
Multifunctional SEI-like structure coating stabilizing Zn anodes at a large current and capacity†
Aosai
Chen
a,
Chenyang
Zhao
a,
Jiaze
Gao
c,
Zhikun
Guo
a,
Xingyuan
Lu
a,
Jiachi
Zhang
a,
Zeping
Liu
a,
Ming
Wang
a,
Nannan
Liu
a,
Lishuang
Fan
ad,
Yu
Zhang
*b and
Naiqing
Zhang
 *ad
*ad
aState Key Laboratory of Urban Water Resource and Environment, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin, 150001, China. E-mail: znqmww@163.com
bSchool of Energy Science and Engineering, Harbin Institute of Technology, Harbin, 150001, China. E-mail: zhangchemistry@hit.edu.cn
cShen-zhen College of International Education, 3 Antuoshan 6th Rd, Futian, Shenzhen, China
dAcademy of Fundamental and Interdisciplinary Sciences, Harbin Institute of Technology, Harbin, 150001, China
First published on 30th November 2022
Abstract
The development and application of aqueous zinc-ion batteries still face some obstacles, such as dendrite growth and side reactions triggered by active water. Here, we constructed a PVA@SR-ZnMoO4 multifunctional coating on the Zn anode with an SEI-like structure (SR refers to SO42− receptor). The PVA@SR outer layer can give the coating some flexibility while enhancing Zn2+ mobility and dispersion. The ZnMoO4 inner layer can effectively inhibit dendrite growth and side reactions. Importantly, a fast Zn2+ migration pathway can be established between PVA and ZnMoO4 in the inorganic–organic composite inner layer, ensuring high-current operation of the Zn anode. Thanks to the “mutual cooperation”, the Zn anode exhibits a high Coulomb efficiency (CE) of 99.42% and good cycling stability of 1700 h at 5 mA cm−2 and 5 mA h cm−2. Moreover, the Zn/α-MnO2 full cell can maintain a residual capacity of 141.7 mA h g−1 without capacity decay after 1000 cycles at 1.0 A g−1.
Broader contextAqueous zinc ion batteries have been emerged as strong candidates for the deployment of large-scale energy storage devices due to their high level of safety and environmental friendliness. Zn anodes are of great interest due to their high capacity, high storage capacity and low reduction potential. In recent years, great progress has been made in the modification of Zn anodes, including in electrolyte additives, electrode structure design and artificial electrode surface modification. Inorganic coating surface modification is one of the most promising practical strategies and has received widespread attention. However, the rigidity and low ionic conductivity of the inorganic coating limit the anode cycling at high current and capacity. Here, we designed a PVA@SR-ZnMoO4 SEI-like structure coating, where the gel outer layer can enhance the coating stability and ZnMoO4 in the inner layer can inhibit dendrites and hydrogen evolution. Importantly, a fast Zn2+ migrating pathway can be established between ZnMoO4 and PVA in the inner layer and it promotes Zn2+ desolvation. Furthermore, the ion transport ability of the migration channel was found to be positively correlated with the ionic affinity of the inorganic material through further studies. This work provides a reasonable solution for stabilizing Zn anodes with high current and capacity cycles, and provides new ideas for anode surface modification with organic–inorganic composite design. |
Introduction
As an aqueous energy storage system, zinc ion batteries (ZIBs) have become a powerful substitute to lithium ion batteries (LIBs) in practical applications due to their high safety, low cost and environmental friendliness.1–8 However, their further development is restricted by the instability of metallic zinc anodes. As shown in Scheme 1, the growth of dendrites is supported by kinetics and thermodynamics, and becomes the main factor affecting the cell lifespan.9–12 In addition, the H2O decomposition and a series of related side reactions in weak acid electrolytes become other unavoidable problems according to the Pourbaix diagram.13–17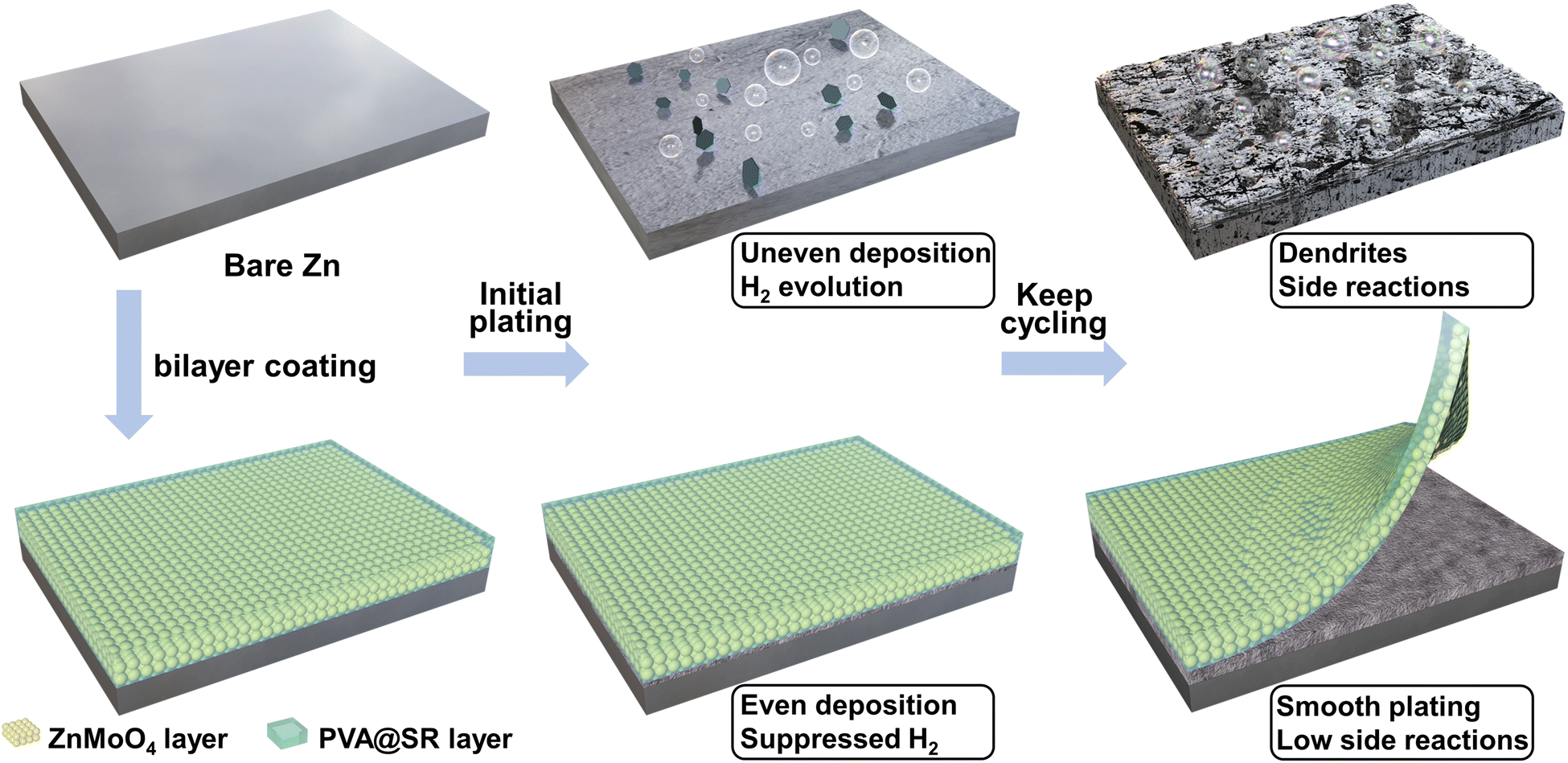 | ||
| Scheme 1 Cycling performance of bare Zn and PVA@SR-ZnMoO4 SEI-like structure coating modified Zn. | ||
In recent years, some progress has been made in anode/electrolyte interface modification,18–22 electrolyte design23–25 and collector optimization26–29 to suppress zinc dendrites. Interfacial inorganic coating modification has become one of the most promising practical strategies and has received wide attention. For example, inorganic coating modification can construct uniform ion transport channels30 or zincophilic sites31 to guide dendrite-free cycling. However, inorganic coating with rigidity does not cope well with volume changes of the anode during cycling, especially under large capacity cycling.32,33 The coatings can crack or even fall off and it is difficult to maintain lasting protection. In addition, the low Zn2+ conductivity of the coatings limits the performance of the anode for high current cycling.34
In view of the defects of inorganic coatings as well as the practical application requirements of high current and capacity cycling of ZIBs,35 we introduced an organic component into the coating to construct a PVA@SR-ZnMoO4 protective layer with an SEI-like structure (only similar in structure, as shown in Scheme 1). The PVA@SR outer layer can act as glue to give the coating some flexibility to improve the coating stability under large-capacity cycling.36,37 Meanwhile, PVA@SR can trap SO42− in the electrolyte and enhance the mobility and dispersion of the counterion Zn2+.9 As for the inner layer, ZnMoO4 can effectively suppress dendrites and side reactions.34 Importantly, unlike the reported mechanisms for high ion migration of organic–inorganic composites,38–40 ZnMoO4 can also establish a fast Zn2+ migrating pathway between the PVA gel based on high zinc affinity, improving the sluggish ion conduction of ZnMoO4 and breaking the ion migration limit of single components that make tzn2+ as high as 0.81. Based on the design of this versatile SEI-like structure coating, the Zn anode can guarantee a high CE of 99.42% and stable cycling at high current and large capacity (1700 h at 5 mA cm−2 and 5 mA h cm−2). This work provides a reasonable solution to stabilizing Zn anodes, the design of an SEI-like structure of the coating provides a new idea for anode surface modification. And the investigation of a new mechanism for rapid Zn2+ migration opens up new directions for the design of anode surface coatings and organic–inorganic composites.
Results and discussion
For high current and capacity stable cycling of ZIBs, the surface coating of the Zn anode needs to meet the following requirements. First, the coating can stabilize the Zn anode without dendrites and side reactions. Second, the surface coating supports rapid Zn2+ migration for high current cycling. Finally, it is also necessary to ensure that the surface coating adheres tightly to the anode and remains intact during cycling, especially under large capacity cycling. However, it is difficult for a single inorganic coating to meet the above three requirements at the same time. Fortunately, inspired by the structure of the solid electrolyte interface (SEI) of LIBs, the design of the modified layer with an SEI-like structure containing both inorganic and organic components can compensate for the shortcomings of the inorganic coating while exploiting and amplifying their own advantages. In previous reports, ZnMoO4 has been proven to effectively inhibit dendrite growth and hydrogen evolution, making it an ideal choice for inorganic components. Considering its acceptable Zn2+ transport and coating stability, PVA gel was chosen as the organic component. Furthermore, the SR was added to bind SO42− and accelerate the migration of the counterion Zn2+. Thus, the constructed SEI-like structure coating is expected to stabilize the Zn anode at high current and capacity.As shown in Fig. 1a, the SEI-like structure coating was obtained by two successive blade castings, where the morphology and hydrophilicity of ZnMoO4 and SR are indicated in Fig. S1–S3 (ESI†). Notably, the PVA gel needs further molding through three freeze–thaw cycles.41 As shown in Fig. 1c, ZnMoO4 exhibits an anorthic crystal system XRD pattern (JCPDS: 00-035-0765) with a coating thickness of about 8 µm (informed from Fig. 1b). Considering the PVA gel shrinks with water loss, the overall thickness of the SEI-like structure coating does not exceed 15 µm (refer to the coating preparation parameters).
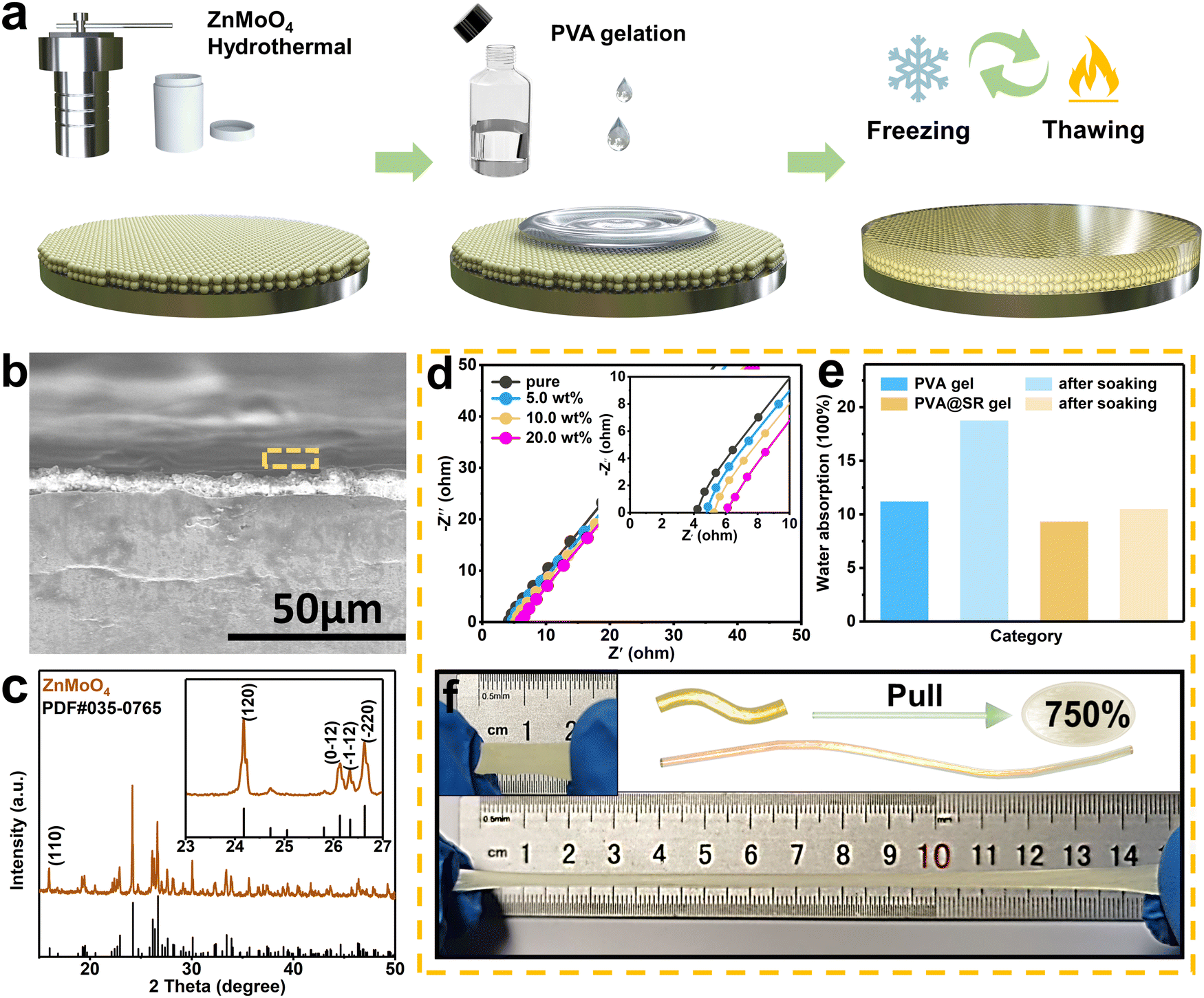 | ||
| Fig. 1 (a) Schematic illustration of the fabrication process of the SEI-like structure coating modified anode. (b) The SEM image of the SEI-like structure coating. Inorganic component: (c) XRD pattern of ZnMoO4. Organic component: (d) EIS spectra of PVA gel with different SR addition, (e) water absorption of pure PVA and 10 wt% SR modified PVA. (f) Ductility of the PVA@SR gel. | ||
For the PVA organic component, the addition of SR significantly affects the gel pore structure. As shown in Fig. S4 (ESI†), the pore size of the gel gradually decreases and the pore wall changed from membrane-like to rod-like with the increased addition. When the addition reached 10 wt%, the pore density reached the peak with an intact pore structure, which facilitated the smooth flow of Zn2+. Furthermore, when the addition continued, the pore structure collapsed and hindered Zn2+ transport.42 Thus, the following studies all used 10 wt% PVA@SR as the organic component. Importantly, as the filler of PVA gel, SR exhibits good compatibility. As shown in Fig. S5 (ESI†), the PVA@SR Raman spectrum shows the coupling of SR signal peaks with a PVA characteristic peak (located at 2950 cm−1), indicating that SR retains the function of binding SO42− and accelerating counterion Zn2+ migration. Based on this, the PVA@SR organic component exhibits a surprisingly high ion transport capacity. As shown in Fig. 1d and Fig. S6 (ESI†), the ionic conductivity of the PVA gel gradually increases with the addition of SR and peaks at 10 wt% (1.419 × 10−2 S cm−1), even higher than that of the 3.0 M ZnSO4 water-based electrolyte (1.182 × 10−2 S cm−1). The negligible ionic conductivity change of the 10 wt% SiO2-modified PVA gel further confirms the important role played by SR (Fig. S7, ESI†).
Besides, the addition of SR also affects the hygroscopicity and ductility of the PVA gel. As shown in Fig. 1e, the water contents of the pure and modified PVA gels were almost comparable in the initial state. After soaking in deionized water for 7 days, the water content of the pure PVA gel increased significantly, and the resulting swelling could seriously affect the coating stability. In contrast, the modified PVA gel remained almost unchanged. As shown in Fig. S8 (ESI†), even after 10 days immersion in electrolyte, the modified layer can still tightly coat the anode and has good anode passivation resistance. Importantly, the ductility of the PVA gel was also somewhat improved as it increased from 500% to 750%, further demonstrating the potential of the modified PVA gel as an organic component (Fig. 1f and Fig. S9, ESI†). This is also confirmed in Fig. S10 (ESI†), where the modified coating adapts well to changes in twisting and bending of the substrate.
In view of the fast ion transport capability of the gel organic component, the symmetric cells were further assembled and the high-current operation supportability was investigated. As shown in Fig. 2a, at a scanning speed of 1 mV s−1, the bare Zn shows a distinct diffusion-limited current plateau just at −0.286 V with a plateau current of only −0.013 A. On the other hand, the modified Zn returns to normal after a brief high current plateau (−0.038 A) between −0.394 V and −0.535 V. This indicates that the SEI-like structure coating inherits the high ion transport ability of the gel component and exhibits the potential for high current cycling.43
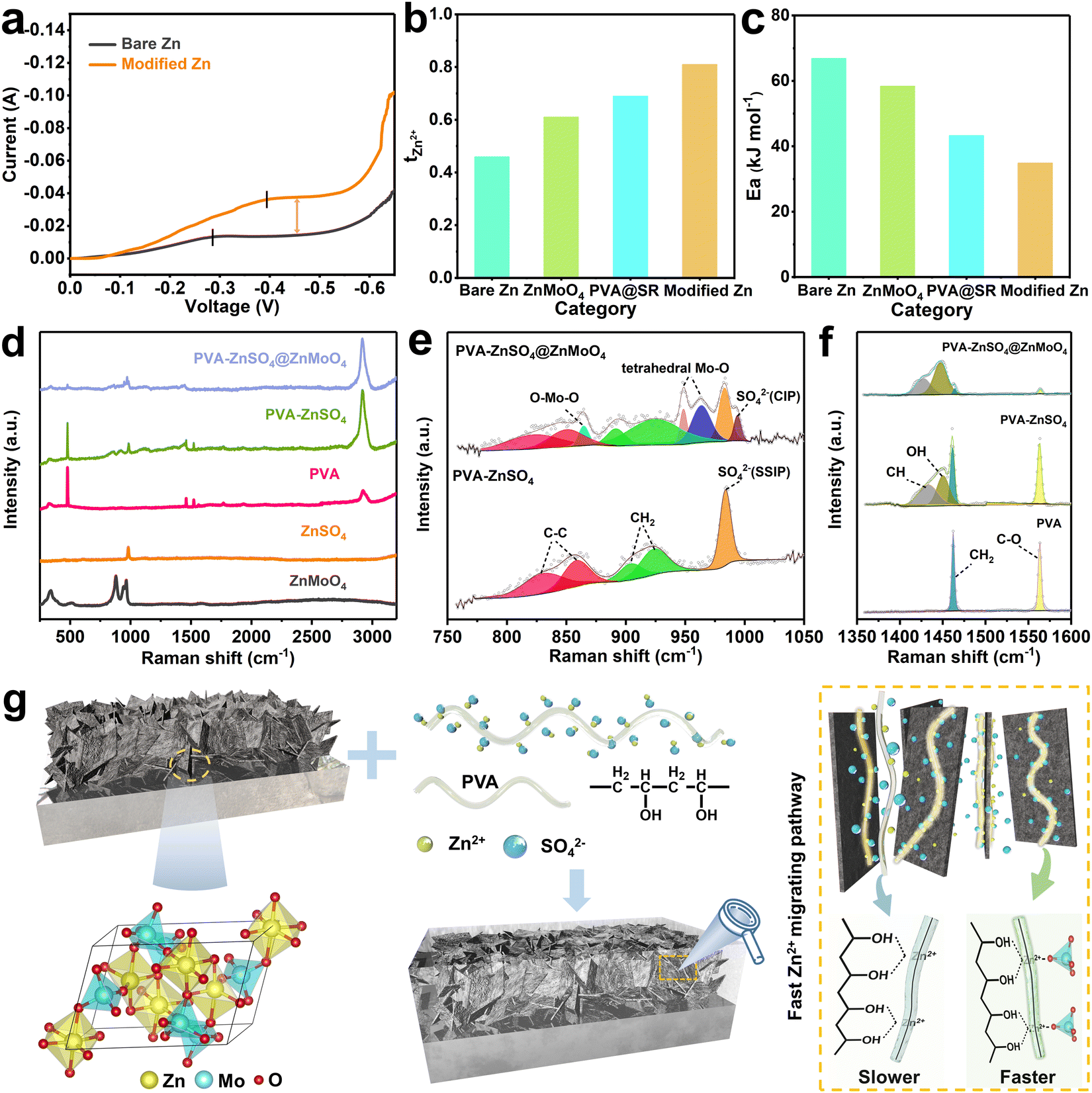 | ||
| Fig. 2 (a) Voltammetry of symmetric cells with 1 M ZnSO4 electrolyte at a scan rate of 1 mV s−1. (b) Zn2+ transference number and (c) activation energy of the zinc anode with different modifications. (d–f) Raman spectra. (g) Schematics of zinc ion migration in the migrating pathway at the ZnMoO4/PVA interface in the hybrid inner layer. | ||
Zn2+ transference number tests were further performed to investigate the Zn2+ migration contribution of the ZnMoO4 inorganic component. As shown in Fig. 2b and Fig. S11 (ESI†), the bare Zn cell exhibits a slow Zn2+ migration with a tzn2+ of only 0.46 due to the presence of hydrated Zn2+ with six coordinated water molecules and counterion SO42− in the electrolyte.44 Differently, the high binding energy of ZnMoO4 to zinc adatoms and the capture of SR to SO42− increased the tzn2+ of electrolyte to 0.61 and 0.69 respectively. Unusually, the tzn2+ of the SEI-like coating modified Zn cell was as high as 0.81, breaking the ion migration limit of the single coating modified Zn cells and showing an unexpected effect of “one plus one is greater than two”. This is also supported by Fig. S12, S13 and Table S1 (ESI†), where the ion diffusion coefficient of the coating can be obtained using the Nernst–Einstein framework.45,46 It can be seen that the ion diffusion coefficient of the SEI-like coating is significantly higher than that of the single coatings.
The same phenomenon was also observed in the activation energy (Ea) test. As shown in Fig. S14 and S15 (ESI†), the charge transfer resistance (Rct) of the modified Zn is significantly lower than that of bare Zn over a wide temperature range and is even almost half that of bare Zn at room temperature. Consequently, the Ea of modified Zn is only 34.9 kJ mol−1, which is much lower than that of bare Zn (66.9 kJ mol−1), showing high electrochemical reactivity and even better than inorganic layer modified Zn and organic layer modified Zn, breaking the reaction kinetic limit of a single coating (Fig. 2c). It has been proved that the main factor affecting the Rct is the Zn2+ desolvation as they enter the electric double layer. This process consumes most of the energy and becomes the rate-controlling step of the whole electrochemical process, directly affecting the Ea.47,48 Thus, the SEI-like structure coating can accelerate the desolvation of Zn2+, which not only facilitates large current cycling, but also suppresses a series of side reactions caused by the active water to some extent.
To explore the mechanism of Zn2+ rapid migration in the SEI-like structure coating, Raman spectroscopy was used to investigate the intrinsic link between the inorganic and organic components. Fig. 2d shows the complete Raman spectra of PVA–ZnSO4, PVA–ZnSO4@ZnMoO4 and each monomer. The detailed spectra in different bands are shown in Fig. 2e and f and Fig. S16 (ESI†), and the spectral analysis is presented in Table S2 (ESI†).49–52 In the Raman shift range of 1350 cm−1 to 1600 cm−1, the appearance of C–H (1432 cm−1) and O–H (1450 cm−1) signal peaks after the introduction of ZnSO4 into PVA demonstrates the migration form of Zn2+ in PVA with the –OH group as the site.53 After further introduction of ZnMoO4, the C–O bond bending vibration peak (478 cm−1 in Fig. S14a, ESI† and 1563 cm−1 in Fig. 2f) and the CH2 shear mode signal peak (1462 cm−1, Fig. 2f) of PVA almost disappear, proving the existence of the interaction between PVA and ZnMoO4.54 Correspondingly, the signal peaks of PVA stretching vibration νcc (822 cm−1 and 852 cm−1) and rocking vibration γCH2 (893 cm−1 and 922 cm−1) in PVA–ZnSO4@ZnMoO4 are redshifted in the range of 750 cm−1 to 1050 cm−1 compared to PVA–ZnSO4. As for ZnMoO4, the signal peaks about the O–Mo–O symmetric stretching bond (880 cm−1) and tetrahedral Mo–O bond (945 cm−1 and 965 cm−1) also present changes in area and position.55 These changes are caused by the interaction between the –OH group of PVA and the Mo–O tetrahedra of ZnMoO4. Consequently, the stretching vibration mode of SO42− in PVA–ZnSO4@ZnMoO4 shows an obvious shoulder peak (993 cm−1), implying weakening of the Zn2+ hydration shell and the transformation of the solvation structure from solvent separated ion pair (SSIP) to contact ion pair (CIP), which is also consistent with the activation energy test results.56,57 In addition, Fig. S17 (ESI†) shows that SR is not involved in the construction of the interfacial Zn2+ fast migrating pathway. Based on the above experimental phenomena, it can be inferred that the interaction between ZnMoO4 and PVA in the hybrid inner layer changes their interfacial environment and constructs a fast Zn2+ migrating pathway between –OH and Mo–O, which accelerates the migration of Zn2+ and promotes the desolvation of Zn2+.54,58 As a result, the establishment of an interfacial fast migrating pathway in the hybrid inner layer can effectively compensate for the lack of Zn2+ conductivity of ZnMoO4 and its hindrance to electrolyte flow (as shown in Fig. S18, ESI†).
Furthermore, to explore the mechanism of ZnMoO4, TiO2-doped PVA gels with different crystal plane orientations were prepared and tested (the SEM images, XRD patterns and BET results are shown in Fig. S19, ESI†). From a previous report, it is known that the zinc affinity of faceted TiO2 (f-TiO2) is weaker than that of commercial TiO2 (c-TiO2) with large (100) crystal face occupancy.59 As a result, the PVA@c-TiO2 exhibited higher ionic conductivity (Fig. S20b, ESI†) and tzn2+ (Fig. S21, ESI†) compared to PVA@f-TiO2. These arise from the different zinc affinities of c-TiO2 and f-TiO2. In Fig. S20a (ESI†), the PVA signal peak at 478 cm−1 associated with the intensity of synergy exhibits different intensities in the c-TiO2 and f-TiO2 systems. The weak intensity indicates that the synergy is strong, and the synergy is stronger in the c-TiO2 system. Fig. S22 (ESI†) reflects the effect of synergy difference on cycle performance. Under cycling conditions of 2 mA cm−2 and 1 mA h cm−2, the voltage of the Zn/c-TiO2 cell was relatively smooth and increased the stable cycle time to 1480 h compared to the drastic voltage fluctuation of the Zn/f-TiO2 cell. Similarly, the variation in the ionic conductivity of the composites brought about by the affinity difference of the inorganic component was also confirmed in Li+–PVA. These provide strong evidence for a positive correlation between the ionic affinity of the inorganic components and the synergic effect (Fig. S23 and S24, ESI†). Based on the above experimental results, it can be speculated that the inorganic component with a high ion binding energy can attract ions and support their interfacial migration (Fig. 2g). Together with the special interfacial environment generated by the interaction between the inorganic component and PVA, it can support fast ion migration. Unlike the existing mechanisms of surface vacancy,38 defect39 and Lewis base,40 this work firstly finds that the inorganic components with high ionic binding energy can effectively enhance the ionic conductivity of organic–inorganic composites and broaden their design ideas.
Based on the promotion of Zn2+ migration by the SEI-like structure coating, its inhibitory effect on dendrites and side reactions were further investigated. As shown in Fig. 3a, b and Fig. S25, S26 (ESI†) the zinc deposition process was monitored in real time using an in situ optical microscope to visualize the modification effect. At the initial stage of deposition (before 1 min), visible bubbles and sharp protrusions (marked by a dashed circle) appeared on the bare Zn and SiO2-coated Zn. As the deposition continues, the uneven deposition gradually deteriorates, showing distinct dendritic protrusions accompanied by severe hydrogen evolution. In contrast, the dendrite growth and hydrogen evolution were significantly inhibited on the monocomponent-layer modified Zn. As for the SEI-like structure coating modified Zn, the zinc deposition layer remained relatively flat without dendrite even with a plating time reaching 8 minutes (deposition capacity equivalent to 6.67 mA h cm−2), indicating the possibility of a stable high current and capacity cycle.
 | ||
| Fig. 3 In situ optical microscopy of (a) bare Zn and (b) modified Zn at a current density of 50 mA cm−2. SEM images of single deposition: (c and d) bare Zn at 0.25 mA cm−2 and 5.0 mA h cm−2; (e–g) modified Zn at 0.25 mA cm−2 and 5.0 mA h cm−2 and related EDS-mapping (h). (i) Chronoamperometry (CA) plots. (j) XRD patterns after cycling (removing the modification layer of the modified Zn anode). | ||
The ex situ SEM images also showed similar results. As shown in Fig. 3c and d, the bare Zn exhibits an obvious dendritic morphology with a deposition capacity of only 0.25 mA h cm−2. In contrast, the surface of modified Zn still remains flat even with a deposition capacity of 5 mA h cm−2 as shown in Fig. 3e–h. Notably, the deposition morphology of zinc changes from flake to block, reducing the possibility of forming hazardous dendrites. Moreover, the relatively dense deposited layer also indicates a suppressed hydrogen evolution.
Both in situ and ex situ studies of zinc plating morphology indicate that the SEI-like structure coating can effectively regulate Zn2+ deposition and inhibit hydrogen generation. However, further electrochemical tests are needed to gain insight into the cycling process. The chronoamperometry (CA) tests were performed to understand the nucleation behavior. At a constant polarization potential (−150 mV), the deposition of Zn2+ can induce changes in the anode surface area, and the nucleation process can be inferred from the response current. As shown in Fig. 3i, the continuous current change indicates the presence of severe Zn2+ 2D diffusion in the bare Zn. In this case, Zn2+ migrates along the anode surface and accumulates at high surface energies, triggering dendrite formation and leading to a rough deposition morphology. Differently, due to the high zinc affinity of ZnMoO4, the Zn2+ is directly reduced to Zn0in situ without location selection. As a result, Zn2+ shifts to stable 3D diffusion after brief 2D diffusion, resulting in dense nucleation sites and smooth deposition morphology, which is consistent with the above morphology results. In addition, the zinc nucleation energy barrier also changed from 63 mV to 30 mV due to the high zinc affinity and Zn2+ conductivity of the SEI-like structure coating (Fig. S27, ESI†).
Tafel curve and LSV plot tests were further performed to investigate the side reactions. As shown in Fig. S28 (ESI†), the hydrogen evolution overpotential of both monocomponent and SEI-like structure coating modified Zn increased significantly compared to bare Zn in the LSV test, where the overpotential of the SEI-like structure coating modified Zn even changed from −0.037 V to −0.118 V, exhibiting an excellent HER inhibitory performance. In addition, the positive shift of corrosion potential and the reduction of corrosion current in the Tafel test also indicate the side reaction suppression by the SEI-like structure coating. As shown in Fig. 3j, the bare Zn exhibited an obvious (ZnSO4)(Zn(OH)2)3(H2O)4 side reaction after 100 cycles, while the modified Zn showed no signal peak for the side product even after 200 cycles. The above tests show that the SEI-like structure coating can effectively inhibit dendrites, hydrogen evolution and related side reactions.
The above tests have shown that the SEI-like structure coating can effectively improve the Zn2+ deposition/dissolution process. Here, the long-cycle protection of the SEI-like structure coating is further investigated. The Zn/Ti asymmetric cells were assembled to study the long-term CE performance as shown in Fig. 4a and Fig. S29 (ESI†). As expected, the bare Ti cell exhibited poor cycling stability with a significant drop in CE within 50 cycles at 0.5 mA cm−2 and 0.5 mA h cm−2. Differently, both inorganic and organic layer modified cells can maintain an average CE of about 96.5% over 150 cycles, and the SEI-like structure coating modified Ti can further increase the CE to 99.42% with stable operation for 500 cycles. Moreover, the SEI-like structure coating modified Ti has the lowest polarization voltage as shown in Fig. S30 (ESI†). These demonstrate the inhibition of side reactions by the SEI-like structure coating and the effectiveness of the organic–inorganic composite modification.
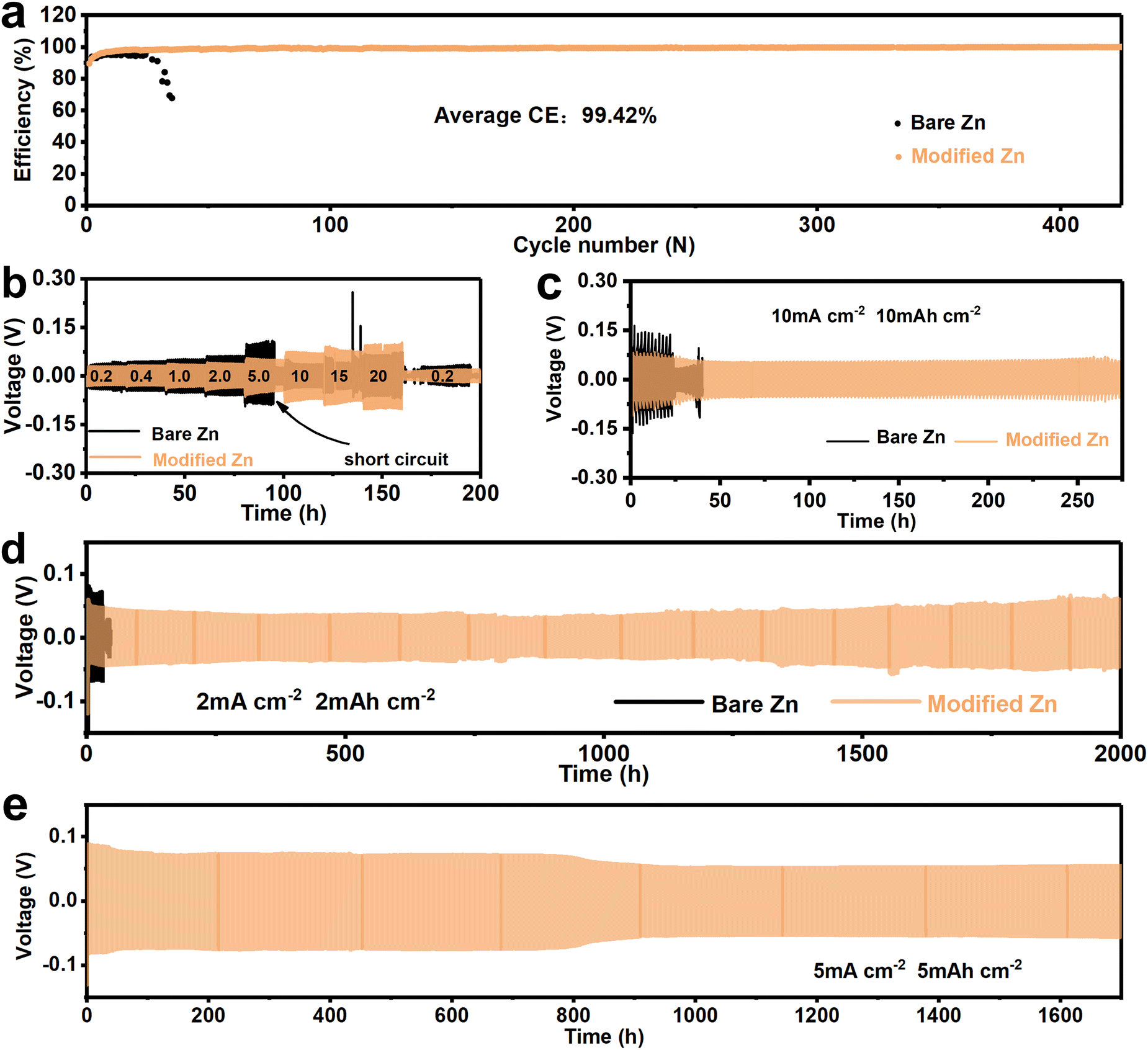 | ||
| Fig. 4 (a) CE performance of bare Zn and modified Zn cycled at 0.5 mA cm−2 and 0.5 mA h cm−2. (b) Rate performance of symmetrical cells. (c) The voltage profiles of Zn symmetric cells at 10 mA cm−2 with 10 mA h cm−2. Long-term galvanostatic cycling performance of symmetrical cells: (d) 2 mA cm−2 with 2 mA h cm−2, and (e) 5 mA cm−2 with 5 mA h cm−2. | ||
Fig. 4b and Fig. S31 and S32 (ESI†) demonstrate the rate performance of symmetric cells, where the current density gradually increased from 0.2 mA cm−2 to 20 mA cm−2. Obviously, the hysteresis voltage of the modified Zn cell is smaller than that of the bare Zn due to the accelerated Zn2+ migration and enhanced deposition/dissolution activity. In addition, as the current density increases, the bare Zn cell rapidly short-circuits and fails at 5.0 mA cm−2. Oppositely, the hysteresis voltage of modified Zn still maintains relative stability even at a high current density of 20 mA cm−2. As shown in Fig. 4c, the modified Zn cell was able to cycle stably for 275 h under harsh cycling conditions of 10 mA cm−2 and 10 mA h cm−2. In contrast, the bare Zn cell showed a short circuit with a sharp voltage drop after 30 h.
Similarly, the modified Zn cell also showed excellent long-term cycle stability. As shown in Fig. 4d and Fig. S33 (ESI†), the bare Zn cell exhibited poor cycling performance and shorted within 50 h at 2 mA cm−2 with 2 mA h cm−2. Differently, both the monocomponent coating and SEI-like structure coating modified Zn exhibited excellent cycle stability, where the composite-modified Zn increased the cycle time to 2000 h with a relatively stable hysteresis voltage. As shown in Fig. S34 (ESI†), the bulging of the cell and the penetrated zinc in the separator suggest that the failure of bare Zn is attributed to H2 evolution and dendrite growth. The corresponding optical images as well as SEM images (Fig. S35, ESI†) also confirm the long-lasting inhibition of zinc dendrites by the modified layer and strongly indicate the high adhesion of the coating to the substrate as well as the high structural stability of the coating itself. Excitingly, the positive effect of the modified layer on the Zn anode was demonstrated thoroughly with 5 mA cm−2 and 5 mA h cm−2. As shown in Fig. 4e, the modified Zn cell can cycle 1700 h with a relatively stable voltage, showing sufficient potential for practical applications, surpassing most previous reports (as shown in Table S3, ESI†). To further understand the enhancement of coating stability by the PVA outer layer on top of the SEI-like structure coating, modified Zn symmetric cells without the PVA outer layer were assembled and tested. As shown in Fig. S36 (ESI†), the coating showed zinc dendrite penetration with 2 mA h cm−2 deposited zinc, and the symmetric cell only cycled for about 500 h at 5 mA cm−2 and 5 mA h cm−2. The above experimental results all indicate that the additional PVA outer layer can further enhance the coating stability and greatly improve the Zn anode cycling stability.
In view of the improvement of the stability of the zinc anode by the SEI-like coating, the cycle performance of the full cell as the entirety will also be correspondingly improved. In detail, full cells were assembled using nanofibrous α-MnO2 as the cathode material (material characterizations are shown in Fig. S37, ESI†) and performed related electrochemical tests. As shown in Fig. 5a, the cyclic voltammetry curves (CV) both exhibit two pairs of redox peaks (A1, C1 and A2, C2), representing the embedding and de-embedding of H+ and Zn2+ in the α-MnO2 cathode material, respectively.60,61 In addition, the consecutive cycle curves of both cells matched well respectively, indicating the reversibility of the electrochemical reaction process. Noteworthily, the modified Zn cell has a smaller redox peak potential gap, thus exhibits a smaller electrochemical polarization and higher reactivity compared to the bare Zn cell. The electrochemical impedance spectra (EIS) in Fig. 5c also clearly shows that the modified Zn cell has a more rapid charge transfer process. Moreover, the peak current density of the modified Zn cell is higher than that of the bare Zn, indicating that the SEI-like structure coating modification of the Zn anode can improve the charge and discharge capacity. This conclusion is further confirmed by Fig. 5d, where the modified Zn cell has a discharge capacity of 255.21 mA h cm−2 compared to 246.82 mA h cm−2 for the bare Zn at 0.2 A g−1. Based on the high reactivity and high discharge capacity of the modified Zn cell, it exhibits a better rate performance. As shown in Fig. 5b, the discharge capacity of the modified Zn cell is higher than that of bare Zn at different current densities with less capacity fluctuation. Importantly, the modified Zn cell exhibits excellent long cycle stability and can maintain a residual capacity of 141.7 mA h g−1 after 1000 cycles at 1.0 A g−1, while the bare Zn cell only has a capacity retention rate of 59.1% (Fig. 5e and Fig. S38, ESI†). As shown in Fig. S39 (ESI†), we further analysed the source of the performance improvement of the modified Zn cell. It can be seen that in addition to the improvement of the anode stability, the dissolution of the α-MnO2 cathode material is also improved to some extent, which may be attributed to the inhibition of water decomposition (see the ESI† for specific analysis). Finally, considering the role of the PVA gel outer layer as a hydrogel electrolyte, we separately assembled the symmetric cell and the full cell and tested the rate performance without additional liquid electrolyte addition. As shown in Fig. S40 (ESI†), both the gel symmetric cell and the gel full cell exhibit relatively excellent rate performances, reflecting the feasibility of epitaxy of the outer PVA gel of the SEI-like coating as a gel electrolyte.
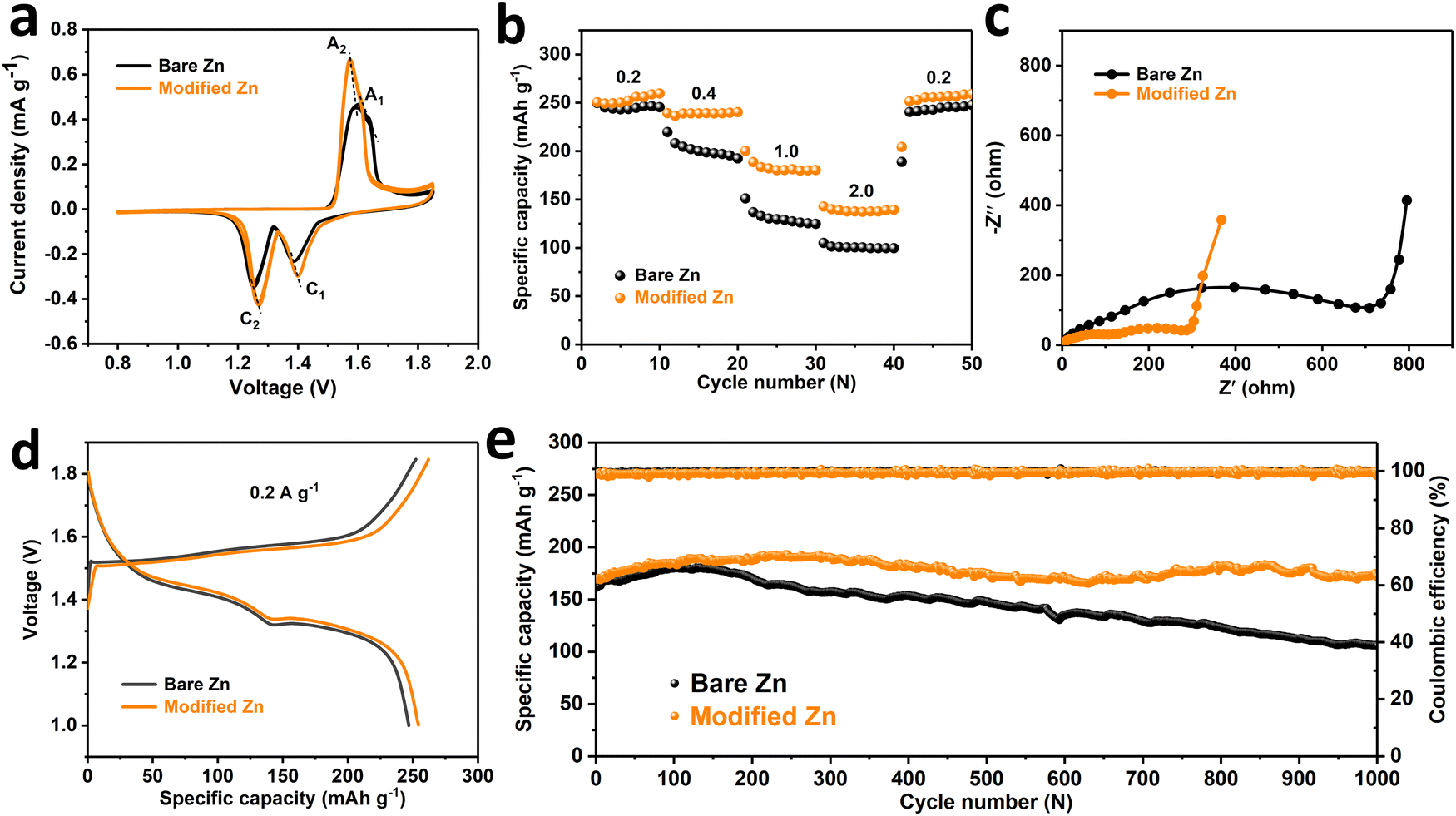 | ||
| Fig. 5 Electrochemical performances of Zn/α-MnO2 batteries with bare Zn and modified Zn anode: (a) CV curves at 0.1 mV s−1, (b) rate performance, (c) EIS curves before cycling, and (d) the charge–discharge curve at 0.2 A g−1 after activation. (e) Long-term cycling performance at 1.0 A g−1. | ||
Conclusion
In summary, the construction of a PVA@SR-ZnMoO4 coating with a SEI-like structure can effectively stabilize the Zn anode. The PVA@SR outer layer with good flexibility can compensate for the rigidity of the inorganic component and improve the coating stability under large capacity cycling. In addition, PVA@SR can bind SO42− and enhance the migration and dispersion of counterion Zn2+. And the inorganic ZnMoO4 in the inner composite layer can effectively suppress dendrites and side reactions. Importantly, the interaction of ZnMoO4 with PVA in the hybrid inner layer can establish a fast Zn2+ migrating pathway and promote the desolvation of Zn2+. Based on these, the Zn anode exhibits a high Coulomb efficiency of 99.42% and a high current and capacity cycling performance of 1700 h at 5 mA cm−2 and 5 mA h cm−2. This work provides a practical design strategy for Zn anode surface modification, opens up the possibility of achieving stable rapid large-capacity cycling and broadens the design ideas for high ionic conductivity composites.Author contributions
A. Chen, Y. Zhang and N. Zhang designed the research. A. Chen, J. Gao, C. Zhao, Z. Guo, X. Lu and N. Liu carried out the synthesis, electrochemical measurements, and data analysis. A. Chen, Y. Zhang, L. Fan and N. Zhang co-wrote the manuscript. N. Zhang supervised the research. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22078078), the Natural Science Foundation of Heilongjiang Province (No. LH2020B008), and the State Key Laboratory of Urban Water Resource and Environment (Harbin Institute of Technology) (No. 2022TS20).Notes and references
- H. Kim, D. A. Boysen, J. M. Newhouse, B. L. Spatocco, B. Chung, P. J. Burke, D. J. Bradwell, K. Jiang, A. A. Tomaszowska, K. Wang, W. Wei, L. A. Ortiz, S. A. Barriga, S. M. Poizeau and D. R. Sadoway, Chem. Rev., 2013, 113, 2075–2099 CrossRef CAS.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed., 2012, 51, 933–935 CrossRef CAS.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. S. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS.
- L. Hong, L. Y. Wang, Y. Wang, X. Wu, W. Huang, Y. Zhou, K. X. Wang and J. S. Chen, Adv. Sci., 2022, 9, e2104866 CrossRef.
- Z. Zhao, R. Wang, C. Peng, W. Chen, T. Wu, B. Hu, W. Weng, Y. Yao, J. Zeng, Z. Chen, P. Liu, Y. Liu, G. Li, J. Guo, H. Lu and Z. Guo, Nat. Commun., 2021, 12, 6606 CrossRef CAS PubMed.
- Y. Zhang, X. Li, L. S. Fan, Y. Shuai and N. Q. Zhang, Cell Rep. Phys. Sci., 2022, 3, 100824 CrossRef CAS.
- N. Liu, X. Wu, L. Fan, S. Gong, Z. Guo, A. Chen, C. Zhao, Y. Mao, N. Zhang and K. Sun, Adv. Mater., 2020, 32, e1908420 CrossRef PubMed.
- A. Chen, C. Zhao, Z. Guo, X. Lu, J. Zhang, N. Liu, Y. Zhang and N. Zhang, Adv. Funct. Mater., 2022, 2203595 CrossRef CAS.
- H. F. Li, L. T. Ma, C. P. Han, Z. F. Wang, Z. X. Liu, Z. J. Tang and C. Y. Zhi, Nano Energy, 2019, 62, 550–587 CrossRef CAS.
- Q. Zhang, J. Luan, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 13180–13191 CrossRef CAS.
- Z. Guo, L. Fan, C. Zhao, A. Chen, N. Liu, Y. Zhang and N. Zhang, Adv. Mater., 2022, 34, e2105133 CrossRef.
- A. Bayaguud, Y. Fu and C. Zhu, J. Energy Chem., 2022, 64, 246–262 CrossRef CAS.
- L. Ma, S. Chen, N. Li, Z. Liu, Z. Tang, J. A. Zapien, S. Chen, J. Fan and C. Zhi, Adv. Mater., 2020, 32, e1908121 CrossRef.
- W. H. Yang, X. F. Du, J. W. Zhao, Z. Chen, J. J. Li, J. Xie, Y. J. Zhang, Z. L. Cui, Q. Y. Kong, Z. M. Zhao, C. G. Wang, Q. C. Zhang and G. L. Cui, Joule, 2020, 4, 1557–1574 CrossRef CAS.
- L. Zhang, B. Zhang, T. Zhang, T. Li, T. F. Shi, W. Li, T. Shen, X. X. Huang, J. J. Xu, X. G. Zhang, Z. Y. Wang and Y. L. Hou, Adv. Funct. Mater., 2021, 31, 2100186 CrossRef CAS.
- R. Li, Y. Li, P. Yang, P. Ren, D. Wang, X. Lu, R. Xu, Y. Li, J. Xue, J. Zhang, M. An, J. Ma, B. Wang, H. Liu and S. Dou, Appl. Catal., B, 2022, 318, 121834 CrossRef CAS.
- X. Liu, F. Yang, W. Xu, Y. Zeng, J. He and X. Lu, Adv. Sci., 2020, 7, 2002173 CrossRef CAS.
- X. S. Xie, S. Q. Liang, J. W. Gao, S. Guo, J. B. Guo, C. Wang, G. Y. Xu, X. W. Wu, G. Chen and J. Zhou, Energy Environ. Sci., 2020, 13, 503–510 RSC.
- W. X. Wang, G. Huang, Y. Z. Wang, Z. Cao, L. Cavallo, M. N. Hedhili and H. N. Alshareef, Adv. Energy Mater., 2022, 12, 2102797 CrossRef CAS.
- C. Zhao, Y. Du, Z. Guo, A. Chen, N. Liu, X. Lu, L. Fan, Y. Zhang and N. Zhang, Energy Storage Mater., 2022, 53, 322–330 CrossRef.
- X. Lu, C. Zhao, A. Chen, Z. Guo, N. Liu, L. Fan, J. Sun and N. Zhang, Chem. Eng. J., 2023, 451, 138772 CrossRef CAS.
- F. Ming, Y. Zhu, G. Huang, A. H. Emwas, H. Liang, Y. Cui and H. N. Alshareef, J. Am. Chem. Soc., 2022, 144, 7160–7170 CrossRef CAS.
- W. Y. Chen, S. Guo, L. P. Qin, L. Y. Li, X. X. Cao, J. Zhou, Z. G. Luo, G. Z. Fang and S. Q. Liang, Adv. Funct. Mater., 2022, 32, 2112609 CrossRef CAS.
- R. Z. Qin, Y. T. Wang, M. Z. Zhang, Y. Wang, S. X. Ding, A. Y. Song, H. C. Yi, L. Y. Yang, Y. L. Song, Y. H. Cui, J. Liu, Z. Q. Wang, S. N. Li, Q. H. Zhao and F. Pan, Nano Energy, 2021, 80, 105478 CrossRef CAS.
- Y. Zeng, X. Zhang, R. Qin, X. Liu, P. Fang, D. Zheng, Y. Tong and X. Lu, Adv. Mater., 2019, 31, e1903675 CrossRef PubMed.
- W. C. Du, E. H. X. Ang, Y. Yang, Y. F. Zhang, M. H. Ye and C. C. Li, Energy Environ. Sci., 2020, 13, 3330–3360 RSC.
- Z. Wang, J. H. Huang, Z. W. Guo, X. L. Dong, Y. Liu, Y. G. Wang and Y. Y. Xia, Joule, 2019, 3, 1289–1300 CrossRef CAS.
- Q. Li, Y. B. Wang, F. N. Mo, D. H. Wang, G. J. Liang, Y. W. Zhao, Q. Yang, Z. D. Huang and C. Y. Zhi, Adv. Energy Mater., 2021, 11, 2003931 CrossRef CAS.
- L. T. Kang, M. W. Cui, F. Y. Jiang, Y. F. Gao, H. J. Luo, J. J. Liu, W. Liang and C. Y. Zhi, Adv. Energy Mater., 2018, 8, 1801090 CrossRef.
- P. C. Liang, J. Yi, X. Y. Liu, K. Wu, Z. Wang, J. Cui, Y. Y. Liu, Y. G. Wang, Y. Y. Xia and J. J. Zhang, Adv. Funct. Mater., 2020, 30, 1908528 CrossRef CAS.
- N. Zhang, S. Huang, Z. Yuan, J. Zhu, Z. Zhao and Z. Niu, Angew. Chem., Int. Ed., 2021, 60, 2861–2865 CrossRef CAS PubMed.
- L. B. Dong, W. Yang, W. Yang, H. Tian, Y. F. Huang, X. L. Wang, C. J. Xu, C. Y. Wang, F. Y. Kang and G. X. Wang, Chem. Eng. J., 2020, 384, 123355 CrossRef CAS.
- A. Chen, C. Zhao, Z. Guo, X. Lu, N. Liu, Y. Zhang, L. Fan and N. Zhang, Energy Storage Mater., 2022, 44, 353–359 CrossRef.
- X. Li, Q. Li, Y. Hou, Q. Yang, Z. Chen, Z. Huang, G. Liang, Y. Zhao, L. Ma, M. Li, Q. Huang and C. Zhi, ACS Nano, 2021, 15, 14631–14642 CrossRef CAS PubMed.
- R. Xu, X.-Q. Zhang, X.-B. Cheng, H.-J. Peng, C.-Z. Zhao, C. Yan and J.-Q. Huang, Adv. Funct. Mater., 2018, 28, 1705838 CrossRef.
- Y. T. He, Y. H. Zhang, X. F. Li, Z. Lv, X. J. Wang, Z. G. Liu and X. Q. Huang, Energy Storage Mater., 2018, 14, 392–401 CrossRef.
- N. Wu, P. H. Chien, Y. Qian, Y. Li, H. Xu, N. S. Grundish, B. Xu, H. Jin, Y. Y. Hu, G. Yu and J. B. Goodenough, Angew. Chem., Int. Ed., 2020, 59, 4131–4137 CrossRef CAS.
- J. X. Zhang, N. Zhao, M. Zhang, Y. Q. Li, P. K. Chu, X. X. Guo, Z. F. Di, X. Wang and H. Li, Nano Energy, 2016, 28, 447–454 CrossRef CAS.
- N. Wu, P. H. Chien, Y. Li, A. Dolocan, H. Xu, B. Xu, N. S. Grundish, H. Jin, Y. Y. Hu and J. B. Goodenough, J. Am. Chem. Soc., 2020, 142, 2497–2505 CrossRef CAS PubMed.
- C. Wu, X. Lu, L. Peng, K. Xu, X. Peng, J. Huang, G. Yu and Y. Xie, Nat. Commun., 2013, 4, 2431 CrossRef PubMed.
- J. Bae, Y. Li, J. Zhang, X. Zhou, F. Zhao, Y. Shi, J. B. Goodenough and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 2096–2100 CrossRef CAS.
- J. Zhi, S. Li, M. Han and P. Chen, Sci. Adv., 2020, 6, eabb1342 CrossRef CAS.
- J. Hao, B. Li, X. Li, X. Zeng, S. Zhang, F. Yang, S. Liu, D. Li, C. Wu and Z. Guo, Adv. Mater., 2020, 32, e2003021 CrossRef PubMed.
- R. Guo and B. M. Gallant, Chem. Mater., 2020, 32, 5525–5533 CrossRef CAS.
- D. T. Boyle, Y. Li, A. Pei, R. A. Vila, Z. Zhang, P. Sayavong, M. S. Kim, W. Huang, H. Wang, Y. Liu, R. Xu, R. Sinclair, J. Qin, Z. Bao and Y. Cui, Nano Lett., 2022, 22, 8224–8232 CrossRef CAS PubMed.
- H. Liu, J.-G. Wang, W. Hua, L. Ren, H. Sun, Z. Hou, Y. Huyan, Y. Cao, C. Wei and F. Kang, Energy Environ. Sci., 2022, 15, 1872–1881 RSC.
- Z. Y. Miao, M. Du, H. Z. Li, F. Zhang, H. C. Jiang, Y. H. Sang, Q. F. Li, H. Liu and S. H. Wang, EcoMat, 2021, 3, e12125 CrossRef CAS.
- S. Krimm, C. Y. Liang and G. B. B. M. Sutherland, J. Chem. Phys., 1956, 25, 549–562 CrossRef CAS.
- I. Y. Prosanov and A. A. Matvienko, Phys. Solid State, 2010, 52, 2203–2206 CrossRef CAS.
- M. Hema, S. Selvasekarapandian, G. Hirankumar, A. Sakunthala, D. Arunkumar and H. Nithya, Spectrochim. Acta, Part A, 2010, 75, 474–478 CrossRef CAS PubMed.
- Y. A. Badr, K. M. A. El-Kader and R. M. Khafagy, J. Appl. Polym. Sci., 2004, 92, 1984–1992 CrossRef CAS.
- S. Alipoori, S. Mazinani, S. H. Aboutalebi and F. Sharif, J. Storage Mater., 2020, 27, 101072 Search PubMed.
- O. Sheng, C. Jin, J. Luo, H. Yuan, H. Huang, Y. Gan, J. Zhang, Y. Xia, C. Liang, W. Zhang and X. Tao, Nano Lett., 2018, 18, 3104–3112 CrossRef CAS PubMed.
- G. D. Saraiva, W. Paraguassu, A. J. R. de Castro, F. F. de Sousa, J. G. da Silva Filho, V. O. S. Neto, J. A. Lima Jr., A. M. R. Teixeira and P. T. C. Freire, Spectrochim. Acta, Part A, 2020, 239, 118501 CrossRef CAS.
- W. W. Rudolph, G. Irmer and G. T. Hefter, Phys. Chem. Chem. Phys., 2003, 5, 5253–5261 RSC.
- H. Yang, Z. Chang, Y. Qiao, H. Deng, X. Mu, P. He and H. Zhou, Angew. Chem., Int. Ed., 2020, 59, 9377–9381 CrossRef CAS PubMed.
- Y. Z. Hu, L. G. Li, H. F. Tu, X. H. Yi, J. Wang, J. J. Xu, W. B. Gong, H. Z. Lin, X. D. Wu and M. N. Liu, Adv. Funct. Mater., 2022, 2203336 CrossRef CAS.
- Q. Zhang, J. Luan, X. Huang, Q. Wang, D. Sun, Y. Tang, X. Ji and H. Wang, Nat. Commun., 2020, 11, 3961 CrossRef CAS PubMed.
- J. Wang, J. G. Wang, X. Qin, Y. Wang, Z. You, H. Liu and M. Shao, ACS Appl. Mater. Interfaces, 2020, 12, 34949–34958 CrossRef CAS.
- Y. Zhong, X. Xu, J. P. Veder and Z. Shao, iScience, 2020, 23, 100943 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee02931f |
| This journal is © The Royal Society of Chemistry 2023 |