
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRing-opening yields and auto-oxidation rates of the resulting peroxy radicals from OH-oxidation of α-pinene and β-pinene†
Ben H. Lee a,
Siddharth Iyerb,
Theo Kurténc,
Jonathan G. Varelasd,
Jingyi Luod,
Regan J. Thomsond and
Joel A. Thornton*a
a,
Siddharth Iyerb,
Theo Kurténc,
Jonathan G. Varelasd,
Jingyi Luod,
Regan J. Thomsond and
Joel A. Thornton*a
aDepartment of Atmospheric Sciences, University of Washington, Seattle, WA, USA. E-mail: joelt@uw.edu
bAerosol Physics Laboratory, Tampere University, Tampere, Finland
cDepartment of Chemistry, University of Helsinki, Helsinki, Finland
dDepartment of Chemistry, Northwestern University, Evanston, IL, USA
First published on 5th January 2023
Abstract
Atmospheric oxidation of monoterpenes (C10H16) contributes to ambient particle number and mass concentrations due, in part, to the resulting peroxy radicals undergoing auto-oxidation to low-volatility highly oxygenated molecules (HOMs). Only one of the structural isomers of the first-generation hydroxy peroxy radical (C10H17O3), that formed with an opening of the cyclobutyl ring, is thought to be responsible for the majority of HOMs derived from OH-initiated oxidation of α and β-pinene. We constrain the yield and auto-oxidation rate of this isomer by using a unique combination of isotopically labeled precursors, such as D3-α-pinene, and direct measurements of peroxy radicals and closed-shell products after a reaction time of only ∼0.7 s. Using competitive rate studies and quantum chemical calculations, we find that the yields of these ring-opened peroxy radicals are 0.47 (0.2, 0.6) for α-pinene and 0.66 (0.4, 0.8) for β-pinene, and the rates of auto-oxidation are 2.9 (1.1, 4.7) s−1 and 0.8 (0.4, 1.6) s−1, respectively, where values in the parentheses represent upper and lower bounds based on experimental and mechanistic uncertainties. These rates and yields are in general agreement with HOM yields determined from independent methods and in a range where OH driven monoterpene HOM formation will be atmospherically important for a wide set of conditions.
Environmental significanceAuto-oxidation of monoterpene (MT) derived organic peroxy radicals leading to HOMs is a key process for prompt formation of SOA observed in the troposphere and in atmospheric simulation chamber experiments. MT SOA is a significant contributor to fine particulate mass concentrations, affecting climate and human health. OH-oxidation of α and β-pinene is a major fate for MT. The fraction of the associated peroxy radicals which undergo auto-oxidation, and the rates of the auto-oxidation compared to bimolecular reactions are key metrics towards quantitative mechanistic predictions of SOA mass yields. We present experimental results for these two metrics, for which there is large variance in literature, that will allow for better descriptions of spatial and temporal variations in MT SOA production. |
1 Introduction
Global emissions of biogenic volatile organic compounds (BVOCs) approach nearly a petagram of carbon per year (Pg C year−1).1–3 Subsequent atmospheric oxidation, initiated by reactions with ozone (O3), hydroxy radicals (OH), or nitrate radicals (NO3), leads to products of varying volatility that can affect the burden and composition of organic aerosol (OA), which in many regions of the world is the dominant component of atmospheric fine particulate matter (PM2.5).4–6 OA formation and growth on the timescales observed in forested environments and in laboratory chamber experiments indicate that organic peroxy radical (RO2) auto-oxidation involving intramolecular RO2 radical propagation with one or more O2 additions is a key mechanism leading to the prompt formation of highly oxygenated molecules (HOMs), which have low to extremely low volatility.7Monoterpene oxidation is a major contributor to SOA in forested regions even where isoprene emissions far exceed those of monoterpenes, such as over the Southeast United States,8–10 as well as in urban areas given that monoterpenes account for a large fraction of anthropogenic volatile chemical products.11,12 Loss pathways of α-pinene (AP) due to reactions with OH and O3 are comparable, whereas OH oxidation is about an order magnitude faster than ozonolysis for β-pinene (BP) under typical conditions of the daytime when BVOC emissions are highest. Theoretical and experimental work have indicated HOM formation only from the first-generation RO2 which have undergone opening of the cyclobutyl (or C-4) ring after OH addition to the less substituted (tertiary for AP and primary for BP) C of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond.13–18 Retainment of the C-4 ring of the first-generation RO2 prohibits intramolecular H-shifts due to steric hindrance of the ring itself and due to the absence of a CC double bond (present only in the C-4 ring-opened isomer) that facilitates the intramolecular H-shifts.15
C double bond.13–18 Retainment of the C-4 ring of the first-generation RO2 prohibits intramolecular H-shifts due to steric hindrance of the ring itself and due to the absence of a CC double bond (present only in the C-4 ring-opened isomer) that facilitates the intramolecular H-shifts.15
We report on experimental determinations of the yields of the C-4 ring-opened RO2 from OH reactions with AP and BP, as well as the rates of auto-oxidation of the resulting first (C10H17O3) and second generation (C10H17O5) peroxy radicals by employing a chemical box model to constrain observations made using a fast flow tube coupled to a long time of flight mass spectrometer (L-ToF) employing ammonium-adduct (NH4+) ionization to directly measure the suite of peroxy radicals and closed-shell products formed after ∼0.7 seconds of initiating OH-oxidation, over a range of nitric oxide (NO) levels. Accurate mechanistic understanding of the yields of these peroxy radicals through auto-oxidation can better help quantitatively explain SOA mass yields, which are used in atmospheric chemical transport and climate models to describe OA formation.19
2 Methods
Experiments were conducted in a room temperature (21 ± 2 °C), atmospheric pressure (1016 ± 15 mbar) flow reactor with a 2.54 cm outer diameter. The entire 8.2 standard liters per minute (slpm) output from the flow reactor passes through a custom-built transverse ion–molecule reaction region (IMR) maintained at atmospheric pressure, and coupled to a Tofwerk AG long time of flight mass spectrometer (LToF-MS). The IMR, refined from that used by Zhao et al.,20 has an inner diameter matching that of the flow reactor to maintain laminar flow and constant flow velocity. A schematic of the transverse IMR is shown in Fig. S1.† NH4+ reagent ions are generated by flowing 1.8 slpm UHP N2 over the headspace of an ammonium hydroxide (NH4OH) solution and through an inline 210Po (NRD) ionizer. The exit of the 210Po source is axially aligned with the entrance capillary of the LToF-MS and orthogonal to the reactor flow direction. Ions enter the sample flow from the outer wall and traverse the 2.2 cm across the sample flow to the LToF-MS capillary aided by a ∼1.5 kV cm−1 electric field applied between the 210Po source and the capillary. Reaction products are detected as NH4+ adducts.21,22Hydroxyl (OH) and hydroperoxyl (HO2) radicals were generated by flowing 1 slpm of humidified UHP N2 through a quartz housing containing a 185 nm radiation Hg lamp (PenRay). Water vapor (H2O) is photolyzed to generate HOx without detectable O3, given the absence of molecular oxygen (O2). Zero-air (Teledyne) containing AP or BP with or without NO was added concentrically around the HOx source output via a modified 0.25 inch PTFE (polytetrafluoroethylene) T-union (Swagelok) to the flow reactor in slight excess of 8.2 slpm.
Mixing ratios of OH in the flow reactor were quantified by titration with isopropanol added to the flow reactor in the same manner as AP and BP (see below) while measuring the resulting acetone. The acetone sensitivity (0.11 cps ppt−1) was determined using a gravimetrically calibrated custom permeation device (see SI†). Thus, acetone detected from isopropanol quantifies the effective OH concentration in the flow reactor that mixes with the VOC. Mixing ratios of HO2 were quantified by incrementally adding nitric oxide (NO) from a 9.8 ppm (±2%) cylinder (Praxair) to convert the HO2 to OH in the presence of excess isopropanol, generating additional acetone. NO was added at 15 incremental steps resulting in 0 to 127 ppb in the flow reactor. Using a kinetic model of isopropanol oxidation, we find good agreement between observed and modeled acetone production as a function of NO added, assuming 0.86 acetone is formed for every isopropanol oxidized by OH23,24 and that the output of the HOx source contains 25 ppt and 125 ppt of OH and HO2, respectively (Fig. S2†).
For the AP or BP oxidation experiments, excess parent VOC was introduced by flowing 5 sccm of UHP N2 over the headspace of a sample vial containing room temperature AP or BP and into the zero-air carrier flow. Assuming the headspace in the vial saturates with AP or BP, we estimate a few ppm of the precursor VOC in the flow tube, though only a very small fraction reacts to form RO2 (see below). Synthesized D3-labeled AP, which is kept at −30 °C when not in use, was introduced by dipping a 1.5 mm OD glass capillary in the pure compound and placing the capillary in the sample vial. The D3-labeled AP was synthesized from β-pinene in nine steps according to the reported literature procedure.25 Calibrations to acetone were performed prior to each experiment of a given monoterpene by adding acetone to the flow tube setup, which was operated identically as during the oxidation experiments.
The HOx source flow and main carrier flow containing VOC and NO (when added) meet and travel 25 cm before passing through the reagent ions crossing the flow to the LToF-MS sample capillary. Given the flow reactor inner diameter and total volumetric flow rate, we maintain laminar flow (Re ∼510) and estimate a residence time of ∼0.7 seconds of the reaction mixture prior to detection. We chose 0.7 seconds reaction time for this study to place a constraint on RO2 auto-oxidation timescales while also allowing for sufficient extent of reaction to generate detectable product concentrations. We estimate that the above conditions lead to a total of ∼60 ppt of monoterpene reacted with OH in the absence of NO, and up to ∼610 ppt at maximum NO (127 ppb, Fig. S3†).
Energy barriers for the alkoxy isomerization reactions were calculated using a similar approach to the one detailed in Møller et al.26 Briefly, the Spartan ’18 program27 was used to perform a systematic conformer sampling of the reactants and transition states using the Merck Molecular Force Field (MMFF) method with a neutral charge enforced on the radical atom. Initial geometry optimizations were carried out at the B3LYP/6-31+G(d) level of theory, and conformers within 2 kcal mol−1 in relative electronic energies were optimized and their frequencies calculated at the ωB97X-D/aug-cc-pVTZ level.28–30 The Gaussian 16 program was used to carry out these optimizations and frequency calculations. On the lowest electronic energy reactant and transition state conformers at the ωB97X-D/aug-cc-pVTZ level, single-point calculations were done at the ROCCSD(T)-F12a/VDZ-F12 level using the Molpro 2019.2 program.31 The stability of the wavefunction was checked at the CCSD(T) stage by running HF calculations (cc-pVDZ basis set using ORsCA 4.2.1 program32) with 15 HOMOs and 15 LUMOs switched 10 times randomly and generating 100 input files with the orbital rotations applied.33 This indicated that no lower-lying wavefunction relative to the default solution was neglected for any of the reactants and transition states. For the products of the alkoxy isomerization reactions, only ωB97X-D/aug-cc-pVTZ optimizations were performed without a systematic conformer sampling step. This is because while the subsequent RRKM simulations are sensitive to the reactant and transition state energies, they are not sensitive to that of the products. However, products nevertheless need to be defined to run the simulations.
The master equation solver for multi-energy well reactions (MESMER34) program was used to carry out the RRKM simulations to determine the fate of alkoxy radicals. The isomerization reactions were treated using the SimpleRRKM method with Eckart tunneling. The reactant alkoxy radicals were assigned as “modelled” in the simulations and given Lennard-Jones potentials sigma = 6.5 Å and epsilon = 600 K, and a ΔEdown value of 225 cm−1, which are identical to those used previously for similar systems.35,36 The resulting quantum chemical output files are available here (https://zenodo.org/record/7214075#.Y3QrjezMJqs).
3 Results and discussion
A suite of products are detected from the OH-initiated oxidation of AP or BP in the presence of NO. Under low NO conditions and after ∼0.7 s of reaction time between OH and the monoterpenes, we detect a series of products containing 10 C and 16, 17, or 18 H atoms, one or no N atoms, with odd and even numbers of O (Fig. 1). The products with odd numbers of H and O atoms, particularly C10H17Ox where x = 3,5,7 dominate the reaction mixture spectrum at short reaction time and low NO. Additionally, groups of compounds with similar molecular formulae (but differing number of O atoms) generally exhibit distinct trends as a function of NO. For instance, compounds with molecular formulae C10H17O3,5,7 initially increase in abundance with NO as the reaction between NO and HO2 (present in the HOx source and also generated for every closed-shell carbonyl product formed) yields OH, which promptly react with the excess AP or BP. The abundances of these C10H17O3,5,7 species eventually decrease with additional NO. Based on their composition and behavior as a function of NO, we conclude these are peroxy radicals (RO2), as their reactions with NO dominate in competition with auto-oxidation. In comparison, products of the reactions between NO and RO2 including multi-functional alkyl nitrates (C10H17NO4–10), carbonyls (C10H16O2–6), and acetone continue to increase with NO, and then approach a plateau. The dominant products observed with increasing NO are, as expected, C10H16O2 (likely pinonaldehyde) for AP and C9H14O (nopinone) for BP, produced from the reaction between NO and the first generation peroxy radical (C10H17O3), as shown in Fig. 1. The sensitivity of NH4+ adduct ionization for most of the 70 identified species is unknown, as such, we show the increase in their ion signals (in units of counts per second, cps) in the presence of HOx. | ||
| Fig. 1 Maximum signal increase encountered relative to when oxidation is off, for each of the 70 identified compounds during OH-initiated oxidation of (a) AP and (b) BP over the course of NO addition from 0 to 127 ppb. | ||
As illustrated in Fig. 2, three structural isomers of the hydroxy peroxy radical (C10H17O3) are formed upon the addition of OH to the CC double bond of AP37,38 or BP.39 The three H-abstraction channels together account for 0.12 of the fraction of OH-initiated oxidation of AP37,38 and 0.099 for BP.39 For AP, OH-addition to the double bond at the tertiary carbon (fA) followed by breaking of the C-4 ring (fC) is of interest (Fig. 2), given the ability of the resulting RO2 to undergo auto-oxidation.13,15,18 For BP, the breaking of the C-4 ring can occur following the OH addition to the primary position.39 The reaction between this ring-opened RO2 isomer of AP and NO produces acetone with reported yields between zero and unity (Table 1). Acetone is also thought to form from two other OH-oxidation pathways of AP + OH: OH addition to the double bond at the more substituted (secondary) carbon (fB) followed by eight generations of O2 addition and NO reaction, and one of the three known H-abstraction pathways (fH-abs) followed by three generations of O2 addition and NO reaction.37 These reaction pathways to acetone under high NO conditions are highlighted in Fig. 2.
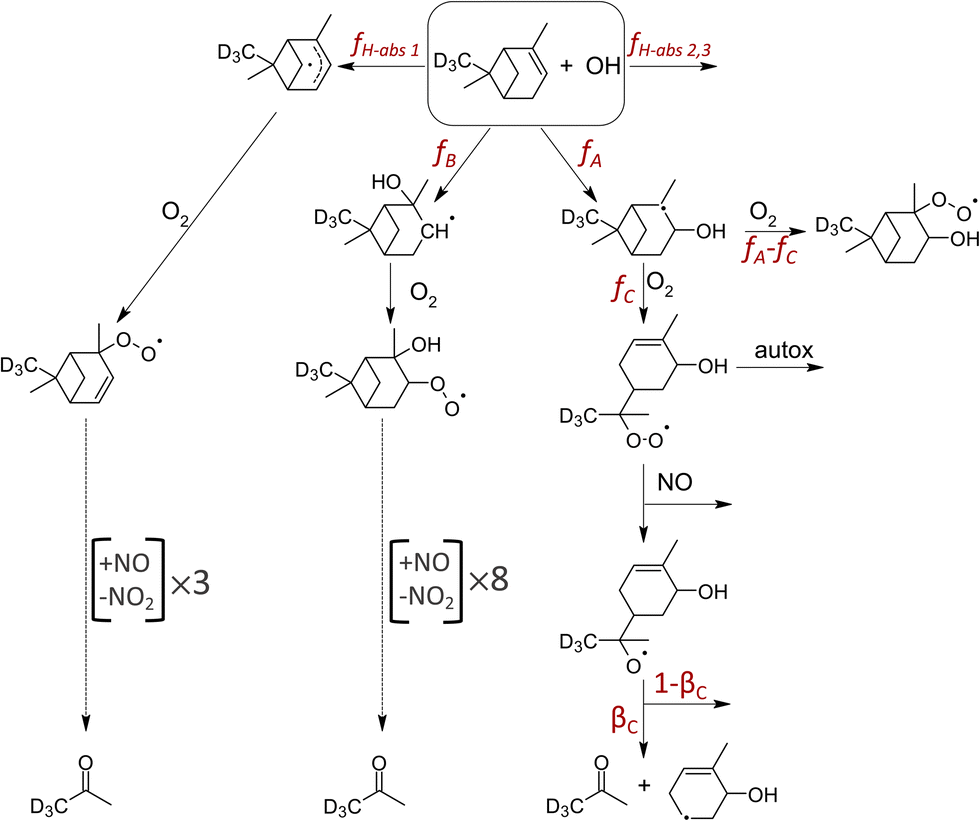 | ||
| Fig. 2 Schematic of the pathways from the OH-initiated oxidation of AP, adapted from Peeters et al.37 and Vereecken et al.16 The three potential pathways to acetone production and auto-oxidation are highlighted. | ||
| f C | β C | α C | |
|---|---|---|---|
| a Xu et al.18 report the quantum calculated branching ratio, or BRring-open (0.32 for AP and 0.44 for BP), which is the fraction of C-4 ring-opened peroxy radical formed following the OH-addition to the secondary position for AP and primary position for BP. We calculate fC using their reported values of BRring-open and the fractions of OH-addition (0.88 for AP and 0.901 for BP) and fractions to OH-addition to the tertiary (0.5 for AP) or primary (0.925 for BP) positions as reported by Vereecken et al.16 and Vereecken and Peeters.39 b Quantum calculations performed for AP, assumed to be identical for this work for BP. | |||
| AP | |||
| MCM23,24 | 0.075 | 1.0 | 0.125 |
| Vereecken and Peeters38 | 0.26 | 0.4 | 0.17 |
| Peeters et al.37 | 0.22 | 0.4 | 0.11 |
| Vereecken et al.16 | 0.22 | 0.0 | 0.11 |
| Xu et al.18 | 0.14a (=0.88 × 0.5 × 0.32) | ||
| Rolletter et al.42 | 0.83 | ||
| Piletic and Kleindienst41 | 0.19 | ||
| This study | 0.47 (0.2, 0.6) | 0.125 ± 0.08 | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| BP | |||
| Xu et al.18 | 0.36a (=0.90 × 0.925 × 0.44) | ||
| Vereecken et al.39 | 0.58 | ||
| Piletic and Kleindienst41 | 0.14 | ||
| This study | 0.66 (0.4, 0.8) | 0.125 ± 0.08b | |
Results from OH-oxidation of isotopically labeled AP that possess three deuterium atoms instead of three hydrogen atoms on one of the two methyl groups attached to the bridge of the C-4 ring (C10H13D3) and that from unlabeled AP (C10H16) are illustrated in Fig. 3a and b. The D3-labeled AP produces D3-labeled acetone, detected as C3H3D3ONH4+ (m/Z = 79.0945), while unlabeled acetone, detected as C3H6ONH4+ (m/Z = 76.0757), is unchanged relative to when oxidation is turned off (Fig. 3a). OH oxidation of unlabeled-AP is accompanied by a comparable increase in unlabeled acetone with no D3-acetone detected (Fig. 3b). These results are consistent with proposed mechanisms of AP and BP that report acetone is essentially only formed from the three C atoms that comprise the C-4 bridge.
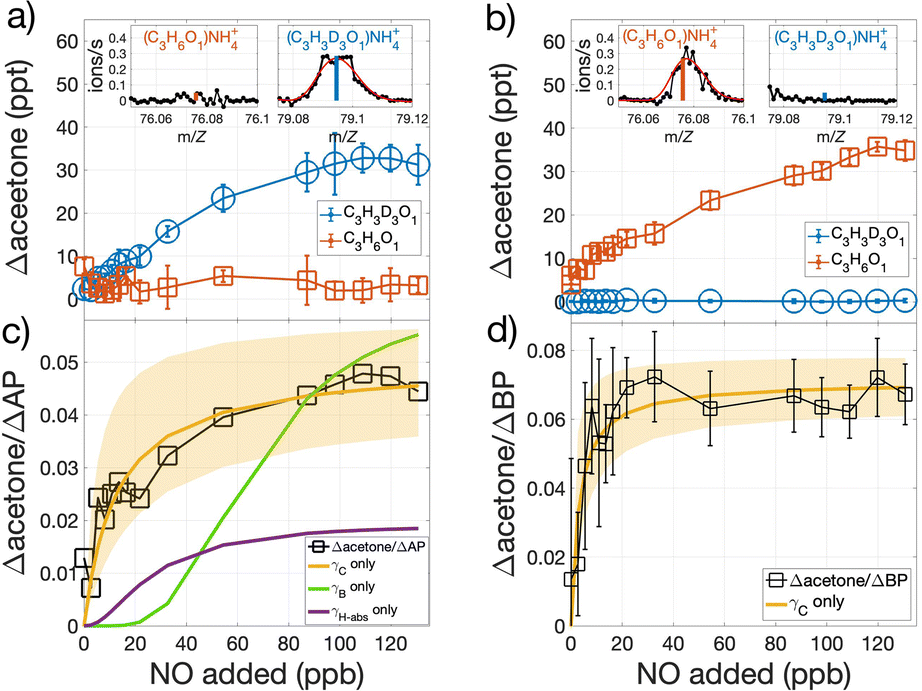 | ||
| Fig. 3 Mixing ratio of acetone produced during OH-oxidation of (a) D3-labeled and (b) unlabeled AP, as a function of NO added. The insets in (a) and (b) show the corresponding mass spectra of D3-labeled and unlabeled acetone in the presence of high NO. The ratio of observed acetone produced per modeled loss of (c) AP and (d) BP, as a function of NO added. (c) The slow rise in Δacetone/ΔAP at low NO values when assuming acetone is produced from only the fB pathway (γB only) or only the fH-abs pathway (γH-abs only), suggest negligible contribution compared to prompt formation from the fC pathway. Non-linear least-squares fits of Δacetone/ΔMT with respect to NO allows for the explicit determination of γC (=fC × βC) and kauto, utilizing eqn (3). Shading in (c) and (d) represent the range of Δacetone/ΔMT with respect to NO given the uncertainties of γC (0.059 (0.5, 0.6) for AP and 0.083 (0.8, 0.9) for BP) and kauto (2.9 (1.1, 4.7) s−1 for AP and 0.8 (0.4, 1.6) s−1 for BP). Values in parentheses represent range of values based on experimental and mechanistic uncertainties (see text). | ||
The isotopic labeling together with short (0.7 s) reaction timescales allow us to distinguish acetone formed promptly upon the reaction between NO and the ring-opened RO2versus those formed from the other two pathways, which require either three (fH-abs) or eight (fB) generations of O2 addition and NO reaction (Fig. 2). The observed acetone produced per AP reacted (Δacetone/ΔAP) as a function of NO is shown in Fig. 3c, alongside that modeled if the H-abstraction pathway (fH-abs) was the only acetone-producing pathway at a yield of 2.1% (γH-abs),16 and if OH-addition to the secondary position (fB) solely produced acetone at a yield of 6% (γB).16 The modeled evolution of Δacetone/ΔAP as a function of NO for neither comes close to simulating the observed Δacetone/ΔAP (Fig. 3c), due to the time required for multiple generations of NO reaction following the initial OH reaction. This result indicates that prompt formation of acetone as observed does not arise from either of these two pathways. In addition, the relative importance of the fB pathway as an acetone source is likely lower than currently reported.
Observations in Fig. 3c follow the trends expected if acetone is a product predominantly of the reaction between the ring-opened RO2 isomer (fC) and NO, which is in competition with a fast unimolecular reaction, as shown in eqn (1)–(3). In eqn (1), the acetone formation rate is defined by the reaction between NO and the ring-opened RO2 isomer where αC is the corresponding alkyl nitrate yield, and βC is the branching ratio of the resulting RO that undergo C–C scission to yield acetone (Fig. 2). Chemical loss of acetone by OH oxidation is negligible given the 0.7 s reaction time and because the AP or BP is in excess.
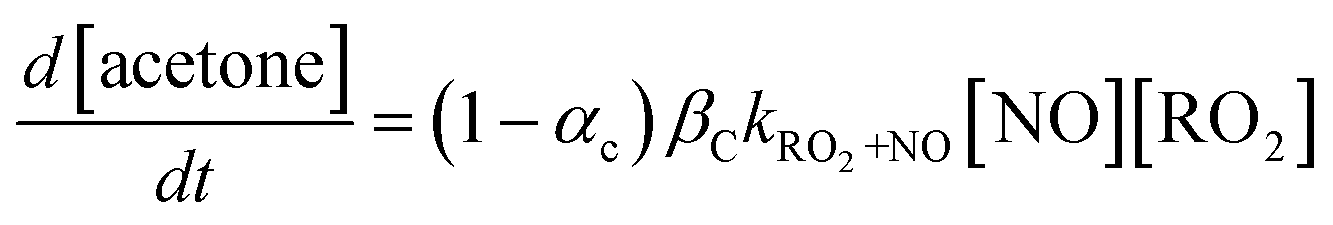 | (1) |
Eqn (2) shows the mass balance for the ring-opened RO2 isomer, produced at a fraction, fC, per reaction of OH with MT, and lost to bimolecular and unimolecular reactions, such as reaction with NO and auto-oxidation, respectively. Reactions with RO2 and HO2 become negligible with the addition of NO.
 | (2) |
Thus, Δacetone/ΔAP increases with NO and then plateaus as shown in eqn (3).
 | (3) |
Assuming negligible acetone contributions from the fB and fH-abs pathways, the absolute value that Δacetone/ΔMT approaches at high NO constrains the yield, γC (=fC × βC), of acetone produced per OH-oxidation of MT, for a given value of αC, for which we assume 0.15. The modeled variability in Δacetone/ΔMT (Fig. S3†) given the variability in reported αC values (Table 1) is within the uncertainties of γC derived from the non-linear least squares fit, shown shaded in Fig. 3c. The alkyl nitrate yields of all other RO2 — which can affect the production of closed-shell carbonyls, and hence, HO2, which promptly react with NO to form OH that then reacts with the excess AP or BP — likewise do not significantly affect Δacetone/ΔMT (Fig. S4†).
Assuming unity for βC, as currently reported in the MCM,23,24fC would be defined by γC, which are 0.059 (0.05, 0.07) and 0.083 (0.08, 0.09) for AP and BP, respectively (Fig. 3c and d). To provide an independent estimate of fC, we conducted quantum calculations to assess the relative importance of the five possible fates of the alkoxy radical (RO) stemming from the C-4 ring-opened isomer of AP, including the C–C bond scission path to yield acetone. These calculations show that the acetone-producing bond scission branching, βC, is either 0.07 (OH up) or 0.18 (OH down), depending on the diastereomer formed. The formation of an endocyclic ether and an intra-molecular H-shift dominate the RO fate (Fig. S5†). As our technique is unable to distinguish structure (OH up or down), we use the mean of the two calculated alkoxy radical branching ratio, βC (=0.125). As such, our results indicate fC of the C-4 ring-opening isomers are 0.47 (0.2, 0.6) for AP, and 0.66 (0.4, 0.8) (assuming identical βC for BP), where the uncertainty range indicated in the parentheses is due mainly to the fate of the alkoxy radical, not experimental variance which is of order 0.08. Contribution of acetone from H-abstraction pathway (fH-abs) and/or the OH-addition to the secondary position pathway (fB) would lower our calculated fC values. Though Xu et al.18 calculated C-4 ring opening branching ratio of 0.32 for the tertiary alkyl radical resulting from OH-addition to to AP, that ratio is 0.97 if determined from their observations of the three hydroxynitrate isomers (C10H17NO4). Our determination of fC of 0.47 and an OH-abstraction fraction of 0.12,16 imply OH-addition to form the secondary alkyl radical is at most 0.40.
In addition to allowing the resolution of different RO2 formation pathways, our short reaction time study constrains the unimolecular auto-oxidation rate (kauto), which is defined by the slope of Δacetone/ΔMT as a function of NO, per eqn (3). We perform a non-linear least-squares fit utilizing eqn (3) on the observed Δacetone/ΔMT versus NO to determine kauto and its uncertainty. We find kauto for the first generation C-4 ring-opened C10H17O3 peroxy radicals of AP and BP are 2.9 (1.1, 4.7) s−1 and 0.8 (0.4, 1.6) s−1, respectively where the uncertainty range in parentheses stems from least squares fit parameters. These determinations are independent of the calculations of fC and βC. The auto-oxidation rate for AP is comparable to those previously published. That for BP is on the lower end, though there is greater variance in published values for BP (Table 2). One possible reason for the discrepancy is that experimental studies such as ours and that of Xu et al.18 cannot distinguish diastereomers, which is relevant for AP.15,40 The ring-opened hydroxy peroxy radical of BP is not affected by stereoselectivity as the hydroxyl functional group is not attached to the 6-member ring. Even amongst computational studies, however, there is not perfect agreement on the rates and, at times, the relative importance of the different pathways. Such difficulties highlight the challenging nature of elucidating chemical mechanisms. For instance, a few computational studies have determined that the C-4 ring opened hydroxy peroxy radicals preferentially form a 6-member endocyclic peroxide (endo-6, see Table 2), in particular, for BP. The resulting C10H17O5 peroxy radical now with two O atoms within the newly-formed 6-member ring is, however, slow to undergo additional auto-oxidation given that it is ring-constrained.13,41
| APa | |
|---|---|
| a Stereoselectivity does not apply to the C-4 ring-opened hydroxy peroxy radical of BP given that the hydroxyl (-OH) functional group is not on the 6-member ring, whereas those of AP can exist as anti (-OH on the 6-member ring relative to the t-butyl peroxy group) and syn. | |
| Xu et al.18 | 4 (2, 6) s−1 |
| Møller et al. (anti)15 | 1.8 s−1 1.1 (1,5) + 0.37 (1,6) + 0.35 (endo-6) |
| Møller et al. (syn)15 | 2.5 s−1 0.16 (1,5) + 2.3 (endo-6) |
| Piletic and Kleindienst41 | 2.4 to 12.2 s−1 2.3 to 11.9 (1,5) + 0.08 to 0.31 (endo-6) |
| Vereecken et al. (anti)17 | 0.6 s−1 (endo-6) |
| Vereecken et al. (syn)17 | 2.6 s−1 (endo-6) |
| Berndt et al. (anti)13 | 1.7 s−1 0.66 (1,5) + 0.96 (1,6) + 0.079 (endo-6) |
| This study | 2.9 (1.1, 4.7) s−1 |
|
|
| BPa | |
| Xu et al.18 | 16 (11, 21) s−1 |
| Møller et al.15 | 5.7 s−1 1.4 (1,5) + 0.28 (1,6) + 4.0 (endo-6) |
| Piletic and Kleindienst41 | 4.5 to 21 s−1 2.7 to 14.9 (1,5) + 1.8 to 6.1 (endo-6) |
| Vereecken et al.17 | 0.6 s−1 (endo-6) |
| This study | 0.8 (0.4, 1.6) s−1 |
To determine whether the first generation C10H17O3 radicals undergo allylic or endocyclic auto-oxidation, we constrain kauto for the subsequent second generation peroxy radicals (C10H17O5) by comparing the modeled and observed evolution of C10H17O5 as a function of NO. In particular, we use the inflection point in NO at which the RO2 abundance starts to decrease, testing its rate of auto-oxidation versus reaction with NO. As shown in Fig. 4, kauto for the AP-derived C10H17O5 exceeds 20 s−1, as increasing the modeled rate above 20 s−1 does not result in significant improvement in model-observation agreement. For the BP-derived C10H17O5, kauto is 11.1 (6.2, 16) s−1. That the kauto of C10H17O5 are much greater than those of C10H17O3 suggests that the first generation C10H17O3 peroxy radicals preferentially undergo allylic and not endocyclic auto-oxidation. These observations are consistent with the mechanisms proposed by Berndt et al.,13 which report theoretical calculations that C10H17O5 (more so than C10H17O3) undergo rapid H-shifts to form the endo-cyclic peroxy radicals (C10H17O7), which are much slower to undergo an additional H-shift given the endo-peroxide ring. Our observational estimates of kauto for the third generation peroxy radicals (C10H17O7), however, are limited by the fact that their abundances approach instrument detection limits at moderate to high NO levels.
 | ||
| Fig. 4 Normalized traces of the second generation C10H17O5 peroxy radicals observed during the OH-oxidation of (a) AP and (b) BP, as a function of NO. Conducting numerous simulations to optimize model-observation agreements allowed us to define an optimal range of kauto for each, which for AP exceeds 20 s−1 and 11.1 (6.2, 16) s−1 for BP. Three simulated traces of C10H17O5 with widely varying rates of their auto-oxidation are shown. | ||
Lastly, recent studies offer contrasting reports on the importance of the H-abstraction pathway. Berndt,43 utilizing a fast flow tube coupled to an ethylaminium ionization mass spectrometer observed negligible products of H-abstraction by OH from α-pinene, whereas Shen et al.44 using an atmospheric simulation chamber coupled to a nitrate ionization mass spectrometer found that the dominant fraction of the products, namely HOMs, from OH oxidation of α-pinene is from H-abstraction, not OH-addition. At low NO, we observed an H-abstraction product (C10H15O4) only for AP, but not BP (Fig. S6†). The amount of C10H15O4, assuming comparable sensitivities to the C10H17Ox RO2, suggest H-abstraction yield of ∼3%, similar to the theoretical work (9%) of Vereecken et al.16 Moreover, at low NO, the C10H17Ox RO2 are dominated by those possessing 3, 5, and 7 oxygen atoms (Fig. S6†), consistent with the observations of Berndt.43 A more detailed analysis of the H-abstraction pathway is beyond the scope of this paper.
4 Conclusions
We present a novel transverse atmospheric pressure ammonium adduct ionization mass spectrometer coupled to a fast flow tube to directly observe OH-initiated oxidation products of AP and BP, including peroxy radicals and those formed from their reaction with NO. By systematically adding NO to probe the fate of these peroxy radicals in competition with auto-oxidation over short timescales (∼0.7 s), with and without isotopically labeled precursors, we distinguish prompt acetone formation from the C-4 ring-opening pathway versus those that require multiple generations of NO reactions. Using quantum calculations to constrain the branching ratio of acetone formed from the ring-opened alkoxy radical (0.125), we find that the fractions of OH-oxidation of AP and BP that form the ring-opened peroxy radicals are 0.47 (0.2, 0.6) and 0.66 (0.4, 0.8), respectively. We also determine a narrow range for the auto-oxidation rates of these first-generation ring-opened peroxy radicals as well as of their associated auto-oxidation products, thereby providing a more complete description of HOM production channels from OH oxidation of AP and BP.Author contributions
JAT: conceptualization. SI and TK: theoretical calculations. JGV, JL, and RJT: synthesis of D3-α-pinene. BHL: experimentation, analysis, writing.Conflicts of interest
“There are no conflicts to declare”.Acknowledgements
This work was funded by National Science Foundation Environmental Chemical Sciences (grant no. CHE-1807204), and the European Research Council under the European Union's Horizon 2020 research and innovation programme under Grant No. 101002728. The authors acknowledge Ezra Wood (Drexel University) for his instructions on HOx generation, and Henrik G. Kjærgaard (University of Copenhagen) and Kristian H. Møller (University of Copenhagen) for their suggestions on theoretical calculations.Notes and references
- A. Guenther, T. Karl, P. Harley, C. Wiedinmyer, P. I. Palmer and C. Geron, Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 2006, 6, 3181–3210, DOI:10.5194/acp-6-3181-2006.
- P. Messina, J. Lathiere, K. Sindelarova, N. Vuichard, C. Granier, J. Ghattas, A. Cozic and D. A. Hauglustaine, Global biogenic volatile organic compound emissions in the ORCHIDEE and MEGAN models and sensitivity to key parameters, Atmos. Chem. Phys., 2016, 16, 14169–14202, DOI:10.5194/acp-16-14169-2016.
- T. E. Pierce, BEIS3 Version 0.9, Environmental Protection Agency, Triangle Park, N.C, 2001, Available via anonymous ftp at http://ftp://ftp.epa.gov/amd/asmd/beis3v09/ Search PubMed.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Evolution of Organic Aerosols in the Atmosphere, Science, 2009, 326(5959), 1525–1529, DOI:10.1126/science.1180353.
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. Van Dingenen, B. Ervens, A. Nenes, C. J. Nielsen, E. Swietlicki, J. P. Putaud, Y. Balkanski, S. Fuzzi, J. Horth, G. K. Moortgat, R. Winterhalter, C. E. L. Myhre, K. Tsigaridis, E. Vignati, E. G. Stephanou and J. Wilson, Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 2005, 5, 1053–1123, DOI:10.5194/acp-5-1053-2005.
- Q. Zhang, J. L. Jimenez, M. R. Canagaratna, J. D. Allan, H. Coe, I. Ulbrich, M. R. Alfarra, A. Takami, A. M. Middlebrook, Y. L. Sun, K. Dzepina, E. Dunlea, K. Docherty, P. F. DeCarlo, D. Salcedo, T. Onasch, J. T. Jayne, T. Miyoshi, A. Shimono, S. Hatakeyama, N. Takegawa, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, P. Williams, K. Bower, R. Bahreini, L. Cottrell, R. J. Griffin, J. Rautiainen, J. Y. Sun, Y. M. Zhang and D. R. Worsnop, Geophys. Res. Lett., 2007, 34, L13801, DOI:10.1029/2007GL029979.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipila, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, F. Lopez-Hilfiker, S. Andres, I.-H. Acir, M. Rissanen, T. Jokinen, S. Schobesberger, J. Kangasluoma, J. Kontkanen, T. Nieminen, T. Kurten, L. B. Nielsen, S. Jorgensen, H. G. Kjaergaard, M. Canagaratna, M. Dal Maso, T. Berndt, T. Petaja, A. Wahner, V.-M. Kerminen, M. Kulmala, D. R. Worsnop, J. Wildt and T. F. Mentel, A large source of low-volatility secondary organic aerosol, Nature, 2014, 506, 476–479, DOI:10.1038/nature13032.
- B. Lee, E. L. D’Ambro, F. D. Lopez-Hilfiker, S. Schobesberger, C. Mohr, M. A. Zawadowicz, J. M. Liu, J. E. Shilling, W. W. Hu, B. B. Palm, J. L. Jimenez, L. Q. Hao, A. Virtanen, H. F. Zhang, A. H. Goldstein, H. O. T. Pye and J. A. Thornton, Resolving Ambient Organic Aerosol Formation and Aging Pathways with Simultaneous Molecular Composition and Volatility Observations, ACS Earth Space Chem., 2020, 4(3), 391–402, DOI:10.1021/acsearthspacechem.9b00302.
- L. Xu, S. Suresh, H. Guo, R. J. Weber and N. L. Ng, Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates, Atmos. Chem. Phys., 2015, 15, 7307–7336, DOI:10.5194/acp-15-7307-2015.
- H. F. Zhang, L. D. Yee, B. H. Lee, M. P. Curtis, D. R. Worton, G. Isaacman-VanWertz, J. H. Offenberg, M. Lewandowski, T. E. Kleindienst, M. R. Beaver, A. L. Holder, W. A. Lonneman, K. S. Docherty, M. Jaoui, H. O. T. Pye, W. W. Hu, D. A. Day, P. Campuzano-Jost, J. L. Jimenez, H. Y. Guo, R. J. Weber, J. de Gouw, A. R. Koss, E. S. Edgerton, W. Brune, C. Mohr, F. D. Lopez-Hilfiker, A. Lutz, N. M. Kreisberg, S. R. Spielman, S. V. Hering, K. R. Wilson, J. A. Thornton and A. H. Goldstein, Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, Proc. Natl. Acad. Sci. U.S.A., 2018, 115(9), 2038–2043, DOI:10.1073/pnas.1717513115.
- M. M. Coggon, G. I. Gkatzelis, B. C. McDonald, J. B. Gilman, R. H. Schwantes, N. Abuhassan, K. C. Aikin, M. F. Arend, T. A. Berkoff, S. S. Brown, T. L. Campos, R. R. Dickerson, G. Gronoff, J. F. Hurley, G. Isaacman-VanWertz, A. R. Koss, M. Li, S. A. McKeen, F. Moshary, J. Peischl, V. Pospisilova, X. R. Ren, A. Wilson, Y. H. Wu, M. Trainer and C. Warneke, Volatile chemical product emissions enhance ozone and modulate urban chemistry, Proc. Natl. Acad. Sci. U.S.A., 2021, 118(32) DOI:10.1073/pnas.2026653118.
- B. C. McDonald, J. A. de Gouw, J. B. Gilman, S. H. Jathar, A. Akherati, C. D. Cappa, J. L. Jimenez, J. Lee-Taylor, P. L. Hayes, S. A. McKeen, Y. Y. Cui, S. W. Kim, D. R. Gentner, G. Isaacman-VanWertz, A. H. Goldstein, R. A. Harley, G. J. Frost, J. M. Roberts, T. B. Ryerson and M. Trainer, Volatile chemical products emerging as largest petrochemical source of urban organic emissions, Science, 2018, 359(6377), 760–764, DOI:10.1126/science.aaq0524.
- T. Berndt, S. Richters, T. Jokinen, N. Hyttinen, T. Kurten, R. V. Otkjaer, H. G. Kjaergaard, F. Stratmann, H. Herrmann, M. Sipila, M. Kulmala and M. Ehn, Nat. Commun., 2016, 7, 13677, DOI:10.1038/ncomms13677.
- N. C. Eddingsaas, C. L. Loza, L. D. Yee, J. H. Seinfeld and P. O. Wennberg, α-pinene photooxidation under controlled chemical conditions – Part 1: Gas-phase composition in low- and high-NOx environments, Atmos. Chem. Phys., 2012, 12, 6489–6504, DOI:10.5194/acp-12-6489-2012.
- K. H. Moller, R. V. Otkjaer, J. Chen and H. G. Kjaergaard, Double Bonds Are Key to Fast Unimolecular Reactivity in First-Generation Monoterpene Hydroxy Peroxy Radicals, J. Phys. Chem. A, 2020, 124(14), 2885–2896, DOI:10.1021/acs.jpca.0c01079.
- L. Vereecken, J. F. Muller and J. Peeters, Low-volatility poly-oxygenates in the OH-initiated atmospheric oxidation of α-pinene: impact of non-traditional peroxyl radical chemistry, Phys. Chem. Chem. Phys., 2007, 9, 5241–5248, 10.1039/b708023a.
- L. Vereecken and J. Peeters, Nontraditional (Per)oxy Ring-Closure Paths in the Atmospheric Oxidation of Isoprene and Monoterpenes, J. Phys. Chem. A, 2004, 108, 5197–5204, DOI:10.1021/jp049219g.
- L. Xu, K. H. Moller, J. D. Crounse, R. V. Otkjwr, H. G. Kjaergaard and P. O. Wennberg, Unimolecular Reactions of Peroxy Radicals Formed in the Oxidation of α-Pinene and β-Pinene by Hydroxyl Radicals, J. Phys. Chem. A, 2019, 123(8), 1661–1674, DOI:10.1021/acs.jpca.8b11726.
- R. C. Xu, J. A. Thornton, B. Lee, Y. X. Zhang, L. Jaegle, F. D. Lopez-Hilfiker, P. Rantala and T. Petaja, Global simulations of monoterpene-derived peroxy radical fates and the distributions of highly oxygenated organic molecules (HOMs) and accretion products, Atmos. Chem. Phys., 2022, 22, 5477–5494, DOI:10.5194/acp-22-5477-2022.
- Y. Zhao, J. K. Chan, F. D. Lopez-Hilfiker, M. A. McKeown, E. L. D’Ambro, J. G. Slowik, J. A. Riffell and J. A. Thornton, An electrospray chemical ionization source for real-time measurement of atmospheric organic and inorganic vapors, Atmos. Meas. Tech., 2017, 10, 3609–3625, DOI:10.5194/amt-10-3609-2017.
- A. Hansel, W. Scholz, B. Mentler, L. Fischer and T. Berndt, Detection of RO2 radicals and other products from cyclohexene ozonolysis with NH4+ and acetate chemical ionization mass spectrometry, Atmos. Environ., 2018, 186, 248–255, DOI:10.1016/j.atmosenv.2018.04.023.
- A. Zaytsev, M. Breitenlechner, A. R. Koss, C. Y. Lim, J. C. Rowe, J. H. Kroll and F. N. Keutsch, Using collision-induced dissociation to constrain sensitivity of ammonia chemical ionization mass spectrometry (NH4+ CIMS) to oxygenated volatile organic compounds, Atmos. Meas. Tech., 2019, 12, 1861–1870, DOI:10.5194/amt-12-1861-2019.
- M. E. Jenkin, S. M. Saunders and M. J. Pilling, The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 1997, 31(1), 81–104, DOI:10.1016/S1352-2310(96)00105-7.
- S. M. Saunders, M. E. Jenkin, R. G. Derwent and M. J. Pilling, Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 2003, 3, 161–180, DOI:10.5194/acp-3-161-2003.
- M. A. Upshur, H. M. Chase, B. F. Strick, C. J. Ebben, L. Fu, H. F. Wang, R. J. Thomson and F. M. Geiger, Vibrational Mode Assignment of α-Pinene by Isotope Editing: One Down, Seventy-One To Go, J. Phys. Chem. A, 2016, 120(17), 2684–2690, DOI:10.1021/acs.jpca.6b01995.
- K. H. Moller, R. V. Otkjaer, N. Hyttinen, T. Kurten and H. G. Kjaergaard, Cost-Effective Implementation of Multiconformer Transition State Theory for Peroxy Radical Hydrogen Shift Reactions, J. Phys. Chem. A, 2016, 120(51), 10072–10087, DOI:10.1021/acs.jpca.6b09370.
- Wavefunction, Inc., Spartan’18 version 1.4.5, Irvine, CA Search PubMed.
- J. D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620, 10.1039/B810189B.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90(1007) DOI:10.1063/1.456153.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions, J. Chem. Phys., 1992, 96(6796) DOI:10.1063/1.462569.
- H. J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Hesselmann, D. Kats, A. Kohn, T. Korona, D. A. Kreplin, Q. L. Ma, T. F. Miller, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, The Molpro quantum chemistry package, J. Chem. Phys., 2020, 152, 144107, DOI:10.1063/5.0005081.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The ORCA quantum chemistry program package, J. Chem. Phys., 2020, 152, 224108, DOI:10.1063/5.000460.
- A. C. Vaucher and M. Reiher, Steering Orbital Optimization out of Local Minima and Saddle Points Toward Lower Energy, J. Chem. Theory Comput., 2017, 13(3), 1219–1228, DOI:10.1021/acs.jctc.7b00011.
- D. R. Glowacki, C. H. Liang, C. Morley, M. J. Pilling and S. H. Robertson, MESMER: An Open-Source Master Equation Solver for Multi-Energy Well Reactions, J. Phys. Chem. A, 2012, 116(38), 9545–9560, DOI:10.1021/jp3051033.
- S. Iyer, M. P. Rissanen, R. Valiev, S. Barua, J. E. Krechmer, J. Thornton, M. Ehn and T. Kurten, Molecular mechanism for rapid autoxidation in α-pinene ozonolysis, Nat. Commun., 2021, 12, 878, DOI:10.1038/s41467-021-21172-w.
- T. Kurten, K. H. Moller, T. B. Nguyen, R. H. Schwantes, P. K. Misztal, L. P. Su, P. O. Wennberg, J. L. Fry and H. G. Kjaergaard, Alkoxy Radical Bond Scissions Explain the Anomalously Low Secondary Organic Aerosol and Organonitrate Yields From α-Pinene + NO3, J. Phys. Chem. Lett., 2017, 8(13), 2826–2834, DOI:10.1021/acs.jpclett.7b01038.
- J. Peeters, L. Vereecken and G. Fantechi, The detailed mechanism of the OH-initiated atmospheric oxidation of α-pinene: a theoretical study, Phys. Chem. Chem. Phys., 2001, 3, 5489–5504, 10.1039/b106555f.
- L. Vereecken and J. Peeters, Theoretical Study of the Formation of Acetone in the OH-Initiated Atmospheric Oxidation of α-Pinene, J. Phys. Chem. A, 2000, 104(47), 11140–11146, DOI:10.1021/jp0025173.
- L. Vereecken and J. Peeters, A theoretical study of the OH-initiated gas-phase oxidation mechanism of β-pinene (C10H16): first generation products, Phys. Chem. Chem. Phys., 2012, 14, 3802–3815, 10.1039/c2cp23711c.
- K. H. Moller, E. Praske, L. Xu, J. D. Crounse, P. O. Wennberg and H. G. Kjaergaard, Stereoselectivity in Atmospheric Autoxidation, J. Phys. Chem. Lett., 2019, 10(20), 6260–6266, DOI:10.1021/acs.jpclett.9b01972.
- I. R. Piletic and T. E. Kleindienst, Rates and Yields of Unimolecular Reactions Producing Highly Oxidized Peroxy Radicals in the OH-Induced Autoxidation of α-Pinene, β-Pinene, and Limonene, J. Phys. Chem. A, 2022, 126(1), 88–100, DOI:10.1021/acs.jpca.1c07961.
- M. Rolletter, M. Kaminski, I. H. Acir, B. Bohn, H. P. Dorn, X. Li, A. Lutz, S. Nehr, F. Rohrer, R. Tillmann, R. Wegener, A. Hofzumahaus, A. Kiendler-Scharr, A. Wahner and H. Fuchs, Investigation of the α-pinene photooxidation by OH in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 2019, 19, 11635–11649, DOI:10.5194/acp-19-11635-2019.
- T. Berndt, Peroxy Radical Processes and Product Formation in the OH Radical-Initiated Oxidation of α-Pinene for Near-Atmospheric Conditions, J. Phys. Chem. A, 2021, 125(41), 9151–9160, DOI:10.1021/acs.jpca.1c05576.
- H. Shen, L. Vereecken, S. Kang, I. Pullinen, H. Fuchs, D. Zhao and T. F. Mentel, Unexpected significance of a minor reaction pathway in daytime formation of biogenic highly oxygenated organic compounds, Sci. Adv., 2022, 8(42) DOI:10.1126/sciadv.abp8702.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ea00133k |
| This journal is © The Royal Society of Chemistry 2023 |