
Engineering a local potassium cation concentrated microenvironment toward the ampere-level current density hydrogen evolution reaction†
Lei
Gao‡
a,
Feixiang
Bao‡
a,
Xin
Tan
*b,
Mengfan
Li
a,
Zhen
Shen
c,
Xuli
Chen
a,
Ziyi
Tang
a,
Wenchuan
Lai
a,
Yangfan
Lu
d,
Peifeng
Huang
a,
Chao
Ma
a,
Sean C.
Smith
e,
Zhizhen
Ye
d,
Zheng
Hu
 *c and
Hongwen
Huang
*af
*c and
Hongwen
Huang
*af
aCollege of Materials Science and Engineering, State Key Laboratory of Advanced Design and Manufacturing for Vehicle Body, Hunan University, Changsha, Hunan 410082, P. R. China. E-mail: huanghw@hnu.edu.cn
bInstitute for Carbon Neutralization, College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, Zhejiang 325035, P. R. China. E-mail: xintan@wzu.edu.cn
cKey Laboratory of Mesoscopic Chemistry of MOE and Jiangsu Provincial Lab for Nanotechnology, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, Jiangsu 210023, P. R. China. E-mail: zhenghu@nju.edu.cn
dState Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, P. R. China
eIntegrated Materials Design Laboratory, Department of Materials Physics, Research School of Physics, The Australian National University, Canberra, ACT 2601, Australia
fShenzhen Research Institute of Hunan University, Shenzhen, Guangdong 518055, P. R. China
First published on 6th December 2022
Abstract
Finding an active and robust non-platinum catalyst toward the alkaline hydrogen evolution reaction (HER) operating at an ampere-level current density is important for emerging anion exchange membrane (AEM) water electrolysis but challenging. Here we report a nanocone-assembled Ru3Ni (NA-Ru3Ni) catalyst that exhibits a low overpotential of 168 mV at 1000 mA cm−2 and a high turnover frequency of 26.5 s−1 at an overpotential of 100 mV, with a Ru3Ni loading of only 0.08 mg cm−2. Moreover, the catalyst could stably operate at 1000 mA cm−2 over 2000 h in a practical AEM electrolyser at 60 °C, showing the best overall performance among ever-reported catalysts. The theoretical simulations and experimental results confirm that the sharp-tip concentrated K+ cations contribute to such remarkable alkaline HER activity by intensifying the polarization of the H–OH bond of interfacial water and decreasing the energy barrier for water dissociation, where the non-covalent interaction is considered as the intrinsic driving force. The present work provides general guidance for the rational design of industrially relevant alkaline HER catalysts.
Broader contextAlkaline anion exchange membrane (AEM) electrolysers show significant advantages in low system cost associated with the less-corrosive media and large current density (generally 1–2 A cm−2) for H2 production, where active and stable alkaline hydrogen evolution reaction (HER) catalysts operating at an ampere-level current density are urgently needed. However, the additional water dissociation step involved in the supply of adsorbed H (*H) reactants largely hinders the high-current-density alkaline HER kinetics. Traditionally, a number of structure-regulation strategies via synergy between the active center and the oxophilic site were developed to accelerate water dissociation, but suffered from unsatisfactory alkaline HER activity at an industrially relevant current density due to the limited interface for water dissociation. Herein, we report the construction of a local potassium cation concentrated microenvironment over a nanocone-assembled Ru3Ni (NA-Ru3Ni) catalyst to overcome the issue of water dissociation. As a result, the NA-Ru3Ni catalyst exhibits a low overpotential of 168 mV at 1000 mA cm−2 and a high turnover frequency of 26.1 s−1 at an overpotential of 100 mV, with a Ru3Ni loading of only 0.08 mg cm−2. Moreover, the catalyst could stably operate at 1000 mA cm−2 over 2000 h in a practical AEM electrolyser at 60 °C. This work not only provides an efficient and robust alkaline HER catalyst for practical AEM electrolysers, but also affords a new perspective for the design of industrially relevant electrocatalysts. |
Introduction
Electrochemical water splitting represents an ideal technology to produce high-purity hydrogen (H2) via the cathodic hydrogen evolution reaction (HER), while simultaneously storing electric energy from intermittent renewable energy sources, e.g., wind and solar.1–4 With great advances in anion exchange membranes (AEMs), alkaline electrolysers based on AEMs have recently received increasing attention because of the significant advantages of low system cost associated with the less-corrosive media and a large current density (generally 1–2 A cm−2) for H2 production.5–9 To efficiently produce H2 in an AEM electrolyser, finding an active and stable alkaline HER catalyst to meet the demand of industrial AEM water electrolysis is the central task for the research community, whose performance should satisfy low overpotential and long lifespan at an ampere-level current density.8–14 However, besides the scarce and expensive Pt, the discovery of active and robust electrocatalysts for the ampere-level-current-density alkaline HER is yet a great challenge.Generally, the HER kinetics in alkaline media is much more sluggish than that in acidic media because of the additional water dissociation step involved in the supply of adsorbed H (*H).8,15–17 When aiming at the high-current-density alkaline HER, promoting the water dissociation to kinetically favor the production of *H reactants, namely the Volmer step, is thus more important. So far, a number of strategies with the basic principles involving adsorption energy optimization15,18 and multisite synergy8,12,19,20 have been explored to accelerate water dissociation, generally in the form of oxophilic element doping,21 phase control,22 strain engineering,23 and interfacial construction.10,24,25 In a notable example, structuring Pt on a metal hydroxide has been well documented to improve the kinetics of water dissociation through the synergy between Pt and oxophilic metal sites.26 Despite the great achievements, the effects of these structure-regulation strategies on accelerating the water dissociation are generally restricted by the limited interface for water dissociation, resulting in unsatisfactory alkaline HER activity at an industrially relevant current density. Accordingly, searching for an alternative mode to substantially increase the supply of *H reactants in alkaline media is urgently desired.
Engineering the interfacial microenvironment is emerging as an alternative and powerful way to regulate the electrocatalytic kinetics through the non-covalent interaction among intermediates/species in an electrical double layer (EDL).27,28 Typically, the alkaline metal cations in the EDL have recently been demonstrated to play significant roles in dictating the reaction kinetics of many electrocatalysis processes, including the alkaline hydrogen evolution reaction (HER).29–31 For example, Liu et al. reported that increasing the cation concentration would promote the alkaline HER, which was attributed to the catalytic role of the OHad–water–alkaline metal cation adducts in accelerating the water dissociation process.30 Monteiro et al. observed the concentration- and electrode-dependent cation effects for the alkaline HER, proposing two-sided roles of the cations.31 Very recently, by employing in situ Raman spectroscopy, Wang et al. demonstrated that alkali Na+ may facilitate water dissociation on the Pd surface by inducing an ordered interfacial water structure that had a shorter Pd–H distance and strengthened the Pd–H boding interaction.32 Despite these advances, the great complexity in understanding the cation effects and challenges in constructing a favorable local environment have largely impeded the exploration of the cation effects to create a practically important catalytic system for the alkaline HER.
Herein, we take a Ru-based catalyst as a model, which is widely considered as a promising alternative to Pt due to the much cheaper cost (490 $ per oz for Ru versus 914 $ per oz for Pt) and similar bonding strength of Ru–H with Pt–H (ca. 65 kcal mol−1),33,34 to showcase a tip-concentrated hydrated K+ strategy for achieving the ampere-level-current-density alkaline HER. Specifically, we build a nanocone-assembled Ru3Ni (NA-Ru3Ni) catalyst that exhibits a low overpotential of 168 mV at 1000 mA cm−2 and a high turnover frequency of 26.5 s−1 at an overpotential of 100 mV. Moreover, the catalyst could stably operate at 1000 mA cm−2 over 2000 h in a practical AEM electrolyser at 60 °C. Finite element simulations and experimental results reveal that the nanotips of the NA-Ru3Ni catalyst (curvature radius of 1.3 nm) enable an enhanced local electric field, and in turn, increase the interfacial hydrated K+ concentration. Further mechanistic studies indicate that the locally increased hydrated K+ concentration promotes the polarization of the H–OH bond of interfacial water via non-covalent interactions, resulting in the reduced energy barrier for the water dissociation process and thus accounting for the greatly improved activity for the alkaline HER.
Results and discussion
Synthesis and characterization
Typically, NA-Ru3Ni was synthesized by the co-reduction of ruthenium acetylacetonate (Ru(acac)3) and nickel acetylacetonate (Ni(acac)2) in a mixed solution containing benzyl alcohol and resorcinol at 180 °C for 1 h (see Methods for details, Fig. S1, ESI†). The representative transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images evidence the formation of a nanocone-assembled structure with high purity and uniformity (Fig. 1a, b and Fig. S2, ESI†). The assembled structure has an average size of 36.2 nm (Fig. S2b, ESI†) and the average curvature radius of the cone tip is around 1.3 nm (Fig. 1c, d and Fig. S3, ESI†). The atomic-resolution HAADF-STEM image recorded along the [010] zone axis clearly shows the periodic ABAB stacking mode, corroborating the hexagonal close-packed (HCP) structure (Fig. 1e). The energy dispersive spectroscopy (EDS) line-scanning profiles (Fig. 1f) and the elemental map (Fig. 1g) indicate a homogenous distribution of Ru and Ni through the whole structure. The atomic ratio of Ru:Ni was further estimated to be about 3.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0 according to inductively coupled plasma mass spectrometry (ICP-MS, Fig. 1h) and STEM-EDS (Fig. S4, ESI†). Besides, the X-ray photoelectron spectroscopy (XPS) survey spectrum verifies the coexistence of Ru and Ni elements (Fig. S5, ESI†). The surface atomic ratio of Ru:Ni obtained from XPS is in line with that from ICP-MS and STEM-EDS. As shown in Fig. 1i, the high-resolution Ru 3p and Ni 2p spectra indicate that most of the surface Ru and Ni are in the metallic state. Notably, the binding energy of the Ru 3p peak for the as-synthesized Ru3Ni is negatively shifted by 0.32 eV relative to that of commercial Ru/C, indicating the electron transfer from Ni to Ru in NA-Ru3Ni.
:1.0 according to inductively coupled plasma mass spectrometry (ICP-MS, Fig. 1h) and STEM-EDS (Fig. S4, ESI†). Besides, the X-ray photoelectron spectroscopy (XPS) survey spectrum verifies the coexistence of Ru and Ni elements (Fig. S5, ESI†). The surface atomic ratio of Ru:Ni obtained from XPS is in line with that from ICP-MS and STEM-EDS. As shown in Fig. 1i, the high-resolution Ru 3p and Ni 2p spectra indicate that most of the surface Ru and Ni are in the metallic state. Notably, the binding energy of the Ru 3p peak for the as-synthesized Ru3Ni is negatively shifted by 0.32 eV relative to that of commercial Ru/C, indicating the electron transfer from Ni to Ru in NA-Ru3Ni.
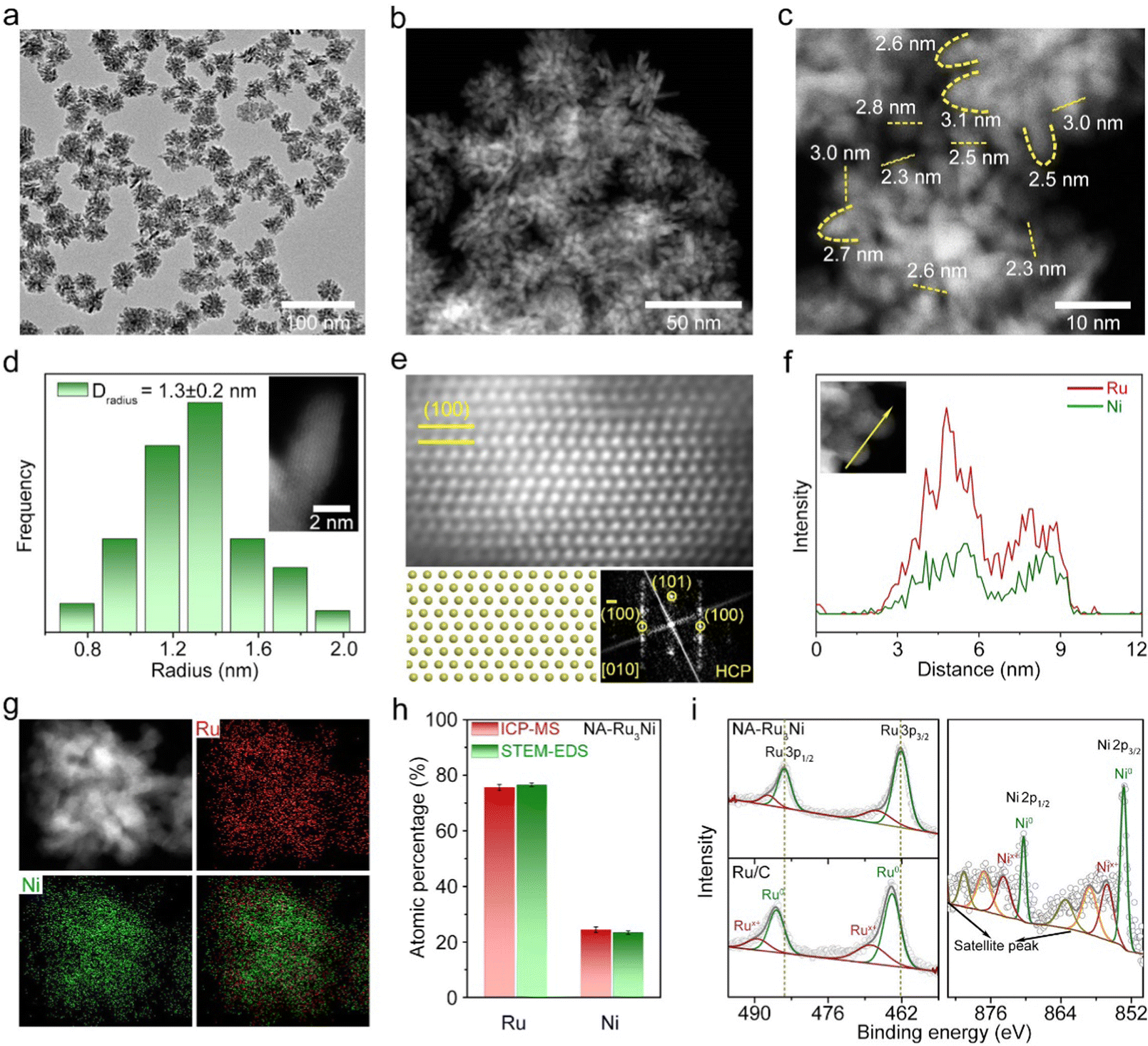 | ||
| Fig. 1 Structural characterization of NA-Ru3Ni. (a) TEM image (b) and (c) HAADF-STEM images. (d) Curvature radius distribution for nanocone tips. The inset shows the HAADF-STEM image of a single nanocone tip. (e) Atomic-resolution aberration-corrected HAADF-STEM image, the corresponding structural models and the FFT pattern. (f) EDS line-scanning elemental profile. (g) EDS elemental mapping images of Ru and Ni. (h) Elemental composition obtained by ICP-MS (red bar) and STEM-EDS (green bar). (i) High-resolution XPS spectra of Ru 3p and Ni 2p, with XPS spectra of Ru/C as a reference. | ||
Electrocatalytic performance toward the alkaline HER
We then evaluated the HER catalytic performance of NA-Ru3Ni in 1 M KOH. The Ni/C, Pt/C, and Ru/C catalysts were employed as benchmark catalysts. Prior to HER measurements, the as-synthesized NA-Ru3Ni was loaded on carbon support to form the NA-Ru3Ni/C catalyst. For all electrochemical tests, we first calibrated the Hg/HgO reference electrode with respect to the reversible hydrogen electrode (Fig. S6, ESI†). Fig. 2a shows the linear sweep voltammetry (LSV) curves of three catalysts with iR-correction at a scan rate of 5 mV s−1. Compared to Ni/C, Pt/C, and Ru/C catalysts, NA-Ru3Ni/C shows the fastest rise in the current density with an increase of the overpotential, demonstrating the highest HER activity. Specifically, NA-Ru3Ni/C presents the lowest overpotential of 14 ± 0.8 mV to deliver a current density of 10 mA cm−2. Even for a current density of 100 mA cm−2, the overpotential of NA-Ru3Ni/C is only 54 ± 0.6 mV, which is much lower than that of commercial Pt/C (88 ± 1.5 mV) and Ru/C (125 ± 1.0 mV) catalysts (Fig. 2b). The overpotentials of NA-Ru3Ni/C at different current densities were further compared with those state-of-the-art Ru-based catalysts (Fig. 2c, Tables S1 and S2, ESI†), showing the superiority in reducing the overpotential for the alkaline HER. Besides, we noted that NA-Ru3Ni/C presents a very low overpotential of 168 mV at an industrial-level current density of 1000 mA cm−2. More impressively, only a low catalyst loading amount of 0.08 mgmetal cm−2 was used to achieve such high current density, highlighting the great potential of the catalyst for practical AEM electrolysis applications (Table S3, ESI†). To obtain the optimum composition of the NA-RuNi for HER, we further explored the HER activities of NA-RuNi with three different Ru/Ni atomic ratios. As shown in Fig. S7 (ESI†), the NA-Ru3Ni catalyst possessed the highest HER activity, indicating the definite impact of the Ru/Ni ratio on HER activity. We conjectured that the synergies between Ru and Ni sites determined by the ratio of Ru/Ni (through the so-called bifunctional mechanism, where the Ru acts as the site for hydrogen adsorption and Ni acts as the site for H2O dissociation) are the dominant reason for the volcano-type relationship between the HER activity and the Ru/Ni ratio.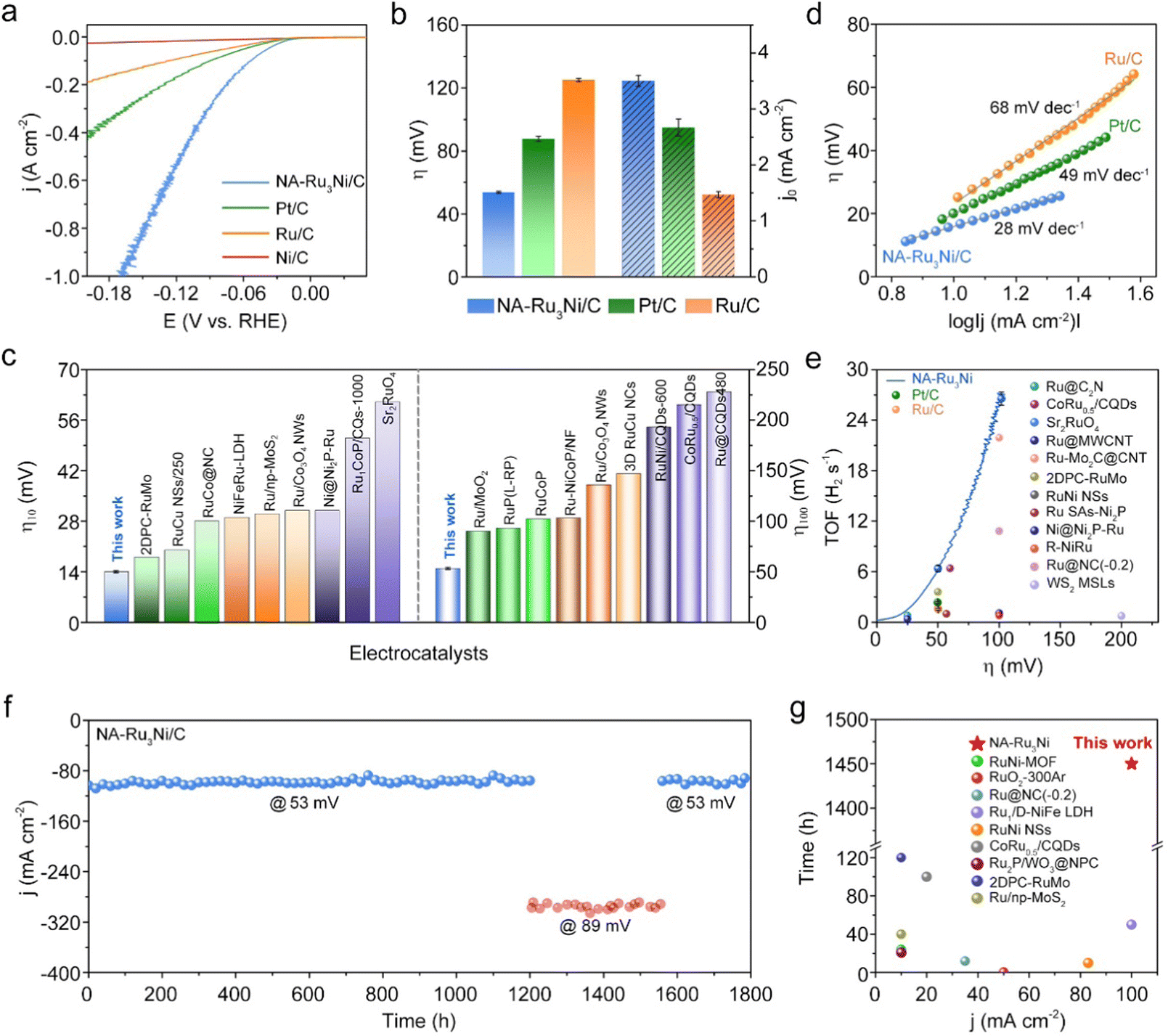 | ||
| Fig. 2 Electrocatalytic alkaline HER performance. (a) Polarization curves (with iR-correction) recorded in 1 M KOH solutions at a sweep rate of 5 mV s−1. (b) Values of overpotential at 100 mA cm−2 and exchange current densities of different catalysts. (c) Comparison of η = 10 mA cm−1 (left) and 100 mA cm−1 (right) over the NA-Ru3Ni/C catalyst with other catalysts reported in the literature. (d) Tafel plots. (e) Comparison of the TOF values. (f) Chronoamperometry curve of the NA-Ru3Ni/C catalyst operated at 53mV (blue) and 89mV (pink) overpotentials for 1800 h. (g) Comparison of long-term stability over the NA-Ru3Ni/C catalyst with ever-reported alkaline HER electrocatalysts. | ||
The specific activity and mass activity of each catalyst were then derived by normalizing the current density against the electrochemically active surface area (ECSA) and metal loading amount (Fig. S8, ESI†), respectively, where the ECSA of each catalyst was estimated by CO stripping voltammetry (Fig. S9 and Table S4, ESI†). As expected, NA-Ru3Ni/C shows greatly enhanced specific activity and mass activity for the alkaline HER compared to the other two catalysts. Consistent with the activity trend, the Tafel plots present the smallest Tafel slope of 28 mV dec−1 for the NA-Ru3Ni/C catalyst, in contrast to the 49 mV dec−1 for commercial Pt/C and 68 mV dec−1 for commercial Ru/C catalysts (Fig. 2d). And the remarkable Tafel slope of the reported NA-Ru3Ni/C surpasses most of those reported Ru-based electrocatalysts (Table S1, ESI†). Even in the strongly polarized region (300–500 mA cm−2), the Tafel slope value (103 mV dec−1) for the NA-Ru3Ni/C catalyst is also much smaller than that for commercial Pt/C (235 mV dec−1) and Ru/C (643 mV dec−1) catalysts (Fig. S10, ESI†), meaning that NA-Ru3Ni/C could maintain the fastest kinetics at a high current density.13
To further evaluate the inherent activity for the alkaline HER, the exchange current density and turnover frequency (TOF) were calculated for the catalysts. As shown in Fig. 2b, NA-Ru3Ni/C presents the highest exchange current density of 3.5 ± 0.1 mA cm−2, 1.3 and 2.3 times higher than those of commercial Pt/C (2.7 ± 0.2 mA cm−2) and Ru/C (1.5 ± 0.1 mA cm−2) catalysts, respectively. The overpotential-dependent TOF for NA-Ru3Ni/C was also plotted in Fig. 2e, evidencing the improved intrinsic activity compared to those ever-reported catalysts. Notably, a high TOF value of 26.5 ± 0.9 H2 s−1 at an overpotential of 100 mV over NA-Ru3Ni/C represents a very high-level intrinsic activity, surpassing most of the reported catalysts (Table S5, ESI†).
Since the operation durability of the catalyst is a crucial parameter for practical application, we subsequently examined the long-term durability of the NA-Ru3Ni/C catalyst for the alkaline HER. After 10000 (10 k) cycles of the accelerated durability tests (ADTs), a negligible shift was observed for the polarization curve of the NA-Ru3Ni/C catalyst (Fig. S11, ESI†), affirming the excellent catalytic stability toward the alkaline HER. More strikingly, the alkaline HER activity of the NA-Ru3Ni/C catalyst is almost constant during the 1800 h operation at a large current density of around 100 (lasting for 1450 h at 53 mV overpotential) or 300 mA cm−2 (lasting for 350 h at 89 mV overpotential) (Fig. 2f). Consistent with the great stability, the NA-Ru3Ni/C catalyst well maintains the morphology after 1800 h of operation (Fig. S12, ESI†). The composition (Ru:Ni = 3.1:1.0) and chemical valence state of the NA-Ru3Ni/C catalyst further verify the outstanding structural stability (Fig. S13, ESI†), which together well rationalize the long-term durability of the NA-Ru3Ni/C catalyst. The long-term durability of NA-Ru3Ni/C catalyst exceeds most reported catalysts for the alkaline HER (Fig. 2g and Table S6, ESI†). All these results together indicate that the NA-Ru3Ni/C catalyst exhibits remarkable alkaline HER performance.
Structural origins for the high HER activity of the NA-Ru3Ni/C catalyst
We further attempted to reveal the structural origins of the greatly improved alkaline HER activity of the NA-Ru3Ni/C catalyst. In principle, the catalytic activity is determined by the surface composition and geometric structure via the ligand and geometric effect.35,36 To explore the effect of surface composition, we changed the surface composition of NA-Ru3Ni by electrochemical dealloying treatment, which was performed with cyclic voltammetry (CV) at a scan rate of 100 mV s−1 at an applied potential from 0.0591 V to 0.909 V (versus reversible hydrogen electrode (RHE)) in 0.1 M HClO4 to remove surface Ni atoms. As observed from the CV curves of NA-Ru3Ni/C recorded before and after dealloying treatment, the disappearance of the Ni2+/Ni3+ redox pair around 1.3 VRHE testifies the successful removal of surface Ni atoms (Fig. S14, ESI†). The polarization curve collected after the dealloying treatment shows a negative shift of 21.7 mV at a current density of 100 mA cm−2, proving the promoting effect of surface Ni for the alkaline HER (Fig. 3a). Note that no amorphous materials were present after electrochemical dealloying treatment, as demonstrated by HAADF-STEM of NA-Ru3Ni upon removal of surface Ni atoms (Fig. S15, ESI†). The accelerated water dissociation associated with the surface oxophilic Ni sites is hypothesized as the main reason for such a promoting effect.37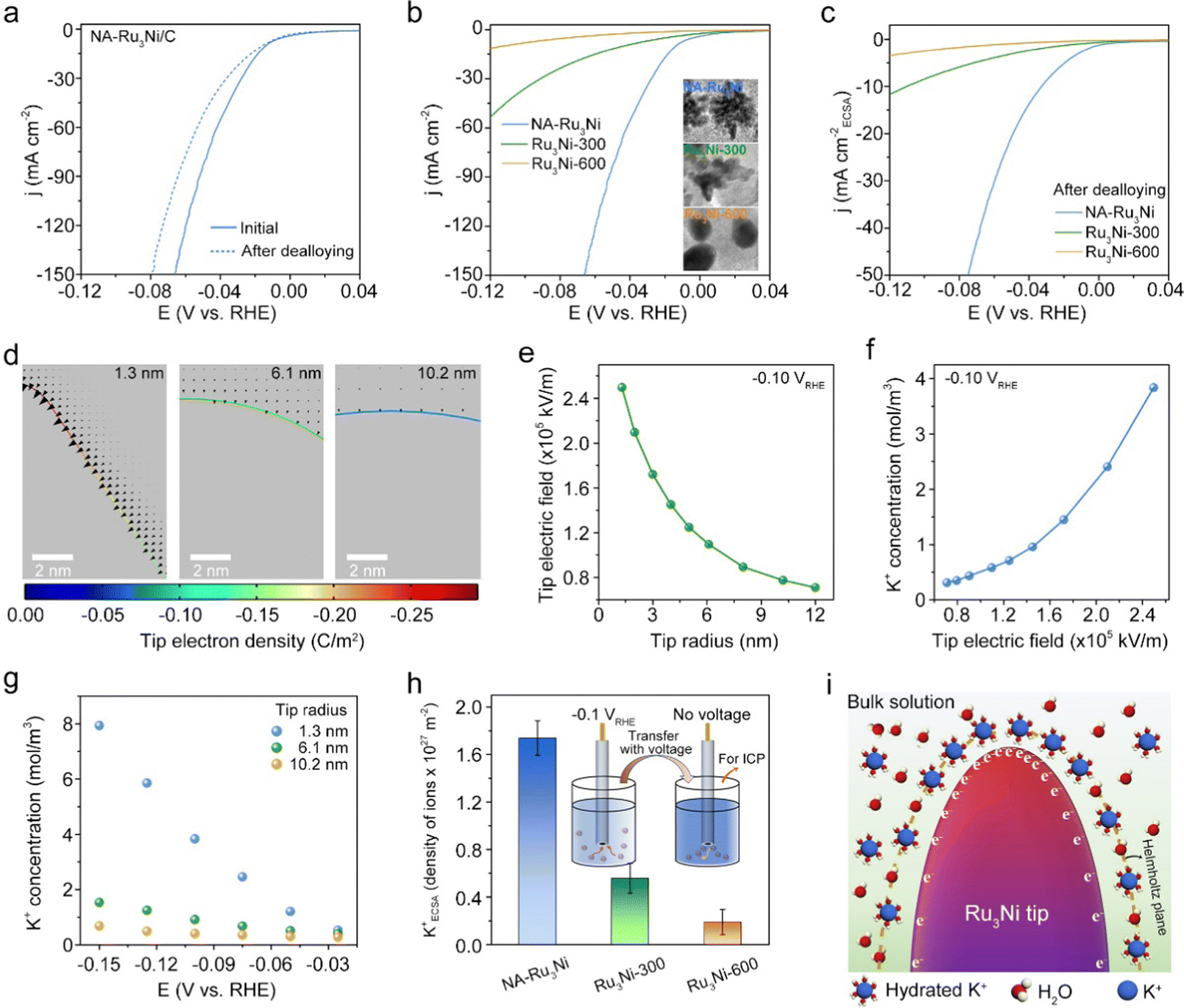 | ||
| Fig. 3 Structural origin for the improved alkaline HER activity of the NA-Ru3Ni catalyst. (a) Polarization curves of the NA-Ru3Ni catalyst recorded before and after the dealloying process. (b) Polarization curves of NA-Ru3Ni, Ru3Ni-300, and Ru3Ni-600 catalysts. The inset shows the TEM images of the catalysts. (c) Polarization curves of NA-Ru3Ni, Ru3Ni-300, and Ru3Ni-600 after the dealloying process. (d) Colour maps of the electron density distribution on the surfaces of different electrodes. Black arrows represent the magnitude and direction of the electric field distribution around the electrode. The curvature radius of the Ru3Ni tips in each panel is 1.3 nm (stands for NA-Ru3Ni, left), 6.1 nm (Ru3Ni-300, middle) and 10.2 nm (Ru3Ni-600, right), respectively. (e) The plot showing the relationship between the tip radius and the local electric field. (f) The plot showing the relationships between the local electric field and the K+ concentration. (g) The applied potential-dependent local K+ concentration on different curved tips. (h) Comparison of tip-induced K+ concentration on different curved Ru3Ni. The density of K+ was measured by ICP-MS. The schematic diagram of insertion shows the testing of the adsorbed K+. (i) Schematic of the interfacial model of NA-Ru3Ni. | ||
We also investigated the impacts of morphological structure on the alkaline HER. Specifically, we modulated the catalyst morphology while keeping the composition unchanged by annealing NA-Ru3Ni at different temperatures, where the as-obtained samples were denoted as Ru3Ni-X (X stands for the annealing temperature, X °C). NA-Ru3Ni with sharp nanotips gradually evolves into a round nanoparticle upon increasing the annealing temperature (inset in Fig. 3b and Fig. S16, ESI†), with growing curvature radii of 6.1 nm for Ru3Ni-300 and 10.2 nm for Ru3Ni-600. The X-ray diffraction analysis indicates the identical phase structure and composition of the samples, as evidenced by the absence of shifts in the diffraction peaks (Fig. S17, ESI†). By examining the LSV curves of Ru3Ni catalysts with different morphologies, it is found that the initial NA-Ru3Ni catalyst possesses overwhelming HER activity than those after annealing (Fig. 3b). From CO stripping, the ECSA of the NA-Ru3Ni catalyst was estimated to be 41.3 m2 g−1, larger than those of Ru3Ni-300 (32.8 m2 g−1) and Ru3Ni-600 (15.2 m2 g−1) catalysts (Fig. S9, S18 and Table S7, ESI†). The above results also agree with the estimations from electrochemical double-layer capacitance (Cdl) at different scan rates (Fig. S19 and S20, ESI†). Based on the evaluated ECSAs, the ECSA-normalized LSV curve was further derived for each catalyst, confirming the highest intrinsic activity of the initial NA-Ru3Ni catalyst for the alkaline HER (Fig. S21, ESI†). To deconvolute the impacts of surface Ni sites, we further compared the ECSA-normalized activity of NA-Ru3Ni, Ru3Ni-300, and Ru3Ni-600 catalysts after dealloying treatment (Fig. 3c, Fig. S22–S24 and Table S7, ESI†). The corresponding HER activities of three catalysts with different curvature radii follow the order of NA-Ru3Ni > Ru3Ni-300 > Ru3Ni-600, hinting at the important role of curvature radius in the alkaline HER activity. In addition, the accelerated alkaline HER kinetics over NA-Ru3Ni is also confirmed by electrochemical impedance spectroscopy (EIS), which exhibits the lowest value (1.5 Ω) of charge transfer resistance (RCT) for NA-Ru3Ni/C when compared with those of Ru3Ni-300/C (20.8 Ω) and Ru3Ni-600/C (28.6 Ω) catalysts at an overpotential of 100 mV (Fig. S25 and S26, ESI†).
All the preceding analyses demonstrate that the Ni alloying and unique nanotip structure can both benefit the alkaline HER performance. Further closer examination of the magnitude in reducing the overpotential over these catalysts indicates that the nanotip structure with a small curvature radius (high-curvature structure) is the regnant factor in achieving the remarkable HER activity of the NA-Ru3Ni/C catalyst (see Fig. 3a–c). In this case, we therefore put our attention below to understand how the high-curvature nanotips boost the alkaline HER.
Insights into how high-curvature nanotips boost the alkaline HER
In principle, the high-curvature structure could concentrate the reagents on the EDL by a tip-enhanced local electrical field, which may impact the reaction kinetics through microenvironment engineering.38,39 To show the tip effect on the local electrical field, COMSOL Multiphysics finite-element-based simulations were performed (see Methods for details). According to our experimental results, nanotips with radii (curvature radii) of 1.3, 6.1, and 10.2 nm were modelled to represent NA-Ru3Ni, Ru3Ni-300, and Ru3Ni-600, respectively. As shown in Fig. 3d, the electron density at the tip increases with curvature. Induced by the greatly enhanced electron density, the locally enhanced electrostatic field is generated, as illustrated by the arrows in Fig. 3d. Quantitatively, the strength of the local electric field presents a 3.6-fold increment with the curvature radius decreasing from 10.2 to 1.3 nm (the field strength increases from 0.7 × 105 to 2.5 × 105 kV m−1) at −0.1 VRHE (Fig. 3e). Driven by the locally enhanced electrostatic field, the hydrated K+ cations would be concentrated on the outer Helmholtz layer in the EDL, which was quantitatively mapped using a Gouy-Chapman-Stern model (Fig. S27, ESI†). The results indicate that the surface K+ concentration at the 1.3 nm-radius tip is 6.6 and 12.7 times higher than those at the 6.1 nm- and 10.2 nm-radius tips, respectively (Fig. 3f). The potential-dependent surface K+ concentration plots reveal that the sharp-tip enhancement effect could dominate the surface K+ concentration relative to the impacts of applied potential (Fig. 3g), implying the great advantages of the sharp-tip structure in modulating the interfacial microenvironment.In addition, we experimentally verified the tip-induced K+ accumulation by comparing the amounts of K+ on three Ru3Ni samples at a bias voltage of −0.1 VRHE. Specifically, the electrode loaded with the catalyst was rapidly transferred from the electrolyte to pure water while keeping voltage, and voltage was later shut to release K+ for ICP analysis (inset in Fig. 3h). The result shows that the value of the ECSA-normalized K+ density of NA-Ru3Ni is 3.2 and 9.2-fold enhancements relative to those of Ru3Ni-300 and Ru3Ni-600, respectively, confirming the tip-induced K+ concentration (Fig. 3h). Combining all these simulations and experimental results together, we thus confirmed this tip-concentrated local hydrated K+ behavior, as schematically illustrated in Fig. 3i.
A remaining question that needs to be addressed is how tip-concentrated local hydrated K+ improves the alkaline HER activity. To this end, we performed density functional theory (DFT) calculations to examine the roles of an enhanced local electric field and concentrated K+ cation in modulating the energy barriers of alkaline HER pathways, namely the Volmer step for water dissociation and Tafel step for H2 generation.16,40 In the light of the experimental results and previously reported Ru-based alloy catalysts,41,42 Ru was identified as the specific active site of NA-Ru3Ni. Accordingly, the slabs were modelled based on the Ru3Ni(0001) surface given the thermodynamically favored stability of the (0001) surface.22,43 A layer of water molecules without K+ (denoted as No K+) and with K+ at 1/12 monolayer (ML) coverage (denoted as with K+ at 1/12 ML coverage) was constructed on the Ru3Ni(0001) surface, respectively, to explore the effect of the K+ cation (see DFT calculations, Fig. 4a). In addition, the Ru3Ni(0001) surface that is negatively charged with one electron but without K+ in the layer of water molecules (denoted as No K+, N = −1) was also simulated for elucidating the impacts of an electric field (Fig. 4a). In general, the key reaction steps in the alkaline HER were considered to consist of the adsorption of H2O, dissociation of H2O to form adsorbed H (H*), a combination of reaction H* to form adsorbed H2, and desorption of H2 from the catalyst surface.22,43 Therefore, Fig. 4b shows the Gibbs free energy (ΔG) diagrams for the alkaline HER on three different models, where the corresponding water dissociation energy barrier (ΔGW) and hydrogen adsorption free energy (ΔGH*) are shown in Fig. 4c. These results show that ΔGW is much larger than ΔGH* in three models, indicating that the water dissociation process to produce H* is the rate-determining step. For water dissociation (the models are shown in Fig. S28–S30, ESI†), the lowest energy barrier (ΔGW = 0.67 eV) is observed on the Ru3Ni (With K+ at 1/12 ML coverage) slab in comparison with the Ru3Ni (No K+, N = −1) slab (ΔGW = 0.73 eV) and Ru3Ni (No K+) slabs (ΔGW = 0.77 eV), suggesting the leading role of interfacial hydrated K+ cations in facilitating the water dissociation step and in turn accelerating the HER kinetics. Note that the effect of different K+ concentrations on ΔGW and ΔGH* was further explored, as shown in Fig. S31 (ESI†). These results thus well support the greatly improved alkaline HER activity over NA-Ru3Ni.
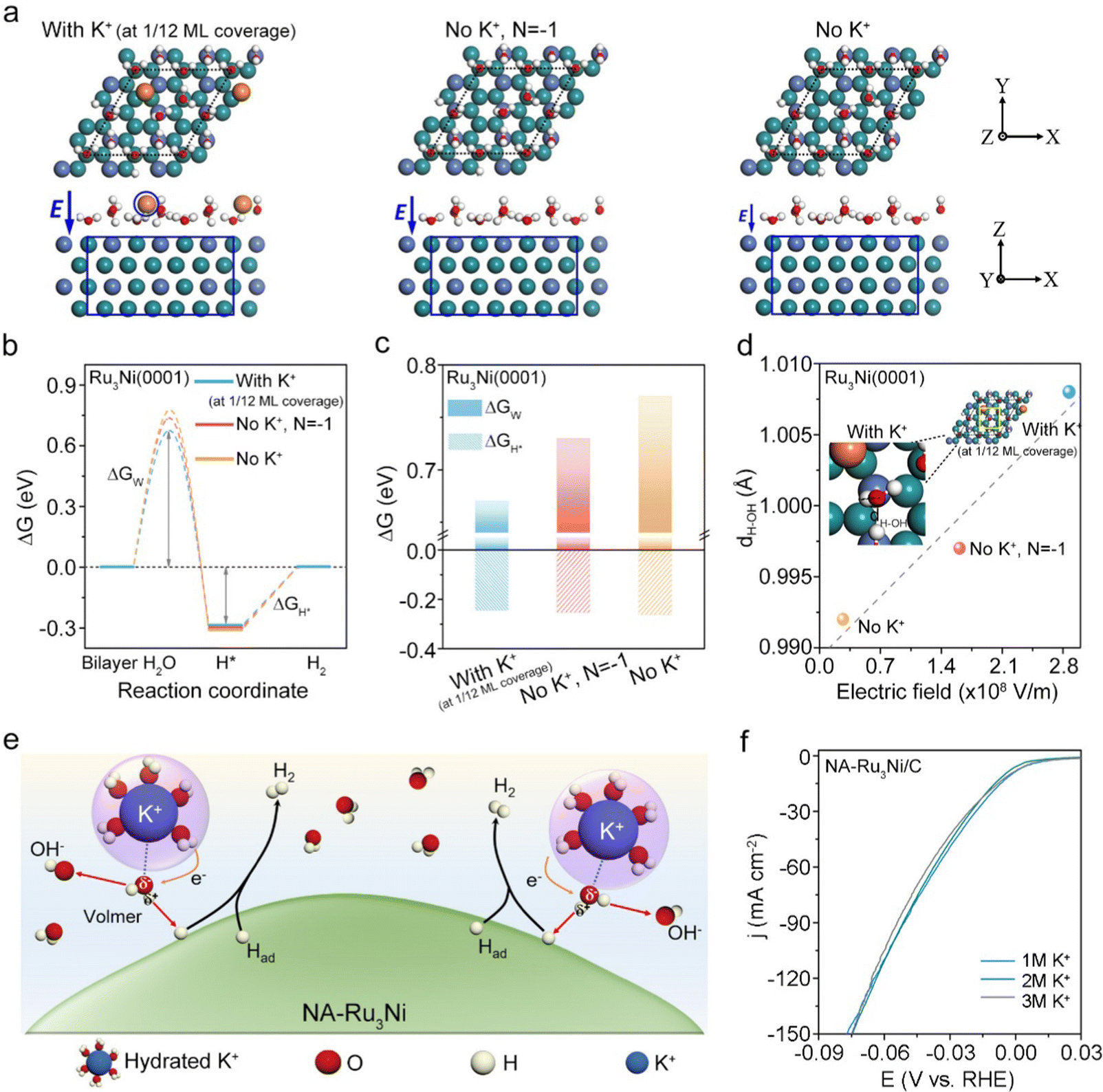 | ||
| Fig. 4 Mechanistic understanding. (a) Theoretical models of the Ru3Ni(0001) surface with a K cation (with K+ at 1/12 ML coverage), the 1e− negatively charged Ru3Ni(0001) surface without a K cation (no K+, N = −1), and the neutral Ru3Ni(0001) surface without a K cation (No K+). The local electric field E at the interface between the metal surface and the electrolyte solution are shown. (b) Gibbs free energy diagrams of the alkaline HER on different systems including reactant initial state, intermediate state, final state, and an additional transition state representing water dissociation. ΔGH* and ΔGW represent the hydrogen adsorption free energy and water dissociation free energy barrier, respectively. (c) The values of ΔGH* and ΔGW on different systems. (d) The measured bond length of the H–OH bond on different slabs. The inset shows atomic configurations of the water dissociation step on the NA-Ru3Ni(0001) surface (With K+ at 1/12 ML coverage), which defines the bond length of the H–OH bond. (e) A schematic showing how K+ promotes interfacial water dissociation on the NA-Ru3Ni surface. (f) Polarization curves of the NA-Ru3Ni/C catalyst in an alkaline electrolyte with different bulk K+ concentrations. The dark cyan, blue, orange, red, and white spheres represent Ru, Ni, K, O, and H atoms, respectively. | ||
We further turned our attention to seeking the understanding of how interfacial hydrated K+ cations favor the water dissociation step. In principle, the hydrated K+ cations at the outer Helmholtz plane would exert the influence on H2O molecules through the electrostatic interaction due to the presence of H2O dipoles, wherein the electric field strength is a crucial factor to determine the magnitude of interaction force.39,44,45 We thus approximately estimated the electrostatic field between each slab and the electrolyte interface by using a parallel plate capacitor model (see Methods for details), wherein the charge was determined by Bader analysis (Table S8, ESI†). It should be mentioned that on the Ru3Ni(0001) surface (No K+), 0.17 e− spontaneously transfers from the H2O layer to the Ru3Ni(0001) surface, which causes the negatively charged metal surface and positively charged surface H2O layer. From the estimations, the Ru3Ni(0001) surface with K+ (at 1/12 ML coverage) shows the enhanced electric field E (2.87 × 108 V m−1) when compared with the other two slabs (Fig. 4d). Under the electric field, the H–OH bond of the H2O molecules at the inner Helmholtz plane would be polarized by the *H–OHδ−–M+ interaction according to the prior fundamental studies,30,32,46 which is also verified using DFT calculations. As shown in Fig. 4d, the largest bond length of H–OH is observed on the Ru3Ni(0001) surface with K+ (at 1/12 ML coverage), verifying the function of K+ in facilitating H2O polarization. Based on the above analyses, Fig. 4e schematically illustrates how hydrated K+ promotes the water dissociation step, which is supposed to be favored by the *H–OHδ−–K+ interaction as reported in prior studies.31
We further experimentally examined the promotional role of K+ in alkaline HER activity by varying the bulk K+ concentration. Clearly, the alkaline HER activity is improved over Ru3Ni-300 and Ru3Ni-600 catalysts upon increasing the K+ concentration, confirming the promotional role of K+ (Fig. S32, ESI†). Intriguingly, the activity of the NA-Ru3Ni/C catalyst is insensitive to the bulk K+ concentration, showing no obvious changes in alkaline HER activity at different bulk K+ concentrations (Fig. 4f). Besides, when using different NaOH concentrations as the electrolyte, a similar effect to that of K+ was obtained (Fig. S33 and S34, ESI†). Therefore, we assume that tip-induced K+ accumulation dominates the real hydrated K+ concentration at the EDL interface, which results in our unusual observation. This unusual phenomenon indeed highlights the dominant role of nanotips in regulating the local microenvironment. Taking all these together, the greatly improved alkaline HER kinetics over the NA-Ru3Ni/C catalyst can be attributed to the tip-concentrated local hydrated K+, which stretches the H–OH bond of interfacial water molecules via non-covalent interactions and in turn lowers ΔGW, ultimately promoting water dissociation.
Long-term durability in an AEM electrolyser
Inspired by the remarkable HER activity and stability of the NA-Ru3Ni/C catalyst in a three-electrode system, we further evaluated the performance of NA-Ru3Ni/C as a cathode with a loading amount of 0.048 mg cm−2 in a practical AEM electrolyser operating at 60 °C. Since the oxygen evolution reaction (OER) activity of NA-Ru3Ni/C was higher than that of the commercial IrO2/C catalyst (Fig. S35, ESI†), herein we constructed an AEM electrolyser with the cathodic and anodic reactions supported by NA-Ru3Ni/C catalysts on carbon paper, as illustrated in Fig. 5a. The polarization curve shows that the NA-Ru3Ni/C||NA-Ru3Ni/C couple possess a small cell voltage (2.048 V) at 1 A cm−2 for water-splitting activity, being superior to commercial Pt/C||IrO2/C (Fig. 5b). Moreover, the gas products and the corresponding faradaic efficiency in 1.0 M KOH were analyzed by gas chromatography (Fig. 5c). This result reflects a nearly 100% faradaic efficiency of H2 production, i.e., almost all electrons contribute to the desired HER during electrocatalysis. Besides, we further performed the chronopotentiometry test of the NA-Ru3Ni/C||NA-Ru3Ni/C couple to evaluate the long-term operation stability for AEM electrolysis. Fig. 5d shows that the voltage of the NA-Ru3Ni/C||NA-Ru3Ni/C couple shows almost no change at a current density of 1.0 A cm−2 over 2000 h of continuous operation, while Pt/C||IrO2, as a comparison, shows an increase of voltage within only 40 h under the same test conditions (Fig. S36, ESI†). The overall performance (including the overpotential at 1000 mA cm−2, TOF, and long-term durability) for the alkaline HER reported herein represents a record-high performance to date (Table S9, ESI†). Such remarkable performance of the NA-Ru3Ni/C catalyst is expected for potential commercialization. | ||
| Fig. 5 The performance of the NA-Ru3Ni/C catalyst for the AEM electrolyser at 60 °C. (a) Schematic diagram of an AEM electrolyser (GDL, gas diffusion layer). (b) Polarization curve of the NA-Ru3Ni/C||NA-Ru3Ni/C couple in an AEM electrolyser. (c) Faradaic efficiency, and theoretical and experimental results of H2 production of NA-Ru3Ni/C. (d) Chronopotentiometry curve for AEM electrolysis using NA-Ru3Ni/C as cathode and anode catalysts operating at 1 A cm−2. The inset shows the photographs of the AEM electrolyser. | ||
Conclusion
In summary, we have developed a very active and stable nanocone-assembled Ru3Ni catalyst that exhibits a low overpotential of 168 mV at 1000 mA cm−2 and a high turnover frequency of 26.5 s−1 at an overpotential of 100 mV. Moreover, the catalyst could stably operate at 1000 mA cm−2 over 2000 h in a practical AEM electrolyser at 60 °C. Finite element simulations and experimental results reveal that the nanotips of the Ru3Ni catalyst enable an enhanced local electric field, and in turn substantially increase the interfacial K+ concentration. Further mechanistic studies indicate that the locally increased hydrated K+ concentration promotes the polarization of the H–OH bond of interfacial water, resulting in the reduced energy barrier for water dissociation and thus accounting for greatly improved alkaline HER activity. The present work not only provides a highly efficient and robust alkaline HER catalytic system for practical AEM electrolysers, but also paves a new way for designing industrially relevant electrocatalytic systems.Author contributions
L. G., F. B., and M. L. performed the sample synthesis, carried out the electrochemical measurements, and analyzed the experimental data. C. M. conducted the TEM and EDX characterization studies. X. T. and S. S. conducted the DFT simulation and theoretical analyses. Z. T. and P. H. conducted the COMSOL Multiphysics simulations. Z. S., X. C., and W. L. helped with the analysis and discussion of experimental data. Y. L. performed the XPS experiments. Z. Y., Z. H., and H. H. conceived the idea and wrote the manuscript. All the authors were involved in the discussion and analysis of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2021YFA1500900 and 2021YFA1502000), the NSFC (No. 21832003, U2032149, 52071174, and 22102052), the Science and Technology Innovation Program of Hunan Province (No. 2021RC3065 and 2021RC2053), the Hunan Provincial Natural Science Foundation of China (No. 2020JJ3001), the Shenzhen Science and Technology Program (No. JCYJ20210324120800002), and the Natural Science Foundation of Jiangsu Province, Major Project (Grant No. BK20212005). This research was also undertaken with the assistance of resources provided by the Pawsey and the National Computing Infrastructure (NCI) facility at the Australian National University, allocated through both the National Computational Merit Allocation Scheme supported by the Australian Government and the Australian Research Council grant LE190100021 (sustaining and strengthening merit-based access at NCI, 2019-2021).Notes and references
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- H. Liu, G. Zhang, X. Zheng, F. Chen and H. Duan, Int. J. Extreme Manuf., 2020, 2, 042001 CrossRef CAS.
- Y. Wang, Y. Jiao, H. Yan, G. Yang, C. Tian, A. Wu, Y. Liu and H. Fu, Angew. Chem., Int. Ed., 2022, 61, e202116233 CAS.
- N. Du, C. Roy, R. Peach, M. Turnbull, M. Thiele and C. Bock, Chem. Rev., 2022, 122, 11830–11895 CrossRef CAS PubMed.
- P. Chen and X. Hu, Adv. Energy Mater., 2020, 10, 2002285 CrossRef CAS.
- W. E. Mustain and P. A. Kohl, Nat. Energy, 2020, 5, 359–360 CrossRef CAS.
- H. Q. Fu, M. Zhou, P. F. Liu, P. Liu, H. Yin, K. Z. Sun, H. G. Yang, M. Al-Mamun, P. Hu, H.-F. Wang and H. Zhao, J. Am. Chem. Soc., 2022, 144, 6028–6039 CrossRef CAS PubMed.
- X. Yan, J. Biemolt, K. Zhao, Y. Zhao, X. Cao, Y. Yang, X. Wu, G. Rothenberg and N. Yan, Nat. Commun., 2021, 12, 4143 CrossRef CAS PubMed.
- Y. Yao, Y. Zhu, C. Pan, C. Wang, S. Hu, W. Xiao, X. Chi, Y. Fang, Y. Yang, H. Deng, S. Xiao, J. Li., Z. Luo and Y. Guo, J. Am. Chem. Soc., 2021, 143, 8720–8730 CrossRef CAS PubMed.
- X. Wu, Z. Wang, D. Zhang, Y. Qin, M. Wang, Y. Han, T. Zhan, B. Yang, S. Li, J. Lai and L. Wang, Nat. Commun., 2021, 12, 4018 CrossRef CAS PubMed.
- L. Gao, Z. Yang, T. Sun, X. Tan, W. Lai, M. Li, J. Kim, Y.-F. Lu, S.-I. Choi, W. Zhang, C. Ma, S. C. Smith, Y.-G. Zhou and H. Huang, Adv. Energy Mater., 2022, 12, 2103943 CrossRef CAS.
- W. Liu, X. Wang, F. Wang, K. Du, Z. Zhang, Y. Guo, H. Yin and D. Wang, Nat. Commun., 2021, 12, 6776 CrossRef CAS PubMed.
- Y.-N. Zhou, H.-J. Liu, Z.-N. Shi, J.-C. Zhou, B. Dong, H.-Y. Zhao, F.-G. Wang, J.-F. Yu and Y.-M. Chai, Nano Res., 2022, 15, 5873–5883 CrossRef CAS.
- B. Mao, P. Sun, Y. Jiang, T. Meng, D. Guo, J. Qin and M. Cao, Angew. Chem., Int. Ed., 2020, 59, 15232–15237 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, A. Vasileff and S.-Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 7568–7579 CrossRef CAS PubMed.
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. T. Miller, L. Sun and J. Hou, Nat. Commun., 2021, 12, 4587 CrossRef CAS PubMed.
- I. T. McCrum and M. T. M. Koper, Nat. Energy, 2020, 5, 891–899 CrossRef CAS.
- X. Jiang, H. Jang, S. Liu, Z. Li, M. G. Kim, C. Li, Q. Qin, X. Liu and J. Cho, Angew. Chem., Int. Ed., 2021, 60, 4110–4116 CrossRef CAS PubMed.
- Y. Zhu, H. A. Tahini, Z. Hu, J. Dai, Y. Chen, H. Sun, W. Zhou, M. Liu, S. C. Smith, H. Wang and Z. Shao, Nat. Commun., 2019, 10, 149 CrossRef PubMed.
- J. Kim, H. Jung, S.-M. Jung, J. Hwang, D. Y. Kim, N. Lee, K.-S. Kim, H. Kwon, Y.-T. Kim, J. W. Han and J. K. Kim, J. Am. Chem. Soc., 2021, 143, 1399–1408 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef CAS PubMed.
- W. Li, Y. Zhao, Y. Liu, M. Sun, G. I. Waterhouse, B. Huang, K. Zhang, T. Zhang and S. Lu, Angew. Chem., Int. Ed., 2021, 60, 3290–3298 CrossRef CAS PubMed.
- L. Deng, F. Hu, M. Ma, S.-C. Huang, Y. Xiong, H.-Y. Chen, L. Li and S. Peng, Angew. Chem., Int. Ed., 2021, 60, 22276–22282 CrossRef CAS PubMed.
- M. Liu, J.-A. Wang, W. Klysubun, G.-G. Wang, S. Sattayaporn, F. Li, Y.-W. Cai, F. Zhang, J. Yu and Y. Yang, Nat. Commun., 2021, 12, 5260 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- Z. Luo, H. Zhang, Y. Yang, X. Wang, Y. Li, Z. Jin, Z. Jiang, C. Liu, W. Xing and J. Ge, Nat. Commun., 2020, 11, 1116 CrossRef CAS PubMed.
- H. Tan, B. Tang, Y. Lu, Q. Ji, L. Lv, H. Duan, N. Li, Y. Wang, S. Feng, Z. Li, C. Wang, F. Hu, Z. Sun and W. Yan, Nat. Commun., 2022, 13, 2024 CrossRef CAS PubMed.
- M. M. Waegele, C. M. Gunathunge, J. Li and X. Li, J. Chem. Phys., 2019, 151, 160902 CrossRef PubMed.
- E. Liu, J. Li, L. Jiao, H. T. T. Doan, Z. Liu, Z. Zhao, Y. Huang, K. M. Abraham, S. Mukerjee and Q. Jia, J. Am. Chem. Soc., 2019, 141, 3232–3239 CrossRef CAS PubMed.
- M. C. O. Monteiro, A. Goyal, P. Moerland and M. T. Koper, ACS Catal., 2021, 11, 14328–14335 CrossRef CAS PubMed.
- Y.-H. Wang, S. Zheng, W.-M. Yang, R.-Y. Zhou, Q.-F. He, P. Radjenovic, J.-C. Dong, S. Li, J. Zheng, Z.-L. Yang, G. Attard, F. Pan, Z.-Q. Tian and J.-F. Li, Nature, 2021, 600, 81–85 CrossRef CAS PubMed.
- J. Mahmood, F. Li, S.-M. Jung, M. S. Okyay, I. Ahmad, S.-J. Kim, N. Park, H. Y. Jeong and J.-B. Baek, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS PubMed.
- C. Rong, X. Shen, Y. Wang, L. Thomsen, T. Zhao, Y. Li, X. Lu, R. Amal and C. Zhao, Adv. Mater., 2022, 34, 2110103 CrossRef CAS PubMed.
- J. A. Rodriguez and D. W. Goodman, Science, 1992, 257, 897–903 CrossRef CAS PubMed.
- L. Gao, X. Li, Z. Yao, H. Bai, Y. Lu, C. Ma, S. Lu, Z. Peng, J. Yang, A. Pan and H. Huang, J. Am. Chem. Soc., 2019, 141, 18083–18090 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- D. Liu, X. Li, S. Chen, H. Yan, C. Wang, C. Wu, Y. A. Haleem, S. Duan, J. Lu, B. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- J. Zhang, T. Wang, P. Liu, Z. Liao, S. Liu, X. Zhuang, M. Chen, E. Zschech and X. Feng, Nat. Commun., 2017, 8, 15437 CrossRef CAS PubMed.
- J. Mao, C.-T. He, J. Pei, W. Chen, D. He, Y. He, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 4958 CrossRef PubMed.
- W. Li, Y. Zhao, Y. Liu, M. Sun, G. I. N. Waterhouse, B. Huang, K. Zhang, T. Zhang and S. Lu, Angew. Chem., Int. Ed., 2021, 133, 3327–3335 CrossRef.
- H. Yan, Y. Xie, A. Wu, Z. Cai, L. Wang, C. Tian, X. Zhang and H. Fu, Adv. Mater., 2019, 31, 1901174 CrossRef PubMed.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS.
- Y. Zhou, Y. Liang, J. Fu, K. Liu, Q. Chen, X. Wang, H. Li, L. Zhu, J. Hu, H. Pan, M. Miyauchi, L. Jiang, E. Cortés and M. Liu, Nano Lett., 2022, 22, 1963–1970 CrossRef PubMed.
- A. Goyal and M. T. M. Koper, Angew. Chem., Int. Ed., 2021, 60, 13452–13462 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, TEM and STEM images, XPS and XRD spectra, TEM-EDS spectrum, CV and LSV curves, CO stripping voltammogram, Nyquist plots, theoretical model structures, and tables. See DOI: https://doi.org/10.1039/d2ee02836k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |