang and J.-Y. Wang, ACS Omega, 2019, 4, 21567 CrossRef CAS PubMed.
- Also see: A. Das, K. Watanabe, H. Morimoto and T. Ohshima, Org. Lett., 2017, 19, 5794 CrossRef CAS PubMed.
- H. Zhang, X.-Y. Zhan, Y. Dong, J. Yang, S. He, Z.-C. Shi, X.-M. Zhang and J.-Y. Wang, RSC Adv., 2020, 10, 16942 RSC.
- (a) M. Kimura, M. Futamata, R. Mukai and Y. Tamaru, J. Am. Chem. Soc., 2005, 127, 4592 CrossRef CAS PubMed; (b) B. Mouhsine, A. Karim, C. Dumont, A. S. Pol, I. Suissse and M. Sauthier, Eur. J. Org. Chem., 2022, e202200042 CAS. Examples for C2-allylation of indoles: (c) P. Ullrich, J. Schmauck, M. Brauns, M. Mantel, M. Breugst and J. Pietruszka, J. Org. Chem., 2020, 85, 1894 CrossRef CAS PubMed; (d) A. A. Atia and M. Kimura, Tetrahedron, 2021, 90, 132213 CrossRef CAS.
- (a) J. W. J. Kennedy and D. G. Hall, Angew. Chem., Int. Ed., 2003, 42, 4732 CrossRef CAS PubMed; (b) S. Dutta, T. Bhattacharya, D. B. Werz and D. Maiti, Chem, 2021, 7, 555 CrossRef CAS.
- A. Ariafard, ACS Catal., 2014, 9, 2896 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, optimisation tables, NMR spectra, DFT data. See DOI: https://doi.org/10.1039/d3dt00745f. |
| This journal is © The Royal Society of Chemistry 2023 |