
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparison of group 4 and thorium M(IV) substituted cyclopentadienyl silanide complexes†
Benjamin L. L.
Réant
 ,
Dukula
De Alwis Jayasinghe
,
Ashley J.
Wooles
,
Stephen T.
Liddle
* and
David P.
Mills
*
,
Dukula
De Alwis Jayasinghe
,
Ashley J.
Wooles
,
Stephen T.
Liddle
* and
David P.
Mills
*
Department of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, U.K. E-mail: stephen.liddle@manchester.ac.uk; david.mills@manchester.ac.uk
First published on 12th May 2023
Abstract
We report the synthesis and characterisation of a series of M(IV) substituted cyclopentadienyl hypersilanide complexes of the general formula [M(CpR)2{Si(SiMe3)3}(X)] (M = Hf, Th; CpR = Cp′, {C5H4(SiMe3)} or Cp′′, {C5H3(SiMe3)2-1,3}; X = Cl, C3H5). The separate salt metathesis reactions of [M(CpR)2(Cl)2] (M = Zr or Hf, CpR = Cp′; M = Hf or Th, CpR = Cp′′) with equimolar K{Si(SiMe3)3} gave the respective mono-silanide complexes [M(Cp′)2{Si(SiMe3)3}(Cl)] (M = Zr, 1; Hf, 2), [Hf(Cp′′)(Cp′){Si(SiMe3)3}(Cl)] (3) and [Th(Cp′′)2{Si(SiMe3)3}(Cl)] (4), with only a trace amount of 3 presumably formed via silatropic and sigmatropic shifts; the synthesis of 1 from [Zr(Cp′)2(Cl)2] and Li{Si(SiMe3)3} has been reported previously. The salt elimination reaction of 2 with one equivalent of allylmagnesium chloride gave [Hf(Cp′)2{Si(SiMe3)3}(η3-C3H5)] (5), whilst the corresponding reaction of 2 with equimolar benzyl potassium yielded [Hf(Cp′)2(CH2Ph)2] (6) together with a mixture of other products, with elimination of both KCl and K{Si(SiMe3)3}. Attempts to prepare isolated [M(CpR)2{Si(SiMe3)3}]+ cations from 4 or 5 by standard abstraction methodologies were unsuccessful. The reduction of 4 with KC8 gave the known Th(III) complex, [Th(Cp′′)3]. Complexes 2–6 were characterised by single crystal XRD, whilst 2, 4 and 5 were additionally characterised by 1H, 13C{1H} and 29Si{1H} NMR spectroscopy, ATR-IR spectroscopy and elemental analysis. In order to probe differences in M(IV)–Si bonds for d- and f-block metals we studied the electronic structures of 1–5 by density functional theory calculations, showing M–Si bonds of similar covalency for Zr(IV) and Hf(IV), and less covalent M–Si bonds for Th(IV).
Introduction
Transition metal (TM) silicon chemistry is well-established,1 with applications ranging from homogeneous (hydro)silylation catalysts2–5 to TM silicides in catalysis, ceramics and microelectronics.6 f-Block silicon chemistry is currently underdeveloped in comparison,1,7 but potential uses include lanthanide (Ln) silanide ({SiR3}) complexes in olefin polymerisation catalysts,8,9 Ln silicides as additives in low-alloy steels,10 and high-density actinide (An) silicide nuclear fuels.11–14 Given that the physical and chemical properties of Ln and An complexes can differ markedly from each other and related early TM complexes,15,16 it follows that comparative studies of their electronic structures and the relative amount of covalency in M–Si bonds are crucial for the development of novel applications.Since the discovery nearly 40 years ago that group 4 TM bent metallocenes can promote the dehydrogenative polymerisation of silanes,17 M(IV) silanide complexes of the general formula [M(CpR)2(SiR3)(X)] (M = Ti, Zr, Hf; CpR = substituted cyclopentadienyl, {C5R5}; X = anion) have been studied extensively;1 however, f-block analogues of these complexes have not previously been isolated. As the +4 oxidation state is limited for organometallic Ln complexes15 and given the increased radiological hazard and decreased stability of An(IV) complexes across the series,16 we targeted a Th(IV) silanide complex for comparison with group 4 M(IV) analogues. Whilst there are >130 structurally authenticated examples of Ti, Zr and Hf complexes containing one or more M–Si bonds, and >90 complexes containing Ln–Si bonds, for An silicon chemistry there are only 1 Th–Si and 6 U–Si bonds reported to date.18 Some of us recently reported the synthesis and characterisation of the sole thorium silanide complex, [Th(Cp′)3{Si(SiMe3)3}] (Cp′ = {C5H4(SiMe3)}) and showed by a variety of calculated covalency metrics that the Th–Si bond was less covalent than the corresponding U–Si bond of the isostructural U(IV) complex [U(Cp′)3{Si(SiMe3)3}].19 Our previously reported attempt to synthesise the heteroleptic U(IV) complex “[U(Cp′′)2{Si(SiMe3)3}(Cl)]” (Cp′′ = {C5H3(SiMe3)2-1,3}) led to metal reduction by the group 1 silanide transfer reagent to give the U(III) complex [U(Cp′′)2{Si(SiMe3)3}]; this complex showed greater covalency over homologous Ln(III) complexes [Ln(Cp′′)2{Si(SiMe3)3}] (Ln = La, Ce, Nd), but was less covalent than the group 4 TM(III) complexes [M(Cp′′)2{Si(SiMe3)3}] (M = Ti, Zr).20
Here we report the synthesis of a series of M(IV) substituted cyclopentadienyl hypersilanide complexes of general formula [M(CpR)2{Si(SiMe3)3}(X)] (M = Hf, Th; CpR = Cp′ or Cp′′; X = Cl or C3H5) by salt metathesis protocols. All complexes were characterised by single crystal XRD, and for samples with elemental analysis results in accord with expected values we additionally collected ATR-IR and 1H, 13C{1H} and 29Si{1H} NMR spectra. Density functional theory (DFT) calculations were performed in order to compare the electronic structures of the Hf(IV) and Th(IV) complexes herein with a previously reported Zr(IV) analogue [Zr(Cp′)2{Si(SiMe3)3}(Cl)];21 the Th–Si bonds were shown to be less covalent than the Zr–Si and Hf–Si bonds, which showed similar covalency.
Results and discussion
Synthesis
The M(IV) bis-substituted cylopentadienyl dichloride precursors [M(CpR)2(Cl)2] (M = Zr or Hf, CpR = Cp′; M = Hf or Th, CpR = Cp′′) were synthesised by separate salt metathesis reactions of parent [MCl4(S)2] (M = Zr or Hf, S = THF; M = Th, S = DME) with two equivalents of MCpR (M = Li, K) followed by work-up and recrystallisation, by following literature procedures.20,22–24 The separate salt elimination reactions of [M(CpR)2(Cl)2] with equimolar K{Si(SiMe3)3}25 gave the respective mono-silanide complexes [M(Cp′)2{Si(SiMe3)3}(Cl)] (M = Zr, 1; Hf, 2), [Hf(Cp′′)(Cp′){Si(SiMe3)3}(Cl)] (3) and [Th(Cp′′)2{Si(SiMe3)3}(Cl)] (4) (Scheme 1). We note that the synthesis of 1 by the salt metathesis reaction of [Zr(Cp′)2(Cl)2] with Li{Si(SiMe3)3} has previously been disclosed by Imori et al.21 Complexes 1, 2 and 4 were obtained in 59–75% yields following work-up and recrystallisation from pentane, whilst 3 formed in <1% crystalline yield, precluding the collection of a full set of characterisation data. We attribute the low yield of 3 to a silyl cleavage reaction occurring, likely by sigmatropic and silatropic shifts, as previously seen in the reactions of MCl4 (M = Zr, Hf) with Cp′H and Cp′′H.26 This side reaction highlights issues associated with attempting to install two bulky Cp′′ ligands at highly Lewis acidic Zr(IV) and Hf(IV) centres by salt elimination protocols. We were unable to prepare [Th(Cp′)2(Cl)2] by a salt metathesis reaction of [Th(Cl)4(DME)2] with two equivalents of LiCp′ due to facile ligand scrambling and the formation of [Th(Cp′)3(Cl)];27 this precluded the isolation of a Th(IV) hypersilanide complex that is isostructural to 1 and 2 to enable a more direct comparison.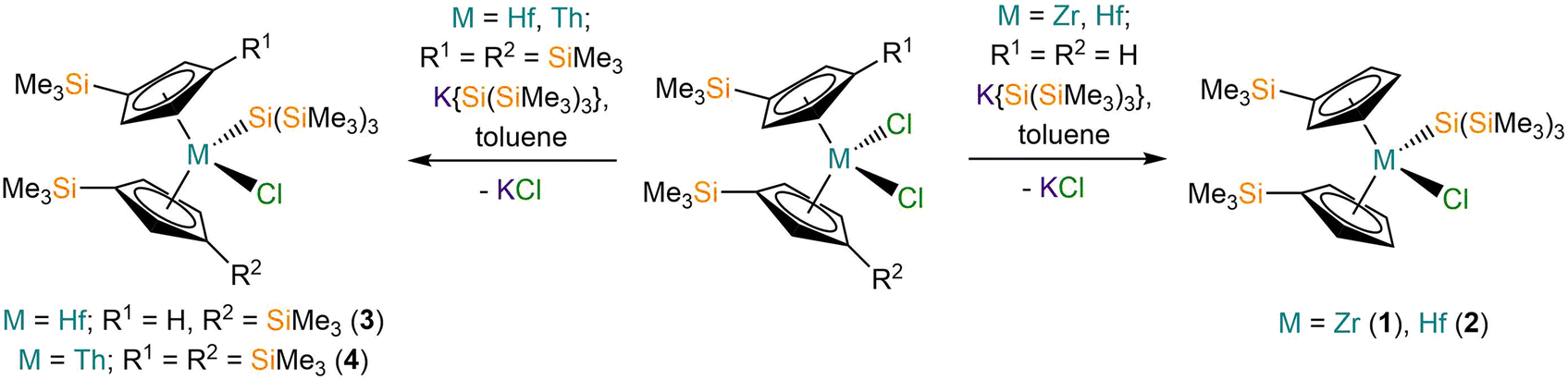 | ||
| Scheme 1 Synthesis of 1–4. | ||
In order to install additional synthetic functionality, the salt metathesis reaction of 2 with allylmagnesium chloride in a mixture of toluene and THF was performed to give [Hf(Cp′)2{Si(SiMe3)3}(η3-C3H5)] (5) in 63% yield following work-up and recrystallisation from pentane to remove the MgCl2 byproduct (Scheme 2). In contrast, the reaction of 2 with benzyl potassium28 in toluene gave several crystals of [Hf(Cp′)2(CH2Ph)2] (6) as part of a mixture of products, presumably by the elimination of both KCl and K{Si(SiMe3)3} (Scheme 2). We attribute the outcome of the latter reaction to a combination of the low solubility of KCH2Ph in toluene, coupled with the proposed intermediate “[Hf(Cp′)2{Si(SiMe3)3}(CH2Ph)]” likely being more reactive towards KCH2Ph than 2, though we cannot discount ligand scrambling processes occurring. A small-scale reaction of 2 with benzyl potassium in C6D6 monitored by 29Si{1H} NMR spectroscopy exhibited a single resonance at −86 ppm after 15 min of reaction time, indicating that a product with a Hf–Si bond is present in the reaction mixture, but this could not be structurally authenticated; the product distribution of this small-scale reaction is likely not representative of the scaled-up version due to differences in solvent, substrate concentration and reaction time.
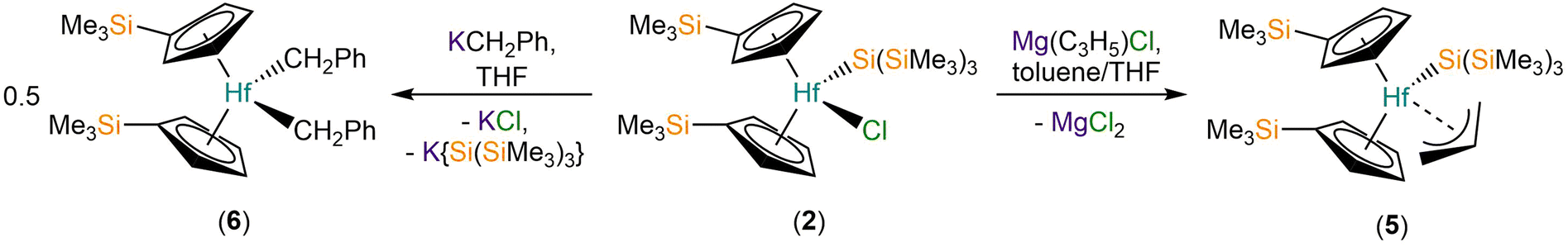 | ||
| Scheme 2 Synthesis of 5 and 6. | ||
In efforts to prepare isolated [M(CpR)2{Si(SiMe3)3}]+ cations by adapting literature abstraction methodologies, complex 4 was treated with [CPh3][Al{OC(CF3)3}4]29 and two equivalents of Et3SiH (in order to form a silylium cation in situ that abstracts Cl− to form strong Si–Cl bonds as an enthalpic driving force)30 in benzene, and complex 5 was treated with [NEt3H][BPh4]31 in either toluene or THF (in order to provide an entropic driving force by the formation of propene and trimethylamine via a protonolysis reaction). No product could be isolated from the former reaction, whilst no reaction was observed between 5 and the ammonium reagent in both solvent systems investigated. Finally, in an attempt to target the Th(III) complex “[Th(Cp′′)2{Si(SiMe3)3}]”, which would be structurally analogous to previously reported Ti, Zr, La, Ce, Nd and U analogues,20 complex 4 was reacted with 1.2 eq. of KC8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32 in DME. Following work-up and recrystallisation from pentane the previously reported homoleptic Th(III) complex [Th(Cp′′)3]33,34 was isolated in 59% crystalline yield, presumably by ligand scrambling of “[Th(Cp′′)2(X)]” following elimination of KX (X = Cl, {Si(SiMe3)3}) in accord with the previously reported reduction of [Th(Cp′′)2(Cl)2] in THF over excess Na/K alloy.32
32 in DME. Following work-up and recrystallisation from pentane the previously reported homoleptic Th(III) complex [Th(Cp′′)3]33,34 was isolated in 59% crystalline yield, presumably by ligand scrambling of “[Th(Cp′′)2(X)]” following elimination of KX (X = Cl, {Si(SiMe3)3}) in accord with the previously reported reduction of [Th(Cp′′)2(Cl)2] in THF over excess Na/K alloy.32
Elemental analysis results obtained for 1, 2, 4 and 5 were in reasonable agreement with expected values, and their ATR-IR spectra (see ESI Fig. S39–S42†) exhibit absorption features that are similar to each other and those previously reported for the closely related M(III) complexes [M(Cp′′)2{Si(SiMe3)3}] (M = Ti, Zr, La, Ce, Nd, U).20 The ATR-IR spectrum of 5 additionally contains the expected resonance at 1531 cm−1 for a Hf(IV) η3-allyl group, which is at a higher frequency to that previously reported for [Hf(Cp*)(C4H4BNiPr2)(η3-C3H5)] (ῦ = 1504 cm−1).35
NMR spectroscopy
Complexes 1, 2, 4 and 5 were characterised by 1H, 13C{1H} and 29Si{1H} NMR spectroscopy (see Table 1 and ESI Fig. S1–S38†). 1H and 13C{1H} NMR chemical shifts were previously reported for 121 and the M(IV) precursors [M(CpR)2(Cl)2],20,22–24 but in order to consistently use C6D6 as the solvent we include these data for [M(Cp′)2(Cl)2] herein, along with their 29Si{1H} NMR spectral data for completeness. The spectral assignments were confirmed by correlation experiments for 1, 2, 4 and 5.
| Complex | Environment | δ H/ppm | δ C/ppm | δ Si/ppm |
|---|---|---|---|---|
| a 1H and 13C{1H} NMR data originally reported in CDCl3 but recollected here in C6D6. b Not observed/obscured by solvent resonance. c Not reported previously, collected herein. | ||||
| [Zr(Cp′)2(Cl)2]22 | SiMe 3-Cp′ | 0.33, s, 18Ha | 0.15a | −6.40 |
| 3,4-Cp′ | 5.95, m, 4Ha | 115.36a | — | |
| 2,5-Cp′ | 6.40, m, 4Ha | 126.12a | — | |
| 1-Cp′ | — | —b | — | |
| [Hf(Cp′)2(Cl)2]22 | SiMe 3-Cp′ | 0.32, s, 18Ha | 0.21a | −6.53 |
| 3,4-Cp′ | 5.89, m, 4Ha | 114.28a | — | |
| 2,5-Cp′ | 6.31, m, 4Ha | 124.94a | — | |
| 1-Cp′ | — | —b | — | |
| [Hf(Cp′′)2(Cl)2]20 | SiMe 3-Cp′′ | 0.40, s, 36H | 0.29 | −6.97 |
| 4,5-Cp′′ | 6.44, m, 4H | 119.48 | — | |
| 2-Cp′′ | 7.20, m, 2H | 142.34 | — | |
| 1,3-Cp′′ | — | —b | — | |
| [Th(Cp′′)2(Cl)2]24 | SiMe 3-Cp′′ | 0.37, s, 36H | 0.23 | −8.83 |
| 4,5-Cp′′ | 6.95, m, 4H | 139.86 | — | |
| 2-Cp′′ | 7.24, m, 2H | 140.47 | — | |
| 1,3-Cp′′ | — | —b | — | |
|
121 |
SiMe 3-Cp′ | 0.26, s, 18H | 0.19 | −6.13c |
| 3,4-Cp′ | 5.02, m, 2H; 5.82, m, 2H | 111.22, 112.46 | — | |
| 2,5-Cp′ | 6.76, m, 2H; 7.67, m, 2H | 114.85, 120.46 | — | |
| 1-Cp′ | — | 129.47 | — | |
| Si(SiMe3)3 | 0.48, s, 27H | 5.22 | −6.61c | |
| Si(SiMe3)3 | — | — | −80.74c | |
| 2 | SiMe 3-Cp′ | 0.26, s, 18H | 0.23 | −6.12 |
| 3,4-Cp′ | 5.00, m, 2H; 5.87, m, 2H | 109.99, 112.59 | — | |
| 2,5-Cp′ | 6.59, m, 2H; 7.50, m, 2H | 115.00, 119.07 | — | |
| 1-Cp′ | — | —b | — | |
| Si(SiMe3)3 | 0.49, s, 27H | 5.43 | −5.66 | |
| Si(SiMe3)3 | — | — | −77.11 | |
| 4 | SiMe 3-Cp′′ | 0.31, s, 18H; 0.34, s, 18H | 0.96, 1.18 | −7.49, −7.67 |
| 4,5-Cp′′ | 7.06, m, 2H; 7.46, m, 2H | 129.70, 131.12 | — | |
| 2-Cp′′ | 7.32, m, 2H | 132.76 | — | |
| 1,3-Cp′′ | — | 140.17, 141.58 | — | |
| Si(SiMe3)3 | 0.60, s, 27H | 6.71 | −0.71 | |
| Si(SiMe3)3 | — | — | −66.31 | |
| 5 | SiMe 3-Cp′ | 0.16, s, 18H | 0.24 | −6.20 |
| 3,4-Cp′ | 4.88, m, 2H; 5.36, m, 2H | 102.69, 104.55 | — | |
| 2,5-Cp′ | 5.62, m, 2H; 6.06, m, 2H | 105.10, 110.27 | — | |
| 1-Cp′ | — | 115.23 | — | |
| Si(SiMe3)3 | 0.52, s, 27H | 6.47 | −5.29 | |
| Si(SiMe3)3 | — | — | −108.82 | |
| CH(CH2)2 | 2.58, d, 4H, 3JHH = 12.0 Hz | 55.86 | — | |
| CH(CH2)2 | 4.03, pent, 1H, 3JHH = 12.0 Hz | 112.67 | — | |
The 1H NMR spectra of [M(Cp′)2(Cl)2] exhibited three major signals in the expected ratio of 4:4:18, with typical second order AA′BB′ four spin systems for the two Cp′ ring proton environments and a singlet for the trimethylsilyl groups. The 1H NMR spectra of 121 and 2 show six major signals in a ratio of 2:2:2:2:27:18, with two sets of AA′BB′ patterns for the Cp′ ring protons; the inequivalent 2,5- and 3,4-Cp′ ring proton resonances arise from the diminished symmetry in 1 and 2 compared with parent [M(Cp′)2(Cl)2],22 which have mirror planes bisecting the metal and Cp′ centroids. The 1H NMR spectrum of 5 shows a similar pattern of signals to 1 and 2, as well as the expected doublet (4H) signal for the methylene and quintet (1H) resonance for the methine of the allyl ligand. This AX4 pattern is characteristic of fluxional behaviour, interconverting between η3- and η1-bound forms faster than the NMR timescale at this temperature; previously reported mono-allyl Hf(IV) complexes can show inequivalent syn- and anti-methylene groups in an AM2X2 pattern due to stronger η3-binding locking this conformation in solution, e.g. [Hf(Cp*)(C4H4BNiPr2)(η3-C3H5)] (Cp* = C5Me5; δH = 6.99 ppm, m, 1H, CH; 3.66 ppm, d, 2H, 3JHH = 15.4 Hz, CH2-anti; 1.70 ppm, d, 2H, 3JHH = 9.0 Hz, CH2-syn)35 and [Hf(Cp′′)(C4H4Me2-2,3)(η3-C3H5)] (δH = 5.65 ppm, m, 1H, CH; 1.64 ppm, m, 2H, CH2-syn; −0.83 ppm, d, 2H, 3JHH = 8.4 Hz, CH2-anti).36 The 1H NMR spectrum of 4 differs slightly, with six signals integrating to 2:2:2:27:18:18; two resonances are observed for the protons at the 4,5-Cp′′ ring positions due to the absence of a mirror plane in 4 compared with parent [Th(Cp′′)2(Cl)2].24 The 13C{1H} NMR spectra of [M(Cp′)2(Cl)2], 1 and 2 contain the same number of resonances as their respective 1H NMR spectra, with no resonance observed for the quaternary ring carbon atoms; by contrast resonances for all ring carbon environments are present in the 13C{1H} NMR spectra of 4 and 5. The silyl group resonances in the 13C{1H} NMR spectra of all complexes studied exhibit satellites from coupling to 29Si nuclei, with 1JSiC ≈ 53 Hz for Cp′/Cp′′ and 1JSiC ≈ 42 Hz for hypersilanide silyl groups. The allyl group signals in the 13C{1H} NMR spectrum of 5 are best compared to those of [Hf(Cp′′)(C4H4Me2-2,3)(η3-C3H5)] (δC = 127.87 ppm, CH; 57.13 ppm, CH2).36
The 29Si{1H} NMR spectra of 1, 2, 4 and 5 all exhibit signals for the silyl groups of CpR rings and the hypersilanide ligands. Most importantly, resonances for the metal-bound silicon atoms were observed for each of these complexes, with the δSi values of structurally analogous 1 (−80.74 ppm) and 2 (−77.11 ppm) being similar to each other and previously reported Zr and Hf hypersilanide complexes, e.g. [M(Cp*)(Cp){Si(SiMe3)3}(Cl)] (M = Zr, δSi = −87.30 ppm;37 M = Hf, δSi = −77.87 ppm38), [M(Cp)2{Si(SiMe3)3}(Cl)] (M = Zr, δSi = −85.5 ppm; M = Hf, δSi = −79.7 ppm).39,40 The substitution of chloride with allyl in 5 leads to a significant upfield shift (δSi = −108.82 ppm), highlighting the sensitivity of silanide chemical shifts to the metal coordination environment. Finally, the δSi value of the metal-bound silicon atom in the Th hypersilanide complex 4 (−66.32 ppm) is significantly downfield to that of the only other known Th(IV) silanide complex, [Th(Cp′)3{Si(SiMe3)3}] (−108.92 ppm).19
Single crystal XRD
Complexes 2–6 and [Hf(Cp′)2(Cl)2] were studied by single crystal XRD; the solid-state structures of 121 and other M(IV) precursors [M(CpR)2(Cl)2]20,22–24 have all been characterised by this method previously (see Fig. 1 and 2 for depictions of 2–6, and Table 2 for selected bond distances and angles; see ESI Fig. S43† for a depiction of [Hf(Cp′)2(Cl)2], together with Tables S1–S3† for additional crystallographic data). All complexes exhibit bent metallocene geometries with typical M⋯CpRcent distances for the metals and CpR ligands present;18 [Hf(Cp′)2(Cl)2] exhibits a similar solid-state structure to both its Zr congener22 and the related Hf complex [Hf(Cp′′)2(Cl)2],20 and as its metrical parameters are unremarkable we do not comment on this structure further.
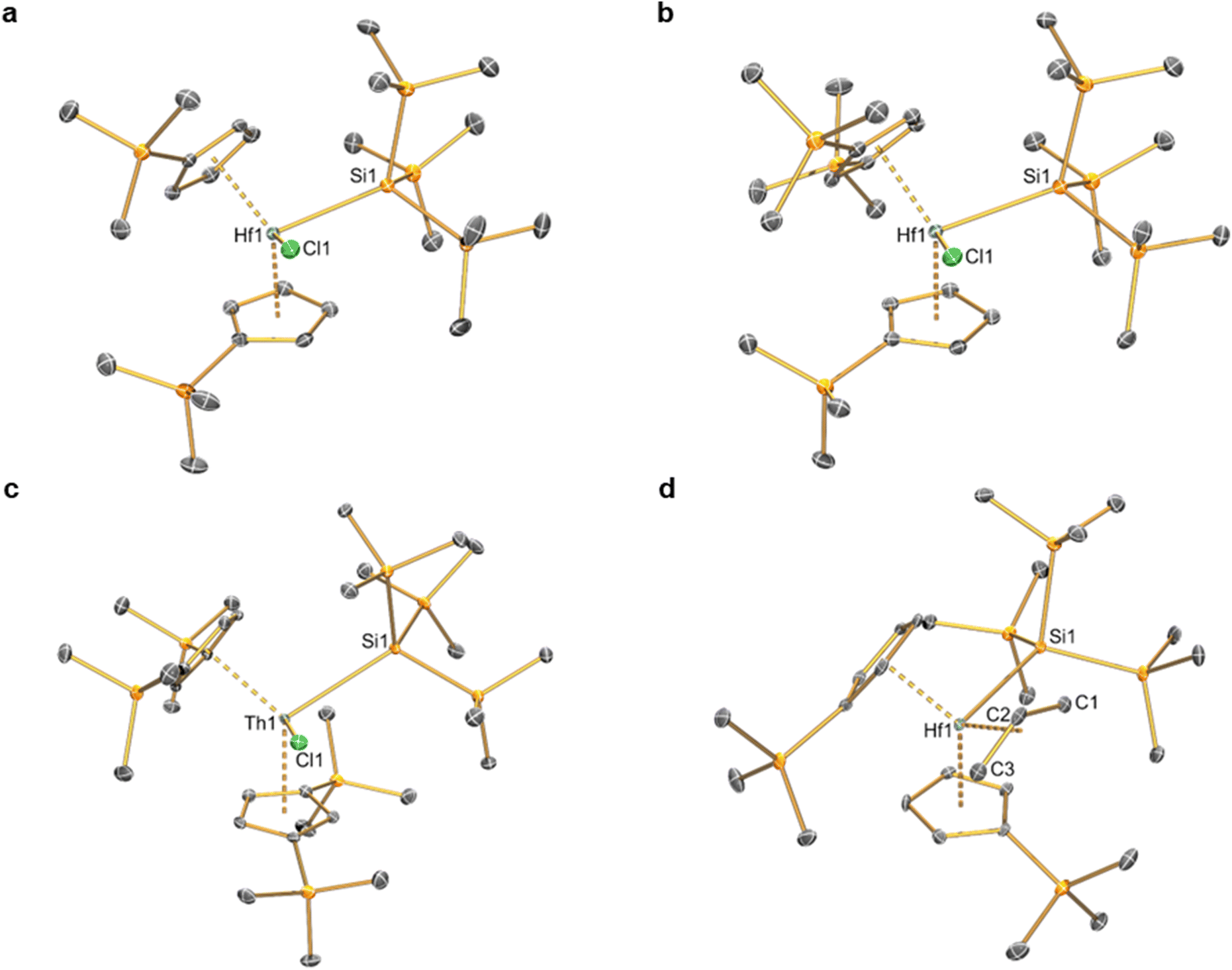 | ||
| Fig. 1 Single crystal XRD structure of (a) [Hf(Cp′)2{Si(SiMe3)3}(Cl)] (2), (b) [Hf(Cp′′)(Cp′){Si(SiMe3)3}(Cl)]·0.5C5H12 (3·0.5C5H12), (c) [Th(Cp′′)2{Si(SiMe3)3}(Cl)] (4) and (d) [Hf(Cp′)2{Si(SiMe3)3}(η3-C3H5)] (5) with selective atom labelling. Displacement ellipsoids set at 30% probability level and hydrogen atoms, lattice solvent and modelled disorder components omitted for clarity. | ||
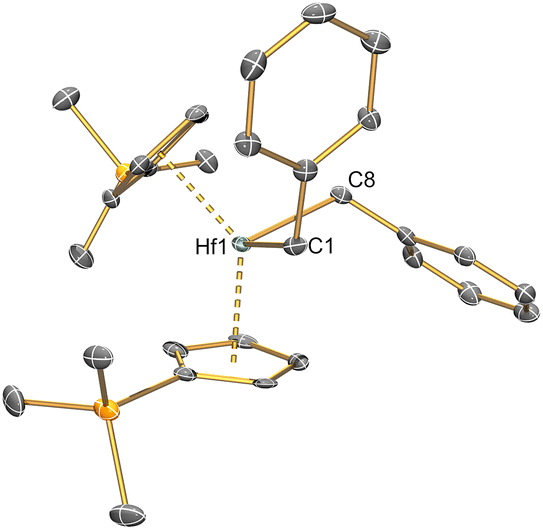 | ||
| Fig. 2 Single crystal XRD structure of [Hf(Cp′)2(CH2Ph)2] (6) with selective atom labelling. Displacement ellipsoids set at 30% probability level and hydrogen atoms omitted for clarity. | ||
| Parameter |
121 |
2 | 3·0.5C5H12 | 4 | 5 |
6a |
|---|---|---|---|---|---|---|
| a Two independent molecules in asymmetric unit, parameters shown for one of these molecules. b Three allyl Hf⋯C distances for 5 and two Hf–C alkyl bond lengths for 6. | ||||||
| M–Si | 2.833(3) | 2.7901(9) | 2.8057(8) | 3.1053(13) | 2.871(2) | — |
| M–Cl | 2.430(2) | 2.4041(7) | 2.3977(7) | 2.6118(11) | — | — |
| Hf–Cb | — | — | — | — | 2.426(5), 2.501(5), 2.555(5) | 2.281(6), 2.268(6) |
| Mean M⋯CpRcent | 2.21(1) | 2.190(3) | 2.198(2) | 2.520(4) | 2.211(4) | 2.198(4) |
| Si–M–Cl | 91.5(1) | 91.20(3) | 91.10(2) | 89.25(6) | — | — |
| CpRcent⋯M⋯CpRcent | 128.3(4) | 128.37(5) | 131.70(5) | 124.33(12) | 127.78(10) | 131.68(11) |
The solid-state structure of 2 is analogous to that of 1, though the bond distances are all shorter in the former complex, as exemplified by the Hf–Si distance (2.7901(9) Å) being shorter than the corresponding Zr–Si bond reported for 1 (2.833(3) Å);21 this is in accord with the greater covalent radii of Zr (1.54 Å) vs. Hf (1.52 Å).41 The structure of 3 is comparable to that of 2, though the additional steric buttressing provided by the additional silyl group in the Cp′′ ring leads to a slightly longer Hf–Si bond (2.8057(8) Å) and a more obtuse CpRcent⋯Hf⋯CpRcent angle (131.70(5)° c.f. 128.37(5)° for 2). Similarly, the Hf–Si bond (2.871(2) Å) in 5 is elongated due to the additional steric bulk of the allyl ligand compared to the chloride in 2; this distance is still shorter than that previously seen for [Hf(Cp*)(Cp){Si(SiMe3)3}(Cl)] (2.881(4) Å), due to increased steric buttressing by Cp* vs. Cp′′.38 The Hf⋯Callyl distances of 5 (range: 2.426(5)–2.555(5) Å) are relatively long in comparison with previously reported Hf allyl complexes,18e.g. [Hf(Cp′′)(C4H4Me2-2,3)(η3-C3H5)] (Hf⋯Callyl range: 2.409(6)–2.452(6) Å);36 this is in accord with the relatively weak binding of the η3-allyl in 5 indicated by solution 1H NMR studies (see above). The Th–Si distance of 4 (3.1053(13) Å) is shorter than that of [Th(Cp′)3{Si(SiMe3)3}] (3.1191(8) Å),19 as expected from the decreased combined steric requirements of one halide and two Cp′′ ligands in the former complex vs. the tris-Cp′ motif in the latter. In 1–5 all M–Si distances are longer than the sum of single bond covalent radii for the respective metal and Si (M = Zr, 2.70 Å; Hf, 2.68 Å; Th, 2.91 Å).41 The presence of a bulky hypersilanide ligand in 1–5 does not lead to significant changes in M–Cl and M⋯CpRcent distances compared to parent [M(CpR)2(Cl)2].20,22–24 Finally, the mean Hf–C distances (2.275(8) Å) and Hf–C–Cipso angles (121.4(4) and 124.9(4)°) of 6 are comparable to previously reported Hf benzyl complexes,18 with the most closely related example being [Hf(Cp)2(CH2Ph)2] (mean Hf–C = 2.293(10) Å, Hf–C–Cipso angles = 120.1(5) and 123.3(5)°).42
Quantum chemical calculations
We performed restricted DFT calculations on 1–6, in order to probe the M–Si and M–C σ-bonds in these complexes further (Table 3). As geometry optimisations effectively reproduced the metrical parameters observed by single crystal XRD we posit that these models are representative of the electronic structures of 1–6; although there are minor variations in the steric bulk provided by ancillary ligands, qualitative comparisons can be made (see ESI Tables S4–S9† for atomic coordinates of geometry-optimised structures). For 1–5 the calculated M–Si distances are longer than those determined experimentally but follow the same trend, with the M–Si bond of 2 shorter than that of 1 and the M–Si bond of 5 being longer than that of 1 and 2, but shorter than the Th–Si bond of 4. The Nalewajski–Mrozek bond indices of 1–3 (range: 0.88–0.89) are essentially identical, with the Th–Si bond of 4 (0.84) lower than these values but higher than the Hf–Si bond of 5 (0.75). The Multipole-Derived Charges (MDC-q) vary markedly for different metals. The qSi values for 2, 3 and 5 are consistently −0.16 to −0.17, with higher values for 1 (−0.28) and 4 (−0.68), and whilst the qM values are similar for the d-block complexes 1–3 and 5 (range: 0.66–0.77), the qTh value for 4 is higher (1.78), showing a higher polarity of M–Si bonds for M = Th vs. Zr and Hf. The Hf–Si bond polarity is higher for 5 than for 2 and 3, showing that the allyl ligand is a stronger donor than chloride. The qHf of 6 is 1.15 and the qC is −0.99, indicating that there is poorer orbital energy matching between Hf and C than between Hf and Si. The Hf–C bonds in 6 are therefore more polar than the Hf–Si bonds in 2, 3 and 5, but are more covalent than the corresponding Th–Si bond in 4.| Entrya | Bond length and indexb,c | Charges | Natural bond orbital (NBO) analyses | QTAIMg | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bond | Bond Length | Bond Index | q M | q Si | q C | M [%] | Si/C [%] | M s/p/d/f [%] | C/Si ns/np [%] | ρ(r) | ∇2ρ(r) | H(r) | ε(r) | |
| a All molecules geometry-optimised without symmetry constraints at the LDA VWN BP86 TZP/ZORA level. b Calculated bond of interest. c Nalewajski–Mrozek bond indices. d Multipole Derived Charges (MDC-q) on M. e MDC-q charge on Si. f MDC-q charge on C. g Quantum Theory of Atoms in Molecules (QTAIM) topological electron density [ρ(r)], Laplacian [∇2ρ(r)], electronic energy density [H(r)], and ellipticity [ε(r)] 3, −1 bond critical point data. | ||||||||||||||
| 1 | Zr–Si | 2.8787 | 0.89 | 0.66 | −0.28 | — | 32 | 68 | 17/0/83/0 | 36/64 | 0.05 | −0.03 | −0.02 | 0.05 |
| 2 | Hf–Si | 2.8483 | 0.89 | 0.69 | −0.17 | — | 29 | 71 | 27/0/73/0 | 37/63 | 0.05 | −0.04 | −0.02 | 0.04 |
| 3 | Hf–Si | 2.8652 | 0.88 | 0.71 | −0.16 | — | 28 | 72 | 29/0/71/0 | 37/63 | 0.05 | −0.03 | −0.02 | 0.05 |
| 4 | Th–Si | 3.1759 | 0.84 | 1.87 | −0.68 | — | 19 | 81 | 34/0/54/12 | 39/61 | 0.07 | 0.07 | −0.01 | 0.01 |
| 5 | Hf–Si | 2.9676 | 0.75 | 0.77 | −0.17 | — | 29 | 71 | 27/0/73/0 | 37/63 | 0.04 | 0.01 | −0.01 | 0.09 |
| 6 | Hf–C | 2.3232 | 0.65 | 1.15 | — | −0.99 | 18 | 82 | 27/073/0 | 23/77 | 0.08 | 0.08 | −0.04 | 0.04 |
Natural Bond Orbital (NBO) analyses of the M–Si and M–C bonds in 1–6 show that these are the Highest Occupied Molecular Orbitals (HOMOs) in all cases (see Fig. 3 for Kohn–Sham Molecular Orbital depictions for 1–6, and ESI Fig. S44–S49† for other selected Kohn–Sham Molecular Orbitals and NBOs; N.B. for 6 the HOMO−1 is also a Hf–C σ-bond). The Zr–Si bond in 1 has the most metal character (32%), followed by the Hf–Si bonds in 2, 3 and 5 (28–29%), with the Th–Si bond in 4 showing the least metal contribution (19%) as expected.16 The Hf–C bonds of 6 have a lower Hf contribution (18%) than any of the Hf–Si bonds, but these are all remarkably invariant to each other with respect to orbital character (27–29% 6s:71–73% 5d); the Zr–Si bond in 1 shows a greater metal nd-orbital component (17% 5s:83% 4d), whilst the Th–Si bond in 4 has the most ns, the least nd, and some 5f contribution (34% 7s:54% 6d:12% 5f). The Si orbital contributions to the M–Si bonds in 1–5 do not vary markedly (36–39% 3s:61–64% 3p), whilst there is a greater np character from C in the Hf–C bonds of 6 (2s 23%:2p 77%).
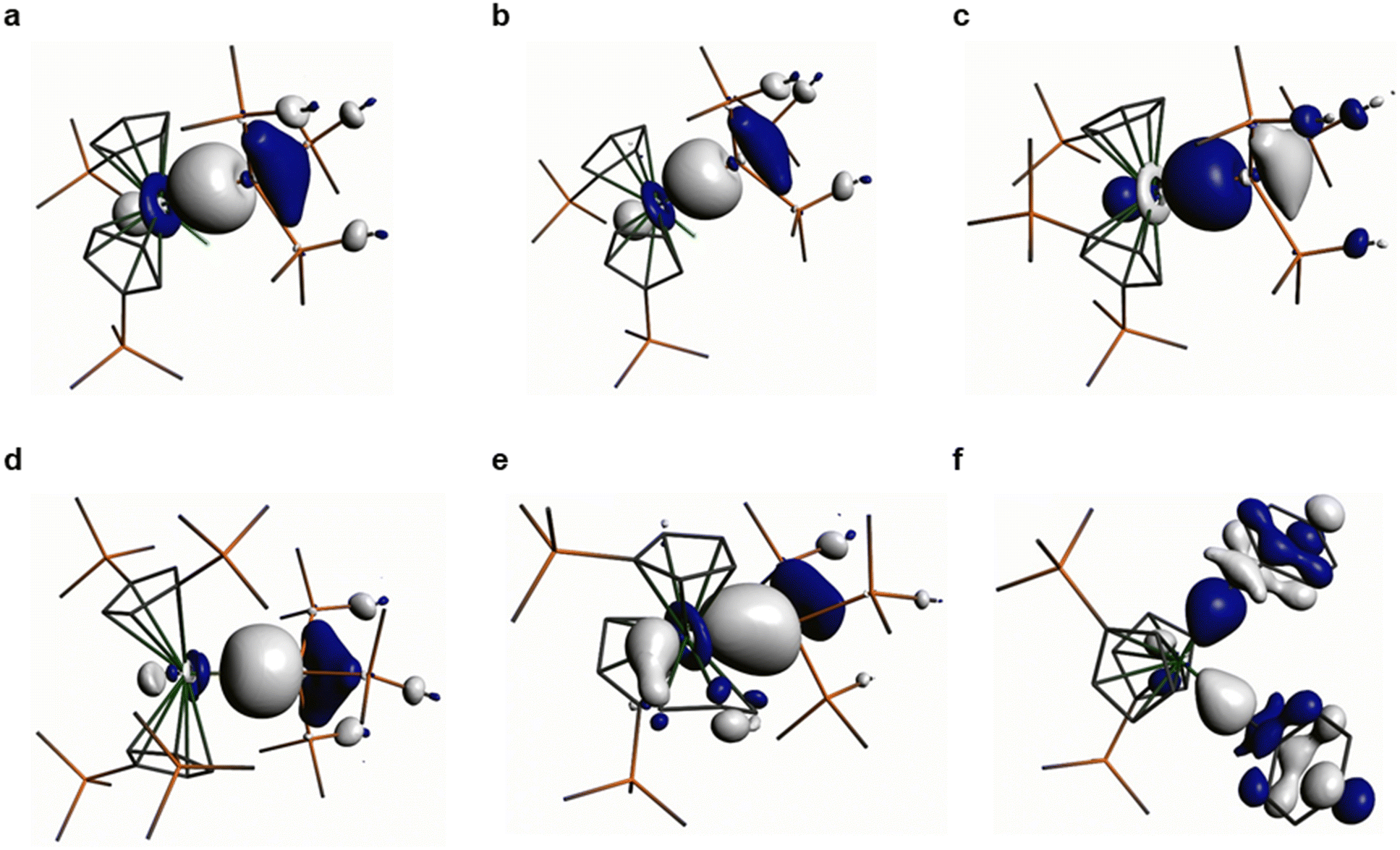 | ||
| Fig. 3 Kohn–Sham Highest Occupied Molecular Orbitals (HOMOs) representing the M–Si or M–C σ-bonds in 1–6. (a) 1 (−4.866 eV); (b) 2 (−4.919 eV); (c) 3 (−4.884 eV); (d) 4 (−4.616 eV); (e) 5 (−4.571 eV); (f) 6 (−4.800 eV). Hydrogen atoms are omitted for clarity. | ||
Quantum Theory of Atoms in Molecules (QTAIM) analyses of 1–6 reveals highly polarised (range of topological electron densities, ρ(r): 0.05–0.08) and cylindrical (range of bond ellipticities, ε(r): 0.01–0.09) M–Si and M–C σ-bonds. Electronic energy densities, H(r), are all negative as expected and for heavy atoms the Laplacian values (∇2ρ(r), range: 0.04–0.07) are insignificant, as we have commented on previously in DFT analyses of diamagnetic f-block silanide complexes.19,43,44 The QTAIM parameters extracted are consistent with the NBO data and bond indices and a visual evaluation of the HOMOs, which all indicate that the Th–Si bond of 4 is less covalent than the Zr–Si bond of 1 and the Hf–Si bonds of 2, 3 and 5. Conversely, the metal-bound silanide resonance in the 29Si NMR spectrum of 4 (−66.31 ppm) is downfield to that of 1 (−80.74 ppm) and 2 (−77.11 ppm), which would indicate lower polarity of the Th–Si bond. This discrepancy could be attributed to Heavy Atom on Light Atom (HALA) spin–orbit effects;45this is also in-line with the larger ρ(r) value of 4 compared to 1–3 perhaps reflecting the more diffuse nature of 6d orbitals, but we note the smaller H(r) value for 4vs. 1–3. A quantitative comparison of the DFT-calculated parameters for 4 with the previously reported Th(IV) hypersilanide complex [Th(Cp′)3{Si(SiMe3)3}] is not possible as these were performed at a different level of theory,19 but qualitatively the Th–Si bond of 4 appears to be more polarised and have a lower metal 6d orbital contribution. This highlights that the replacement of a CpR ligand with a chloride ligand in the Th coordination sphere can have a significant effect on the Th–Si bond, though we caution against making firm conclusions as the calculated Th–Si bond for 4 (3.1759 Å) is 2.3% longer than the distance determined by single crystal XRD (3.1053(13) Å), whereas for [Th(Cp′)3{Si(SiMe3)3}] there was only a 1.6% discrepancy between theory and experiment (3.071 Å calculated vs. 3.1191(8) Å measured).19
Conclusion
We have used salt metathesis protocols to synthesise a series of M(IV) hypersilanide complexes for M = Hf and Th, supported by two substituted cyclopentadienyls and one chloride or allyl ancillary ligand, in order to compare their M–Si bonds with each other and previously reported similar Th(IV)19 and Zr(IV)21 hypersilanide complexes. Attempts to prepare neutral Th(III) or cationic M(IV) (M = Hf, Th) hypersilanide analogues of these complexes by respective reduction or anion abstraction protocols were unsuccessful under the reaction conditions investigated. However, this work has provided the second structurally authenticated example of a Th–Si bond, and has allowed an extended solid-state structural and 29Si{1H} NMR spectroscopic comparison of group 4 metal and Th(IV) silanide complexes. We have also shown the importance of the steric bulk of supporting silyl-substituted cyclopentadienyl ligands in dictating the reaction outcome when installing a hypersilanide ligand, due to differences in M(IV) ion size, Lewis acidity and covalency between these metals. DFT calculations of structurally similar M(IV) hypersilanide complexes for M = Zr, Hf and Th confirmed as expected that the highly polarised Th–Si bonds are less covalent than Zr–Si and Hf–Si bonds, which exhibit similar covalency to each other.Experimental
General methods
All manipulations were performed under strict anaerobic conditions using argon as the inert gas, by using a combination of Schlenk line and glove box techniques. HPLC grade pentane, toluene and THF were passed through alumina columns in a solvent purification system. Benzene, 1,4-dioxane and DME were dried by refluxing over potassium for 24 h followed by distillation. All solvents were degassed under dynamic vacuum and were stored over potassium mirrors (benzene pentane, toluene) or 4 Å molecular sieves (1,4-dioxane, DME, THF) before use. C6D6 was dried by refluxing over potassium for 2 days in a J. Young tap-appended ampoule before trap-to-trap distillation and three freeze–pump–thaw cycles to degas before use. [Zr(Cp′)2(Cl)2] (M = Zr, Hf),22 [M(Cp′′)2(Cl)2] (M = Hf,20 Th24), K{Si(SiMe3)3},25 KCH2Ph,28 [CPh3][Al{OC(CF3)3}4],29 [NEt3H][BPh4]31 and KC832 were prepared by previously reported procedures, whilst complex 1 was prepared by adapting the literature method.21 Details of the revised synthesis and data obtained for 1 are provided below, together with synthetic procedures and experimental data obtained for 2–6. All other starting materials were purchased from commercial sources and were used as received. Caution: Thorium from natural sources is a weak α-emitter; it is recommended that compounds of this element are only manipulated by trained personnel in designated radiochemical laboratories, and that α-counting equipment is routinely available for monitoring the radiochemical hazard.
NMR spectra were recorded on samples in J. Young tap-appended NMR tubes using a Bruker Avance III 400 spectrometer operating at 400.07 (1H), 100.60 (13C) or 79.48 (29Si) MHz. 1H and 13C{1H} NMR spectra were internally referenced to the residual solvent resonance of C6D6, whilst 29Si{1H} NMR spectra were externally referenced to tetramethylsilane; chemical shifts are provided in ppm. NMR data was analysed using the MestReNova software suite.46 ATR-IR spectra of microcrystalline powders were collected on a Bruker Alpha 2 spectrometer. Elemental analyses were carried out by Mr Martin Jennings and Mrs Anne Davies using a Flash 2000 elemental analyser at the Microanalytical service, Department of Chemistry, the University of Manchester. Results generally showed reasonable agreement with expected compositions, though low carbon compositions were reproducibly measured for 4. This is a common occurrence for early metal complexes with high silicon content, including for f-block Cp′′ complexes, where it has been attributed to the formation of silicon carbide, leading to incomplete combustion;47 the observation of low carbon values in elemental analyses of [Th(Cp′)3{Si(SiMe3)3}] was previously ascribed to the same phenomenon.19
Crystallographic methods
Crystals of 1–6 were dispersed in fomblin and single crystals were selected and transferred to MicroMounts™ on a goniometer head under a cryostream for exposure to X-rays. Crystallographic data for [Hf(Cp′)2(Cl)2], 2, 5 and 6 were obtained using an Oxford Diffraction Xcalibur diffractometer equipped with an Agilent Atlas CCD detector with graphite-monochromated Mo Kα (λ = 0.71073 Å) radiation, whilst data for 3·0.5C5H12 and 4 were collected using a Rigaku FR-X diffractometer equipped with a HyPix 6000HE photon counting pixel array detector with mirror-monochromated Cu Kα (1.54184 Å) radiation. Intensities were integrated from data recorded on 0.5° (5) or 1° ([Hf(Cp′)2(Cl)2], 2, 3·0.5C5H12, 4 and 6) frames by ω rotation. Cell parameters were refined from the observed positions of all strong reflections in each data set. A Gaussian grid face-indexed with a beam profile was applied for all structures.48 The SHELXT program was used to solve all structures.49 Datasets were refined by full-matrix least-squares on all unique F2 values,49 with anisotropic displacement parameters for all non-hydrogen atoms, and with constrained riding hydrogen geometries; Uiso(H) was set at 1.2 (1.5 for methyl groups) times Ueq of the parent atom. The largest features in final difference syntheses were close to heavy atoms and were of no chemical significance. The CrysAlisPro program48 was used for control and integration, and the SHELX suite49,50 was employed through the OLEX2 program51 for structure solution and refinement. The programs ORTEP-352 and POV-Ray53 were used in combination to generate depictions of 1–6.Computational methods
The Amsterdam Density Functional (ADF) program (2017 version) was used to perform restricted DFT calculations using standard convergence criteria.54,55 Atomic coordinates determined from single crystal XRD were used as the start points for geometry optimisations, which were performed with no constraints applied. Slater-type orbital TZP polarisation all-electron basis sets from the Dirac and zeroth order regular approximation (ZORA) and triple-zeta with polarisation (TZP) database of the Amsterdam Density Functional (ADF) package were used for DFT geometry optimisations. Scalar relativistic approaches (spin–orbit neglected) were used within the ZORA Hamiltonian to include relativistic effects.56–58 The local density approximation (LDA) with a correlation potential was used in all calculations.59 Generalised gradient approximation corrections were performed using the functionals of Becke and Perdew.60,61 NBO analysis was carried out using the NBO v6.0 program.62 QTAIM analysis63,64 was performed within the ADF suite. The Kohn–Sham MOs and NBOs were visualised using ADFView.Synthetic procedures
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank the University of Manchester for a PhD studentship for B.L.L.R. (Nuclear Endowment) and access to the Computational Shared Facility, and the European Research Council (CoG-816268 and CoG-612724) and the UK EPSRC (EP/M027015/1 and EP/P001386/1) for funding. S. T. L. thanks the Alexander von Humboldt Foundation for a Friedrich Wilhelm Bessel Research Award. We thank Dr George Whitehead and Dr Iñigo Vitorica-Yrezabal for assistance with collection of single crystal XRD data.References
- Organosilicon Compounds: Theory and Experiment (Synthesis), Ed. V. Y. Lee, Academic Press, Elsevier Inc., Amsterdam, 2017 Search PubMed.
- R. Waterman, P. G. Hayes and T. D. Tilley, Acc. Chem. Res., 2007, 40, 712–719 CrossRef CAS PubMed.
- W. Gao, X. Zhang, X. Xie and S. Ding, Chem. Commun., 2020, 56, 2012–2015 RSC.
- A. Walczak, H. Stachowiak, G. Kurpik, J. Kaźmierczak, G. Hreczycho and A. R. Stefankiewicz, J. Catal., 2019, 373, 139–146 CrossRef CAS.
- V. Srinivas, Y. Nakajima, K. Sato and S. Shimada, Org. Lett., 2018, 20, 12–15 CrossRef CAS PubMed.
- X. Chen and C. Liang, Catal. Sci. Technol., 2019, 9, 4785–4820 RSC.
- B. L. L. Réant, S. T. Liddle and D. P. Mills, Chem. Sci., 2020, 11, 10871–10886 RSC.
- I. Castillo and T. D. Tilley, Organometallics, 2001, 20, 5598–5605 CrossRef CAS.
- A. D. Sadow and T. D. Tilley, J. Am. Chem. Soc., 2005, 127, 643–656 CrossRef CAS PubMed.
- F. Pan, J. Zhang, H. L. Chen, Y. H. Su, C. L. Kuo, Y. H. Su, S. H. Chen, K. J. Lin, P. H. Hsieh and W. S. Hwang, Materials, 2016, 9, 417 CrossRef PubMed.
- J. T. White, A. T. Nelson, J. T. Dunwoody, D. D. Byler, D. J. Safarik and K. J. Mcclellan, J. Nucl. Mater., 2015, 464, 275–280 CrossRef CAS.
- T. L. Wilson, E. E. Moore, D. Adorno Lopes, V. Kocevski, E. Sooby Wood, J. T. White, A. T. Nelson, J. W. McMurray, S. C. Middleburg, P. Xu and T. M. Besmann, Adv. Appl. Ceram., 2018, 117, S76–S81 CrossRef CAS.
- U. E. Humphrey and M. U. Khandaker, Renewable Sustainable Energy Rev., 2018, 97, 259–275 CrossRef CAS.
- S. He and J. Cai, Ann. Nucl. Energy, 2020, 140, 107303 CrossRef CAS.
- The Rare Earth Elements: Fundamentals and Applications, Ed. D. A. Atwood, John Wiley & Sons, Ltd, Chichester, 2012 Search PubMed.
- The Chemistry of the Actinide and Transactinide Elements, Ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Dordrecht, 3rd edn, 2006 Search PubMed.
- C. Aitken, J. F. Harrod and E. Samuel, J. Organomet. Chem., 1985, 279, C11–C13 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed . Search date 13th March 2023.
- B. L. L. Réant, V. E. J. Berryman, J. A. Seed, A. R. Basford, A. Formanuik, A. J. Wooles, N. Kaltsoyannis, S. T. Liddle and D. P. Mills, Chem. Commun., 2020, 56, 12620–12623 RSC.
- G. K. Gransbury, B. L. L. Réant, A. J. Wooles, J. Emerson-King, N. F. Chilton, S. T. Liddle and D. P. Mills, Chem. Sci., 2023, 14, 621–634 RSC.
- T. Imori, R. H. Heyn, T. D. Tilley and A. L. Rheingold, J. Organomet. Chem., 1995, 493, 83–89 CrossRef CAS.
- M. F. Lappert, C. J. Pickett, P. I. Riley and P. I. W. Yarrow, J. Chem. Soc., Dalton Trans., 1981, 805–813 RSC.
- A. Antiñolo, M. F. Lappert, A. Singh, D. J. Winterborn, L. M. Engelhardt, C. L. Raston, A. H. White, A. J. Carty and N. J. Taylor, J. Chem. Soc., Dalton Trans., 1987, 1463–1472 RSC.
- P. C. Blake, M. F. Lappert, R. G. Taylor, J. L. Atwood, W. E. Hunter and H. Zhang, J. Chem. Soc., Dalton Trans., 1995, 3335–3341 RSC.
- C. Marschner, Eur. J. Inorg. Chem., 1998, 221–226 CrossRef CAS.
- C. H. Winter, X.-X. Zhou, A. A. Dobbs and M. J. Heeg, Organometallics, 1991, 10, 210–214 CrossRef CAS.
- C. J. Windorff, M. R. MacDonald, J. W. Ziller and W. J. Evans, Z. Anorg. Allg. Chem., 2017, 643, 2011–2018 CrossRef CAS.
- M. Schlosser and J. Hartmann, Angew. Chem., Int. Ed. Engl., 1973, 12, 508–509 CrossRef.
- I. Krossing, H. Brands, R. Feuerhake and S. Koenig, J. Fluor. Chem., 2001, 112, 83–90 CrossRef CAS.
- J. B. Lambert, S. Zhang and S. M. Ciro, Organometallics, 1994, 13, 2430–2443 CrossRef CAS.
- J. K. Peterson, M. R. MacDonald, J. W. Ziller and W. J. Evans, Organometallics, 2013, 32, 2625–2631 CrossRef CAS.
- D. E. Bergbreiter and J. M. Killough, J. Am. Chem. Soc., 1978, 100, 2126–2134 CrossRef CAS.
- P. C. Blake, M. F. Lappert, J. L. Atwood and H. Zhang, J. Chem. Soc., Chem. Commun., 1986, 1148–1149 RSC.
- R. R. Langeslay, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, Chem. Sci., 2015, 6, 517–521 RSC.
- G. J. Pindado, M. Thornton-Pett and M. Bochmann, J. Chem. Soc., Dalton Trans., 1997, 3115–3128 RSC.
- A. Pastor, A. F. Kiely, L. M. Henling, M. W. Day and J. E. Bercaw, J. Organomet. Chem., 1997, 528, 65–75 CrossRef CAS.
- F. H. Elsner, T. D. Tilley, A. L. Rheingold and S. J. Geib, J. Organomet. Chem., 1988, 358, 169–183 CrossRef CAS.
- H.-G. Woo, R. H. Heyn and T. D. Tilley, J. Am. Chem. Soc., 1992, 114, 5698–5707 CrossRef CAS.
- B. K. Campion, J. Falk and T. D. Tilley, J. Am. Chem. Soc., 1987, 109, 2049–2056 CrossRef CAS.
- C. Kayser, D. Frank, J. Baumgartner and C. Marschner, J. Organomet. Chem., 2003, 667, 149–153 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- K. C. Jantunen, B. L. Scott and J. L. Kiplinger, J. Alloys Compd., 2007, 444–445, 363–368 CrossRef CAS.
- B. L. L. Réant, V. E. J. Berryman, A. R. Basford, L. E. Nodaraki, A. J. Wooles, F. Tuna, N. Kaltsoyannis, D. P. Mills and S. T. Liddle, J. Am. Chem. Soc., 2021, 143, 9813–9824 CrossRef PubMed.
- B. L. L. Réant, A. J. Wooles, S. T. Liddle and D. P. Mills, Inorg. Chem., 2023, 62, 137–146 CrossRef PubMed.
- M. Kaupp, O. L. Malkina, V. G. Malkin and P. Pyykkö, Chem. – Eur. J., 1998, 4, 118–126 CrossRef CAS.
- MestReNova v.14.3.1, Mestrelab Research, S. L. Santiago de Compostela, Spain, 2022; https://mestrelab.com/software/mnova/ Search PubMed.
- P. B. Hitchcock, M. F. Lappert, L. Maron and A. V. Protchenko, Angew. Chem., Int. Ed., 2008, 47, 1488–1491 CrossRef CAS PubMed.
- CrysAlis PRO, Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- Persistence of Vision Raytracer Pty. Ltd, Persistence of Vision Raytracer, v.3.7, Williamstown, VIC, Australia, 2013. Retrieved from https://www.povray.org/download/ Search PubMed.
- C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, A. C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783 CrossRef CAS.
- E. van Lenthe, A. E. Ehlers and E. J. Baerends, J. Chem. Phys., 1999, 110, 8943 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO v.6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, USA, 2013; https://nbo6.chem.wisc.edu/ Search PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, New York, 1990 Search PubMed.
- R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and ATR-IR spectra, crystallographic data, and DFT calculations. CCDC 2252356–2252361. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00987d |
| This journal is © The Royal Society of Chemistry 2023 |