
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn autonomous remotely operated gas chromatograph for chemically resolved monitoring of atmospheric volatile organic compounds†
Deborah F.
McGlynn
 ,
Namrata Shanmukh
Panji
,
Graham
Frazier
,
Chenyang
Bi
and
Gabriel
Isaacman-VanWertz
*
,
Namrata Shanmukh
Panji
,
Graham
Frazier
,
Chenyang
Bi
and
Gabriel
Isaacman-VanWertz
*
Department of Civil and Environmental Engineering, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: ivw@vt.edu; Tel: +1 540 231 0011
First published on 3rd January 2023
Abstract
Volatile organic compounds (VOCs) range in their reaction rates with atmospheric oxidants by several orders of magnitude. Therefore, studying their atmospheric concentrations across seasons and years requires isomer resolution to fully understand their impact on oxidant budgets and secondary organic aerosol formation. An automated gas chromatograph/flame ionization detector (GC-FID) was developed for hourly sampling and analysis of C5–C15 hydrocarbons at remote locations. Samples are collected on an air-cooled multibed adsorbent trap for preconcentration of hydrocarbons in the target volatility range, specifically designed to minimize dead volume and enable rapid heating and sample flushing. Instrument control uses custom electronics designed to allow flexible autonomous operation at moderate cost, with automated data transfer and processing. The instrument has been deployed for over two years with samples collected mid-canopy from the Virginia Forest Laboratory located in the Pace research forest in central Virginia. We present here the design of the instrument itself, control electronics, and calibration and data analysis approaches to facilitate the development of similar systems by the atmospheric chemistry community. Detection limits of all species are in the range of a few to tens of ppt and the instrument is suitable for detection of a wide range of biogenic, lightly oxygenated, and anthropogenic (predominantly hydrocarbon) compounds. Data from calibrations are examined to provide understanding of instrument stability and quantify uncertainty. In this work, we present challenges and recommendations for future deployments, as well as suggested adaptions to decrease required maintenance and increase instrument up-time. The presented design is particularly suitable for long-term and remote deployment campaigns where access, maintenance, and transport of materials are difficult.
Environmental significanceAtmospheric volatile organic compounds (VOCs) are emitted from both natural and anthropogenic sources and play a major role in the formation of air pollutants like ozone and organic aerosol. However, the rate of their chemical degradation in the atmosphere vary by orders of magnitude and long-term trends in their composition and emission are uncertain, particularly in a changing climate. Long term monitoring of VOCs is necessary to understand and predict changes in atmospheric composition, but few long-term monitoring sites exist. This work outlines the development and operation of an automated gas-chromatography flame ionization detector to measure VOCs with high chemical and temporal detail, seeking to facilitate expanded availability of these critical data. By describing and making available automated data processing and calibration procedures, this manuscript further decreases the time required to process the data from such an automated system. |
1 Introduction
VOCs are emitted from both anthropogenic and natural sources, though natural sources contribute ∼90% to VOC emissions.1 VOCs include a wide range of species, with rate constants and mixing ratios varying by several orders of magnitude, and highly variable impacts on the formation of secondary organic aerosol. Furthermore, in the case of biogenic emissions, concentrations and the chemical characteristics of species emitted is dependent on vegetation type, seasonality, and a range of external stimuli.1–3 These factors make understanding their role in oxidant and aerosol budget difficult. In particular, understanding the impact of changing climate on various timescales requires monitoring of their emissions and concentrations on both short and long timescales. Furthermore, due to the widely variable physical and chemical properties of these compounds, it is important to understand these temporal and spatial patterns not only for compound classes as a whole, but for individual compounds. Unfortunately, limitations in instrument capabilities make long-term, chemically resolved datasets of VOCs relatively scarce.A wide range of methods for detecting and monitoring VOCs are available. They vary in their ability to detect speciated VOCs and to run autonomously for extended periods of time. Additionally, they vary in their ability to detect species at low concentration, high volatility, and/or low volatility. For example, offline methods such as sampling into canisters or adsorbent tubes for later analysis on gas chromatography/mass spectrometry systems have been used for many years. This type of sampling allows for speciated analysis, but requires substantial manual labor and on-site access.4 Therefore, the temporal resolution of the resulting data from these types of offline measurements is at the discretion of the researcher, but typically on the order of daily to weekly for long-term sampling.5,6 There is also a long history of sampling campaigns using offline GC-FID for VOC detection.7–11 While these campaigns significantly increased our understanding of VOCs, they did not have the benefit of technological advances utilizing automation and advanced data processing techniques.
There are instruments available to measure VOCs in real-time (time resolution of minutes, seconds, or faster). For example, proton transfer reaction mass spectrometry (PTR-MS) offers high temporal resolution and has seen wide use in the field of atmospheric chemistry for fast measurements of a wide range of VOCs.4,12–16 Like PTR-MS, other forms of direct-sampling mass spectrometric instruments have recently seen substantial increase in use in field measurements of gas-phase organic compounds.17–19 These instruments have significantly advanced understanding of VOC composition by providing time resolution sufficiently high to enable emission and deposition flux measurements through eddy-covariance techniques (which require multiple measurements per second). However, instruments such as these that rely on mass spectrometry for chemical resolution and cannot provide resolution of isomers with the same chemical formula, impacting their utility for understanding impacts on oxidant and aerosol budgets. Furthermore, instruments relying on mass spectrometry often require significant maintenance and tuning, which complicates their use for reliable deployment longer than a few weeks to a few months.4,7,13
In contrast, gas chromatographic tools provide high chemical resolution by enabling resolution of individual compounds, though at the cost of lower temporal resolution (typically ∼hourly). These tools may be coupled to a mass spectrometer for high chemical resolution, though this introduces the same difficulty in long-term deployments for any mass spectrometric tool. A GC may also be coupled to a flame ionization detector (FID) or other single-channel detector that provides no additional chemical resolution.20 While the latter decreases the capabilities of chemical resolution, they offer a stable, predictable response over long periods of time with relatively little maintenance.21,22 These features make them an attractive detector for automated, remote, and online measurement campaigns.
Due to their higher chemical resolution, many modern field deployable gas chromatographic systems rely on mass spectrometric detection,7,9,23–26 but such an approach frequently requires higher degrees of maintenance and operator intervention than is ideal for long-term, remote measurement sites. Many modern reported systems focus on high volatility hydrocarbons (C2–C10) for weeks to months at a time without cryogen trapping and with reported detection limits between 5 and 150 ppt.11,24,27 For the most part, these systems rely on similar physical features such as varied adsorbent trap composition to capture compounds that vary in volatility and Nafion dryers to remove condensation. The reported detection limit of these systems ranged from ∼1 ppt to ∼0.4 ppb.11 Additional works detail the development of a system to increase the range of detected hydrocarbon species to C2–C12.23,28 Both systems had two separate preconcentration traps to capture high volatility and intermediate volatility hydrocarbons.23,28 Despite the difficulty in long-term deployment of a mass spectrometry based system, there has been some success in achieving multiyear speciated BVOC measurements. For example, Hellén et al.29 deployed a GC-MS for a few months at a time in three years (2011, 2015, and 2016), including an 8 month period in 2016. This highly detailed data provided significant insight into the concentrations and atmospheric impacts of BVOCs. Interestingly, Hellén et al. observed β-caryophyllene to be an important contributor to ozone reactivity at their research site in the boreal forest, in contrast to measurements by the instrument described here, which did not observe significant concentrations of the compound.36 Though current measurement campaigns such as these are lacking, they can significantly improve model outcomes and our understanding of the implications of climate change on BVOC emissions.
Additional work in the area of automated GC for BVOC detection include a portable GC-PID (photoionization detection) for detection of isoprene and a handful of selected VOCs30 and aircraft deployable GC-MS for fast detection of halogens,31 and halocarbons, hydrocarbons, and a handful of oxygenated VOCs, amongst others.32 These advances have sought to make chromatographic systems more field-deployable for long-term measurements.
In this work we present an automated, online, GC-FID system capable of detecting hydrocarbons and lightly oxygenated hydrocarbons between C5–C15 designed in particular to capture major biogenic VOC classes including isoprene, monoterpenes, and sesquiterpenes, and other anthropogenic hydrocarbon compounds within this target range. This work relies on a preconcentrator trap previously described in the literature33 that efficiently collects high volatility species such as pentane and isoprene as well as intermediate volatility compounds. We present details of the design of the instrument and trap configuration, the custom electronics, and the calibration and data processing approach, with the goal of expanding the availability of long-term chemically resolved VOC measurements by facilitating the collection of such measurements by other researchers. We also detail automated data processing approaches implemented in the freely available TERN software program in IGOR34 to decrease the time required to integrate the amount of data produced from an automated GC.
2 Methods
2.1 Instrument overview
An overview of the instrument is provided in Fig. 1. Air is pulled by a pump through a multi-bed adsorbent trap (described in detail below) at the maximum flow rate enabled by the trap, roughly 150 sccm. During sampling, the GC column is directly supplied helium through a 6-port valve within a valve oven which is held at 150 °C. For analysis, the 6-port valve position is switched to connect the trap to the column. The trap is purged for 30 seconds with helium carrier gas and then desorbed by heating to 165 °C (±10 °C) in a helium flow for a period of 2 minutes. Previous work by Wernis et al. (2021) found improved peak shape of the earliest eluting peaks (i.e., pentane, isoprene) when heated has rapidly as possible during desorption. Heating is therefore uncontrolled (i.e., not managed by a PID controller), prioritizing rapid heating to or beyond the desired setpoint as opposed to stable maintenance at a given setpoint. In spite of this, the trap heating profile is very consistent between runs, with a nearly identical rapid heating period in every run, followed by some oscillation above the setpoint of 165 °C (Fig. 2).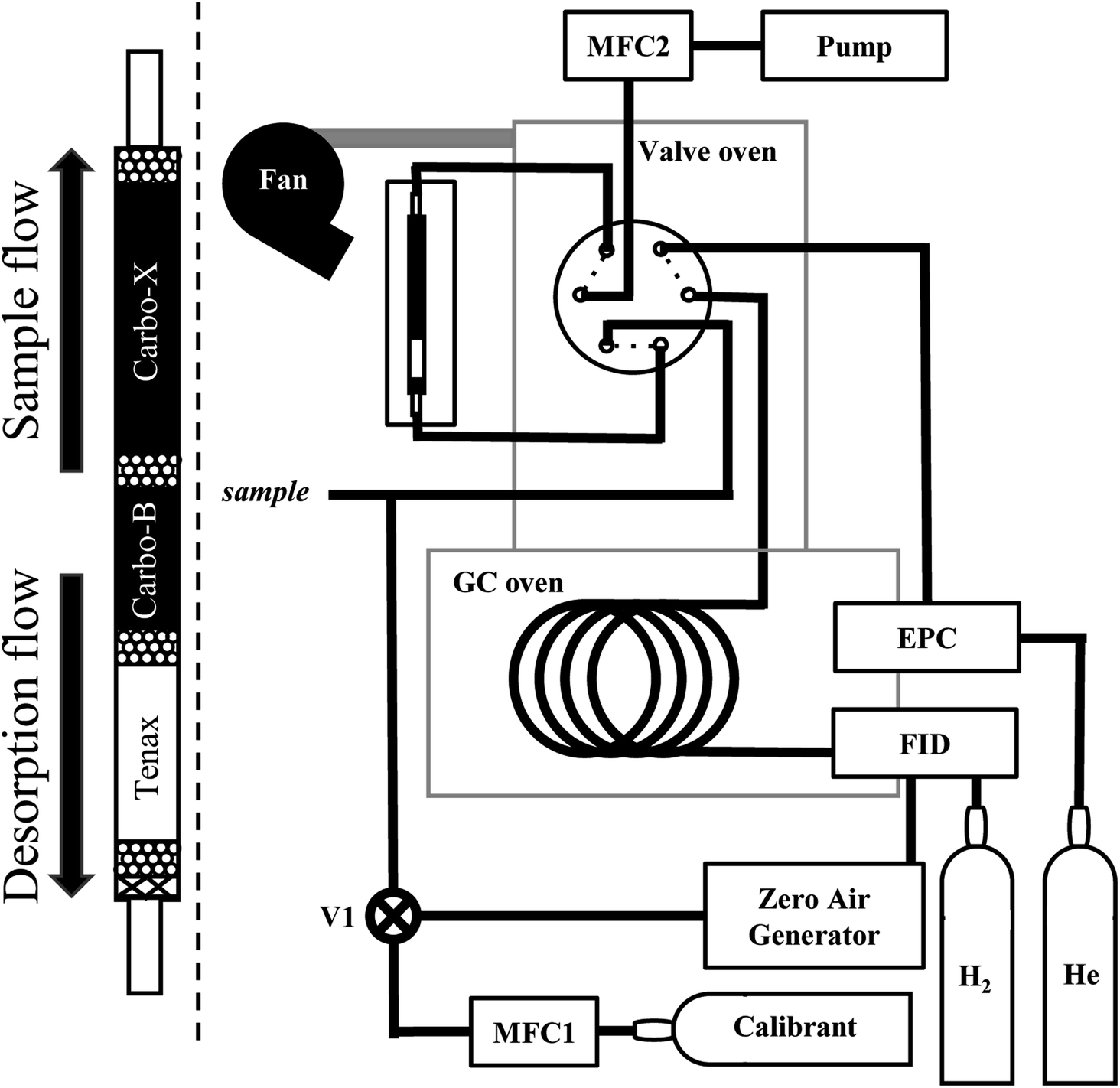 | ||
| Fig. 1 Instrument overview. Instrument overview depicting the gas chromatograph (GC) connections and plumbing. The trap is encased within a metal housing containing cartridge heaters and cooled by a fan; a zoomed in representation of the trap, which is connected to the 6-port valve, is shown left of the dashed line. The 6-port valve is located within a valve oven situated on-top of the GC oven, connected to the column through the oven walls. The flame ionization detector (FID) and column are supplied by gases using electronic pressure controllers (EPC) controlled by the GC. Sample and calibrant flows are controlled by mass flow controllers (MFC). A valve (V1) is used for calibrants and zero air. | ||
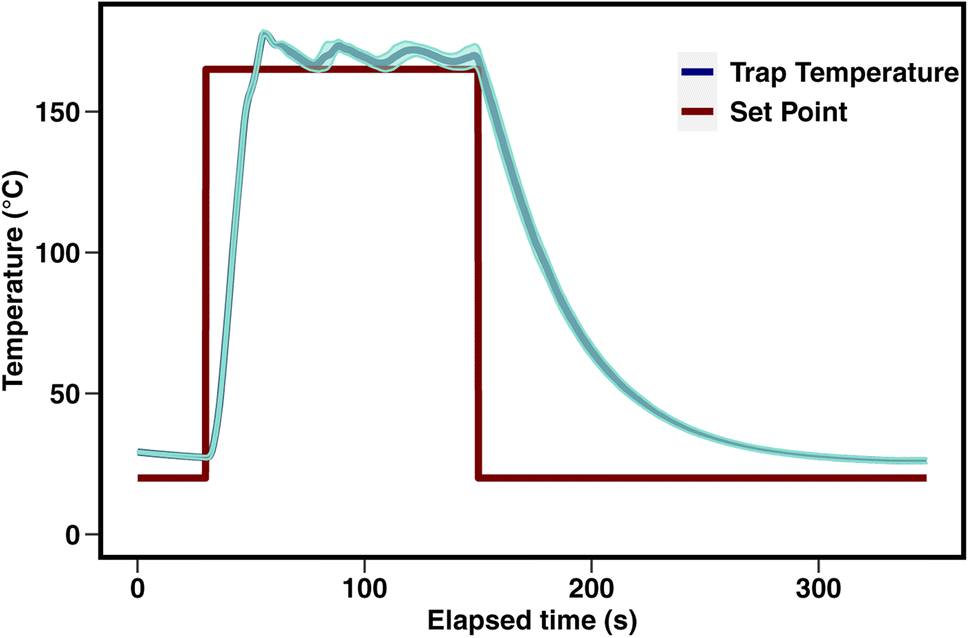 | ||
| Fig. 2 Trap heating and cooling profile for sample desorption. The red line depicts the set point, and the cyan line depicts the mean and standard deviation for all runs over a representative three week period in August 2021. | ||
Following the heating period, the trap is fan-cooled for a period of 3 minutes. The entire desorption, heat, and cool time of the trap takes 5.5 minutes, after which the valve is actuated back to the sampling position to isolate the GC operation and begin the collection of a new sample. The GC oven temperature is increased from 35 °C to 250 °C at 6 °C min−1 with a 10 minute hold at the maximum temperature, making the sample analysis 46.33 minutes, followed by an oven cooling period of about 9 minutes prior to the start of the next analysis. A sample is collected for the duration of the GC analysis and cooling, providing ∼54.5 minutes of sampling (∼8 liters of sampled air) when operating with hourly time resolution. Analytes are separated using a mid-polarity GC column (Rtx-624, 60 m × 0.32 mm × 1.8 μm, Restek Inc.) and detected by a flame ionization detector (FID).
The instrument is built on a commercially available GC-FID, in this case an Agilent 7820A or Agilent 7890B outfitted with an FID (both have been tested in this work). GC flow is controlled by an electronic pressure controller (EPC) on-board the GC. The heated valve oven and 6-port valve may be integrated into and controlled by the GC or may be an independently controlled oven and valve (e.g., commercially available through Vici Valco); both configurations have been tested in this work. Positioning of the valve box directly on top of the GC oven provides a contiguous heated zone for the analyte to pass into the oven with no connections or cold spots (Fig. 1). The carrier and make up gas used for this setup is helium while hydrogen fuels the FID, both gases are supplied by AirGas (5.0 grade). Air for the FID is generated onsite at 125 psig with a zero-air generator (Parker 75-83NA).
The instrument as configured for this work includes a single trap, column, and detector. However, inclusion of a second channel operating in parallel would require minimal modification by adding an additional trap and valve, and outfitting the GC with a second column and FID. Though not yet implemented here, the instrument electronics, described in detail below, are designed to enable expansion to a two-channel system with minimal modification.
2.2 Adsorbent trap and desorption program
The trap is designed to capture species with a range in volatilities by using multiple layered beds of adsorbents with different adsorptive strength following previously demonstrated designs.33,35 The trap consists of, in order of upstream to downstream flow, 10 mg of Tenax® TA (60–80 mesh), 20 mg of Carbopack™ B (60–80 mesh), and at least 50 mg of Carbopack™ X (60–80 mesh) with ∼10 mg glass beads (50–70 sieve) on either side of each adsorbent (Fig. 1, left). Adsorbents are held in place by packing the outlet of the trap with glass beads (50–70 sieve) and then glass wool.Because the system is not a two-step purge-and-trap system and has no focusing between the sample trap and the head of the GC column, any compound not strongly retained by the column is immediately mobile once desorbed. Desorption of the trap consequently affects peak shape of high volatility compounds, and the rapid heating and flushing of the trap is a critical instrument design. To minimize retention in the trap during desorption, the trap (manufactured by Aerosol Dynamics Inc.) consists of a 1/8′′ (3.2 mm) outer diameter, thin walled (0.13 mm) passivated metal tube, partially flattened to a thickness of roughly 1/16′′ (1.6 mm) to minimize swept volume.33 The trap is heated using two 1/8′′ diameter, 100 watt cartridge heaters sandwiched within a low thermal mass manifold composed of two aluminum blocks (3 mm × 20 mm × 76 mm, Aerosol Dynamics Inc.). The metal blocks are grooved in the shape of the trap to hold it in place. Due to the low thermal mass of the manifold and isolation from other instrument components, heating of the manifold is achieved in under 30 seconds. The manifold also hosts a 1/32′′ K-type thermocouple which is used to monitor the temperature and control the heating of the trap.
2.3 Electronic design
A moderate cost data acquisition board (LabJack U6 Pro) is the central interface between the hardware and software and is housed within a standalone electronics enclosure (Fig. 3). An 8-channel relay board powered by a 24 VDC power supply controls six switched 24 VDC channels, and two switched 110 VAC channels; all channels are independently fused, accessed by screw terminals on the face of the enclosure, and controlled by digital out (DO) channels. The fan and valve shown in Fig. 1 are controlled by two of the 24 VDC channels. The cartridge heaters are controlled by one of the 110 VAC channels, in which a 24 VDC relay is used to switch two independent solid-state relays (SSRs, Sensata-Crydom) to provide better heat dissipation.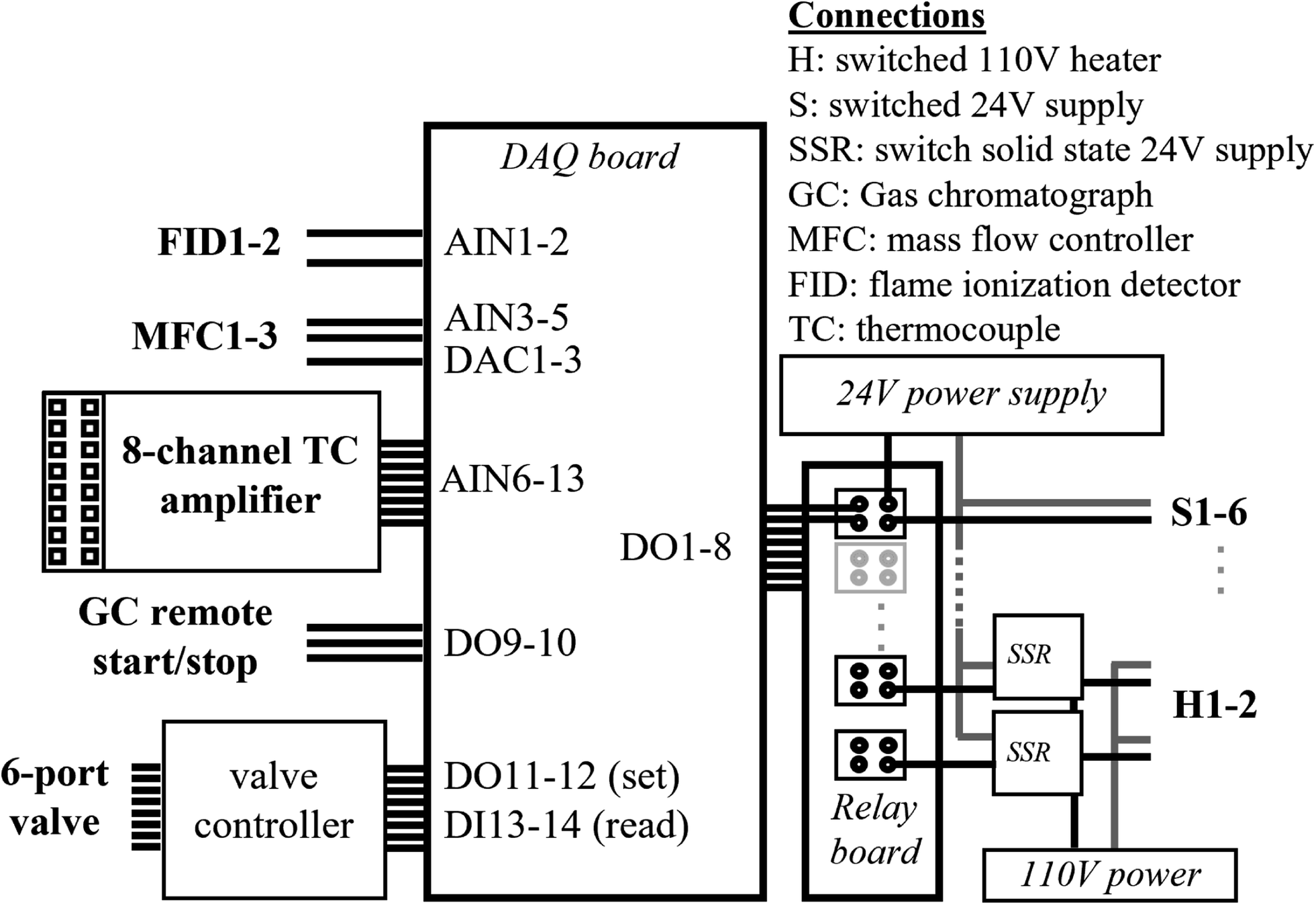 | ||
| Fig. 3 Diagram of the control electronics. Connections between the data acquisition board and controlled instrument components are shown. A 24 V relay board controls 6 directly switched circuits, including those used for the fan and valves, and 2 switched solid state relays (SSR) that control 110 V power for the heats. Additional connections include the 6-port valve, thermocouple (TC) hub, mass flow controllers (MFC), FID signals, and GC start and stop signals. | ||
Additional connections provide an interface with other instrument components. Starting and stopping of the GC-FID is enabled through a DB9 remote start/stop controlled by two digital out channels, which are pulled low to drive a change in state. Additional digital out (DO) and in (DI) channels interface with an independent controller to set and read back the state of the 6-port valve within a heated enclosure (Vici Valco HVE2), situated on top of the GC. Temperatures of the trap, valve oven, GC oven, and two points along the inlet are monitored by thermocouples connected to a commercially available thermocouple hub (HGSI TCA-MS-K-8-A4). The hub is mounted onto the enclosure in order to amplify signals from 8 thermocouples (5 are in use for the current single channel system) and convert to standard (i.e., 0–5 V) analog input (AIN). Thermocouple connections could be made directly to the data acquisition board, but the approach used here simplifies the system by avoiding the need for independent cold junction compensation and the need to separately mount thermocouple connectors on the face of the enclosure. Two mass flow controllers (MFC, model numbers MCW-500SCCM, and MC-100SCCM) are each controlled by digital-to-analog converters (DAC), with flow readback using an AIN channel. An additional AIN is available for flow measurement using a third MFC, though no DAC is available to set this flow on the data acquisition board used in this setup. Signal from up to two FIDs is read by analog inputs configured as differential-ended signals (i.e., two AI channels for detector). Many of these components are not used in the present configuration, and are instead reserved to enable expansion to a two-channel system with no additional changes; these reserved components include: one of the FID readback channels (for a second detector), the MFC readback channel (to measure flow on a second trap), up to four of the 24 VDC switched channels (for a second fan and additional valves), and one of the 110 VAC switched channels (for heating a second trap). All electronics are controlled through custom LabVIEW code (National Instruments), which sets digital outputs on a set schedule and reads back analog inputs to store as datafiles.
2.4 Automated data analysis
A key advance in the autonomous deployment is automated data processing, as the reduction and analysis of chromatographic data is time consuming and labor intensive. Automated data processing is achieved here through several software advances. First, data is automatically uploaded to an online repository using a custom, standalone executable that is configurable and compiled from open-source Python code. The code and executable are available through GitHub (https://github.com/gabrielivw/DOsync). This program monitors a folder on the instrument computer and syncs any new data files to a repository, while the same program running on an analytical computer syncs the repository to a local folder. This code is designed to interface with storage space in Amazon Web Services through the data repository DigitalOcean (https://www.digitalocean.com/), and is referred to for simplicity as “DOsync.” file types, folder locations, and digital repository locations are specified by a configuration file.Files added to the local analytical computer are processed using an updated version of TERN, a freely-available and customizable chromatographic data analysis package in the Igor Pro programming environment (Wavemetrics, Inc.). The most recent publicly available version of this software package is available at https://sites.google.com/site/terninigor/software-download. Several advances to this package since originally described34 have been implemented here to enable automated data processing. A number of new autonomous features include:
(1) Monitoring a data acquisition folder for automated ingestion of newly collected data files.
(2) Ingestion of metadata by monitoring a spreadsheet and/or file names.
(3) Fitting of peaks using default parameters that are allowed to vary between analytes to improve fits.
(4) Preliminary calibration and application to integrated data.
(5) Generation and saving of user-specified plots.
Once a file is downloaded from the repository by DOsync, the new file is automatically ingested into TERN (1) and assigned metadata (i.e., ambient sample, calibrant concentration, etc.) based on its filename (2). Chromatographic peaks in these samples representing known analytes of interest (e.g., those in the calibration standard) are integrated by fitting them to idealized peak shapes (Gaussian or Gaussian convolved with an exponential decay) following the previously published approach,34 with fitting improved by specifying compound-specific default fit parameters (e.g., peak width or fitting window) (3). Integrated data are approximately calibrated using a user-provided response factor (4), and calibrated results are plotted and saved to a folder (5).
In short, data generated by the instrument is automatically processed into preliminary calibrated data and figures for the user to monitor with little operator interaction. This represents a significant step forward in the viability of long-term deployment of GC based instrumentation such as the instrument described here.
2.5 Data collection and calibration
The data used to evaluate and examine the instrument in this work is collected during an ongoing, multi-year field deployment at the Virginia Forest Lab (VFL), located in central Virginia. The VFL sits on the east side of the Blue Ridge Mountains and is about 25 km east-southeast of Charlottesville, VA. The site receives some anthropogenic emissions from nearby towns and roadways. The instrument sits at the base of a 40 meter meteorological tower, inside a climate-controlled, internet-connected lab that is supplied by line power. Samples are collected mid-canopy from a heated inlet with a sample flow of 1350 sccm. The instrument subsamples off this bypass flow at a sample flow rate of ∼150 sccm. Instrument performance is monitored and evaluated by the automated calibration procedure described in Section 2.4. In the present deployment, a calibration sample occurs every seventh hour at one of five different calibrant concentrations. Two of the calibrations performed each day are a zero and tracking standard. The third daily calibrant varies between three rotating calibrant mixing ratios. For each non-zero calibration, a zero-air sample mixes with a multi-component calibrant (Apel-Riemer Environmental Inc.) at one of four different flow rates, generating four different mixing ratios of calibrant mixtures. The composition and pure volume mixing ratio of the multi-component calibrant used for this instrument range between 4.35 and 17.60 ppb for monoterpene, anthropogenic, oxygenated, and sesquiterpene species while isoprene is at 40.3 ppb. The flow rates of 5, 20 (tracking), 50, and 100 sccm dilute the calibrant into ∼1350 sccm of zero air to create the 4 different calibrant levels. Estimated limits of detection for isoprene, oxidation products, monoterpenes, and sesquiterpenes are 20, 4.3, 2.2, and 2.7 ppt, respectively.36Automated calibration began on June 25th, 2020. Prior to this date, the instrument was calibrated manually either through introduction of gas-phase standards (starting May 2020) or injection of liquid standards. Liquid standard injections early in deployment were found to agree within 5% with later manually-controlled gas-phase calibrations and automated gas-phase calibrations, indicating stable instrument response across this period.
2.6 Conversion of integrated FID signal to concentration
The calibration data from each week identify any changes in sensitivity, in most cases due either to temperature-driven changes in trapping efficiency or adsorbent degradation, and aid conversion of the integrated FID signal to concentration (ppb). The concentration (Csamp,cal) of each calibrant compound sampled onto the trap is calculated by adjusting the concentration in the calibrant supply tank (Csupply,cal) by the dilution of calibrant flow (Qcal) into the total sample flow rate (Qtot).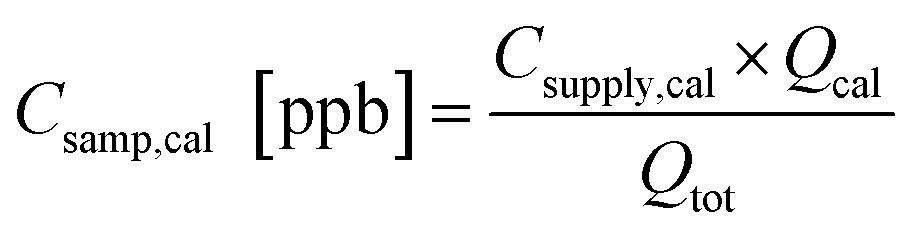 | (1) |
A universal response factor, RF, representing FID signal, S, generated by a unit of reduced carbon can be calculated:
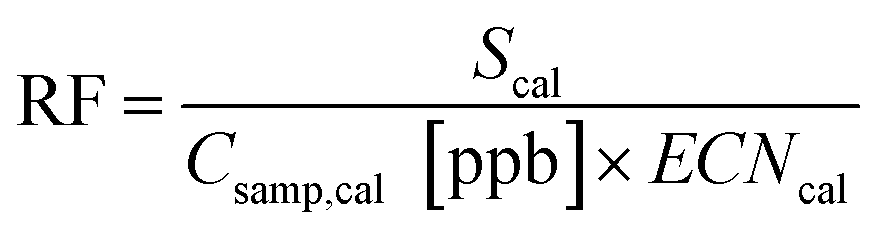 | (2) |
In this equation, effective carbon number, ECN, is equivalent to the number of carbon atoms, NC, for a fully reduced hydrocarbon, and for oxygenates represents the number of hydrocarbon atoms that would yield an equivalent FID signal.21,37 For fully reduced hydrocarbons this response factor is equal to the slope, m, of a linear regression between the integrated FID signal of a given calibrant and the known calibrant concentration in ppbC:
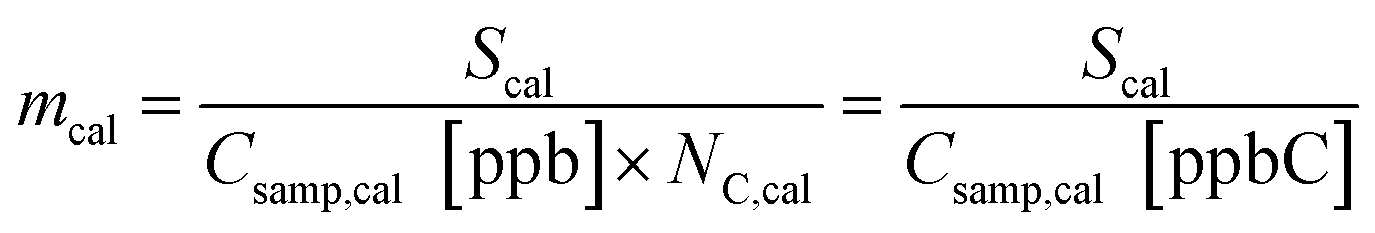 | (3) |
For oxygenates, the response factor can be calculated from the slope based on the ratio of ECN and NC. In theory, all calibrants can therefore be combined to generate a universal response factor per unit carbon, which can be used to calibrate any given compound, i, as long as an ECN is known or can be estimated:
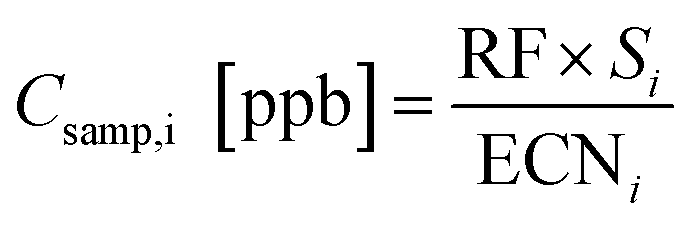 | (4) |
In practice, we show below that it is nevertheless beneficial to calibrate using authentic standards with similar chemical functionality as the analyte of interest. In other words, while RF is theoretically universal, it varies by compound class likely due to instrument limitations unrelated to the FID itself (e.g., transfer losses). In this work, slopes for each calibrant are calculated for each week and used to generate response factors that calibrate all data collected during the week. To evaluate tracking standard concentrations, calibrants are calibrated using the same authentic standard, while for other compounds a response factor is used based on the slope of the most chemically similar calibrant or calibrants (e.g., limonene and α-pinene for other monoterpenes).
3 Results and discussion
3.1 Chemical range accessed
A wide range of species are detected on this instrument, including a number of BVOCs which were previously reported.36 Additional detected compounds include several anthropogenic species such as benzene, toluene, ethylbenzene, and xylenes (BTEX) (Table 1). We also detect a handful of oxygenated species such as eucalyptol and methyl salicylate (Table 1), in addition to those previously reported. An example chromatogram with peak labels can be found in Fig. 4. Additional species beyond those reported in Fig. 4 and Table 1 are detected, those reported here are simply to provide an indication of the range and capability of the instrument and methodology.| Terpenes | Anthropogenic | Oxygenated | |||
|---|---|---|---|---|---|
| Compound | Symbol | Compound | Symbol | Compound | Symbol |
| Isoprene | I | Pentane | A1 | Methacrolein | O1 |
| Thujene | M1 | Carbon tetrachloride | A2 | Methyl vinyl ketone | O2 |
| Tricyclene | M2 | Benzene | A3 | Eucalyptol | O3 |
| α-Pinene | M3 | Pentanal | A4 | Methyl salicylate | O4 |
| Fenchene | M4 | Toluene | A5 | ||
| Camphene | M5 | Hexanal | A6 | ||
| Sabinene | M6 | PCBTF | A7 | ||
| β-Pinene | M7 | Ethylbenzene | A8 | ||
| Limonene | M8 | m,p-Xylene | A9 | ||
| Cymene | M9 | o-Xylene | A10 | ||
| β-Phellandrene | M10 | Decanal | A11 | ||
| γ-Terpinene | M11 | ||||
| α-Cedrene | S1 | ||||
| β-Cedrene | S2 | ||||
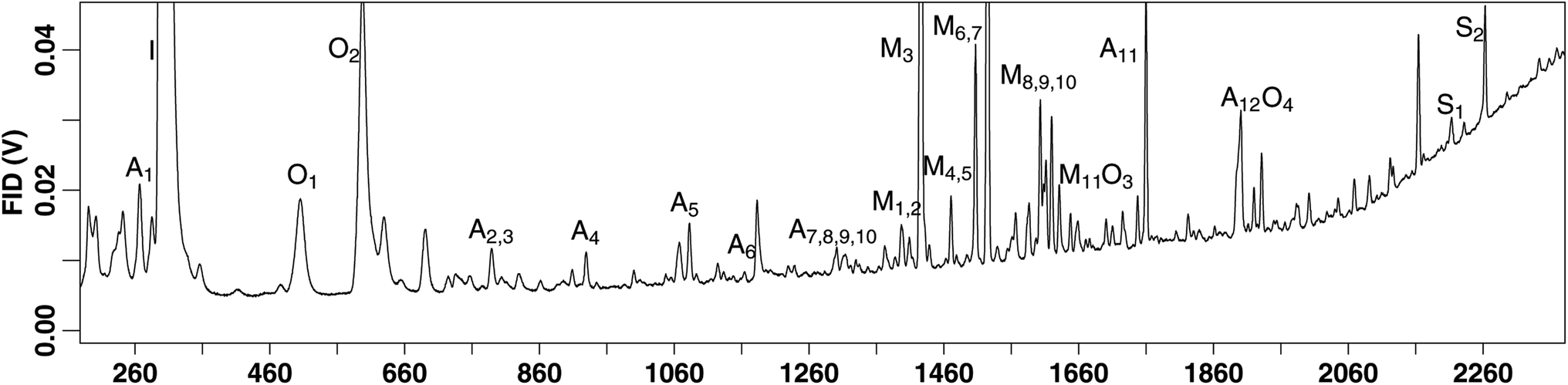 | ||
| Fig. 4 An example chromatogram with a label for a range of compounds detected by the instrument. “A” denotes that the compound is anthropogenic, “I”, “M”, “S” are terpenes or isoprene, monoterpene, and sesquiterpene, and “O” are oxygenated species. | ||
3.2 Calibration curve
All hydrocarbon compounds in the calibration tank fit a linear regression of FID signal as a function of carbon concentration (ppbC) (Fig. 5). For compounds present in ambient samples, this average slope can be used to convert peak integrations into mixing ratios in ppbC for any hydrocarbon analyte, which can then be converted to ppb using a known or estimated effective carbon number for each compound21,22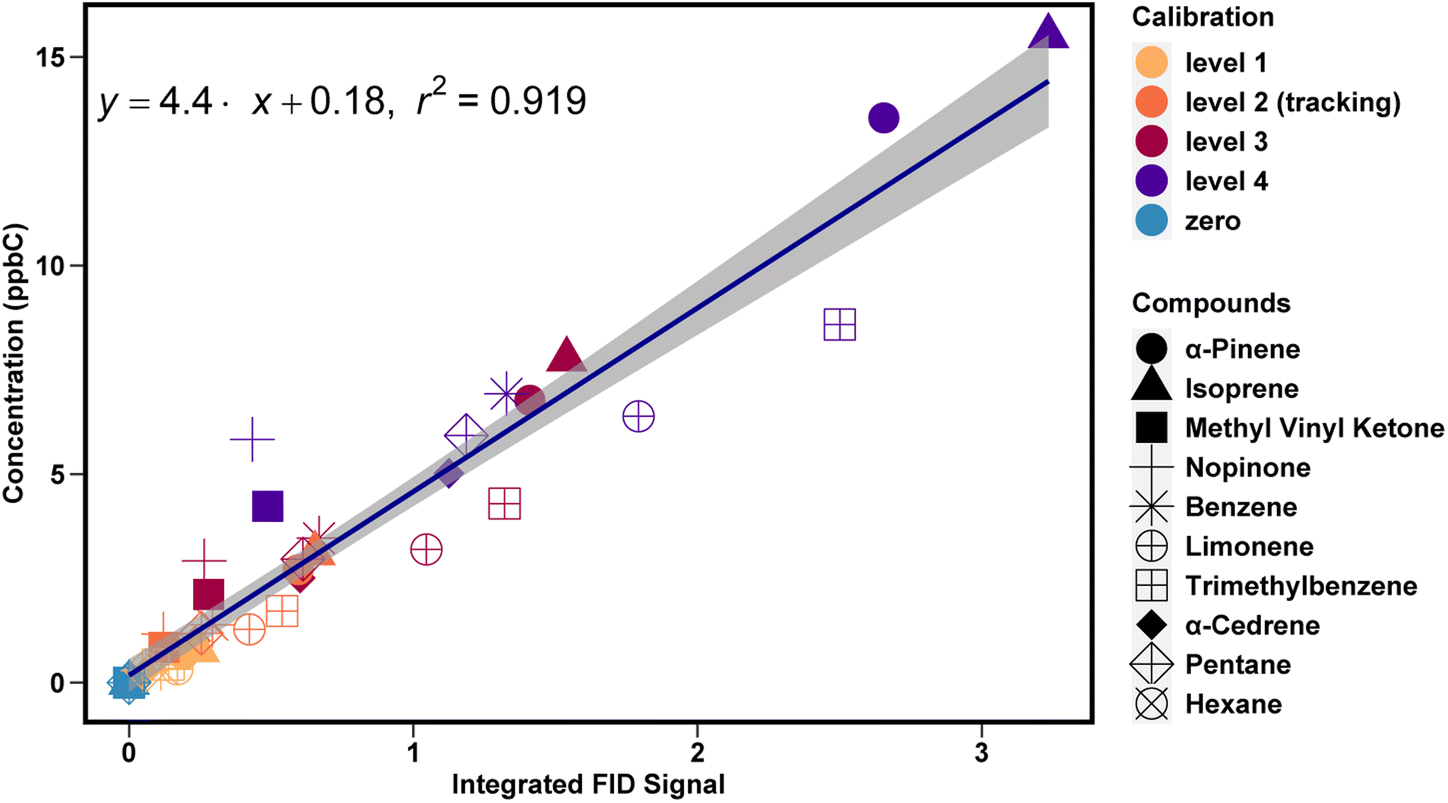 | ||
| Fig. 5 A linear regression plot between the integrated FID signal and the calibrant concentration for one week of calibrations. The slope from this linear regression represents a response factor per unit carbon that is used to convert instrument response to concentration. The shading represents the uncertainty of the slope of the linear regression. Colors represent different calibration levels (i.e., different dilution flows) and symbols represent calibrants. | ||
The use of a unified calibration curve allows for simple calibration of compounds for which no authentic standard is available, enabling quantification with low uncertainty for a wide range of ambient compounds. However, compounds without authentic standards will have somewhat higher uncertainty in accuracy. Note in Fig. 5 the lower slope for limonene and trimethylbenzene. Faiola et al. 2012 report measured effective carbon numbers of 9.5 for limonene and 9.22 for trimethylbenzene, but 9.76 for α-pinene, so some difference in slope is expected, though this does not fully explain the observed differences. Nonetheless, calibrating these compounds using the unified slope method results in a bias of ∼20% (Fig. S1†). Similarly, the unified calibration method does not perform well for oxygenated compounds. The slope for MVK and nopinone is found to be 11 (Fig. S2†). The ECNs of these compounds are roughly 25% (MVK) and 10% (nopinone) lower than their carbon number, so some reduced signal per ppbC (yielding a higher calibration slope) is expected, but this issue alone does not account for the larger difference in observed slope. Instead, reduced signal from oxygenates is likely due to partial removal by the ozone scrubber, an issue that has been previously observed38 and suggests that oxygenates should be calibrated using the response of an oxygenated calibrant and not simply the unified calibration curve.
Once response factors are calculated they are used to convert the integrated FID signal (V s) to concentration (ppb). Response factors for compounds in the calibration tank that are chemically similar are generally different by less than 20%. Calibrants that are observed to meet this similarity criteria (e.g., hydrocarbons in the middle region of the chromatogram) are combined into a unified curve used to calibrate compounds of that chemical class; while this approach introduces up to 10% uncertainty (average difference between individual and unified calibration curves), it also mitigates uncertainties introduced in the integrations or responses of any individual calibrant. For example, a combined hydrocarbon curve is used to calibrate monoterpenes, while the methyl vinyl ketone curve is used to calibrate methyl vinyl ketone and methacrolein.
3.3 Stable calibration and uncertainty
To evaluate the long-term stability and precision of this instrument, concentrations measured in the tracking standard are compared to expected values (Fig. 6). Response factors for each individual compound are used in order to assess instrument precision without the confounding issue of potential biases or errors in accuracy due to using a unified calibration curve, as discussed above. Across a 4 month period examined here, the concentration measured is in good agreement with the expected value, shown in Fig. 6 for a biogenic VOC (limonene), an anthropogenic VOC (1,3,5-trimethylbenzene), and a compound at the low-volatility edge of the instrument capabilities (α-cedrene). For compounds in the middle of the instrument range (e.g., limonene and trimethylbenzene), nearly all points fall within 15% of the expected value. For compounds at the edges of instrument capability have somewhat lower precision, roughly 20%, likely due to adsorption onto inlet lines and/or incomplete transfer through the valve oven.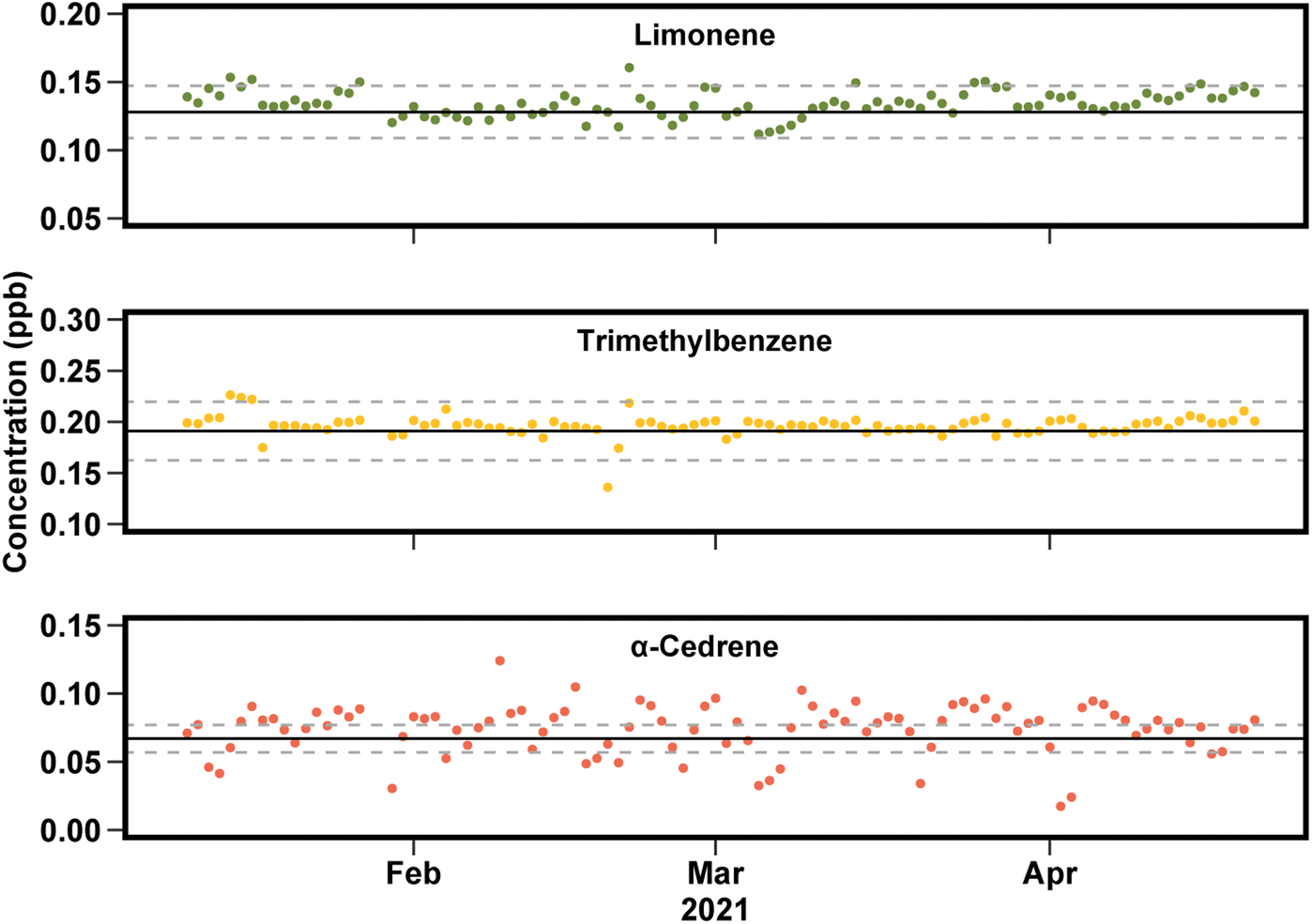 | ||
| Fig. 6 Measured concentrations in tracking standard for limonene, trimethylbenzene, and α-cedrene each day between January and May 2021. Solid line is known concentration in the calibration, with ±10% in dashed lines. | ||
Overall, the data presented in Fig. 5 and 6 suggest instrument uncertainty of roughly 10% for most compounds, at least during periods of near-optimal instrument operation. Harder-to-measure compounds may suffer somewhat higher uncertainty, and compounds for which no authentic standard or known response factor is available suffer an additional 20% in accuracy. These values are consistent with known uncertainties in chromatographic peak integration (impacting precision) and effective carbon number (impacting accuracy for uncalibrated compounds).
Monitoring tracking standards of compounds in multiple chemical classes can help elucidate a number of hardware issues. For example, monitoring calibrant response can elucidate failures of the zero air generator (e.g., high signals may indicate ambient air infiltration into zero air) and degradation of trap materials (e.g., reduction in signal for higher volatility components). Response of tracking standards is used to guide maintenance schedules such as replacing the trap, which has been observed to be necessary once or twice per year to maintain stable trapping efficiency for all analytes.
Unlike a mass spectrometer, an FID does not provide any chemical resolution of the eluting sample, so analytes are more likely to co-elute with neighboring peaks which could increase uncertainty. As long as a saddle-point exists between two neighboring peaks, deconvolution of the two peaks does not introduce significant uncertainty. In more poorly resolved peaks, in particular when no saddle exists between two peaks, co-elution can significantly increase uncertainty and also lead to significant broadening of the peak fit.34 To address this issue, peak integration uses peak width as a validation metric. If the peak width deviates too far from expectations, the peak is either not fit or is flagged as potentially incorrect and re-fitting occurs through manually tweaking the fit. This minimizes the impact of co-elution on quantitative uncertainty, so while poorly resolved peaks may have somewhat higher uncertainty, it is not expected to be significant; rather, if the fit is uncertain or poor, an integration is simply not reported.
An additional potential source of uncertainty for any trap-based analytical system such as this instrument is incomplete collection of sampled analytes, particularly volatile species. The trap used in this work has been previously extensively examined. Wernis et al., 2021 determined that at the operating temperature of this instrument (30 °C), 15 mg of Carbopack X (the most retentive adsorbent) provided sufficient trapping efficiency for the most volatile analytes reported here (breakthrough volume substantially greater than the sample volume), while the instrument reported here uses at least 50 mg of Carbopack X. We consequently do not anticipate significant uncertainty from incomplete trapping.
3.4 Long-term speciated measurements
This automated method of data capture, calibration, and integration has enabled over two years of speciated data collection, ranging from C5–C15. In Fig. 7 two years of isoprene and total monoterpenes are presented, as well as the breakdown of species contribution for total monoterpenes over the two years. Looking at the total time series, isoprene is detected at levels of 20 ppb at times but is often between 5 and 10 ppb in the warmest months of the year. Isoprene is largely emitted by deciduous trees with leaves and is therefore not emitted between October and May. Monoterpenes range between 0.1 and 2 ppb throughout the year. Concentrations for these species are highest in the warmest months, much like isoprene, but emissions of these compounds from coniferous trees cause them to be present throughout the year. The largest contribution to total monoterpenes is α-pinene, followed by β-pinene, camphene, and limonene (Fig. 7C).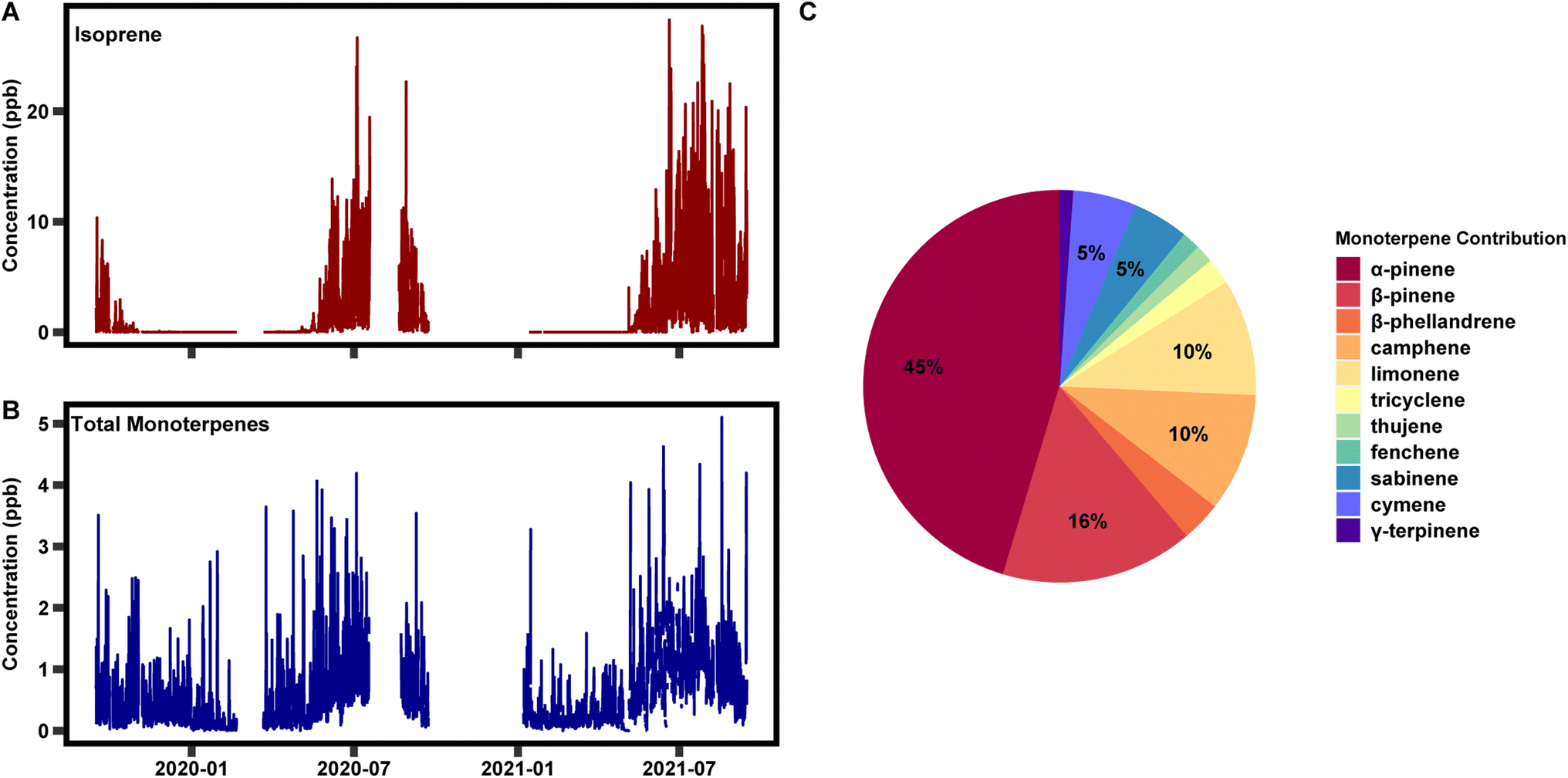 | ||
| Fig. 7 A two year time series of (A) isoprene and (B) total monoterpenes, and (C) the average break down of individual monoterpene species over the two year period. | ||
Previously published work with this instrument has focused on temporal variability on diurnal, seasonal, and interannual timescales and quantified the impact of individual species on atmospheric reactivity.36,39 This work found that isoprene was a large contributor to OH reactivity when emitted. Monoterpenes contributed the most to ozone and nitrate reactivity year-round and OH reactivity in the cooler months. Monoterpenes also exhibit variability within their chemical class on all three timescales. Most notably, a subset of monoterpenes are emitted at higher levels in the summer months due to the presence of light; they are emitted in a light dependent manner, much like isoprene. This variability had impacts on atmospheric reactivity on both diurnal and seasonal timescales. Additionally, interannual variability was observed in the ratio of composition at the site on both diurnal and seasonal time scales, but additional years of data are be necessary to determine the driver(s). These measurements were taken alongside a suite of ecological, meteorological, and chemical variables enabling the determination of the driver of the variability. These data are presented here as an example of the information that can be learned through the long-term measurements enabled by this instrument. As of the time of publication, data collection is ongoing.
3.5 Additional recommendations for improved long-term deployment
Several components in this system require regular maintenance due to constant use, and represent weak spots in the autonomous remote operation of a GC instrument for VOC measurements. Particular failure points in this system include the oil-free vacuum pump used to pull sample flow, the zero-air generator, and the computer. Regular maintenance on the schedules recommended by the suppliers, if not more frequently can mitigate some failures and reduce downtime. We nevertheless identify here the issues surrounding maintenance and component failure for which backup and/or maintenance plans should be considered in developing long-term VOC measurements as described here.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hours. Downtime due to pump failure has been mitigated in our system by including a backup pump in parallel, with a three-way electronic valve controlling which pump is used and a relay-controlled outlet providing power to the backup pump (commercially available or custom-built). One of the 24 VDC channels can be remotely activated to switch the electronic valve and turn on the backup pump, allowing continued sample flow without the need for an onset visit; in principle, activation of this relay could be an automated function of measured sample flow. This level of complexity is in practice unnecessary as long as the generated data are being regularly monitored and accessing the site within a couple days of pump failure is feasible.
000 hours. Downtime due to pump failure has been mitigated in our system by including a backup pump in parallel, with a three-way electronic valve controlling which pump is used and a relay-controlled outlet providing power to the backup pump (commercially available or custom-built). One of the 24 VDC channels can be remotely activated to switch the electronic valve and turn on the backup pump, allowing continued sample flow without the need for an onset visit; in principle, activation of this relay could be an automated function of measured sample flow. This level of complexity is in practice unnecessary as long as the generated data are being regularly monitored and accessing the site within a couple days of pump failure is feasible.
4 Conclusion
Remote and online monitoring of C5–C15, including lightly oxygenated species was achieved through the development of an automated gas chromatograph using a flame ionization detector. Sample preconcentration is achieved using a recently developed adsorbent trap carefully designed to minimize swept volumes and thermal mass.33 This instrument enables hourly measurement of volatile organic compounds spanning four to five orders of magnitude in volatility, without cryogen or thermoelectric cooling.The instrument described here is designed to make collection of VOCs at high temporal and chemical resolution feasible in a wide range of locations, with the goal of broadening the availability of long-term measurements of reactive carbon. The instrument is highly adaptable and could be expanded and tuned to enable additional detection ranges, for instance through selecting different columns and/or trapping materials. Additionally, making the described minor changes to the instrument in order to operate it as a two-channel system would further broaden the potential use cases by enabling measurements at two inlet locations simultaneously, or onto two separate traps or columns. Development and deployment of instruments similar to the one presented here could vastly improve understanding of spatial and temporal distributions of VOC concentrations and emissions, and have the potential to improve VOC, ozone, and secondary organic aerosol predictions.
Author contributions
D. F. M. developed and deployed the first version of this instrument, maintained the instrument for the first two years, analyzed the data, and wrote the manuscript. N. S. P. developed and deployed the second version of the instrument and is currently maintaining it in the field. G. F. assisted with the first deployment of the instrument and assisted with instrument maintenance. C. B. assisted with instrument troubleshooting. G. I. V. W. developed the instrument plans, managed instrument development, deployed the instrument, manages troubleshooting, and directed the analysis of the data and writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Science Foundation (AGS 2046367, as well as collaborative grants AGS 1837882 and AGS 1837891). Tower maintenance and operation were supported in part by the Pace Endowment. Deborah F. McGlynn is supported in part by Virginia Space Grant Consortium Graduate Research Fellowships.Notes and references
- A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons and X. Wang, Geosci. Model Dev., 2012, 5, 1471–1492 CrossRef.
- G. Schuh, A. Heiden, T. Hoffmann, J. Kahl, P. Rockel, J. Rudolph and J. Wildt, J. Atmos. Chem., 1997, 27, 291–318 CrossRef.
- F. Loreto, A. Förster, M. Dürr, O. Csiky and G. Seufert, Plant, Cell Environ., 1998, 21, 101–107 CrossRef.
- C. Kalogridis, V. Gros, R. Sarda-Esteve, B. Langford, B. Loubet, B. Bonsang, N. Bonnaire, E. Nemitz, A. C. Genard, C. Boissard, C. Fernandez, E. Ormeño, D. Baisnée, I. Reiter and J. Lathière, Atmos. Chem. Phys., 2014, 14, 10085–10102 CrossRef.
- M. W. Holdren, H. H. Westberg and P. R. Zimmerman, J. Geophys. Res., 1979, 84, 5083–5087 CrossRef.
- I. J. Simpson, M. P. Andersen, S. Meinardi, L. Bruhwiler, N. J. Blake, D. Helmig, F. Sherwood Rowland and D. R. Blake, Nature, 2012, 488, 490–494 CrossRef PubMed.
- D. B. Millet, N. M. Donahue, S. N. Pandis, A. Polidori, C. O. Stanier, B. J. Turpin and A. H. Goldstein, J. Geophys. Res.: Atmos., 2005, 110, 1–17 CrossRef.
- G. W. Schade and A. H. Goldstein, J. Geophys. Res.: Atmos., 2001, 106, 3111–3123 CrossRef CAS.
- P. D. Goldan, W. C. Kuster, E. Williams, P. C. Murphy, F. C. Fehsenfeld and J. Meagher, J. Geophys. Res., 2004, 109, D21309 CrossRef.
- G. W. Schade and A. H. Goldstein, Geophys. Res. Lett., 2003, 30, 10–13 CrossRef.
- D. Tanner, D. Helmig, J. Hueber and P. Goldan, J. Chromatogr. A, 2006, 1111, 76–88 CrossRef CAS.
- B. Davison, R. Taipale, B. Langford, P. Misztal, S. Fares, G. Matteucci, F. Loreto, J. N. Cape, J. Rinne and C. N. Hewitt, Biogeosciences, 2009, 6, 1655–1670 CrossRef CAS.
- L. H. Mielke, K. A. Pratt, P. B. Shepson, S. A. Mcluckey, A. Wisthaler and A. Hansel, Anal. Chem., 2010, 82, 8835–8840 CrossRef.
- J.-H. Park, A. H. Goldstein, J. Timkovsky, S. Fares, R. Weber, J. Karlik and R. Holzinger, Science, 2013, 341, 643–647 CrossRef CAS.
- J. de Gouw and C. Warneke, Mass Spectrom. Rev., 2007, 26, 223–257 CrossRef CAS.
- B. Yuan, A. R. Koss, C. Warneke, M. Coggon, K. Sekimoto and J. A. de Gouw, Chem. Rev., 2017, 117, 13187–13229 CrossRef PubMed.
- B. R. Ayres, H. M. Allen, D. C. Draper, S. S. Brown, R. J. Wild, J. L. Jimenez, D. A. Day, P. Campuzano-Jost, W. Hu, J. de Gouw, A. Koss, R. C. Cohen, K. C. Duffey, P. Romer, K. Baumann, E. Edgerton, S. Takahama, J. A. Thornton, B. H. Lee, F. D. Lopez-Hilfiker, C. Mohr, P. O. Wennberg, T. B. Nguyen, A. Teng, A. H. Goldstein, K. Olson and J. L. Fry, Atmos. Chem. Phys., 2015, 15, 13377–13392 CrossRef.
- A. Mehra, J. E. Krechmer, A. Lambe, C. Sarkar, L. Williams, F. Khalaj, A. Guenther, J. Jayne, H. Coe, D. Worsnop, C. Faiola and M. Canagaratna, Atmos. Chem. Phys., 2020, 20, 10953–10965 CrossRef.
- J. Liebmann, N. Sobanski, J. Schuladen, E. Karu, H. Hellén, H. Hakola, Q. Zha, M. Ehn, M. Riva, L. Heikkinen, J. Williams, H. Fischer, J. Lelieveld and J. N. Crowley, Atmos. Chem. Phys., 2019, 19, 10391–10403 CrossRef.
- J. H. Gross, Mass Spectrometry, Springer International Publishing, Cham, Switzerland, 3rd edn, 2004, p. 986 Search PubMed.
- C. L. Faiola, M. H. Erickson, V. L. Fricaud, B. T. Jobson and T. M. Vanreken, Atmos. Meas. Tech., 2012, 5, 1911–1923 CrossRef.
- J. Sternberg, W. Gallaway and D. Jones, in Gas Chromatography: Third International Symposium, ed. N. Brenner, J. E. Callen and M. D. Weiss, Academic Press, New York, NY, 1962 Search PubMed.
- M. Wang, L. Zeng, S. Lu, M. Shao, X. Liu, X. Yu, W. Chen, B. Yuan, Q. Zhang, M. Hu and Z. Zhang, Anal. Methods, 2014, 6, 9424–9434 RSC.
- M. De Blas, M. Navazo, L. Alonso, N. Durana and J. Iza, Sci. Total Environ., 2011, 409, 5459–5469 CrossRef PubMed.
- E. C. Apel, J. Geophys. Res., 2003, 108, 8794 CrossRef.
- B. J. Williams, A. H. Goldstein, N. M. Kreisberg and S. V. Hering, Aerosol Sci. Technol., 2006, 40, 627–638 CrossRef.
- G. Roest and G. Schade, Atmos. Chem. Phys., 2017, 17, 11163–11176 CrossRef.
- A. Panopoulou, E. Liakakou, S. Sauvage, V. Gros, N. Locoge, I. Stavroulas, B. Bonsang, E. Gerasopoulos and N. Mihalopoulos, Atmos. Environ., 2020, 243, 117803 CrossRef.
- H. Hellén, A. P. Praplan, T. Tykkä, I. Ylivinkka, V. Vakkari, J. Bäck, T. Petäjä, M. Kulmala and H. Hakola, Atmos. Chem. Phys., 2018, 18, 13839–13863 CrossRef.
- C. G. Bolas, V. Ferracci, A. D. Robinson, M. I. Mead, M. S. M. Nadzir, J. A. Pyle, R. L. Jones and N. R. P. Harris, Atmos. Meas. Tech., 2020, 13, 821–838 CrossRef.
- S. Sala, H. Bönisch, T. Keber, D. E. Oram, G. Mills and A. Engel, Atmos. Chem. Phys., 2014, 14, 6903–6923 CrossRef.
- E. Bourtsoukidis, F. Helleis, L. Tomsche, H. Fischer, R. Hofmann, J. Lelieveld and J. Williams, Atmos. Meas. Tech., 2017, 10, 5089–5105 CrossRef.
- R. A. Wernis, N. M. Kreisberg, R. J. Weber, Y. Liang, J. Jayne, S. Hering and A. H. Goldstein, Atmos. Meas. Tech., 2021, 14, 6533–6550 CrossRef.
- G. Isaacman-VanWertz, D. T. Sueper, K. C. Aikin, B. M. Lerner, J. B. Gilman, J. A. de Gouw, D. R. Worsnop and A. H. Goldstein, J. Chromatogr. A, 2017, 1529, 81–92 CrossRef PubMed.
- D. R. Gentner, G. Isaacman, D. R. Worton, A. W. H. Chan, T. R. Dallmann, L. Davis, S. Liu, D. A. Day, L. M. Russell, K. R. Wilson, R. Weber, A. Guha, R. A. Harley and A. H. Goldstein, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18318–18323 CrossRef.
- D. F. McGlynn, L. E. R. Barry, M. T. Lerdau, S. E. Pusede and G. Isaacman-VanWertz, Atmos. Chem. Phys., 2021, 21, 15755–15770 CrossRef.
- J. T. Scanlon and D. E. Willis, J. Chromatogr. Sci., 1985, 23, 333–340 Search PubMed.
- J. Pollmann, J. Ortega and D. Helmig, Environ. Sci. Technol., 2005, 39, 9620–9629 CrossRef PubMed.
- D. F. McGlynn and G. Isaacman-VanWertz, In-Canopy Biogenic Volatile Organic Compounds Mixing Ratios at the Virginia Forest Lab, 2022, https://data.mendeley.com/datasets/jx3vn5xxcn/1 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ea00079b |
| This journal is © The Royal Society of Chemistry 2023 |