
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Observed in-plume gaseous elemental mercury depletion suggests significant mercury scavenging by volcanic aerosols†
Alkuin M.
Koenig
 a,
Olivier
Magand
ab,
Clemence
Rose
c,
Andrea
Di Muro
def,
Yuzo
Miyazaki
g,
Aurelie
Colomb
c,
Matti
Rissanen
hi,
Christopher F.
Lee
jk,
Theodore K.
Koenig
jkl,
Rainer
Volkamer
jk,
Jerome
Brioude
m,
Bert
Verreyken
nopq,
Tjarda
Roberts
rs,
Brock A.
Edwards
t,
Karine
Sellegri
c,
Santiago
Arellano
u,
Philippe
Kowalski
de,
Alessandro
Aiuppa
v,
Jeroen E.
Sonke
w and
Aurélien
Dommergue
a
a,
Olivier
Magand
ab,
Clemence
Rose
c,
Andrea
Di Muro
def,
Yuzo
Miyazaki
g,
Aurelie
Colomb
c,
Matti
Rissanen
hi,
Christopher F.
Lee
jk,
Theodore K.
Koenig
jkl,
Rainer
Volkamer
jk,
Jerome
Brioude
m,
Bert
Verreyken
nopq,
Tjarda
Roberts
rs,
Brock A.
Edwards
t,
Karine
Sellegri
c,
Santiago
Arellano
u,
Philippe
Kowalski
de,
Alessandro
Aiuppa
v,
Jeroen E.
Sonke
w and
Aurélien
Dommergue
a
aUniv. Grenoble Alpes, CNRS, INRAE, IRD, Grenoble INP, IGE, 38000 Grenoble, France
bObservatoire des Sciences de l'Univers à La Réunion (OSU-R), UAR 3365, CNRS, Université de La Réunion, Météo France, 97744 Saint-Denis, La Réunion, France
cUniversité Clermont Auvergne, CNRS, Laboratoire de Météorologie Physique (LaMP), 63000 Clermont-Ferrand, France
dObservatoire Volcanologique du Piton de la Fournaise, Institut de physique du globe de Paris, 97418, La Plaine des Cafres, France
eInstitut de physique du globe de Paris, CNRS, Université de Paris, 75005, Paris, France
fUniversité Claude Bernard Lyon1, Observatoire de Lyon, Laboratoire LGL-TPE, Lyon, France
gInstitute of Low Temperature Science, Hokkaido University, Sapporo, 060-0819, Japan
hAerosol Physics Laboratory, Tampere University, FI-33101 Tampere, Finland
iDepartment of Chemistry, University of Helsinki, FI-00014 Helsinki, Finland
jDepartment of Chemistry, University of Colorado Boulder, Boulder, CO 80309, USA
kCooperative Institute for Research in Environmental Sciences, University of Colorado, Boulder, CO 80309, USA
lKey Joint Laboratory of Environmental Simulation and Pollution Control, BIC-ESAT and IJRC, College of Environmental Sciences and Engineering, Peking University, Beijing 100871, China
mLaboratoire de l'Atmosphère et des Cyclones, UMR 8105, CNRS, Université de La Réunion, Météo France, 97744 Saint-Denis, La Réunion, France
nRoyal Belgian Institute for Space Aeronomy, 1180 Brussels, Belgium
oDepartment of Chemistry, Ghent University, 9000 Ghent, Belgium
pLaboratoire de l'Atmosphère et des Cyclones, UMR 8105, CNRS, Université de La Réunion, Météo France, 97744 Saint-Denis, France
qBiosystems Dynamics and Exchanges (BIODYNE), Gembloux Agro-Biotech, University of Liège, 5030 Gembloux, Belgium
rLPC2E, Laboratoire de Physique et de Chimie de l'Environnement et de l`Espace, University Orléans, CNRS, Orléans, France
sLMD/IPSL, ENS, Université PSL, École Polytechnique, Institut Polytechnique de Paris, Sorbonne Université, CNRS, Paris, France
tCentre for Earth Observation Science, Department of Environment and Geography, University of Manitoba, Winnipeg, R3T 2N2, Canada
uDepartment of Space, Earth and Environment, Chalmers University of Technology, Göteborg, Sweden
vDipartimento di Scienze della Terra e del Mare, Università di Palermo, Palermo, Italy
wGéosciences Environnement Toulouse, CNRS/IRD/Université Paul Sabatier Toulouse 3, Toulouse, France
First published on 7th August 2023
Abstract
Terrestrial volcanism is known to emit mercury (Hg) into the atmosphere. However, despite many years of investigation, its net impact on the atmospheric Hg budget remains insufficiently constrained, in part because the transformations of Hg in volcanic plumes as they age and mix with background air are poorly understood. Here we report the observation of complete gaseous elemental mercury (GEM) depletion events in dilute and moderately aged (∼3–7 hours) volcanic plumes from Piton de la Fournaise on Réunion Island. While it has been suggested that co-emitted bromine could, once photochemically activated, deplete GEM in a volcanic plume, we measured low bromine concentrations in both the gas- and particle-phase and observed complete GEM depletion even before sunrise, ruling out a leading role of bromine chemistry here. Instead, we hypothesize that the GEM depletions were mainly caused by gas–particle interactions with sulfate-rich volcanic particles (mostly of submicron size), abundantly present in the dilute plume. We consider heterogeneous GEM oxidation and GEM uptake by particles as plausible manifestations of such a process and derive empirical rate constants. By extrapolation, we estimate that volcanic aerosols may scavenge 210 Mg y−1 (67–480 Mg y−1) of Hg from the atmosphere globally, acting effectively as atmospheric mercury sink. While this estimate is subject to large uncertainties, it highlights that Hg transformations in aging volcanic plumes must be better understood to determine the net impact of volcanism on the atmospheric Hg budget and Hg deposition pathways.
Environmental significanceIt has long been known that volcanoes emit mercury, a potent neurotoxin, into the atmosphere. However, the post-emission behaviour and transformations of mercury in a volcanic plume, an extreme and chemically active environment, remain mostly unknown. Such transformations are of great importance as they determine if mercury is rather transported globally or deposited regionally. Here we present experimental evidence that, within a volcanic plume, mercury can get efficiently converted into forms that sediment quickly and are washed out by rain. This implies that terrestrial volcanism may not only directly emit mercury into the atmosphere but also indirectly remove it. While this would reduce the atmospheric mercury burden, it would also increase mercury deposition and human exposure in volcanically active regions. |
1. Introduction
While terrestrial volcanism is thought to be an important natural primary source of mercury (Hg) to the atmosphere, the magnitude of volcanic Hg fluxes remains highly uncertain. Estimates1 range from 45 Mg y−1 to 2000 Mg y−1, with the most recent study2 suggesting Hg emissions of 179 ± 39 Mg y−1 and 20 ± 20 Mg y−1 (mean ± standard deviation) for passive and eruptive volcanic degassing, respectively. These estimates were mostly obtained by combining the estimated global volcanic SO2 flux with a Hg/SO2 emission ratio. Observed Hg/SO2 emission ratios, however, can vary by several orders of magnitude between different volcanic systems.1,3The speciation of Hg in volcanic plumes is less studied than total Hg emissions and is thus subject to even larger uncertainties.1 This is problematic, as the fate of Hg in volcanic plumes and its environmental impact depends importantly on Hg speciation and in-plume transformations. Gaseous elemental mercury (GEM) is water-insoluble and thus not significantly removed from the atmosphere by wet deposition. Reactive mercury, on the other hand, whether in the gas phase as gaseous oxidized mercury (GOM) or attached to particles as particle-bound mercury (PBM), is considerably more water-soluble than GEM, making it subject to significant wet deposition. In addition, reactive mercury tends to have significantly larger dry deposition velocities than GEM.4 GEM consequently has a relatively long atmospheric lifetime (i.e. e-folding time) of ∼6–12 months which allows for global circulation and dispersal,5,6 while GOM and PBM have much shorter lifetimes (hours to days)7 and thus more regional impacts through localized deposition.
Volcanic plume chemistry is complex, particularly because conditions within a plume can differ greatly from typical atmospheric conditions, for example with regards to temperature, the concentration of radicals, particle loading, acidity, and sulfur and halogen contents.8 As the initial plume cools and mixes with background air, the speciation of atmospheric Hg likely evolves.9,10 Indeed, while emitted Hg is mainly in the form of GEM when it exits the vent,1,9,11,12 partial oxidation of volcanic GEM has been observed to occur within minutes upon plume cooling and dilution.13
As volcanoes can be significant emitters of halogens,14,15 it has been hypothesized that bromine chemistry, more specifically the so-called “bromine explosion”, could cause rapid GEM oxidation in volcanic plumes.10,16,17 The “bromine explosion” is an autocatalytic and heterogeneous photochemical reaction mechanism that causes the rapid generation of bromine radicals,10,16,17 one of the most important GEM oxidants in the atmosphere.5,6,18 This process is regularly observed at polar sunrise,19–21 where it can cause complete depletion of GEM.22,23 Von Glasow (2010)10 suggested that bromine-related in-plume GEM oxidation could significantly reduce the net contribution of volcanism to the atmospheric Hg burden by shortening the atmospheric lifetime of volcanic Hg as well as background Hg entrained during plume dilution. If GEM can indeed be efficiently scavenged by other plume constituents, GEM concentrations in a volcanic plume could conceivably fall below the atmospheric background if the in-plume scavenging outweighs volcanic emission. However, perhaps due to the difficulty of obtaining Hg measurements in an aged volcanic plume, such an event has not yet been reported.
Here we present GEM depletion events in dilute and moderately aged (∼3–7 hours) volcanic plumes from Piton de La Fournaise on La Réunion Island, observed ∼38.5 km away at Maïdo mountain observatory (2160 masl). With the help of a large set of ancillary observations, we thoroughly describe the observed GEM depletions, explore possible drivers, and discuss implications for the atmospheric Hg budget.
2. Methodology
2.1. Maïdo observatory and volcanism on Réunion Island
Maïdo observatory24 (latitude: −21.0792°; longitude: 55.38°) lies at an altitude of 2160 masl on the western flank of La Réunion Island (see Fig. 1 and Table 1), a relatively small tropical island in the Southern Hemisphere Indian ocean. GEM concentrations at Maïdo are mostly below 1 ng m−3, but show a marked and regular diurnal variation that has been attributed to mixing processes and surface photo-reemission.25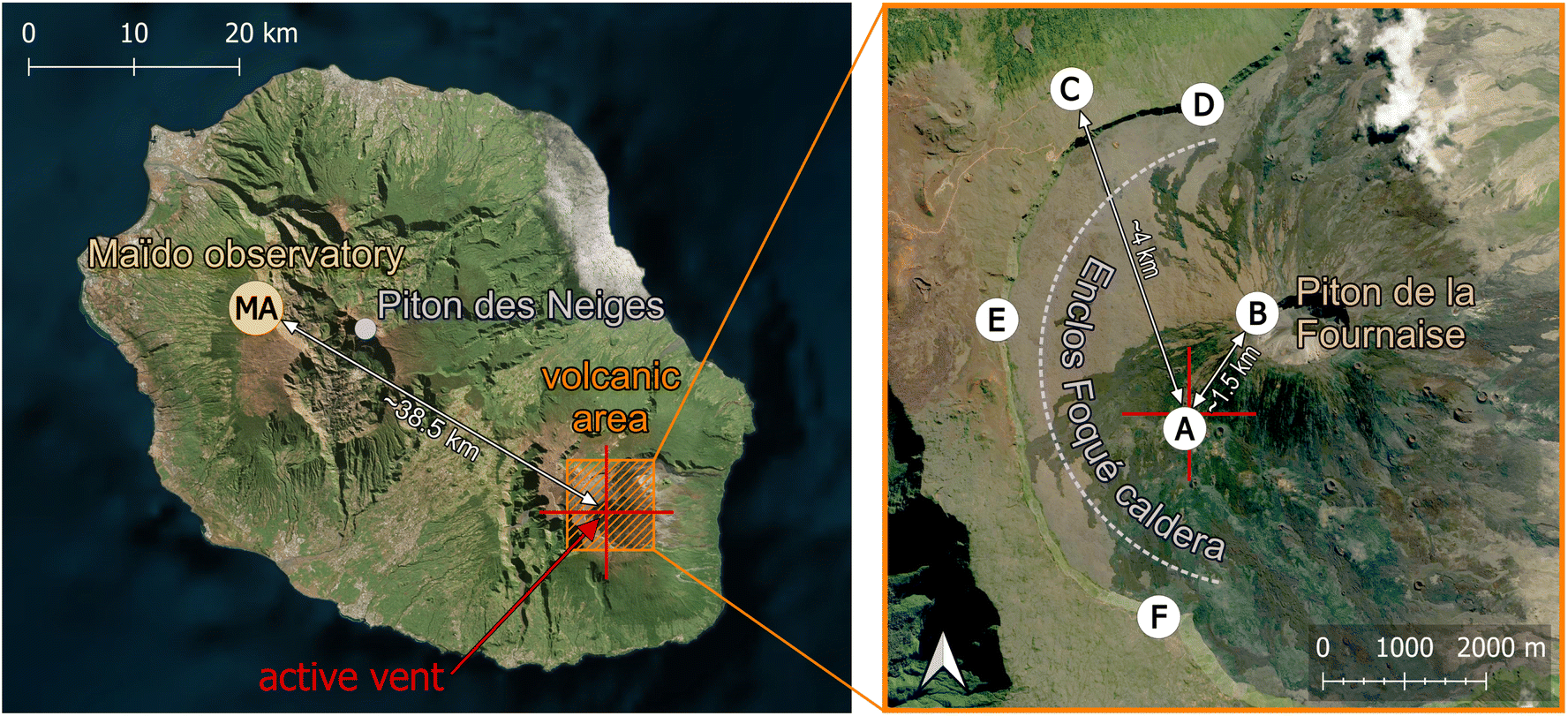 | ||
| Fig. 1 Site overview. See Table 1 for information about instrumentation. Aerial images obtained from Bing maps. | ||
La Réunion has been shaped by volcanic activity and is characterized by its rugged orography. Two volcanoes form most of the island, the dormant Piton de Neiges, whose ∼3070 masl high summit is the highest peak of the island and is located about 10 km east of Maïdo observatory, and the very active Piton de la Fournaise (∼2630 masl), about 40 km south-east of the observatory. Piton de la Fournaise is a basaltic hot spot shield volcano with one effusive eruption every ∼10 months on average.26,27 While no studies investigated the mercury degassing of Piton de la Fournaise, at least one study attributed high soil mercury concentrations on La Réunion Island to the island's volcanic history.28
| Maïdo observatory (MA) | Volcanic area | |||
|---|---|---|---|---|
| Magnitude | Instrument | Location | Magnitude | Instrument |
| GEM | Tekran 2537A | A | GEM | Passive samplers |
| SO2 concentrations | Teledyne API T100U analyser | B | SO2, H2S, CO2, H2O concentrations; pressure and temperature | Multi-GaS analyzer |
| Aerosol size distribution | DMPS | C | Temperature, RH, wind speed and direction | MERCURY weather automatic station |
| Water-soluble inorganic ions and organic carbon | High-volume air sampler & cascade impactor | D, E and F | SO2 flux | Scanning DOAS |
| SO2, BrO column densities | CU MAX-DOAS | |||
| H2SO4 (gas phase) | APi-TOF | |||
| Temperature, RH, wind speed and direction | Vaisala Weather Transmitter WXT520 | |||
| Solar radiation | SPN1 Sunshine Pyranometer | |||
The volcanic episode of Piton de la Fournaise that is of interest for the present study lasted from the 27th of April 2018 to the 1st of June 2018, and GEM depletion was observed on the 29th of April. The main active vent of the eruption was at coordinates of −21.254° (latitude) and 55.700° (longitude), at an elevation of 2229 masl (see red cross in Fig. 1).
2.2. Instrumental
2.2.1.1. Gaseous elemental mercury (GEM). GEM was measured at Maïdo with a 15 minutes time resolution with a Tekran 2537A analyzer, as already described in detail in Koenig et al. (2023).25 The instrument was maintained and regularly calibrated according to standard GMOS (Global Mercury Observation System) procedures. While standard GMOS procedures usually imply working with hourly averages, here we work directly with the 15 minutes resolution data set to better constrain the time evolution of the observed volcanic GEM depletion events, which were of short duration. We report the concentrations of GEM, as well as all other in situ measured magnitudes described further below, at standard conditions of 273.14 K and 1013.25 hPa (STP). Conversion to STP was achieved by calculating the volume of sampled air at 273.14 K and 1013.25 hPa from the volume of sampled air at ambient temperature and pressure, using the ideal gas law.
The operation of the Tekran 2537A is based on mercury amalgamation on a gold trap and subsequent thermal desorption.29 To allow for continuous observations, two different gold traps (here trap “A” and trap “B”) are alternated. As trap “A” was operating at full efficiency during the volcanic episode on the 29th of April 2018, we assign a relative uncertainty of 10% (coverage factor k = 2) to all trap-A-based observations. Trap B, however, was operating with reduced efficiency, leading to several observations being rejected by our strict QA/QC procedures.30 In addition, we assign a larger relative uncertainty of 20% (k = 2) to all trap-B- based observations, even if flagged “valid”.
We assigned half the instrument detection limit to all instrument readings below this limit. Based on the results from Ambrose (2017)29 and Slemr et al. (2016),31 we considered 1 pg of mass on the gold trap as the instrumental detection limit under automated peak calibration. At a 15 minutes sampling interval and the used flow rate of 1.3 L min−1 (STP), this corresponds to a detection limit of 0.051 ng m−3. For uncertainty calculations, we assumed a rectangular (i.e. uniform) distribution between 0.0 ng m−3 and 0.051 ng m−3.
2.2.1.2. Meteorological parameters. Atmospheric pressure, temperature, relative humidity, wind speed, and wind direction were measured with a Vaisala Weather Transmitter WXT520, while solar radiation (global, direct, and diffuse) was measured with a SPN1 Sunshine Pyranometer. Both were configured to acquire one measurement every ∼3 seconds.
2.2.1.3. Sulfur dioxide (SO2) and gas-phase sulfuric acid (H2SO4). Gaseous SO2 was measured at Maïdo observatory with a Teledyne API T100U analyser with a time resolution of 1 minute and a detection limit of 0.05 ppbv.
Gas-phase H2SO4 was measured with an atmospheric pressure interface time-of-flight mass spectrometer (APi-TOF, Aerodyne Research Inc. and TOFWERK AG)32 equipped with a chemical ionization (CI) inlet (CI-APi-TOF)33 employing nitrate reagent ions. All methodological details and calculations concerning the instrumental setup are already described in Rose et al. (2021).34
2.2.1.4. Aerosol size distribution. The aerosol size distribution of aerosols with electrical mobility diameter between 8.5 and 700 nm (14 size bins centered on 10, 13.7, 18.8, 25.7, 35.2, 48.3 66.2, 90.7, 124.2, 170.2, 233.2, 319.6, 437.9, and 600 nm) was measured with a custom-built differential mobility particle sizer (DMPS), which performs one complete scan every ∼460 seconds. This instrument was previously used to study new particle formation on La Réunion Island and has been described in detail elsewhere.35,36 Briefly, particles are first charged to equilibrium using a Ni-63 bipolar charger, after which they enter the DMPS, which includes a TSI-type differential mobility analyzer operating in a closed loop and a TSI 3010 condensation particle counter. The instrument was operated behind a whole air inlet (higher size cut-off of 25 μm for an average wind speed of 4 m s−1), and measurement protocols were defined with respect to the ACTRIS (Aerosol, Clouds and Trace Gases Research Infrastructure) recommendations regarding both the flow rates and relative humidity.37 Based on the intercomparison study from Wiedensohler et al. (2012),37 we assign a relative uncertainty of 10% (k = 2) to the aerosol size distribution in the 20–200 nm size range, and 30% outside this range.
2.2.1.5. Filter-based observations of inorganic ions and organic carbon. Submicron and supermicron aerosols were sampled at Maïdo from the 15th of March 2018 to the 24th of May 2018 in the framework of the OCTAVE (Oxygenated Compounds in the Tropical Atmosphere: Variability and Exchanges) project38 during its intensive observation period, as already described in detail in Simu et al. (2021).39 Briefly, ambient aerosols were collected using two high-volume air samplers at a flow rate of 1130 L min−1 (at local temperature and pressure) for each. One of the samplers performed daytime sampling (running between 07:00 and 18:00 local time), while the other performed nighttime sampling (running between 22:00 and 05:00 local time). Both samplers were turned off between 05:00 and 07:00 and between 18:00 and 22:00 local time, defined as transition periods. A cascade impactor (TE-230, Tish Environmental, Inc.) was attached to each high-volume air sampler to collect size-segregated aerosol samples. All aerosol samples were collected onto quartz-fiber filters. For the submicron size (bottom stage of the impactors; aerodynamic diameter < 0.95 μm) filter changes were performed every 2–3 days and the sampling was performed continuously. For the supermicron size, filters were changed less frequently (every 2–13 days) and sampling was not done continuously, giving rise to significant data gaps (see ESI Fig. S1†). Mass concentrations of supermicron aerosols reported here correspond to the sum of the top four stages (aerodynamic diameter > 0.95 μm) of the impactor.
Samples were analyzed for water-soluble inorganic ions (SO42−, NH4+, K+, Na+, Mg2+, Ca2+, Cl−, Br−, NO2−, NO3−) and organic carbon (both water-soluble and insoluble). Water-soluble aerosols were extracted from the filters using ultrapure water, followed by filtration through a syringe filter (0.22 μm). Inorganic ions in the extract solution were quantified using an ion chromatograph (Model 761 compact IC, Metrohm).
2.2.1.6. BrO, IO, and SO2 slant column densities. The University of Colorado Multi-AXis Differential Optical Absorption Spectroscopy (CU MAX-DOAS) instrument40,41 measures ultraviolet-visible scattered-sunlight spectra in a sequence of elevation angles (angle above the horizon) and an azimuth angle of ∼100° (clockwise from North). The instrument was operational at Maïdo before, during, and after the April–June 2018 eruption of Piton de la Fournaise.42,43 Spectra measured during the volcanic eruption were analyzed with respect to a reference spectrum measured in the absence of the volcanic plume using a DOAS spectral retrieval algorithm to retrieve SO2, BrO, and IO differential slant column densities (dSCDs).
BrO dSCDs were retrieved in a spectral fit window of 328.5 to 359 nm, simultaneously fitting absorption structures from O3, NO2, HCHO, O2–O2, and the Ring effect. IO SCDs were retrieved in a spectral fit window of 417.5 to 438 nm, simultaneously fitting absorption structures from O3, NO2, O2–O2, H2O, and the Ring effect. Finally, SO2 dSCDs were retrieved in a spectral fit window of 323 to 335 nm, simultaneously fitting absorption structures from O3, NO2, BrO, HCHO, and the Ring effect.
Since the spectral fit windows for BrO and SO2 are overlapping, we assume that the observed air mass factors for the two species are approximately the same. BrO/SO2 ratios were calculated using the BrO and SO2 dSCDs observed at various elevation angles (15°, 25°, 33°, 37°, 40°, 42°, 43°, 44°, 45°, 46°, 47°, 48°, 50°, 53°) and ∼100° clockwise from north.
2.2.2.1. GEM passive samplers. Two GEM passive samplers44,45 were deployed between the 28th of April 2018 and the 4th of May 2018 close (∼200 m distance) to the eruptive vent of the April–June 2018 eruption of Piton de la Fournaise (see label “A” in Fig. 1). A larger number of GEM passive samplers was operating all over La Réunion Island between 2018 and 2019. This passive sampler data set is described in detail in Hoang et al. (2023).45
2.2.2.2. Meterological parameters. Wind speed and wind direction were measured at Piton de Partage (Bellecombe station) (latitude: −21.22°; longitude: 55.69°; 2245 masl; see label “C” in Fig. 1), about 4 km north-north-east of the eruptive vent, with a DEOLIA 96 wind sensor integrated into a MERCURY weather automatic station, managed by the French meteorological and climatological service “Météo-France”.
2.2.2.3. SO2 concentrations and SO2 flux. In situ SO2 concentrations were measured at the summit of Piton de la Fournaise (latitude: −21.243°; longitude: 55.709°; 2632 masl; ∼1.5 km from the vent; see label “B” in Fig. 1) with a Multi-GaS analyser performing 4 sets of measurements per day (two daytime, two night-time), each lasting 30 minutes. The Multi-GAS46 is a standard volcanic gas sensing technique that is commonly used for in situ real-time observations (at 0.1 to 1 Hz rate) of SO2 (and other major volcanic gas species like CO2, H2O, H2S) concentrations in near-vent volcanic plumes. SO2 concentrations are measured using a T3STF CiTiceL® specific electrochemical sensor from CityTecnology.
SO2 degassing fluxes from Piton de la Fournaise were observed remotely with three scanning-DOAS instruments, stationed on the cliffs of the Enclos Fouqué caldera (see labels “D”,”E”, and ”F” in Fig. 1). The instruments are part of the Network for Observation of Volcanic and Atmospheric Change (NOVAC)47,48 and have been operated almost continuously since 2007. The flux is derived from measurements of diffused solar radiation in the spectral range between 280 and 430 nm, obtained sequentially by scanning the line of sight through a flat or conical surface that intersects the volcanic plume. Spectra are then analyzed by DOAS to obtain slant column densities of SO2, BrO, and other species relative to the background. Evaluations were done in the 314–327 nm and 331–353 nm for SO2 and BrO, respectively, and included cross-sections for O3 absorption, the Ring-effect, a 5th-degree polynomial, and generic corrections for dark current, offset, stray-light, and wavelength shifts (against a synthetic solar-atlas). The line density obtained by integrating the columns in the cross-section of the plume is multiplied by the normal component of wind velocity at plume height to derive the flux. The height and direction of the center of mass of the plume are calculated by triangulation from simultaneous measurements by two stations. For this study, we used plume velocity data from the ERA5 re-analysis product of the European Centre for Medium-Range Weather Forecasts (ECMWF), with a time resolution of 1 h. One SO2 flux measurement is obtained every 5–10 minutes, depending on light conditions. SO2 fluxes could only be accurately determined for plumes that left the rim of the caldera (Enclos Fouqué) and passed the field of view of at least 2 DOAS instruments.
2.3. Modelled, calculated, and estimated magnitudes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times) drew a random value from the probability distribution of each parameter and re-solved the respective equation. Finally, based on the distribution of results (n = 100000), we estimated the most likely value (or “best guess”) as the median of the distribution, and its 95% confidence interval as the 2.5th to 97.5th percentile of the distribution. A schematized representation of this procedure can be found in ESI Fig. S2.†
000 times) drew a random value from the probability distribution of each parameter and re-solved the respective equation. Finally, based on the distribution of results (n = 100000), we estimated the most likely value (or “best guess”) as the median of the distribution, and its 95% confidence interval as the 2.5th to 97.5th percentile of the distribution. A schematized representation of this procedure can be found in ESI Fig. S2.†
We use the following notation to represent results and their 95% confidence interval: median (2.5th percentile; 97.5th percentile). On the other hand, we use the “±” notation to express the expanded uncertainty of magnitudes at coverage factor of k = 2, unless otherwise indicated.
The DMPS does not detect particles larger than ∼700 nm in diameter and may have lower collection efficiency toward the limits of its detection range.37 As fine particle mass in a volcanic plume often has a mass peak close to ∼1 μm,54–57 PM0.7DMPS most likely underestimates the submicron particle mass in the volcanic plumes. While the total submicron mass was not measured, we count with filter-based measurements of water-soluble inorganic ions in PM0.95, among them sulfate (see Section 2.2.1.5). It has already been reported that sulfate dominated the mass of inorganic ions at Maïdo during the April–June 2018 volcanic episode.39
To estimate variation of PM0.95 sulfate on the 29th of April we construct a PM0.95 sulfate proxy (PM0.95sulfate proxy). We assume that volcanic PM0.95 sulfate can be expressed as a function of observed SO2, a clear volcanic plume tracer and precursor for secondary sulfate, and the DMPS-derived particle volume (PV0.7DMPS), which is proportional to PM0.7. We then constrained the so-defined PM0.95sulfate proxy by comparison to filter-based sulfate measurements, i.e. considering that PM0.95sulfate proxy should be equal to measured PM0.95 sulfate if averaged over the same sampling period. For simplicity and because of the lack of sufficient observational constraint, we did not include the estimated plume age (see Section 2.3.4 below) in the calculation of PM0.95sulfate proxy, even though plume age is expected to influence the sulfate to SO2 ratio. To account for the latter, and because PM0.95sulfate proxy was not directly measured but indirectly determined, we assign to PM0.95sulfate proxy an elevated relative uncertainty of 50% (k = 2). More details about PM0.95sulfate proxy are given in Appendix A.
While sulfate tends to be a dominant compound (by mass) in volcanic submicron particles,56,58 PM0.95sulfate proxy may still significantly underestimate the total submicron particle mass if the observed plumes were rich in primary volcanic ash. While we cannot precisely quantify the contribution of primary volcanic ash because key ash constituents, like silicon oxides, aluminium oxides, or iron oxides, were not measured, its contribution to volcanic submicron particle mass is expected to lie between 0 and 50% after some plume aging has occurred.59,60 In fact, for most eruptions from Piton de la Fournaise very low content of fine ash was reported.61
Upon arrival at Maïdo, we expect the plume to be strongly mixed and diluted by background air. To estimate kinetic rate constants, we assume that the dilution occurred near-instantly (compared to the plume transport time of several hours) after emission from the vent, after which the plume continued travelling without much further dilution until arrival at Maïdo. With this simplification, the mean interaction time between GEM and plume constituents can be set equal to the plume transport time. It should be noted that this simplification likely overestimates the average interaction time between entrained background GEM and plume constituents because real plume dilution might have occurred in several steps or more gradually during the transport. For example, background GEM entrained into the plume shortly before arrival at Maïdo would have had a much shorter interaction time with plume constituents than estimated here. The transport time might also overestimate the interaction time because reactive chemicals or surface adsorption sites on particles might not be instantly available after plume emission but form over time.
If we assume insignificant SO2 scavenging in the plume during the transport from the vent to Maïdo, we can parametrize GEMvolcanic as the product of the SO2 concentration observed in the plume and a GEM/SO2 emission ratio (ER) from the vent (eqn (1)). While gas-phase SO2 is certainly oxidized within volcanic plumes,62,63 this occurs, outside clouds, reportedly at a rate of ∼1–2% h−1 at solar noon.63,64 Assuming a similar SO2 oxidation rate in the plumes observed here, which encountered mostly cloudless conditions, we would expect that >90% of the emitted SO2 remained in the plume after transport times relevant here (∼3–7 hours), making SO2 a fairly conservative tracer for the present purpose.
| GEMno scavenging = GEMbackground + GEMvolcanic ≈ GEMbackground + ER × SO2 | (1) |
We constrained the mean GEM/SO2 ER from Piton de la Fournaise in the April–June 2018 eruptive period by combining GEM passive sampler observations with Multi-GaS SO2 observations from the summit (see Section 2.2.2.3). We obtain an upper limit for the GEM/SO2 ER of 2.4 × 10−6 [ng ng−1], which lies within, but at the lower end of ERs reported in the literature (since the year 2000: median: 5.4 × 10−6; 10th to 90th percentile: 8.4 × 10−7 to 1.6 × 10−5; see ESI Tables S1 and S2†). On the other hand, we use the lowest ER reported in the literature so far (1.5 × 10−7)65 as a conservative lower limit for the ER from Piton de la Fournaise. This is described in detail in Appendix C.
For all ER-related calculations and Monte Carlo simulations, we express the GEM/SO2 ER from Piton de la Fournaise as a uniform distribution constrained between the above-defined upper and lower limits (2.4 × 10−6 and 1.5 × 10−7 [ng ng−1], respectively).
3. Results and discussion
3.1. Description of the volcanic episode and GEM depletion events
Piton de la Fournaise entered an eruptive phase on the 27th of April 2018, as evidenced by large daily average SO2 degassing fluxes of ∼200 t d−1 on the 28th (see Fig. 2). Daily SO2 fluxes peaked on the 30th of April with ∼500 t d−1 and then decreased rapidly in the following days, reaching almost undetectable levels by ∼May 10th. Fluxes remained low and plumes seldomly left the Enclos Fouqué caldera rim for the rest of the eruptive phase which lasted until the 1st of June. These observations agree well with satellite-based observations from the Ozone Monitoring Instrument (OMI) and the Ozone Mapping and Profiling Suite (OMPS), as reported by the NASA Global SO2 Monitoring Homepage (https://so2.gsfc.nasa.gov/). Despite large initial SO2 fluxes, ground-level in situ SO2 concentrations at the summit of Piton de la Fournaise (∼1.5 km from the vent) remained initially unaffected. In contrast, in situ SO2 concentrations already peaked on the 29th of April at Maïdo, which is ∼38.5 km away from the vent. This indicates that the plume rise was initially strong, facilitating atmospheric plume transport by winds aloft. On May 10th SO2 concentrations peaked at the summit of Piton de la Fournaise, indicating that by this time the eruptive plume rise declined enough to allow the plume to be observed at ground level in that area.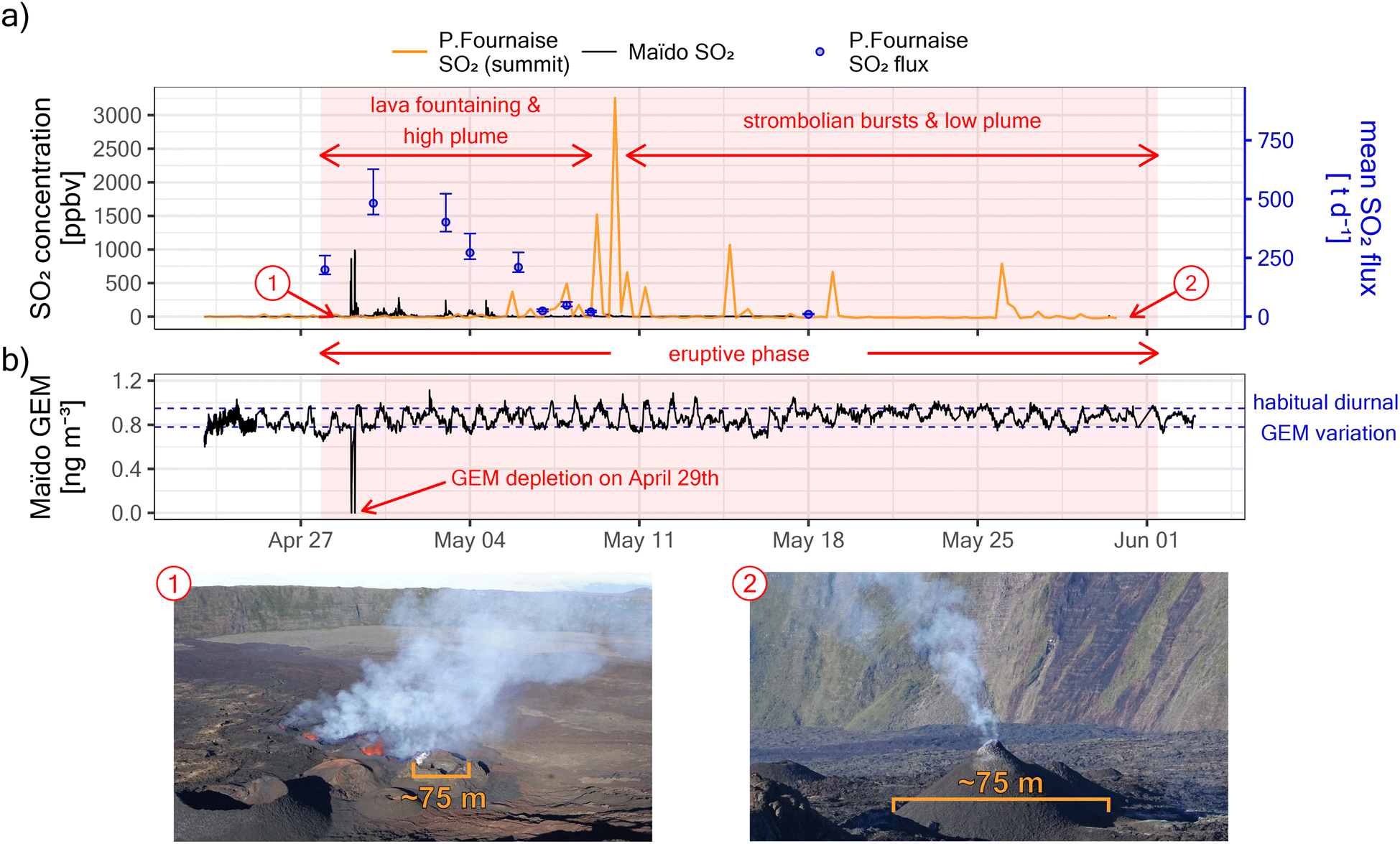 | ||
| Fig. 2 (a) Evolution of the eruptive phase of Piton de la Fournaise between the 27th of April and the 1st of June 2018. The early event was characterized by lava fountaining, large SO2 degassing, and high plume rise (see photography “1”). By – May 10th, the intensity of the eruption had diminished importantly, and the rest of the eruptive phase was characterized by strombolian bursts, low SO2 degassing, and low plume rise (see photography “2”). (b) Maïdo GEM observations during the eruptive phase. GEM depletions were observed only on April 29th, coinciding with the only large peaks in SO2 concentrations observed in situ at Maïdo (see a). The GEM fluctuation before and afterward does not correspond to a volcanic signal, but the habitual diurnal GEM variation at Maïdo.25 | ||
On the 29th of April 2018, i.e. early in the eruptive phase and while plume rise was still significant, we observed at Maïdo GEM depletion in transiting volcanic plumes (Fig. 2b and 3a). GEM depletion was complete (i.e. concentrations below the instrument detection limit) in two instances, and the first complete GEM depletion event was observed before sunrise (solar zenith angle between 96° and 93°, Fig. 3d). All GEM depletion events were clearly related to the passage of volcanic plumes, as evidenced by strongly enhanced concentrations of SO2 (up to ∼900 ppbv, Fig. 3b) and suspended particles (up to ∼28 ± 8 μg m−3 for PM0.7DMPS, up to 570 ± 290 μg m−3 for PM0.95sulfate proxy, Fig. 3c). The onset of the first GEM depletion coincided with a change in transport pathways at ∼6:00 local time (local time = UTC + 4), as indicated by a sharp decrease in wind speed at Maïdo (from ∼18 km h−1 to ∼5 km h−1, Fig. 3e) and a decrease in observed air temperature (from ∼15 °C to ∼10 °C, Fig. 3f). Intriguingly, no GEM depletion was observed before 6:00, even though the first strong volcanic plume had already arrived at around ∼5:20. This will be discussed further below.
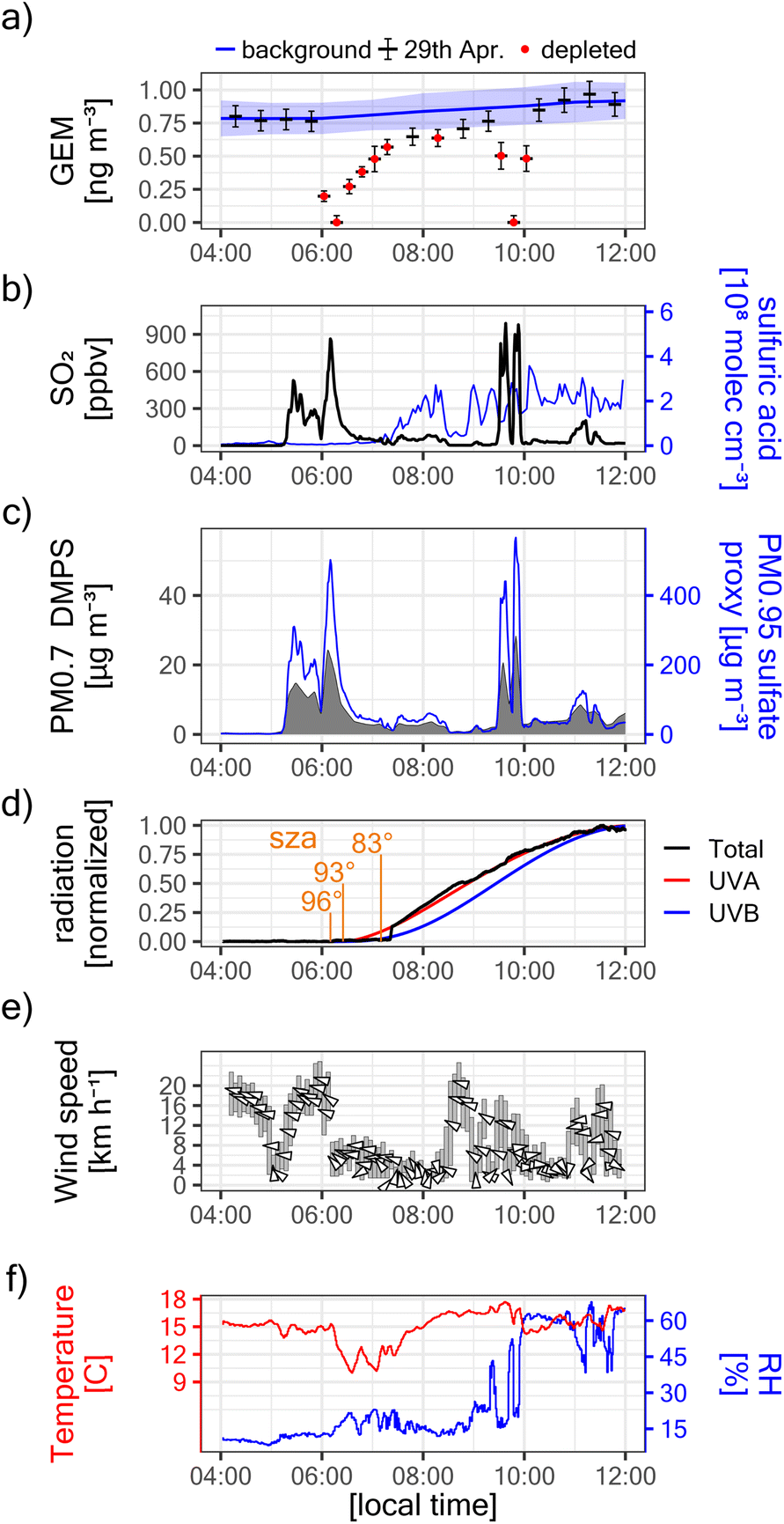 | ||
| Fig. 3 GEM depletion events and ancillary in situ observations at Maïdo observatory on April 29th, 2018. (a) Observed GEM. The beginning and end of each black segment correspond to the sampling interval of the respective data point. Vertical error bars give the 95% confidence interval. The blue line and blue shaded area give the background concentration (mean ± 2 standard deviation). Note that, at STP, 1 ng m−3 corresponds to ∼112 parts-per-quadrillon (ppqv). Red points: significantly GEM-depleted instances. (b) Observed SO2 and gas-phase sulfuric acid. (c) Estimated mass of particles with a diameter of 8.5–700 nm (PM0.7DMPS) and estimated sulfate mass in particles with diameter <0.95 μm (PM0.95sulfate proxy). For better readability, the wide uncertainty intervals are not shown (±30% for PM0.7DMPS; ± 50% for PM0.95sulfate proxy). (d) Normalized solar radiation. Total radiation was measured (1 = 1000 W m−2). UVA and UVB radiation were estimated with the TUV model (1 = 50 W m−2 and 1 = 3.0 W m−2, respectively). The solar zenith angle (sza) is given at selected instances. (e) Observed wind speed and direction. The arrows indicate the direction towards which the wind blows (upwards pointing arrows indicate winds blowing from south to north). The y-axis shows the vector mean wind speed during the averaging interval (5 minutes). The grey bars indicate the 25th to 75th percentile of the instantaneous wind speed during each averaging interval. Note that the arrows can lie outside the grey bars, as wind speeds associated with opposite wind directions cancel each other out upon vector averaging. (f) Observed temperature and relative humidity (RH). | ||
3.2. Aerosol composition
Insight into the composition of volcanic aerosols is gained from filter-based observations at Maïdo (see Section 2.2.1.5). While the high-volume samplers were not operating during the first complete GEM depletion (shortly after 6 AM), which fell into the transition period between nighttime and daytime sampling, all plumes associated with GEM depletion events after 7:00 were captured in a PM0.95 daytime filter. The corresponding filter was sampling from the 25th of April 07:00 to the 29th of April 18:00. As no important volcanic influence was detected at Maïdo before the 29th of April (Fig. 2a), it can be assumed that significant enhancements of mean concentrations in this sampling period (compared to the time before) can be attributed near-exclusively to volcanic plumes on the 29th of April.Between the 25th of April and the 29th of April, the average daytime PM0.95 sulfate concentration was ∼16.7 μg m3, which is extremely elevated compared to the time before the onset of volcanic activity (∼0.7 μg m3; Fig. 4a). The total concentration of crustal element ions (K+, Na+, Mg2+, and Ca2+) was also significantly increased (from ∼0.03 μg m3 to ∼0.2 μg m3). While this suggests some contribution of volcanic primary ash, the enhancement was much less pronounced than for sulfate. Halogen ion (Br− and Cl−) concentrations were not affected by the onset of volcanic activity. Similarly, the concentration of organic carbon was ∼1 μg m3 and thus not significantly enhanced compared to the time before, indicating carbon-poor volcanic particles. In contrast to PM0.95, supermicron particle concentration and composition was largely unaffected by the onset of the volcanic episode (see ESI Fig. S1†), suggesting that particle mass in the volcanic plumes was dominantly in the submicron mode.
 | ||
| Fig. 4 (a) Water-soluble inorganic ions and organic carbon in submicron aerosols (PM0.95) at Maïdo. Concentrations are given at STP. The beginning and end of each horizontal segment correspond to the sampling interval of the respective filter. Note the logarithmic y-axis. The dashed vertical line corresponds to the day of observed GEM depletion (April 29th). Missing values correspond to concentrations below the detection limit. (b) Molar ratio between sulfate and ammonium. (c) Ion charge balance considering all measured ions (SO42−, NH4+, K+, Na+, Mg2+, Ca2+, Cl−, Br−, NO2−, NO3−). A negative charge balance indicates acidic aerosols. | ||
Before the onset of the eruptive phase, the molar ratio between SO42− and NH4+ in PM0.95 was around 0.5 (Fig. 4b), suggesting complete sulfate neutralization by ammonium, and that ammonium sulfate ((NH4)2SO4) was the dominant sulfate species. This changed during the volcanic episode, for which highly elevated SO42−/NH4+ molar ratios of ∼6–9 suggest only a minor contribution of ammonium sulfate, and instead an elevated contribution of other sulfate species. This is most likely sulfuric acid, considering that the negative charge balance of the sampled ionic species (Fig. 4c) suggests highly acidic aerosols.63 It is known that sulfuric acid droplets can make up a large fraction of volcanic particles,55,63,66 and if volcanic ash particles are present in the plume, they are expected to be coated in aqueous sulfuric acid when SO2 and its oxidation products are present.67 As sulfuric acid is extremely hygroscopic, these particles absorb atmospheric moisture even when relative humidity is very low,68 as was the case during the GEM depletion events (RH ∼15–20%, see Fig. 3f).
3.3. The possible role of bromine chemistry
Previous work has suggested that rapid GEM oxidation could occur in volcanic plumes following a gas-phase reaction with co-emitted bromine that has become activated as radicals (see Introduction). However, multiple lines of evidence rule out a leading role for bromine chemistry in the GEM depletions reported here:(1) Plumes from Piton de la Fournaise appear to be halogen-poor. Halogens in volcanic plumes are mostly found in the form of halides like HCl and HBr.14,69–72 Although no data on gas-phase HCl and HBr are available for the 2018 eruption, previous gas observations at Piton de la Fournaise indicate a low-HCl and low-HBr signature for gases emitted from this volcano73,74 and hot-spot volcanoes in general,70 with HBr/SO2 ratios typically being up to one order of magnitude lower than in subduction zone volcanic gases.70,71 In addition, we observed no enhancement of particle phase Cl− or Br− in submicron aerosols when Piton de la Fournaise entered volcanic activity (see Fig. 4a). As HCl and HBr partially adsorb onto aerosols,14,75 the low concentrations of halogen ions in the particle phase also suggest halogen-poor plumes in the gas phase.
(2) We observed GEM depletion before sunrise. Even for bromine-rich volcanic plumes, most bromine is initially in the form of HBr, which does not oxidize mercury. It has been proposed that quick conversion of HBr into radical species may occur in the so-called “effective source region”, the region of the crater near to the vent where hot and oxygen-poor volcanic gases first mix and interact with cool and oxygen-rich background air.8 However, it is unlikely that this process can generate sufficient radical bromine species to completely deplete atmospheric GEM within a few hours.16 For this to happen, a “bromine explosion” would most likely need to occur,14 which is a photochemical process that requires UV radiation. In contrast, we observed the first complete GEM depletion entirely before sunrise (solar zenith angle between 96° and 93°, see Fig. 3d), so that practically no radiation was available (TUV model; UVA < 1 × 10−3 W m−2, UVB < 7 × 10−2 W m−2), and photolysis rates for Br-relevant photochemical reactions were very low (TUV model; k < 1.3 × 10−5 s−1 for Br2 → Br + Br; k < 5.0 × 10−5 s−1 for BrO → Br + O).
(3) There is insufficient BrO to explain appreciable GEM oxidation by bromine chemistry, even in daytime plumes. MAX-DOAS observations at Maïdo captured the volcanic plume passages on the 29th of April after sunrise. No measurements are available before sunrise, since the instrument relies on scattered solar photons as a light source. While observed SO2 column densities were strongly enhanced (maximum: 1.08 ± 0.05 × 1018 mol cm−2), only very minor enhancements in the BrO column densities were observed (see ESI Fig. S3†). BrO/SO2 molar ratios for the daytime plumes on the 29th of April were (1.28 ± 0.07) × 10−5 (N = 2), at the lower end of reported values in volcanic plumes.14,76–78 We estimate the GEM lifetime with regard to bromine-initiated oxidation as roughly 36 h in the daytime plume (see ESI Table S3†). This estimate assumes 1 ppmv SO2, 40 ppbv O3, BrO/SO2 = 1.2 × 10−5, and Br/BrO = 0.1, corresponding to an average Br radical concentration of 1.2 pptv inside the plume. The GEM + Br reaction, thermal decomposition of Br-GEM adduct, and scavenging of Br-GEM + O3 were treated using reaction rate constants from Shah et al. (2021)6 (temperature = 288 K; pressure = 800 hPa). This GEM lifetime is conservative, as SO2 was typically lower in the dilute plume. It was also found to be robust towards variations in O3 (GEM lifetime varies <1 h if O3 was 10 times lower; and remains unchanged at higher O3 in the plume). At the observed low levels of BrO, bromine oxidation could thus explain at best ∼8% GEM oxidation over 3 h, and ∼18% over 7 h, even in the daytime plume. This is congruent with a recent study16 that modelled radical halogen chemistry in volcanic plumes from Mount Etna and, coupled to a gas-phase mercury scheme, obtained very low net GEM oxidation within the plume, even at BrO/SO2 ratios of >1 × 10−4 (i.e. about an order of magnitude higher than observed here).
In summary, the low abundance of gas- and particle-phase halogens coupled with the measured GEM depletion in the unilluminated plume make halogen-induced GEM oxidation extremely unlikely. In lack of a plausible bromine activation mechanism before sunrise, the observation of complete GEM depletion prior to sunrise must have other causes.
3.4. The hypothesis: particle-induced GEM depletion
Ermolin et al. (2018)79 separated volcanic nanoparticles (<∼100 nm in at least one dimension) from settled bulk ash and analysed them for trace metals, finding that they had very elevated Hg mass fractions of 9–36 ppm (1 ppm = 1 μgHg g−1particle), corresponding to an enrichment factor of up to ∼450 with respect to the earth's crust (Hg mass fraction ∼0.08 ppm).80,81 The authors suggested that a strong accumulation of trace elements like Hg occurs in the course of a volcanic eruption and upon plume dilution. In another study, coincident observations of PBM (∼100 pg m−3) and suspended particles below 2.5 μm in diameter (PM2.5; ∼12.5 μg m−3) in a ∼10 days aged volcanic plume suggest a similarly elevated particle Hg mass fraction of ∼8 ppm.82 To our knowledge, no other study investigated Hg in fine particles (defined here as ≤10 μm in diameter) of volcanic origin, be it on settled particles or within a volcanic plume.On the other hand, many studies have explored PBM in urban smog, a similarly fine-particle-loaded and sulfur-rich, albeit less extreme environment. Measurements in polluted urban environments suggest strong Hg enrichment in suspended fine particles, with Hg mass fractions in the ppm range.83–86 While these high Hg contents have been associated with direct PBM emission from industry and traffic,85,87 they may also result from gas–particle-interactions, for example from heterogeneous GEM oxidation and Hg uptake by particles.87–90 As most oxidized Hg species are semi-volatile, they partition between the gas phase and the particulate phase.91
Parallels can also be drawn between volcanic plumes and a similarly extreme environment: industrial flue gas. GEM is commonly removed from flue gas by injection of fly ash or activated carbon, which leads to efficient GEM scavenging within minutes. The underlying process appears to be mainly chemisorption,92–95i.e. irreversible reactive Hg uptake involving a chemical reaction between Hg and active sites (adsorption sites) on the particle surface. Indeed, GEM removal is enhanced if the aerosol was impregnated (i.e. pre-treated) with halogens,96–99 or sulfur.100 Sulfur impregnation greatly increases the abundancy of sulfite and sulfates (especially sulfuric acid) on particle surfaces and within pores, compounds that likely act as Hg adsorption sites to generate HgSO4,92,94,100–103 which is very stable at ambient temperatures.95
We thus hypothesize that the GEM depletions observed here were mainly caused by gas–particle interactions, considering the strongly elevated concentrations of sulfate-rich and highly acidic particles in the volcanic plumes (see Fig. 3c), and the relatively long interaction time (several hours) between particles and gas-phase Hg before plume arrival at Maïdo. In the following sections, we explore heterogeneous GEM oxidation and GEM uptake by particles as possible manifestations of such a gas–particle interaction. However, we highlight that we have no suitable high-resolution observations that allow us to determine whether the “missing” GEM was found in the gas phase (in the form of GOM) or in the particulate phase (in the form of PBM) so that a definite answer about the underlying nature of interaction cannot be achieved here.
| c = c0exp(ktPM) | (2) |
Eqn (2) can be written in a form that allows estimating the kinetic rate constant k with a linear regression (eqn (3)). Note that, despite the form of eqn (3), we do not force the regression through the origin, because it does not significantly affect our results and because the appropriateness of this practice has been subject to discussion.104
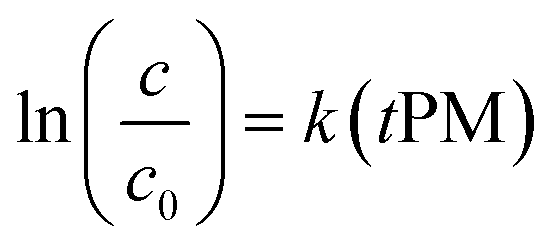 | (3) |
We find that the GEM depletions adjust well to a first-order relationship, for both PM0.7DMPS (Fig. 5a) and PM0.95sulfate proxy (ESI Fig. S4†). Assuming a higher GEM/SO2 ER from Piton de la Fournaise and consequently higher GEM scavenging yields higher kinetic rate constants (i.e. a steeper slope of the fit) (Fig. 5a). It also improves the goodness of fit (R2), mainly because the two clear outliers before 5:55 AM local time, when observed GEM appeared unaffected by the first volcanic plume passage (“A” and “B” in Fig. 5a; see also Fig. 3), move closer to the line of best fit. We hypothesize that these outliers result from a competition between volcanic GEM emission and GEM scavenging in the plume, i.e. GEM depletion at first plume arrival might have been masked by large volcanic GEM emissions that raised GEM concentrations in the freshly diluted plume significantly above the background. More generally, considering that GEM/SO2 ERs from a single vent can vary within minutes,11 some of the scatter around the lines of best fit might be attributable to a fluctuating ER during the event.
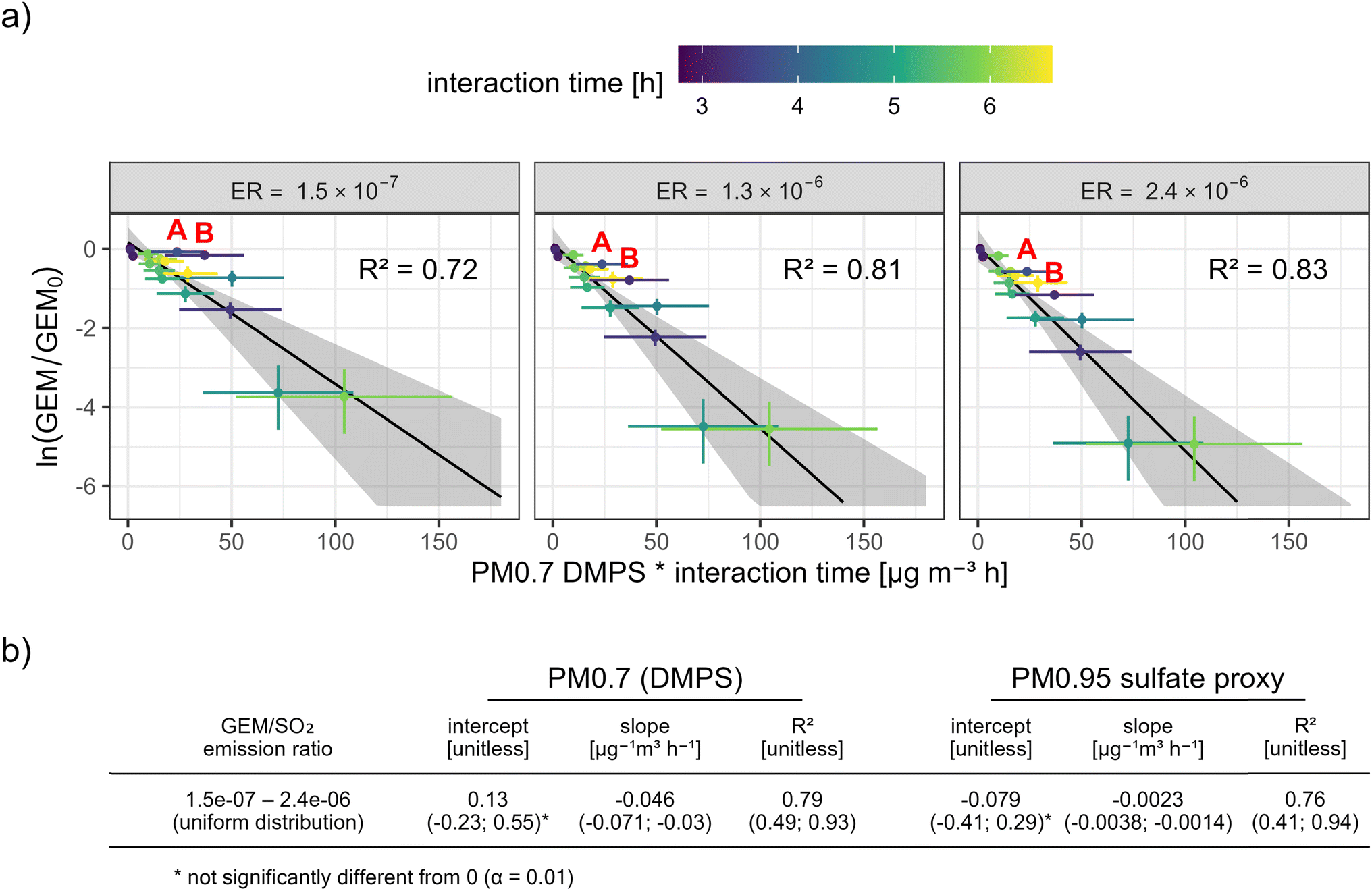 | ||
| Fig. 5 (a) Observed GEM depletion in function of PM0.7DMPS and interaction time, and for three different possible GEM/SO2 emission ratios from Piton de la Fournaise (corresponding to the minimum, median, and maximum of the uniform probability distribution assigned to the GEM/SO2 emission ratio; see Section 2.3.5 and Appendix C). The error bars and the shaded area show 95% confidence intervals. The color scale shows the estimated mean interaction time between volcanic particles and GEM before the plume's arrival at Maïdo. The figure and the fits are based on all observations between 3:55 and 10:10 local time. Data points marked with “A” and “B” indicate the most notable outliers, sampled between 5:10–5:25 and 5:40–5:55 local time, respectively. Table (b) Estimated fit parameters, also for PM0.95sulfate proxy (see ESI Fig. S4†). All fit parameters and their 95% confidence intervals were obtained with Monte Carlo simulations. The slope of the fit corresponds to the estimated interaction rate constant. | ||
Based on a Monte Carlo simulation (see Section 2.3.1) and considering uncertainties in observed GEM, estimated particle mass, estimated transport time, and the estimated GEM/SO2 emission ratio, we obtain empirical GEM scavenging rate constants of −0.046 (−0.071; −0.030) μg−1 m3 h−1 and -0.0023 (−0.0038; −0.0014) μg−1 m3 h−1 for PM0.7DMPS and PM0.95sulfate proxy, respectively (see Fig. 5b). These rate constants should be seen as first approximations with large uncertainties, and they are likely biased low because the real interaction time between GEM and reactive plume constituents was most likely shorter than the plume transport time estimated in Section 2.3.4. Nevertheless, we encourage the use of these rate constants as a first approximation to address particle-related GEM scavenging in aging volcanic plumes. In any case, the conditions for which they were obtained have to be kept in mind, i.e. for 3–7 hours aged volcanic plumes, abundantly present and likely sulfate-dominated (mostly sulfuric acid) submicron volcanic particles, and for temperature and relative humidity ranges of ∼10–15 C and ∼15–20%, respectively. Any extrapolation from these conditions must be approached with care.
The amount of GEM that was scavenged (i.e. is “missing”) in the plume is simply the difference between observed plume GEM concentrations and GEM concentrations that would be expected in the absence of any scavenging (see Section 2.3.5). We estimate that 160 (64; 290) μg of GEM was scavenged per gram of PM0.7DMPS, and 10.4 (3.3; 24) μg of GEM was scavenged per gram of PM0.95sulfate proxy (see Fig. 6a). If we hypothesize that all “missing GEM” was taken up by particles in the volcanic plumes, then these values correspond directly to the resulting Hg mass fractions (in ppm) in the respective particle type (PM0.7DMPS or PM0.95sulfate).
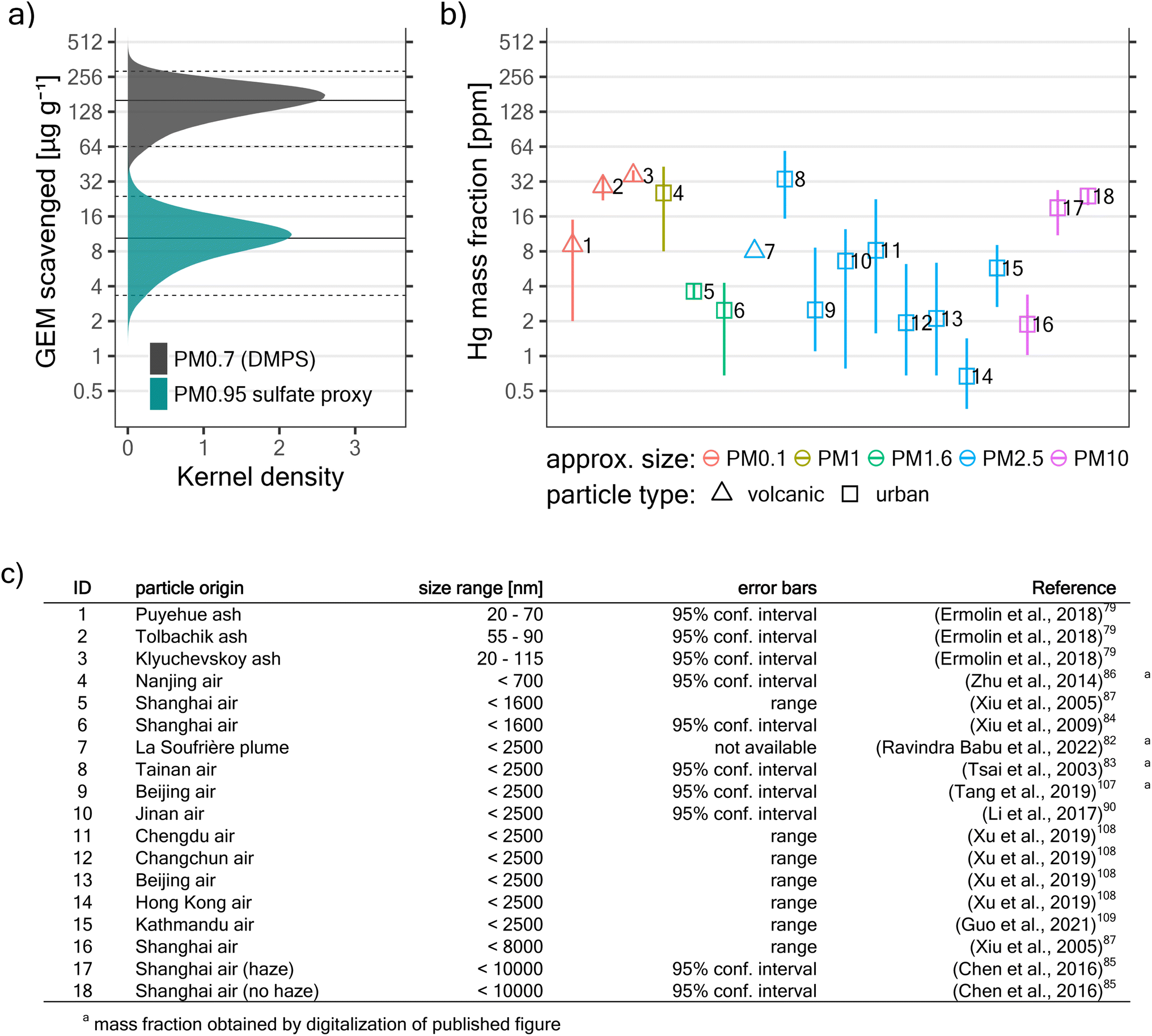 | ||
| Fig. 6 Estimated magnitude of in-plume GEM scavenging and literature review. (a) Micrograms of scavenged GEM per gram of suspended particle mass. This also corresponds to the particle Hg mass fraction (in ppm) if complete uptake of all “missing GEM” by particles is assumed. Distributions were obtained through Monte Carlo simulations. Horizontal lines show the 2.5th, 50th (median), and 97.5th percentile, respectively. Note the logarithmic y-axis. (b) Literature review of Hg mass fractions in volcanic and urban fine particles as directly reported or otherwise derivable from previous studies (see panel (c) for further information). PM0.1, PM1, PM1.6, PM2.5, and PM10 correspond to all suspended particles with a diameter below ∼0.1 μm, 1 μm, 1.6 μm, 2.5 μm, and 10 μm, respectively. | ||
To put these mass fractions into context, we reviewed published work concerning Hg mass fractions in volcanic fine particles and suspended fine particles (<10 μm in diameter) in urban environments79,82–87,90,107–109 (this review does not intend to be exhaustive). Assuming complete uptake of all the “missing” GEM onto PM0.7DMPS leads to Hg mass fractions that are quite large compared to published work (see Fig. 6). On the other hand, if we assume complete uptake onto PM0.95sulfate proxy, which arguably captures a much larger fraction of the suspended submicron particle mass than PM0.7DMPS, Hg mass fractions fall well into the range of values reported elsewhere.
We thus hypothesize that the GEM depletions observed here could result largely from (irreversible) reactive Hg uptake by volcanic submicron particles. These particles might have an elevated capacity for Hg uptake because of their high sulfate content, similar to what has been found for sulfur-impregnated particles in industrial flue gas.92,94,101–103 Besides, noble metals or transition metal oxides in primary volcanic ash, even if only a minor particle constituent, might exert a catalytic function for GEM scavenging.110,111
4. Implications
The observation of GEM depletion in dilute volcanic plumes has important implications for the atmospheric Hg budget and Hg deposition pathways, regardless of the underlying physical and chemical mechanisms, and regardless of whether GEM was converted into GOM or PBM within the plume. As both GOM and PBM are water-soluble and have atmospheric lifetimes significantly shorter than that of GEM, such a conversion would lead to larger Hg deposition and Hg exposure in regions close to degassing volcanoes. This is concerning, as it has been estimated that ∼15% of the world's population lives within less than 100 km of a Holocene volcano (i.e. a volcano that has been active in the last ∼10000 years; Stand 2015).112
These GEM depletions also suggest that terrestrial volcanism does not only act as a source of mercury to the atmosphere through direct Hg degassing. It may also act as a sink for atmospheric mercury due to the simultaneous emission of aerosols that do not only scavenge GEM that was emitted from the volcanic vent but also GEM that was already present in the atmosphere. The source and sink effects may partly balance each other out, and both may have to be adequately estimated to determine the net impact of terrestrial volcanism on the atmospheric Hg reservoir.
Based on the results from Section 3.4.2 (see also Fig. 6), the possible magnitude of Hg scavenging by volcanic aerosols can be constrained: The global mass flux of volcanically-derived sulfate is estimated to be ∼20 × 106 Mg y−1, on average.113,114 Assuming that volcanic aerosols scavenge GEM proportionally to their sulfate content with a relationship of 10.4 (3.3; 24) μgGEM g−1sulfate, the resulting contribution of volcanic aerosols to global GEM scavenging would be 210 (67; 480) Mg y−1.
This estimate is evidently subject to large uncertainties concerning the underlying physical or chemical mechanisms of the gas-particle interactions, the capacity of volcanic particles to take up GEM, their global mass flux, the role of somewhat larger volcanic particles (e.g. PM2.5, PM10), the influence of particle composition (e.g. sulfate and halogen content, acidity, noble metals or transition metal oxides), and so on. Nevertheless, it illustrates that the source and sink effects of terrestrial volcanism on the atmospheric Hg budget may be of similar magnitude and that both require attention.
5. Conclusions
We observed depletion of GEM in dilute and moderately aged (∼3–7 hours) volcanic plumes. While previous work has suggested that such a depletion might occur due to volcanic bromine emissions and reactive bromine chemistry, multiple lines of evidence rule out a mostly bromine-driven process here. Instead, we hypothesized that gas–particle interactions were the underlying driver and explored possible manifestations of such a gas–particle interaction. We derived empirical interaction rate constants and constrained the magnitude of GEM scavenging in function of the volcanic submicron particle mass. Extrapolating our observations and results to the global scale, we estimated that volcanic aerosols might globally scavenge 210 (67; 480) Mg y−1 of Hg from the atmosphere. While subject to significant uncertainties, this is similar in magnitude to estimated Hg emissions from terrestrial volcanism,1,2 highlighting that the fate of Hg in volcanic plumes, as they age, dilute, and entrain background air, requires further attention.Our results have important implications for the role of volcanism in the global atmospheric Hg budget. They show that terrestrial volcanism, generally assumed to be a source of Hg to the atmosphere, may also have a significant sink effect on atmospheric Hg through the co-emission of Hg-scavenging aerosols. While this process would tend to reduce the global atmospheric Hg pool, it would also tend to enhance Hg deposition and the potential for Hg exposure in volcanic regions inhabited by human populations.
6. Recommendations for further research
• Observational constraint of Hg transformations in aging volcanic plumes is needed. To address the physical and chemical nature of gas–particle interactions, as well as their kinetics, simultaneous observations of speciated Hg and ancillary variables such as SO2, O3, BrO, and suspended fine particles (at least <10 μm, better <2.5 μm or <1 μm), ideally both in their concentration and composition, would be highly beneficial.• How the particle composition (e.g. sulfate content, halogen content, noble metals & transition metal oxides) may affect the capacity of volcanic aerosols to scavenge Hg requires further evaluation. This could either be achieved by adequate observations in a sufficiently aged volcanic plume or by the sampling of already settled volcanic particles (see Ermolin et al., 2018).79 In the latter case, it appears essential to size-segregate the particles. At the very least, fine particles (at least <10 μm, but preferentially smaller) should be analysed separately from the bulk ash.
• More generally, we suggest that measurements of PBM should, whenever possible, be complemented by simultaneous measurements of the particle mass in the corresponding particle size range to allow the determination of Hg mass fractions. This would allow constraining Hg uptake capacities and gas-particle partitioning, as well as improve the inter-comparability between studies.
Appendices
A. PM0.95 sulfate proxy
As SO2 is a tracer for plume intensity and plume dilution, one can expect a positive correlation between SO2 concentrations and primary sulfate aerosol in the plume. SO2 is also a precursor for secondary sulfate, which is formed when SO2 is oxidized. For relatively short timescales, when most of the SO2 is not yet consumed in the plume (as was most likely the case for the plumes observed here; see Section 2.3.5), higher SO2 concentrations indicate a higher potential for sulfate generation, and we would expect that secondary sulfate as well correlates with SO2. On the other hand, assuming that a fraction of sulfate is found on particles below 700 nm in diameter, higher PM0.95 sulfate should be directly reflected in higher DMPS-derived particle volume (PV0.7DMPS). We would consequently also expect a positive correlation between PM0.95 sulfate and PV0.7DMPS.We consider the expected correlations with both SO2 and PV0.7DMPS to define PM0.95sulfate proxy, by expressing it as a linear function of the geometric mean of SO2 and the PV0.7DMPS anomaly (eqn (4)). We employed the PV0.7DMPS anomaly, defined as PV0.7DMPS in the plume minus PV0.7DMPS background concentrations, to avoid relating volcanic sulfate to particles that are not of volcanic origin.
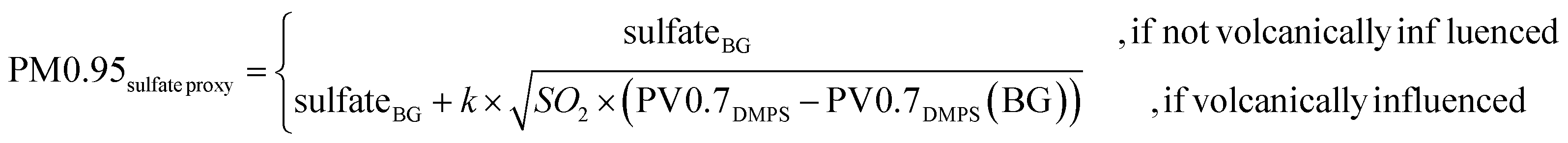 | (4) |
We constrain the fitting parameters sulfateBG and k of PM0.95sulfate proxy by comparison to our filter-based sulfate measurements. More precisely, we compute a linear regression between sulfate measurements and 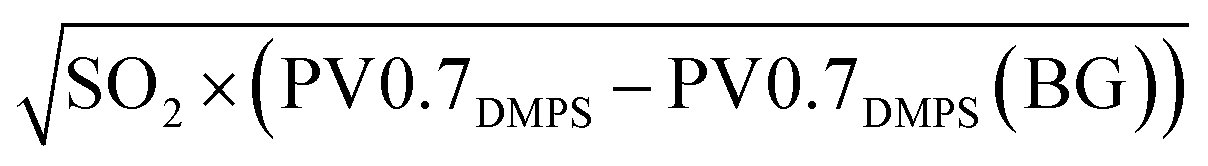 averaged over the same time period. We only use daytime filters for this comparison, as no volcanic plume related to observed GEM depletion events was captured in a nightime filter. We also limit the comparison to filters that were operating between the 1st of April 2018 and the 2nd of May 2018, i.e. before the eruptive phase and early in the eruptive phase (see Fig. 2 and 4), because the relationship between PV0.7DMPS, SO2, and PM0.95 sulfate likely changes as the eruptive period of Piton de la Fournaise evolves. Values of 1.3 ± 2 [μg m−3] and 4500 ± 500 [μgsulfateppbv−1/2 cm−3/2 m−3/2] are obtained for sulfateBG and k, respectively (see Fig. 7). While the linear relationship is strong (R2 = 0.99), we assign an elevated relative uncertainty of ±50% (k = 2) to PM0.95sulfate proxy, for reasons that were already laid out in Section 2.3.3.
averaged over the same time period. We only use daytime filters for this comparison, as no volcanic plume related to observed GEM depletion events was captured in a nightime filter. We also limit the comparison to filters that were operating between the 1st of April 2018 and the 2nd of May 2018, i.e. before the eruptive phase and early in the eruptive phase (see Fig. 2 and 4), because the relationship between PV0.7DMPS, SO2, and PM0.95 sulfate likely changes as the eruptive period of Piton de la Fournaise evolves. Values of 1.3 ± 2 [μg m−3] and 4500 ± 500 [μgsulfateppbv−1/2 cm−3/2 m−3/2] are obtained for sulfateBG and k, respectively (see Fig. 7). While the linear relationship is strong (R2 = 0.99), we assign an elevated relative uncertainty of ±50% (k = 2) to PM0.95sulfate proxy, for reasons that were already laid out in Section 2.3.3.
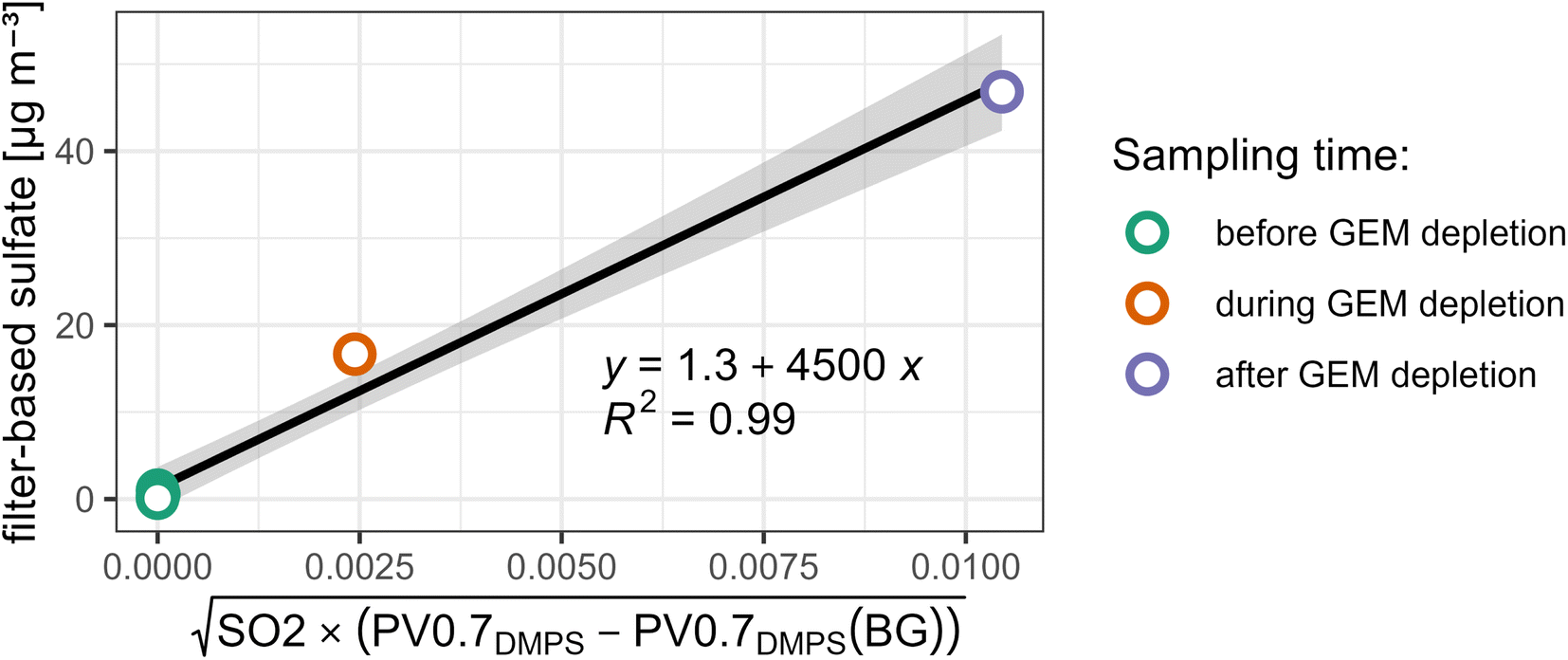 | ||
| Fig. 7 Constraining fitting parameters for PM0.95sulfate proxy with filter-based measurements under consideration that PM0.95sulfate proxy should be equal to measured PM0.95 sulfate, if averaged over the same sampling period. | ||
B. Plume transport time
The plume transport time, i.e. the time between plume emission at Piton de la Fournaise (at time t – transport time) and plume arrival at Maïdo (at time t), was estimated with eqn (5), which we solved numerically, as the transport time is contained in both sides of the equation. This is because the observation that is used to represent wind speed at plume departure is the wind speed at Piton Partage at time t – transport time, i.e. it depends on the estimated transport time itself.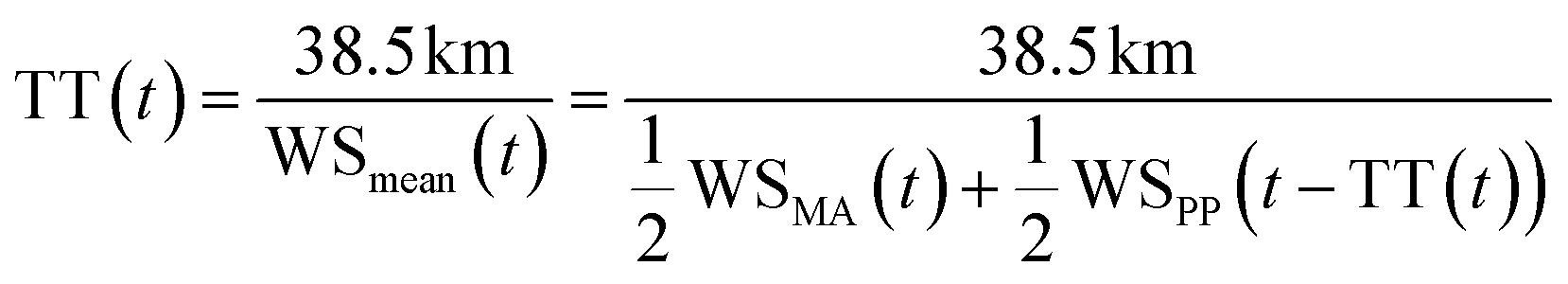 | (5) |
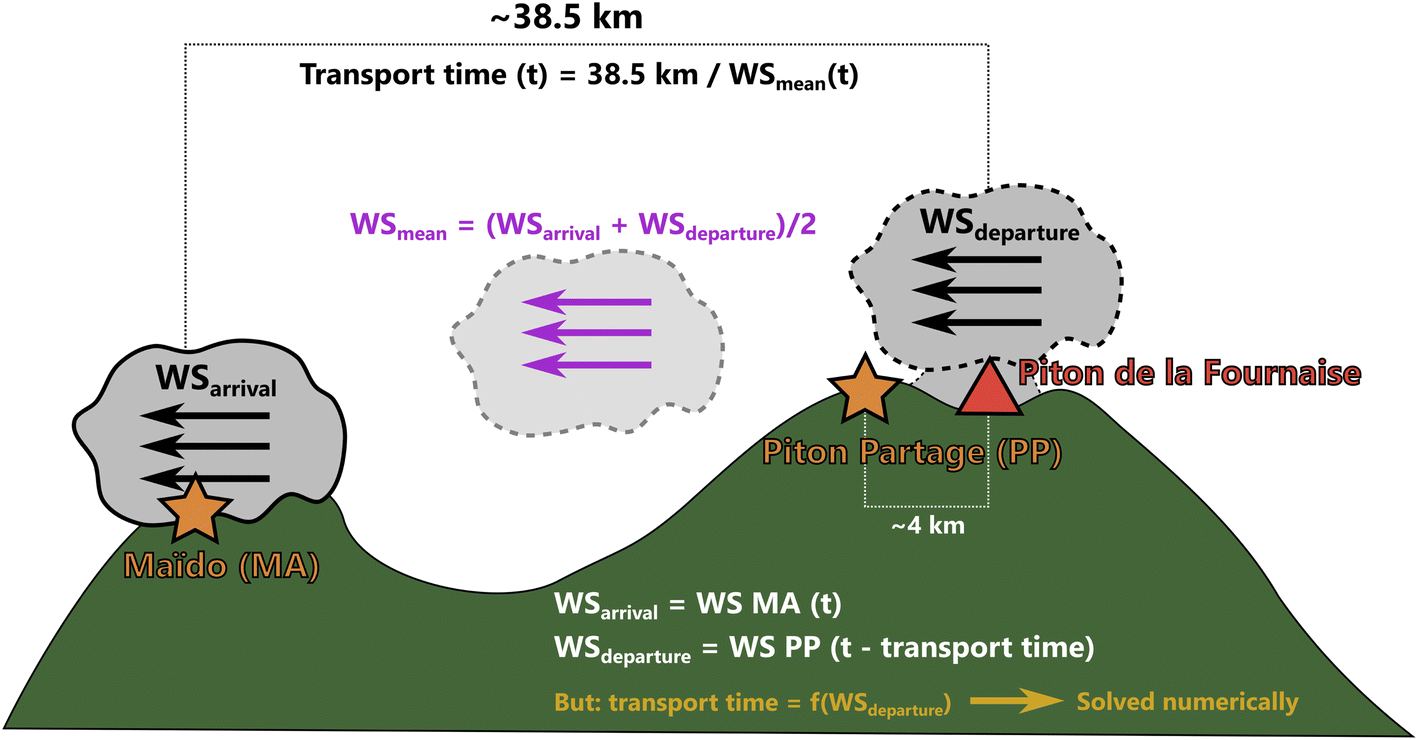 | ||
| Fig. 8 Scheme of transport time estimation (following eqn (5)) for a plume arriving at Maïdo at time “t”. | ||
C. GEM/SO2 emission ratio
The two GEM passive samplers that were operating close to the eruptive vent (∼200 m distance) yielded elevated GEM concentrations (1.44 ± 0.14 ng m−3) compared to the rest of the passive samplers deployed on the island (1.08 ± 0.08 ng m−3). The difference (0.36 ± 0.16 ng m3) is statistically significant at the 95% confidence level (p = 0.02; N = 81; nonparametric Mann–Whitney Rank Sum test), strongly suggesting GEM emission by Piton de la Fournaise. We take 0.52 ng m−3 as an upper limit for the volcanic GEM enhancement (i.e. measured GEM minus background), under consideration of the passive sampler uncertainties as reported in Hoang et al. (2023).45 Although a recent study suggests that passive samplers may underestimate GEM in volcanic conditions,115 this effect is not considered here as it remains difficult to quantify and must be confirmed by further studies.As a lower limit for the mean volcanic SO2 enhancement at the location of the passive samplers, we take the mean SO2 concentrations at the summit over the whole eruptive period (∼76 ppbv). Combining the upper and lower limits for GEM and SO2 enhancements, respectively, one obtains 2.4 × 10−6 [ng ng−1] as an upper limit for the mean ER from Piton de la Fournaise. This is an upper limit because SO2 concentrations at the passive sampler location (∼200 m from the vent) were most likely significantly higher than at the summit (∼1.5 km from the vent). However, the exact difference is difficult to quantify as it depends on parameters such as wind speed, turbulence, vertical plume transport, and so on. To define a conservative lower limit for the GEM/SO2 emission ratio from Piton de la Fournaise, we thus do not use the limited observations here, but adopt instead directly the lowest GEM/SO2 ER reported in the literature so far (1.5 × 10−7 [ng ng−1]).65
In the absence of any additional observations that allow to constrain the probability distribution of the GEM/SO2 ER from Piton de la Fournaise, we express it as a uniform distribution between the above-defined upper and lower limits (2.4 × 10−6 and 1.5 × 10−7 [ng ng−1], respectively). Unless otherwise mentioned, the whole of this distribution is considered for all ER-related calculations in the manuscript (through Monte Carlo simulations).
Data availability
Maïdo L2 GEM data (https://doi.org/10.25326/352) are freely available116 at https://gmos.aeris-data.fr/ (last access: 30 January 2023) from the GMOS-FR data portal coordinated by IGE (Institut des Géosciences de l'Environnement – Grenoble, France; technical PI: Olivier Magand) and maintained by the French national center for atmospheric data and services (AERIS). O2–O2, SO2, BrO, and IO differential slant column densities (dSCDs) measured by the University of Colorado Multi-AXis Differential Optical Absorption Spectroscopy (CU MAX-DOAS) are free available at https://doi.org/10.5281/zenodo.7864745. DMPS data are available from the EBAS Data Centre (https://ebas.nilu.no/; last access April 28th, 2023). MultiGas and DOAS data from OVPF (IPGP) are available upon request.Author contributions
AMK performed the formal analysis, created visualizations, and wrote the manuscript with contributions from all authors. All authors contributed to experimental or statistical methods. OM, CR, ADM, YM, AC, MR, CFL, TKK, RV, KS, SA, PK, AA, and AD were involved in data curation. OM, ADM, YM, AC, and SA performed field work. OM, AD, and JES conceptualized the observational setup and defined research goals. OM, CR, ADM, YM, AC, MR, TKK, RV, JB, KS, JES, and AD acquired funding. YM supervised laboratory analysis of aerosol samples. OM, CR, ADM, YM, AC, MR, CFL, TKK, RV, KS, and SA provided experimental data. AD, JES, and OM supervised the research.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This publication is part of the GMOS-Train project that has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 860497. Support was received from the European Union's Horizon 2020 Research and Innovation programme (ACTRIS TNA; grant agreement no. 654109). The support from the Academy of Finland (331207) is appreciated. R. V. acknowledges funding from the US National Science Foundation (AGS-1620530, AGS-1951514). TJR acknowledges ANR PRC VOLC-HAL-CLIM ANR-18-CE01-0018. S. A acknowledges funding from the Swedish National Space Agency (Dnr. 149/18). Maïdo GEM data were collected via instruments coordinated by the IGE-PTICHA technical platform dedicated to atmospheric chemistry field instrumentation. The authors acknowledge the AERIS data infrastructure for providing access to the GEM data in this study. We acknowledge OPAR (Observatoire de Physique de l'Atmosphère à La Réunion), funded by CNRS-INSU and Université de La Réunion and managed by OSU-R (Observatoire des Sciences de l'Univers à La Réunion, UMS 3365). Jean-Marc Metzger, from OSU-R, and Christelle Barthe, from LACy and Meteo-France, are particularly acknowledged for their support in the implementation of OPAR's instrumentation, and the provision of meteorological data.References
- B. A. Edwards, D. S. Kushner, P. M. Outridge and F. Wang, Fifty years of volcanic mercury emission research: Knowledge gaps and future directions, Sci. Total Environ., 2021, 757, 143800 CrossRef CAS
.
- C. Li, J. E. Sonke, G. Le Roux, N. Piotrowska, N. Van der Putten, S. J. Roberts, T. Daley, E. Rice, R. Gehrels, M. Enrico, D. Mauquoy, T. P. Roland and F. De Vleeschouwer, Unequal Anthropogenic Enrichment of Mercury in Earth's Northern and Southern Hemispheres, ACS Earth Space Chem., 2020, 4, 2073–2081 CrossRef CAS
-
E. Bagnato, G. Tamburello, G. Avard, M. Martinez-Cruz, M. Enrico, X. Fu, M. Sprovieri and J. E. Sonke, Mercury fluxes from volcanic and geothermal sources: an update, Geological Society, London, Special Publications, 2014, vol. 410, pp. 263–285 Search PubMed
- L. Zhang, L. P. Wright and P. Blanchard, A review of current knowledge concerning dry deposition of atmospheric mercury, Atmos. Environ., 2009, 43, 5853–5864 CrossRef CAS
- H. M. Horowitz, D. J. Jacob, Y. Zhang, T. S. Dibble, F. Slemr, H. M. Amos, J. A. Schmidt, E. S. Corbitt, E. A. Marais and E. M. Sunderland, A new mechanism for atmospheric mercury redox chemistry: implications for the global mercury budget, Atmos. Chem. Phys., 2017, 17, 6353–6371 CrossRef CAS
- V. Shah, D. J. Jacob, C. P. Thackray, X. Wang, E. M. Sunderland, T. S. Dibble, A. Saiz-Lopez, I. Černušák, V. Kellö, P. J. Castro, R. Wu and C. Wang, Improved Mechanistic Model of the Atmospheric Redox Chemistry of Mercury, Environ. Sci. Technol., 2021, 1c03160 Search PubMed
- B. Gworek, W. Dmuchowski and A. H. Baczewska-Dąbrowska, Mercury in the terrestrial environment: a review, Environ. Sci. Eur., 2020, 32, 128 CrossRef CAS
- N. Bobrowski, R. Von Glasow, A. Aiuppa, S. Inguaggiato, I. Louban, O. W. Ibrahim and U. Platt, Reactive halogen chemistry in volcanic plumes, J. Geophys. Res., 2007, 112, D06311 CrossRef
- R. S. Martin, M. L. I. Witt, D. M. Pyle, T. A. Mather, S. F. L. Watt, E. Bagnato and S. Calabrese, Rapid oxidation of mercury (Hg) at volcanic vents: Insights from high temperature thermodynamic models of Mt Etna's emissions, Chem. Geol., 2011, 283, 279–286 CrossRef CAS
- R. von Glasow, Atmospheric chemistry in volcanic plumes, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 6594–6599 CrossRef CAS PubMed
- A. Aiuppa, E. Bagnato, M. L. I. Witt, T. A. Mather, F. Parello, D. M. Pyle and R. S. Martin, Real-time simultaneous detection of volcanic Hg and SO 2 at La Fossa Crater, Vulcano (Aeolian Islands, Sicily), Geophys. Res. Lett., 2007, 34, L21307 CrossRef
- E. Bagnato, A. Aiuppa, F. Parello, S. Calabrese, W. D'Alessandro, T. A. Mather, A. J. S. McGonigle, D. M. Pyle and I. Wängberg, Degassing of gaseous (elemental and reactive) and particulate mercury from Mount Etna volcano (Southern Italy), Atmos. Environ., 2007, 41, 7377–7388 CrossRef CAS
- T. Zambardi, J. E. Sonke, J. P. Toutain, F. Sortino and H. Shinohara, Mercury emissions and stable isotopic compositions at Vulcano Island (Italy), Earth Planet. Sci. Lett., 2009, 277, 236–243 CrossRef CAS
- A. Gutmann, N. Bobrowski, T. J. Roberts, J. Rüdiger and T. Hoffmann, Advances in Bromine Speciation in Volcanic Plumes, Front. Earth Sci., 2018, 6, 213 CrossRef
- J. Gliß, N. Bobrowski, L. Vogel, D. Pöhler and U. Platt, OClO and BrO observations in the volcanic plume of Mt. Etna – implications on the chemistry of chlorine and bromine species in volcanic plumes, Atmos. Chem. Phys., 2015, 15, 5659–5681 CrossRef
- L. Surl, T. Roberts and S. Bekki, Observation and modelling of ozone-destructive halogen chemistry in a passively degassing volcanic plume, Atmos. Chem. Phys., 2021, 21, 12413–12441 CrossRef CAS
- T. J. Roberts, C. F. Braban, R. S. Martin, C. Oppenheimer, J. W. Adams, R. A. Cox, R. L. Jones and P. T. Griffiths, Modelling reactive halogen formation and ozone depletion in volcanic plumes, Chem. Geol., 2009, 263, 151–163 CrossRef CAS
- C. D. Holmes, D. J. Jacob, E. S. Corbitt, J. Mao, X. Yang, R. Talbot and F. Slemr, Global atmospheric model for mercury including oxidation by bromine atoms, Atmos. Chem. Phys., 2010, 10, 12037–12057 CrossRef CAS
- L. A. Barrie, J. W. Bottenheim, R. C. Schnell, P. J. Crutzen and R. A. Rasmussen, Ozone destruction and photochemical reactions at polar sunrise in the lower Arctic atmosphere, Nature, 1988, 334, 138–141 CrossRef CAS
- W. R. Simpson, R. von Glasow, K. Riedel, P. Anderson, P. Ariya, J. Bottenheim, J. Burrows, L. J. Carpenter, U. Frieß, M. E. Goodsite, D. Heard, M. Hutterli, H.-W. Jacobi, L. Kaleschke, B. Neff, J. Plane, U. Platt, A. Richter, H. Roscoe, R. Sander, P. Shepson, J. Sodeau, A. Steffen, T. Wagner and E. Wolff, Halogens and their role in polar boundary-layer ozone depletion, Atmos. Chem. Phys., 2007, 7, 4375–4418 CrossRef CAS
- J. W. Halfacre, T. N. Knepp, P. B. Shepson, C. R. Thompson, K. A. Pratt, B. Li, P. K. Peterson, S. J. Walsh, W. R. Simpson, P. A. Matrai, J. W. Bottenheim, S. Netcheva, D. K. Perovich and A. Richter, Temporal and spatial characteristics of ozone depletion events from measurements in the Arctic, Atmos. Chem. Phys., 2014, 14, 4875–4894 CrossRef
- S. Wang, S. M. McNamara, C. W. Moore, D. Obrist, A. Steffen, P. B. Shepson, R. M. Staebler, A. R. W. Raso and K. A. Pratt, Direct detection of atmospheric atomic bromine leading to mercury and ozone depletion, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 14479–14484 CrossRef CAS PubMed
- A. Steffen, T. Douglas, M. Amyot, P. Ariya, K. Aspmo, T. Berg, J. Bottenheim, S. Brooks, F. Cobbett, A. Dastoor, A. Dommergue, R. Ebinghaus, C. Ferrari, K. Gardfeldt, M. E. Goodsite, D. Lean, A. J. Poulain, C. Scherz, H. Skov, J. Sommar and C. Temme, A synthesis of atmospheric mercury depletion event chemistry in the atmosphere and snow, Atmos. Chem. Phys., 2008, 8, 1445–1482 CrossRef CAS
- J.-L. Baray, Y. Courcoux, P. Keckhut, T. Portafaix, P. Tulet, J.-P. Cammas, A. Hauchecorne, S. Godin Beekmann, M. De Mazière, C. Hermans, F. Desmet, K. Sellegri, A. Colomb, M. Ramonet, J. Sciare, C. Vuillemin, C. Hoareau, D. Dionisi, V. Duflot, H. Vérèmes, J. Porteneuve, F. Gabarrot, T. Gaudo, J.-M. Metzger, G. Payen, J. Leclair de Bellevue, C. Barthe, F. Posny, P. Ricaud, A. Abchiche and R. Delmas, Maïdo observatory: a new high-altitude station facility at Reunion Island (21° S, 55° E) for long-term atmospheric remote sensing and in situ measurements, Atmos. Meas. Tech., 2013, 6, 2865–2877 CrossRef
- A. M. Koenig, O. Magand, B. Verreyken, J. Brioude, C. Amelynck, N. Schoon, A. Colomb, B. Ferreira Araujo, M. Ramonet, M. K. Sha, J.-P. Cammas, J. E. Sonke and A. Dommergue, Mercury in the free troposphere and bidirectional atmosphere – vegetation exchanges – insights from Maïdo mountain observatory in the Southern Hemisphere tropics, Atmos. Chem. Phys., 2023, 23, 1309–1328 CrossRef CAS
- G. Roult, A. Peltier, B. Taisne, T. Staudacher, V. Ferrazzini and A. Di Muro, A new comprehensive classification of the Piton de la Fournaise activity spanning the 1985–2010 period. Search and analysis of short-term precursors from a broad-band seismological station, J. Volcanol. Geotherm. Res., 2012, 241–242, 78–104 CrossRef CAS
- L. Michon, A. Di Muro, N. Villeneuve, C. Saint-Marc, P. Fadda and F. Manta, Explosive activity of the summit cone of Piton de la Fournaise volcano (La Réunion island): A historical and geological review, J. Volcanol. Geotherm. Res., 2013, 264, 117–133 CrossRef CAS
- E. Dœlsch, H. Saint Macary and V. Van de Kerchove, Sources of very high heavy metal content in soils of volcanic island (La Réunion), J. Geochem. Explor., 2006, 88, 194–197 CrossRef
- J. L. Ambrose, Improved methods for signal processing in measurements of mercury by Tekran® 2537A and 2537B instruments, Atmos. Meas. Tech., 2017, 10, 5063–5073 CrossRef
- O. Magand, D. Boulanger and A. Dommergue, GMOS-FR (Global Mercury Observation System – FRance) Research Data Management Plan for Atmospheric Mercury Datasets, DOI:10.5281/ZENODO.7406251.
- F. Slemr, A. Weigelt, R. Ebinghaus, H. H. Kock, J. Bödewadt, C. A. M. Brenninkmeijer, A. Rauthe-Schöch, S. Weber, M. Hermann, J. Becker, A. Zahn and B. Martinsson, Atmospheric mercury measurements onboard the CARIBIC passenger aircraft, Atmos. Meas. Tech., 2016, 9, 2291–2302 CrossRef CAS
- H. Junninen, M. Ehn, T. Petäjä, L. Luosujärvi, T. Kotiaho, R. Kostiainen, U. Rohner, M. Gonin, K. Fuhrer, M. Kulmala and D. R. Worsnop, A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 2010, 3, 1039–1053 CrossRef CAS
- T. Jokinen, M. Sipilä, H. Junninen, M. Ehn, G. Lönn, J. Hakala, T. Petäjä, R. L. Mauldin, M. Kulmala and D. R. Worsnop, Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 2012, 12, 4117–4125 CrossRef CAS
- C. Rose, M. P. Rissanen, S. Iyer, J. Duplissy, C. Yan, J. B. Nowak, A. Colomb, R. Dupuy, X.-C. He, J. Lampilahti, Y. J. Tham, D. Wimmer, J.-M. Metzger, P. Tulet, J. Brioude, C. Planche, M. Kulmala and K. Sellegri, Investigation of several proxies to estimate sulfuric acid concentration under volcanic plume conditions, Atmos. Chem. Phys., 2021, 21, 4541–4560 CrossRef CAS
- B. Foucart, K. Sellegri, P. Tulet, C. Rose, J.-M. Metzger and D. Picard, High occurrence of new particle formation events at the Maïdo high-altitude observatory (2150 m), Réunion (Indian Ocean), Atmos. Chem. Phys., 2018, 18, 9243–9261 CrossRef
- C. Rose, B. Foucart, D. Picard, A. Colomb, J.-M. Metzger, P. Tulet and K. Sellegri, New particle formation in the volcanic eruption plume of the Piton de la Fournaise: specific features from a long-term dataset, Atmos. Chem. Phys., 2019, 19, 13243–13265 CrossRef CAS
- A. Wiedensohler, W. Birmili, A. Nowak, A. Sonntag, K. Weinhold, M. Merkel, B. Wehner, T. Tuch, S. Pfeifer, M. Fiebig, A. M. Fjäraa, E. Asmi, K. Sellegri, R. Depuy, H. Venzac, P. Villani, P. Laj, P. Aalto, J. A. Ogren, E. Swietlicki, P. Williams, P. Roldin, P. Quincey, C. Hüglin, R. Fierz-Schmidhauser, M. Gysel, E. Weingartner, F. Riccobono, S. Santos, C. Grüning, K. Faloon, D. Beddows, R. Harrison, C. Monahan, S. G. Jennings, C. D. O'Dowd, A. Marinoni, H.-G. Horn, L. Keck, J. Jiang, J. Scheckman, P. H. McMurry, Z. Deng, C. S. Zhao, M. Moerman, B. Henzing, G. de Leeuw, G. Löschau and S. Bastian, Mobility particle size spectrometers: harmonization of technical standards and data structure to facilitate high quality long-term observations of atmospheric particle number size distributions, Atmos. Meas. Tech., 2012, 5, 657–685 CrossRef CAS
- B. Verreyken, C. Amelynck, N. Schoon, J.-F. Müller, J. Brioude, N. Kumps, C. Hermans, J.-M. Metzger, A. Colomb and T. Stavrakou, Measurement report: Source apportionment of volatile organic compounds at the remote high-altitude Maïdo observatory, Atmos. Chem. Phys., 2021, 21, 12965–12988 CrossRef CAS
- S. A. Simu, Y. Miyazaki, E. Tachibana, H. Finkenzeller, J. Brioude, A. Colomb, O. Magand, B. Verreyken, S. Evan, R. Volkamer and T. Stavrakou, Origin of water-soluble organic aerosols at the Maïdo high-altitude observatory, Réunion Island, in the tropical Indian Ocean, Atmos. Chem. Phys., 2021, 21, 17017–17029 CrossRef CAS
- S. Coburn, B. Dix, R. Sinreich and R. Volkamer, The CU ground MAX-DOAS instrument: characterization of RMS noise limitations and first measurements near Pensacola, FL of BrO, IO, and CHOCHO, Atmos. Meas. Tech., 2011, 4, 2421–2439 CrossRef CAS
- S. Coburn, B. Dix, E. Edgerton, C. D. Holmes, D. Kinnison, Q. Liang, A. ter Schure, S. Wang and R. Volkamer, Mercury oxidation from bromine chemistry in the free troposphere over the southeastern US, Atmos. Chem. Phys., 2016, 16, 3743–3760 CrossRef CAS
- H. Finkenzeller, S. Iyer, X.-C. He, M. Simon, T. K. Koenig, C. F. Lee, R. Valiev, V. Hofbauer, A. Amorim, R. Baalbaki, A. Baccarini, L. Beck, D. M. Bell, L. Caudillo, D. Chen, R. Chiu, B. Chu, L. Dada, J. Duplissy, M. Heinritzi, D. Kemppainen, C. Kim, J. Krechmer, A. Kürten, A. Kvashnin, H. Lamkaddam, C. P. Lee, K. Lehtipalo, Z. Li, V. Makhmutov, H. E. Manninen, G. Marie, R. Marten, R. L. Mauldin, B. Mentler, T. Müller, T. Petäjä, M. Philippov, A. Ranjithkumar, B. Rörup, J. Shen, D. Stolzenburg, C. Tauber, Y. J. Tham, A. Tomé, M. Vazquez-Pufleau, A. C. Wagner, D. S. Wang, M. Wang, Y. Wang, S. K. Weber, W. Nie, Y. Wu, M. Xiao, Q. Ye, M. Zauner-Wieczorek, A. Hansel, U. Baltensperger, J. Brioude, J. Curtius, N. M. Donahue, I. E. Haddad, R. C. Flagan, M. Kulmala, J. Kirkby, M. Sipilä, D. R. Worsnop, T. Kurten, M. Rissanen and R. Volkamer, The gas-phase formation mechanism of iodic acid as an atmospheric aerosol source, Nat. Chem., 2023, 15, 129–135 CrossRef CAS PubMed
- B. Verreyken, C. Amelynck, J. Brioude, J.-F. Müller, N. Schoon, N. Kumps, A. Colomb, J.-M. Metzger, C. F. Lee, T. K. Koenig, R. Volkamer and T. Stavrakou, Characterisation of African biomass burning plumes and impacts on the atmospheric composition over the south-west Indian Ocean, Atmos. Chem. Phys., 2020, 20, 14821–14845 CrossRef CAS
- D. S. McLagan, C. P. J. Mitchell, H. Huang, Y. D. Lei, A. S. Cole, A. Steffen, H. Hung and F. Wania, A High-Precision Passive Air Sampler for Gaseous Mercury, Environ. Sci. Technol. Lett., 2016, 3, 24–29 CrossRef CAS
- C. Hoang, O. Magand, J. Brioude, A. Dimuro, C. Brunet, C. Ah-Peng, Y. Bertrand, A. Dommergue, Y. D. Lei and F. Wania, Probing the limits of sampling gaseous elemental mercury passively in the remote atmosphere, Environ. Sci.: Atmos., 2023, 3, 268–281 CAS
- A. Aiuppa, Chemical mapping of a fumarolic field: La Fossa Crater, Vulcano Island (Aeolian Islands, Italy), Geophys. Res. Lett., 2005, 32, L13309 CrossRef
- B. Galle, M. Johansson, C. Rivera, Y. Zhang, M. Kihlman, C. Kern, T. Lehmann, U. Platt, S. Arellano and S. Hidalgo, Network for Observation of Volcanic and Atmospheric Change (NOVAC)—A global network for volcanic gas monitoring: Network layout and instrument description, J. Geophys. Res., 2010, 115, D05304 CrossRef
- S. Arellano, B. Galle, F. Apaza, G. Avard, C. Barrington, N. Bobrowski, C. Bucarey, V. Burbano, M. Burton, Z. Chacón, G. Chigna, C. J. Clarito, V. Conde, F. Costa, M. De Moor, H. Delgado-Granados, A. Di Muro, D. Fernandez, G. Garzón, H. Gunawan, N. Haerani, T. H. Hansteen, S. Hidalgo, S. Inguaggiato, M. Johansson, C. Kern, M. Kihlman, P. Kowalski, P. Masias, F. Montalvo, J. Möller, U. Platt, C. Rivera, A. Saballos, G. Salerno, B. Taisne, F. Vásconez, G. Velásquez, F. Vita and M. Yalire, Synoptic analysis of a decade of daily measurements of SO2 emission in the troposphere from volcanoes of the global ground-based Network for Observation of Volcanic and Atmospheric Change, Earth Syst. Sci. Data, 2021, 13, 1167–1188 CrossRef
- N. Metropolis and S. Ulam, The Monte Carlo Method, J. Am. Stat. Assoc., 1949, 44, 335–341 CrossRef CAS
- H. Janssen, Monte-Carlo based uncertainty analysis: Sampling efficiency and sampling convergence, Reliab. Eng. Syst. Saf., 2013, 109, 123–132 CrossRef
- NCAR, Tropospheric Ultraviolet and Visible (TUV) Radiation Model v5.3, 2019 Search PubMed.
- J. I. Gmitro and T. Vermeulen, Vapor-liquid equilibria for aqueous sulfuric acid, AIChE J., 1964, 10, 740–746 CrossRef
- C. E. L. Myhre, C. J. Nielsen and O. W. Saastad, Density and Surface Tension of Aqueous H2 SO4 at Low Temperature, J. Chem. Eng. Data, 1998, 43, 617–622 CrossRef CAS
- T. A. Mather, A. G. Allen, C. Oppenheimer, D. M. Pyle and A. J. S. McGonigle, Size-Resolved Characterisation of Soluble Ions in the Particles in the Tropospheric Plume of Masaya Volcano, Nicaragua: Origins and Plume Processing, J. Atmos. Chem., 2003, 46, 207–237 CrossRef CAS
- J. J. Naughton, V. Lewis, D. Thomas and J. B. Finlayson, Fume compositions found at various stages of activity at Kilauea Volcano, Hawaii, J. Geophys. Res., 1975, 80, 2963–2966 CrossRef CAS
- T. J. Roberts, D. Vignelles, M. Liuzzo, G. Giudice, A. Aiuppa, M. Coltelli, G. Salerno, M. Chartier, B. Couté, G. Berthet, T. Lurton, F. Dulac and J.-B. Renard, The primary volcanic aerosol emission from Mt Etna: Size-resolved particles with SO2 and role in plume reactive halogen chemistry, Geochim. Cosmochim. Acta, 2018, 222, 74–93 CrossRef CAS
- T. A. Mather, V. I. Tsanev, D. M. Pyle, A. J. S. McGonigle, C. Oppenheimer and A. G. Allen, Characterization and evolution of tropospheric plumes from Lascar and Villarrica volcanoes, Chile, J. Geophys. Res., 2004, 109, 148–227 CrossRef
- R. S. Martin, T. A. Mather, D. M. Pyle, M. Power, A. G. Allen, A. Aiuppa, C. J. Horwell and E. P. W. Ward, Composition-resolved size distributions of volcanic aerosols in the Mt. Etna plumes, J. Geophys. Res., 2008, 113, D17211 CrossRef
- S. M. Andersson, B. G. Martinsson, J. Friberg, C. A. M. Brenninkmeijer, A. Rauthe-Schöch, M. Hermann, P. F. J. van Velthoven and A. Zahn, Composition and evolution of volcanic aerosol from eruptions of Kasatochi, Sarychev and Eyjafjallajökull in 2008–2010 based on CARIBIC observations, Atmos. Chem. Phys., 2013, 13, 1781–1796 CrossRef
- C. O'Dowd, D. Ceburnis, J. Ovadnevaite, G. Martucci, J. Bialek, C. Monahan, H. Berresheim, A. Vaishya, T. Grigas, S. G. Jennings, P. McVeigh, S. Varghese, R. Flanagan, D. Martin, E. Moran, K. Lambkin, T. Semmler, C. Perrino and R. McGrath, The Eyjafjallajökull ash plume – Part I: Physical, chemical and optical characteristics, Atmos. Environ., 2012, 48, 129–142 CrossRef
- S. Thivet, L. Gurioli, A. Di Muro, J. Eychenne, P. Besson and J.-M. Nedelec, Variability of ash deposits at Piton de la Fournaise (La Reunion Island): insights into fragmentation processes at basaltic shield volcanoes, Bull. Volcanol., 2020, 82, 63 CrossRef
- A. K. Pattantyus, S. Businger and S. G. Howell, Review of sulfur dioxide to sulfate aerosol chemistry at Kīlauea Volcano, Hawaii, Atmos. Environ., 2018, 185, 262–271 CrossRef CAS
- J. H. Kroll, E. S. Cross, J. F. Hunter, S. Pai, L. M. M. Wallace, P. L. Croteau, J. T. Jayne, D. R. Worsnop, C. L. Heald, J. G. Murphy and S. L. Frankel, Atmospheric Evolution of Sulfur Emissions from Kilauea: Real-Time Measurements of Oxidation, Dilution, and Neutralization within a Volcanic Plume, Environ. Sci. Technol., 2015, 49, 4129–4137 CrossRef CAS PubMed
- S. A. Carn, K. D. Froyd, B. E. Anderson, P. Wennberg, J. Crounse, K. Spencer, J. E. Dibb, N. A. Krotkov, E. V. Browell, J. W. Hair, G. Diskin, G. Sachse and S. A. Vay,
In situ measurements of tropospheric volcanic plumes in Ecuador and Colombia during TC4, J. Geophys. Res., 2011, 116, D00J24 CrossRef
- R. Ferrara, B. Mazzolai, E. Lanzillotta, E. Nucaro and N. Pirrone, Volcanoes as emission sources of atmospheric mercury in the Mediterranean basin, Sci. Total Environ., 2000, 259, 115–121 CrossRef CAS PubMed
- R. D. Cadle and E. R. Frank, Particles in the fume from the 1967 Kilauea eruption, J. Geophys. Res., 1968, 73, 4780–4783 CrossRef CAS
- B. Langmann, On the Role of Climate Forcing by Volcanic Sulphate and Volcanic Ash, Adv. Meteorol., 2014, 1–17 Search PubMed
- R. Zhang, A. F. Khalizov, J. Pagels, D. Zhang, H. Xue and P. H. McMurry, Variability in morphology, hygroscopicity, and optical properties of soot aerosols during atmospheric processing, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 10291–10296 CrossRef CAS PubMed
- D. M. Pyle and T. A. Mather, Halogens in igneous processes and their fluxes to the atmosphere and oceans from volcanic activity: A review, Chem. Geol., 2009, 263, 110–121 CrossRef CAS
-
J. D. Webster, D. R. Baker and A. Aiuppa, in The Role of Halogens in Terrestrial and Extraterrestrial Geochemical Processes, ed. D. E. Harlov and L. Aranovich, Springer International Publishing, Cham, 2018, pp. 307–430 Search PubMed
- A. Cadoux, G. Iacono-Marziano, B. Scaillet, A. Aiuppa, T. A. Mather, D. M. Pyle, E. Deloule, E. Gennaro and A. Paonita, The role of melt composition on aqueous fluid vs. silicate melt partitioning of bromine in magmas, Earth Planet. Sci. Lett., 2018, 498, 450–463 CrossRef CAS
- A. Cadoux, S. Tegtmeier and A. Aiuppa, Natural Halogen Emissions to the Atmosphere: Sources, Flux, and Environmental Impact, Elements, 2022, 18, 27–33 CrossRef CAS
-
A. Di Muro, N. Métrich, P. Allard, A. Aiuppa, M. Burton, B. Galle and T. Staudacher, in Active Volcanoes of the Southwest Indian Ocean, ed. P. Bachelery, J.-F. Lenat, A. Di Muro and L. Michon, Springer Berlin Heidelberg, Berlin, Heidelberg, 2016, pp. 203–222 Search PubMed
-
P. Allard, A. La Spina, G. Tamburello, A. Aiuppa, M. Burton, A. Di Muro and T. Staudacher, First measurements of magmatic gas composition and fluxes during an eruption (October 2010) of Piton de la Fournaise hot spot volcano, La Reunion Island, in 11th Gas Workshop, Commission on the Chemistry of Volcanic Gases (CCVG)-IAVCEI-6, Kamchatka, Russia, 2011 Search PubMed
- C. S. Witham, C. Oppenheimer and C. J. Horwell, Volcanic ash-leachates: a review and recommendations for sampling methods, J. Volcanol. Geotherm. Res., 2005, 141, 299–326 CrossRef CAS
- C. Hörmann, H. Sihler, N. Bobrowski, S. Beirle, M. Penning de Vries, U. Platt and T. Wagner, Systematic investigation of bromine monoxide in volcanic plumes from space by using the GOME-2 instrument, Atmos. Chem. Phys., 2013, 13, 4749–4781 CrossRef
- N. Bobrowski, G. Hönninger, B. Galle and U. Platt, Detection of bromine monoxide in a volcanic plume, Nature, 2003, 423, 273–276 CrossRef CAS PubMed
- P. Bani, C. Oppenheimer, V. I. Tsanev, S. A. Carn, S. J. Cronin, R. Crimp, J. A. Calkins, D. Charley, M. Lardy and T. R. Roberts, Surge in sulphur and halogen degassing from Ambrym volcano, Vanuatu, Bull. Volcanol., 2009, 71, 1159–1168 CrossRef
- M. S. Ermolin, P. S. Fedotov, N. A. Malik and V. K. Karandashev, Nanoparticles of volcanic ash as a carrier for toxic elements on the global scale, Chemosphere, 2018, 200, 16–22 CrossRef CAS PubMed
- S. R. Taylor, Abundance of chemical elements in the continental crust: a new table, Geochim. Cosmochim. Acta, 1964, 28, 1273–1285 CrossRef CAS
-
W. F. Fitzgerald and C. H. Lamborg, in Treatise on Geochemistry, Elsevier, 2007, pp. 1–47 Search PubMed
- S. Ravindra Babu, L. S. P. Nguyen, G.-R. Sheu, S. M. Griffith, S. K. Pani, H.-Y. Huang and N.-H. Lin, Long-range transport of La Soufrière volcanic plume to the western North Pacific: Influence on atmospheric mercury and aerosol properties, Atmos. Environ., 2022, 268, 118806 CrossRef CAS
- Y. I. Tsai, S.-C. Kuo and Y.-H. Lin, Temporal characteristics of inhalable mercury and arsenic aerosols in the urban atmosphere in southern Taiwan, Atmos. Environ., 2003, 37, 3401–3411 CrossRef CAS
- G. Xiu, J. Cai, W. Zhang, D. Zhang, A. Büeler, S. Lee, Y. Shen, L. Xu, X. Huang and P. Zhang, Speciated mercury in size-fractionated particles in Shanghai ambient air, Atmos. Environ., 2009, 43, 3145–3154 CrossRef CAS
- X. Chen, R. Balasubramanian, Q. Zhu, S. N. Behera, D. Bo, X. Huang, H. Xie and J. Cheng, Characteristics of atmospheric particulate mercury in size-fractionated particles during haze days in Shanghai, Atmos. Environ., 2016, 131, 400–408 CrossRef CAS
- J. Zhu, T. Wang, R. Talbot, H. Mao, X. Yang, C. Fu, J. Sun, B. Zhuang, S. Li, Y. Han and M. Xie, Characteristics of atmospheric mercury deposition and size-fractionated particulate mercury in urban Nanjing, China, Atmos. Chem. Phys., 2014, 14, 2233–2244 CrossRef
- G. L. Xiu, Q. Jin, D. Zhang, S. Shi, X. Huang, W. Zhang, L. Bao, P. Gao and B. Chen, Characterization of size-fractionated particulate mercury in Shanghai ambient air, Atmos. Environ., 2005, 39, 419–427 CrossRef CAS
- M. Sakata and K. Marumoto, Formation of atmospheric particulate mercury in the Tokyo metropolitan area, Atmos. Environ., 2002, 36, 239–246 CrossRef CAS
- C. Seigneur, H. Abeck, G. Chia, M. Reinhard, N. S. Bloom, E. Prestbo and P. Saxena, Mercury adsorption to elemental carbon (soot) particles and atmospheric particulate matter, Atmos. Environ., 1998, 32, 2649–2657 CrossRef CAS
- Y. Li, Y. Wang, Y. Li, T. Li, H. Mao, R. Talbot, X. Nie, C. Wu, Y. Zhao, C. Hou, G. Wang, J. Zhou and G. Qie, Characteristics and potential sources of atmospheric particulate mercury in Jinan, China, Sci. Total Environ., 2017, 574, 1424–1431 CrossRef CAS PubMed
- H. M. Amos, D. J. Jacob, C. D. Holmes, J. A. Fisher, Q. Wang, R. M. Yantosca, E. S. Corbitt, E. Galarneau, A. P. Rutter, M. S. Gustin, A. Steffen, J. J. Schauer, J. A. Graydon, V. L. St. Louis, R. W. Talbot, E. S. Edgerton, Y. Zhang and E. M. Sunderland, Gas-particle partitioning of atmospheric Hg(II) and its effect on global mercury deposition, Atmos. Chem. Phys., 2012, 12, 591–603 CrossRef CAS
- L. Tong, T. Yue, P. Zuo, X. Zhang, C. Wang, J. Gao and K. Wang, Effect of characteristics of KI-impregnated activated carbon and flue gas components on Hg0 removal, Fuel, 2017, 197, 1–7 CrossRef CAS
- B. Fu, R. Sun, H. Yao, J. C. Hower, J. Yuan, G. Luo, H. Hu, S. M. Mardon and Q. Tang, Mercury stable isotope fractionation during gaseous elemental mercury adsorption onto coal fly ash particles: Experimental and field observations, J. Hazard. Mater., 2021, 405, 124280 CrossRef CAS PubMed
- M. She, C. Q. Jia, Y. Duan and C. Zhu, Influence of Different Sulfur Forms on Gas-Phase Mercury Removal by SO2-Impregnated Porous Carbons, Energy Fuels, 2020, 34, 2064–2073 CrossRef CAS
- S. Wu, Md. A. Uddin, S. Nagano, M. Ozaki and E. Sasaoka, Fundamental Study on Decomposition Characteristics of Mercury Compounds over Solid Powder by Temperature-Programmed Decomposition Desorption Mass Spectrometry, Energy Fuels, 2011, 25, 144–153 CrossRef CAS
- S. B. Ghorishi, R. M. Keeney, S. D. Serre, B. K. Gullett and W. S. Jozewicz, Development of a Cl-Impregnated Activated Carbon for Entrained-Flow Capture of Elemental Mercury, Environ. Sci. Technol., 2002, 36, 4454–4459 CrossRef CAS PubMed
- J. Wu, J. H. Chen, S. B. Zhang, P. He, J. H. Fang and Y. C. Wu, Removal of Gas-Phase Elemental Mercury by Bromine-Impregnated Activated Carbon, AMR, 2011, 356–360, 1660–1663 Search PubMed
- Y. Yao, V. Velpari and J. Economy, In search of brominated activated carbon fibers for elemental mercury removal from power plant effluents, J. Mater. Chem. A, 2013, 1, 12103 RSC
- J. Liu, W. Qu and C. Zheng, Theoretical studies of mercury–bromine species adsorption mechanism on carbonaceous surface, Proc. Combust. Inst., 2013, 34, 2811–2819 CrossRef CAS
- H.-C. Hsi, M. J. Rood, M. Rostam-Abadi, S. Chen and R. Chang, Mercury Adsorption Properties of Sulfur-Impregnated Adsorbents, J. Environ. Eng., 2002, 128, 1080–1089 CrossRef CAS
- E. A. Morris, D. W. Kirk, C. Q. Jia and K. Morita, Roles of Sulfuric Acid in Elemental Mercury Removal by Activated Carbon and Sulfur-Impregnated Activated Carbon, Environ. Sci. Technol., 2012, 46, 7905–7912 CrossRef CAS PubMed
- K.-D. Henning, K. Keldenicht, K. Knoblauch and J. Degel, Impregnated activated carbon for mercury removal, Gas Sep. Purif., 1988, 2, 20–22 CrossRef CAS
- R. D. Vidic, M.-T. Chang and R. C. Thurnau, Kinetics of Vapor-Phase Mercury Uptake by Virgin and Sulfur-Impregnated Activated Carbons, J. Air Waste Manage. Assoc., 1998, 48, 247–255 CrossRef CAS PubMed
- J. G. Eisenhauer, Regression through the Origin, Teach. Stat., 2003, 25, 76–80 CrossRef
- R. F. Pueschel, P. B. Russell, D. A. Allen, G. V. Ferry, K. G. Snetsinger, J. M. Livingston and S. Verma, Physical and optical properties of the Pinatubo volcanic aerosol: Aircraft observations with impactors and a Sun-tracking photometer, J. Geophys. Res., 1994, 99, 12915 CrossRef
- J. Vernier, T. D. Fairlie, T. Deshler, M. Natarajan, T. Knepp, K. Foster, F. G. Wienhold, K. M. Bedka, L. Thomason and C. Trepte,
In situ and space-based observations of the Kelud volcanic plume: The persistence of ash in the lower stratosphere, J. Geophys. Res. Atmos., 2016, 121, 104–118 CrossRef
- Y. Tang, S. Wang, Q. Wu, K. Liu, Z. Li, J. Zou, D. Hou, Y. Wu and L. Duan, Measurement of size-fractionated particulate-bound mercury in Beijing and implications on sources and dry deposition of mercury, Sci. Total Environ., 2019, 675, 176–183 CrossRef CAS PubMed
- H. M. Xu, R. Y. Sun, J. J. Cao, R.-J. Huang, B. Guinot, Z. X. Shen, M. Jiskra, C. X. Li, B. Y. Du, C. He, S. X. Liu, T. Zhang and J. E. Sonke, Mercury stable isotope compositions of Chinese urban fine particulates in winter haze days: Implications for Hg sources and transformations, Chem. Geol., 2019, 504, 267–275 CrossRef CAS
- J. Guo, C. M. Sharma, L. Tripathee, S. Kang, X. Fu, J. Huang, K. L. Shrestha and P. Chen, Source identification of atmospheric particle-bound mercury in the Himalayan foothills through non-isotopic and isotope analyses, Environ. Pollut., 2021, 286, 117317 CrossRef CAS PubMed
- Y. Gao, Z. Zhang, J. Wu, L. Duan, A. Umar, L. Sun, Z. Guo and Q. Wang, A Critical Review on the Heterogeneous Catalytic Oxidation of Elemental Mercury in Flue Gases, Environ. Sci. Technol., 2013, 47, 10813–10823 CrossRef CAS PubMed
- H. Kamata, S. Ueno, N. Sato and T. Naito, Mercury oxidation by hydrochloric acid over TiO2 supported metal oxide catalysts in coal combustion flue gas, Fuel Process. Technol., 2009, 90, 947–951 CrossRef CAS
- S. Freire, A. Florczyk, M. Pesaresi and R. Sliuzas, An Improved Global Analysis of Population Distribution in Proximity to Active Volcanoes, 1975–2015, ISPRS Int. J. Geo-Inf., 2019, 8, 341 CrossRef
- M. F. Hochella, D. W. Mogk, J. Ranville, I. C. Allen, G. W. Luther, L. C. Marr, B. P. McGrail, M. Murayama, N. P. Qafoku, K. M. Rosso, N. Sahai, P. A. Schroeder, P. Vikesland, P. Westerhoff and Y. Yang, Natural, incidental, and engineered nanomaterials and their impacts on the Earth system, Science, 2019, 363, eaau8299 CrossRef PubMed
- C. Anastasio and S. T. Martin, Atmospheric Nanoparticles, Rev. Mineral. Geochem., 2001, 44, 293–349 CrossRef CAS
- B. A. Edwards, M. A. Pfeffer, P. Jóhannsson, P. M. Outridge and F. Wang, An inter-method comparison of mercury measurements in Icelandic volcanic gases, Appl. Geochem., 2023, 105654 CrossRef CAS
- O. Magand and A. Dommergue, Continuous Measurements of Atmospheric Mercury at Maido Observatory (L2) [Dataset], Aeris, DOI:10.25326/352.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ea00063j |
| This journal is © The Royal Society of Chemistry 2023 |