
A tailored electrolyte for safe and durable potassium ion batteries†
Ling
Fan‡
 *a,
Huabin
Xie‡
a,
Yanyao
Hu
a,
Zhuoma
Caixiang
a,
Apparao M.
Rao
b,
Jiang
Zhou
c and
Bingan
Lu
*a
*a,
Huabin
Xie‡
a,
Yanyao
Hu
a,
Zhuoma
Caixiang
a,
Apparao M.
Rao
b,
Jiang
Zhou
c and
Bingan
Lu
*a
aSchool of Physics and Electronics, Hunan University, Changsha, 410083, P. R. China. E-mail: fanling@hnu.edu.cn; luba2012@hnu.edu.cn
bDepartment of Physics and Astronomy, Clemson Nanomaterials Institute, Clemson University, Clemson, SC, USA
cSchool of Materials Science and Engineering, Central South University, Changsha, 410083, P. R. China
First published on 3rd January 2023
Abstract
Electrolytes are critical for the safety and long-term cyclability of potassium ion batteries (PIBs). However, conventional low-concentration electrolytes cannot simultaneously achieve safe and durable PIBs due to their highly flammable solvents or ineffective solid-electrolyte interphase (SEI). Herein, we demonstrate a low-concentration, non-flammable, and weakly solvating electrolyte that simultaneously enhances the safety and cyclability of PIBs. The solvent exhibits non-flammable and weak solvation features, which bolsters the battery safety and the formation of anion-derived SEI even in the low-concentration electrolyte. Specifically, the proposed electrolyte enables a running time of over 200 days and 2 years for K‖Cu and K‖graphite cells, respectively. In addition, the K0.5MnO2‖graphite full cell combined with this proposed electrolyte can operate for 100 cycles with high Coulombic efficiency of 99.7%. This study provides a guiding principle in tailoring electrolytes for achieving safe and long-lasting potassium ion batteries.
Broader contextLow-cost, safety, and long-term cycling stability are key parameters for large-scale stationary energy storage. Compared to other non-lithium based rechargeable batteries, potassium ion batteries (PIBs) possess the merits of high voltage due to the more negative standard reduction potential of potassium (−2.93 V). Meanwhile, the commercial graphite can be used as an anode for PIBs and shows a theoretical capacity of 279 mA h g−1, further increasing the cost effectiveness of PIBs. However, safety concerns and durable stability of PIBs are the main obstacles for their practical applications. Herein, we demonstrate a low-concentration, non-flammable, and weakly solvating electrolyte that simultaneously enhances the safety and cyclability of PIBs. Specifically, the proposed electrolyte enables a running time of over 200 days and 2 years for K‖Cu and K‖graphite cells, respectively. In addition, the K0.5MnO2‖graphite full cell combined with this proposed electrolyte can operate for 100 cycles with high Coulombic efficiency of 99.7%. This study provides a guiding principle in tailoring electrolytes for achieving safe and long-lasting potassium ion batteries and beyond. |
Introduction
Potassium-ion batteries (PIBs) are promising candidates for large-scale stationary energy storage due to their superior cost-effectiveness and high voltage.1–3 More importantly, graphite (a commercial electrode material for LIBs) can be used as an anode for PIBs.4–6 Thus, the utilization of currently mature production technologies of LIBs is feasible for PIBs.7 Although significant achievements have been made on the cathode8–11 and anode12–14 materials, the safety concerns and long-term cycle stability arising from the highly flammable organic electrolytes and ineffective solid-electrolyte interphase (SEI)15 are still hindering the practical applications of PIBs.It is well known that electrolytes significantly impact batteries' security, and utilizing a non-flammable electrolyte solvent is beneficial for realizing high security.16,17 Organic phosphates,18 which are known for their intrinsically non-flammable nature, have been extensively used as solvents in batteries. However, the conventionally low-concentration (1 M) phosphates-based electrolytes cannot form effective SEI,17 resulting in continuous electrolyte decomposition and rapid capacity fading. Though high-concentration (>2.5 M) electrolytes could enhance cycling stability,19–21 they suffer from the disadvantages of high viscosity and high salt cost,22–24 hindering their practical applications. Therefore, it is highly imperative yet challenging to uncover low-concentration electrolytes for achieving durable and safe PIBs.
The long-term cyclability of a battery, as another key parameter for large-scale stationary energy storage, could be tailored by the electrolytes and the resulting SEI properties. It has been confirmed that the formation and composition of SEI are closely associated with the ion solvation structures in electrolytes. Generally, the solvents surrounding cations will result in the formation of solvent-derived SEI,25,26 and the cation-anion clusters in electrolytes are conducive to achieving an anion-derived SEI.19,27 Typically, the anion-derived SEI is beneficial to achieving superior stability,13,28,29 and thus developing low-concentration electrolytes with the solvation structure of cation-anion clusters is preferred. As known, the weakly solvating electrolyte, in which the cation-solvent interactions are weakened while the cation-anion interactions are strengthened,30 is preferred to form an anion-derived SEI even in moderate concentration. Meanwhile, considering the low charge density of K+, the K+-solvent interactions are intrinsically weaker than that of other metal ions (Li+ and Na+)-solvent interactions, which is conducive to achieving weakly solvating electrolytes.
In this work, we report a low-concentration (1 M) electrolyte consisting of a single salt of potassium bis(fluorosulfonyl)imide (KFSI) and a single solvent of tris (2,2,2-trifluoroethyl) phosphate (FTEP) that enables safe and durable PIBs. Benefiting from the weakly solvating ability of FTEP solvent, this electrolyte (1 M KFSI–FTEP) promotes the formation of an anion-derived SEI on the anode surface. Also, it enables the durable cycling stability of K‖graphite (running time for over 2 years) and K‖Cu cells (running time for over 200 days). Interestingly, this electrolyte is also suitable for a high voltage Mn-based Prussian blue analogue (Mn-PBA) cathodes (4.2 V), the K‖Mn-PBA cell delivers good stability for 200 cycles with a capacity retention of 77.3%. Furthermore, the K0.5MnO2‖graphite full cell also delivers superior stability of 100 cycles with high Coulombic efficiency of 99.7%.
Results
Tailoring non-flammable and weakly solvating electrolyte
Safe long-term operation (i.e., devoid of thermal runaway and fire) is indispensable for PIBs before they can be employed in large-scale electric energy storage applications. Generally, electrolyte solvents are undoubtedly one of the main reasons for fire when thermal runaway happens in a cell. The traditional electrolytes, mainly carbonate- and ether-based electrolytes, are highly flammable and hazardous (Fig. 1a). Therefore, to guarantee battery security, it might be better to prepare potassium ion electrolytes with non-flammable solvents (Fig. 1a). We performed the flame tests of the commonly used carbonate and phosphate ester electrolytes to investigate their flammability (Fig. 1b). As expected, we found the carbonate electrolytes with solvents of ethylene carbonate (EC), propylene carbonate (PC), dimethyl carbonate (DMC), ethyl methyl carbonate (EMC), or diethyl carbonate (DEC) are highly flammable. By contrast, the phosphate ester electrolytes with trimethyl phosphate (TMP), triethyl phosphate (TEP), and FTEP solvents are non-flammable. Moreover, the self-extinguishing time (SET) of phosphate ester electrolytes (∼0 s g−1) is significantly lower than that of carbonate electrolytes (>25 s g−1) (Fig. S1, ESI†), further demonstrating the potentially high-security feature of phosphate electrolytes. Besides, it should be mentioned that the high-concentration electrolyte (3 M KFSI in 1,2-dimethoxyethane) and the commonly used potassium ion electrolytes are also highly flammable (Fig. S2, ESI†). These findings demonstrate that the utilization of non-flammable phosphate ester electrolytes could remarkably reduce the fire risk and thus enhance the battery security.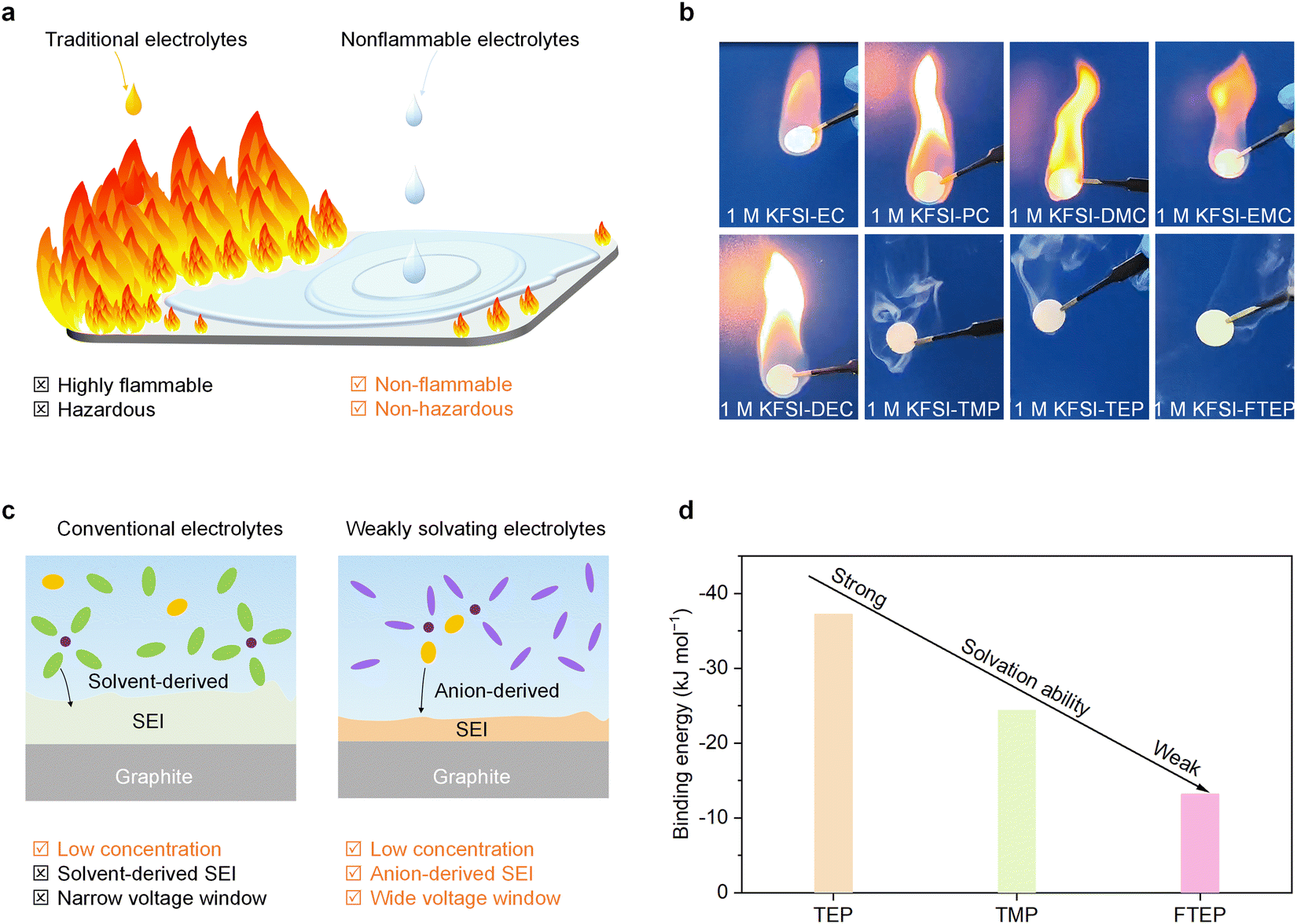 | ||
| Fig. 1 Developing non-flammable and weakly solvating electrolytes. (a) An illustration that compares the hazardous properties of traditional (carbonate- and ether-based) and non-flammable (phosphate ester-based) electrolytes. (b) Flame tests of various electrolytes. (c) The illustrations of SEI formation in low-concentration conventional electrolytes and weakly solvating electrolytes. (d) The binding energies between various solvents and K+. | ||
As mentioned previously, the electrochemical performance of a battery is another key parameter for their practical applications, and the electrolytes certainly play a decisive role in the SEI properties and electrochemical performance.15 Particularly, the ion solvation structures in the electrolyte are believed to dominate the SEI formation and interfacial chemistry, further determining the electrochemical properties. Generally, in low-concentrated conventional electrolytes, the K+ surrounded by solvent molecules are prevalent, leading to solvent-derived SEI and narrow voltage window (Fig. 1c). By contrast, in low-concentrated weakly solvating electrolytes, the K+ is preferred to form K+-anions clusters, which is conducive to achieving anion-derived SEI and wide electrochemical window (Fig. 1c). Therefore, it is important to understand the propensity of K+ to solvate with various solvents for realizing weakly solvating electrolytes. The binding energy, calculated by the density functional theory (DFT) method, is used to elucidate the solvation ability between various non-flammable solvents and K+ (Fig. 1d). Typically, the more negative the binding energy, the stronger the solvation ability. Clearly, the absolute value of binding energy between FTEP and K+ is the lowest among the selected non-flammable solvents, which possibly because of the fluorination of solvents weakened its solvation ability.31 In other words, the solvating ability of FTEP with K+ is the weakest among the selected non-flammable solvents, indicating that the FTEP is more suitable for the preparation of weakly solvating electrolytes.
Electrolyte solvation structures
Considering the similar molecular structures and non-flammable feature of TEP and FTEP, two typical electrolytes of 1 M KFSI–TEP and 1 M KFSI–FTEP are further investigated. The solvation structures of K+ in these electrolytes were studied by DFT molecular dynamics (DFT-MD) simulation, conducted by a pre-equilibrated process for 5 ps followed by a production process for 10 ps (Fig. S3, ESI†). The K+ coordinating environments in the two electrolytes are quite different according to the snapshots of electrolyte chemical structures (Fig. 2a). Meanwhile, the percentage of free FTEP solvent (31.8%) is much higher than that of free TEP solvent (4.3%) (Fig. S, ESI†). Obviously, the existence of a large amount of free FTEP solvents in 1 M KFSI–FTEP electrolyte further verifying the weakly solvating ability of FTEP with K+.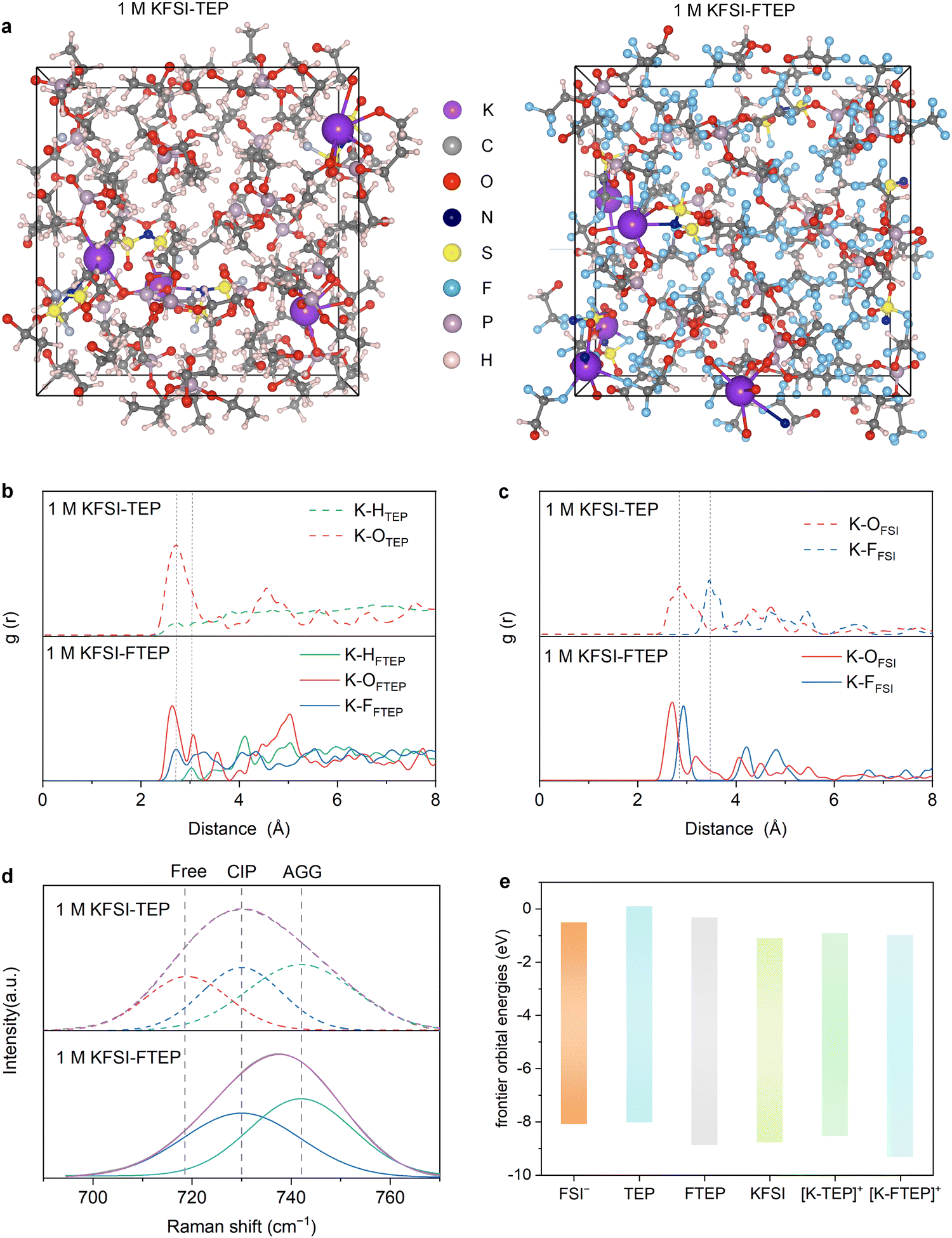 | ||
| Fig. 2 The solvation structures of 1 M KFSI in FTEP or TEP electrolytes. (a) Snapshots of the 1 M KFSI–TEP and 1 M KFSI–FTEP electrolytes obtained from DFT-MD simulations. The radial distribution functions (RDFs) of (b) H, O, and F from solvents and (c) O and F from FSI− anions around K+ in different electrolytes. (d) Raman spectra of the two electrolytes. (e) The energy levels of various molecules or ion clusters. | ||
Furthermore, the radial distribution functions (RDFs) of K–OFTEP exhibits two distinct peaks appear around 2.63 Å and 3.07 Å (Fig. 2b), while there is only one broad peak located at around 2.72 Å for K–OTEP, demonstrating the different solvating behaviors of TEP and FTEP. Meanwhile, a peak is present at around 2.72 Å in both RDFs, corresponding to the K–HTEP or K–FFTEP, indicating that the F atoms in FTEP have participated in the solvation sheath and are beneficial to obtaining F-rich interfaces. Besides, there are significant differences in the RDFs of K–OFSI and K–FFSI for the two electrolytes (Fig. 2c). Especially the RDFs peak of K–OFSI lies at around 2.85 Å in 1 M KFSI–TEP electrolyte, while it shifts to 2.70 Å in 1 M KFSI–FTEP electrolyte. The short distance of K–OFSI in RDFs indicating the stronger interactions between K+ and FSI− anions in 1 M KFSI–FTEP electrolyte. Simultaneously, the RDFs peak of K–FFSI in 1 M KFSI–TEP electrolyte appears at 3.45 Å, which is much higher than that in 1 M KFSI–FTEP electrolyte (2.93 Å). This result further illustrating that the FSI− anions prefer to coordinate with K+ in 1 M KFSI–FTEP electrolyte and the shorter K–F distance is also conducive to obtaining F-rich interfaces.
The unique solvation structures of the two electrolytes are further verified by the Raman spectroscopy (Fig. 2d). Clearly, compared to the 1 M KFSI–TEP electrolyte, an redshift of the S-N-S stretching mode of FSI− anions was observed for 1 M KFSI–FTEP electrolyte. This phenomenon indicates that the cation-anion interactions are significantly strengthened in 1 M KFSI–FTEP electrolyte, as evidenced by the increased proportion of contact ion pairs (CIP) and aggregate (AGG) clusters, consistent with the DFT-MD simulations. Moreover, the frontier molecular orbital energies (Fig. 2e and Fig. S5, ESI†) also reveal that the lowest unoccupied molecular orbital (LUMO) energy levels of FTEP is more negative than that of TEP, indicating the FTEP is preferentially reduced on the anode compared with TEP. Meanwhile, the highest occupied molecular orbital (HOMO) of FTEP is much lower than that of TEP and FSI− anions, demonstrating that the FTEP-based electrolytes may exhibit good oxidative stability.32 In addition, a similar trend in LUMO–HOMO energy levels is achieved for the KFSI, [K–TEP]+, and [K–FTEP]+ clusters. In short, these results have demonstrated that the cation-anion interactions are significantly strengthened in 1 M KFSI–FTEP electrolyte, which is conducive to obtaining anion-derived F-rich SEI and excellent electrochemical performance. At the same time, it should be pointed out that the weakly solvating ability of FTEP will cause the ion cluster and poor ion motions,26 resulting in low ionic conductivity (Table S1, ESI†). Nevertheless, the ionic conductivity of the 1 M KFSI–FTEP is still sufficient for the normal operation of potassium-based batteries.
Enhanced potassium metal and graphite stability
The electrochemical performance of the two typical electrolytes is first evaluated in potassium metal cells. When using 1 M KFSI–TEP electrolyte, the K‖Cu half-cell exhibits poor stability of 33 cycles with a low average CE of 24.1% (Fig. 3a). The inferior performance of K‖Cu cells in 1 M KFSI–TEP electrolyte is likely related to its electrolyte solvation structures, in which the resulting SEI cannot prevent the electrolyte decomposition effectively. Remarkably, when using 1 M KFSI–FTEP electrolyte, the K‖Cu half-cell delivers long stability of 500 cycles with an average CE of 96.5% (Fig. 3a). It should be pointed that the running time of this K‖Cu cell has reached up to over 200 days. Such a long operation time of the K‖Cu cell has rarely been reported in previously literatures30,33–36 (Fig. 3b). The charge–discharge profiles of K‖Cu cell also reveal small polarization voltage and high reversibility when using 1 M KFSI–FTEP electrolyte (Fig. S6, ESI†), such an excellent electrochemical performance is closely associated with the weakly solvation ability of FTEP. Meanwhile, even at a higher areal capacity (2 mA h cm−2) and current density (0.5 mA cm−2), the K‖Cu cell using 1 M KFSI–FTEP electrolyte also exhibits superior stability and CE compared to that using 1 M KFSI–TEP electrolyte (Fig. S7a, ESI†). In addition, the modified Aurbach efficiency37 of K‖Cu cell with the 1 M KFSI–FTEP electrolyte could reach up to 97.6%, whereas failed with the 1 M KFSI–TEP electrolyte (Fig. S7b, ESI†), further highlighting the advantages of the weakly solvating electrolyte (1 M KFSI–FTEP). The overpotential of the two electrolytes in K‖K symmetric cells is further evaluated (Fig. 3c and Fig. S8, ESI†). Clearly, when using 1 M KFSI–FTEP electrolyte, the K‖K symmetric cell exhibits a smaller overpotential, consistent with the results of K‖Cu cells.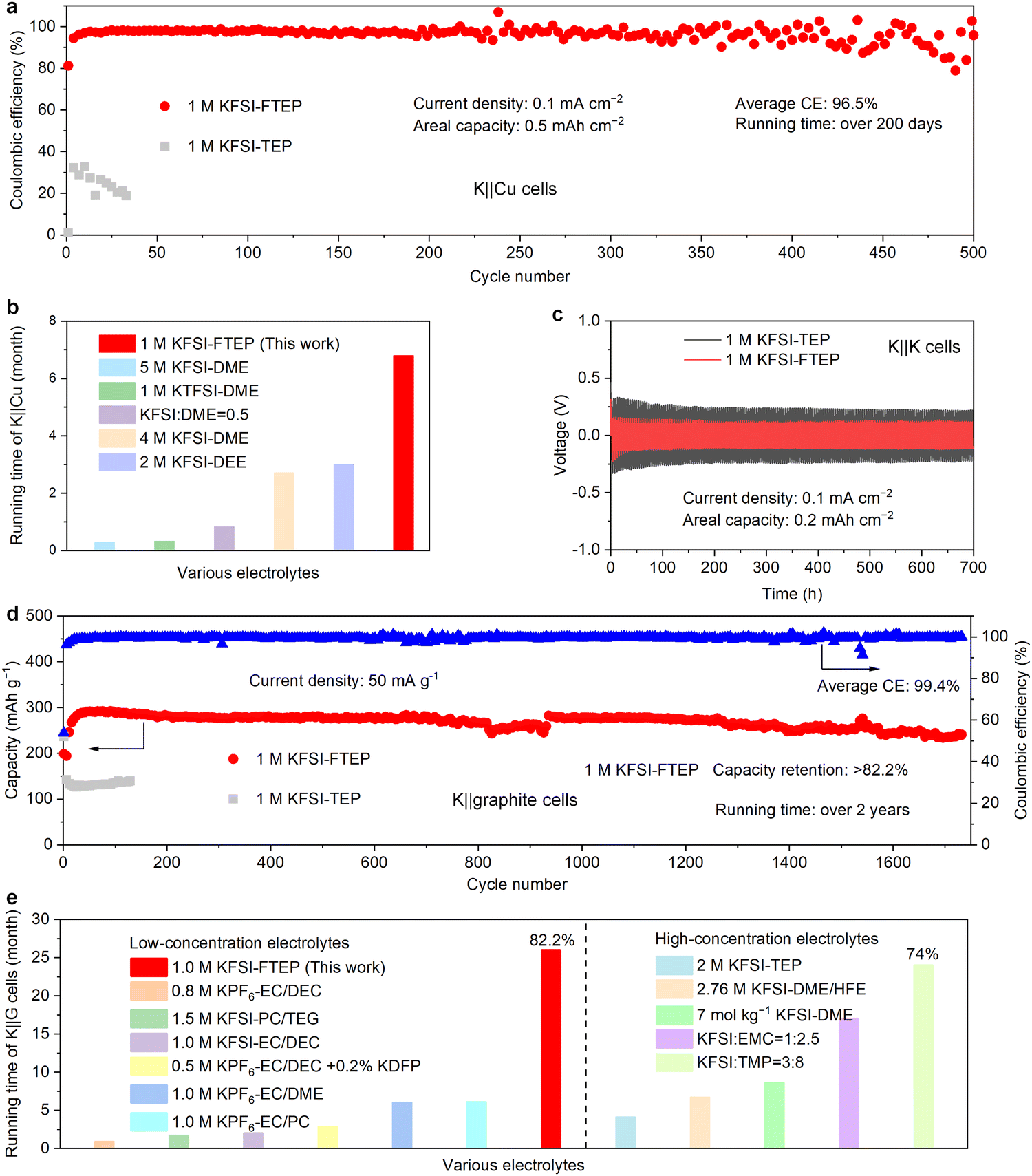 | ||
| Fig. 3 The electrochemical performance of potassium metal and graphite anode with the two electrolytes. (a) Coulombic efficiency and cycle stability of K‖Cu cells. (b) Comparison of K‖Cu cells running times with various electrolytes. (c) Cycling performance of K‖K symmetric cells. (d) Coulombic efficiency and cycle stability of the graphite anode. (e) Comparison of K‖graphite cells running times with various electrolytes. | ||
Considering the low voltage platform and the moderate capacity of graphite (Fig. S9, ESI†), high energy density full cells could be achieved with graphite as anode.7,10,38–47 Therefore, a long-term cycling stability of graphite electrodes is crucial for the practical applications of PIBs. The compatibility of graphite electrodes and the two electrolytes is further investigated (Fig. 3d). Apparently, the graphite anode with 1 M KFSI–TEP electrolyte exhibits inferior capacity. Noticeably, when using 1 M KFSI–FTEP electrolyte, the graphite anode reveals a long stability of over 1700 cycles with a capacity retention of 82.2% based on the highest capacity during cycling. Meanwhile, the graphite electrode with 1 M KFSI–FTEP electrolyte also exhibits overlapped charge–discharge profiles at different cycles, further verifying its good stability (Fig. S10a, ESI†). It is worth mentioning that the operation time of this K‖graphite cell has reached up to over 2 years (26 months), such a long-term stability has rarely been reported previously in low-concentrated electrolytes4,6,48–51 and even high-concentrated electrolytes13,14,17,28,52 (Fig. 3e and Table S2, ESI†). Moreover, when using 1 M KFSI–FTEP electrolyte, the graphite electrode also reveals higher capacity at various current densities (Fig. S10b, ESI†). The good electrochemical performance of graphite electrode in the 1 M KFSI–FTEP electrolyte is also verified by electrochemical impedance spectroscopy (EIS) characterization (Fig. S11, ESI†).
Morphology evolution and SEI characterizations
As discussed above, there are considerable electrochemical differences between the two electrolytes in K‖Cu and K‖graphite cells, which are strongly related to their morphology evolution and SEI compositions. Scanning electron microscopy (SEM) was used to investigate the surface morphology of Cu foil after K metal plating or stripping. Compared to the bare Cu foil (Fig. S12, ESI†), the Cu foil after K plating reveals a uniform and flat surface when using 1 M KFSI–FTEP electrolyte, indicating no dendrite formation (Fig. 4a and Fig. S13a, ESI†). However, when using 1 M KFSI–TEP electrolyte, numerous cracks and porous dendrites could be seen for the Cu foil after K plating (Fig. 4b and Fig. S13b, ESI†), which are detrimental to its electrochemical performance. Furthermore, significant morphological differences could also be observed on Cu foil after K stripping with these two electrolytes. With 1 M KFSI–FTEP as the electrolyte, the low-magnification SEM image of Cu foil reveals a relatively clean surface (Fig. S13c, ESI†), implying the near absence of “dead K”. Meanwhile, the high-resolution SEM image reveals a uniform and dense SEI formed on the Cu surface (Fig. 4c), which should be responsible for its excellent long-term stability and high CE. By contrast, with 1 M KFSI–TEP as the electrolyte, the SEM image of Cu foil shows a rough and porous structure (Fig. 4d and Fig. S13d, ESI†), indicating numerous “dead K” was formed, which could account for its poor cycle stability and low CE in K‖Cu cells.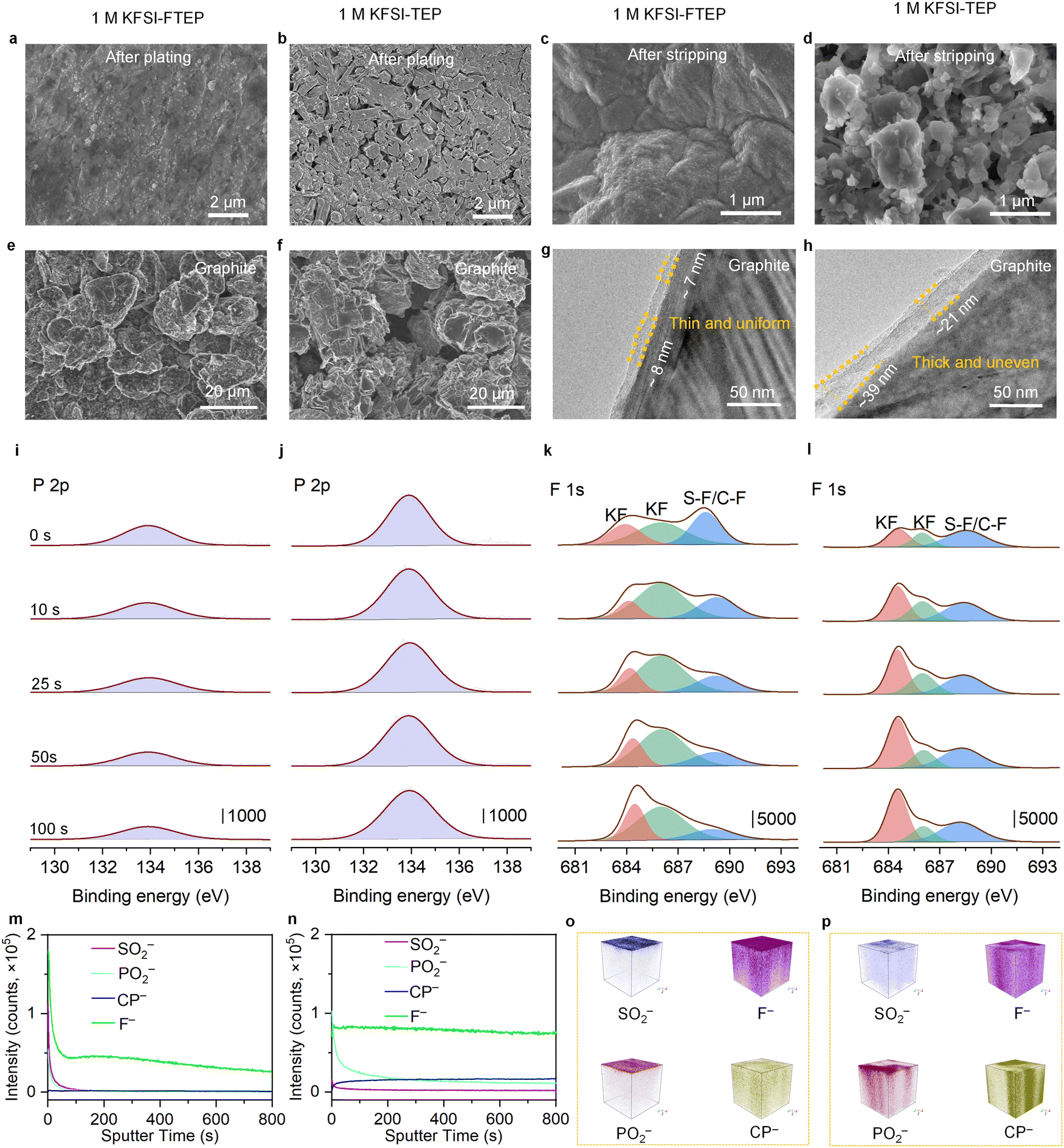 | ||
| Fig. 4 The morphology evolution and SEI characterizations of the anode with two electrolytes after 5 cycles. SEM images of Cu foil after (a and b) plating and (c and d) stripping. (e and f) SEM images and (g and h) TEM images of the cycled graphite electrode. (i and j) P2p and (k and l) F1s XPS depth profiling of the cycled graphite electrode. (m and n) TOF-SIMS depth profiling and (o and p) 3D render region of the selected typical second ion fragments on the cycled graphite electrode. (a, c, e, g, i, k, m and o) 1 M KFSI–FTEP electrolyte. (b, d, f, h, j, l, n and p) 1 M KFSI–TEP electrolyte. | ||
Moreover, the morphology evolution of the graphite electrode was further investigated by SEM. Clearly, compared to the pristine graphite electrodes (Fig. S14a, ESI†), the graphite electrodes with 1 M KFSI–FTEP electrolyte retain their original morphology after cycling (Fig. 4e and Fig. S14b, ESI†), which is also responsible for their superior cycle stability. However, when using 1 M KFSI–TEP electrolyte, the graphite electrodes undergo severely structure degeneration after cycling (Fig. 4f and Fig. S14c, ESI†), which can be used to explain their inferior electrochemical performance. The structures of SEI on graphite electrodes after cycles is further characterized by transmission electron microscopy (TEM). The TEM image of the graphite electrode with 1 M KFSI–FTEP electrolyte reveals a thin and uniform SEI (Fig. 4g). Meanwhile, the elemental mapping images of the graphite electrode show that the formed SEI is thin and contains F-rich compounds (Fig. S15a, ESI†), which is beneficial for achieving excellent cycle stability. However, with 1 M KFSI–TEP electrolyte, the graphite electrode exhibits a relatively thick and uneven SEI (Fig. 4h), which could also be verified by the elemental mapping images (Fig. S15b, ESI†). Besides, the percentage of C and P elements for the graphite electrode with 1 M KFSI–FTEP (vs. 1 M KFSI–TEP) electrolyte are 97.40% (vs. 62.76%) and 0.04% (vs. 5.94%), respectively (Fig. S15c, ESI†). These results confirmed that the graphite electrode with 1 M KFSI–TEP suffered severe electrolyte solvent decomposition, which should be responsible for its poor electrochemical performance.
The elemental compositions of the SEI on graphite electrodes are further investigated by X-ray photoelectron spectroscopy (XPS) and time of-flight secondary ion mass spectrometry (TOF-SIMS). The full survey XPS curves (Fig. S16, ESI†) reveal that the peak intensities of P2p and F1s are quite difference with different electrolytes. Specially, the main peak intensity of P2p XPS at different depth profiling with 1 M KFSI–FTEP electrolyte (Fig. 4i) is much lower than that with 1 M KFSI–TEP electrolyte (Fig. 4j), indicating serious solvent decomposition in 1 M KFSI–TEP electrolyte. A detailed depth profiling indicated that the atomic ratio of the P element on the surface of the graphite electrode is 1.65% with 1 M KFSI–FTEP electrolyte, which decreased slightly to 1.31% after etching time of 100 seconds (Fig. S17a, ESI†). While for the 1 M KFSI–TEP electrolyte, the atomic ratio of the P element reaches up to 5.39% after etching for 100 seconds, slightly higher than that on the electrode surface of 4.79% (Fig. S17a, ESI†). Compared to the 1 M KFSI–FTEP electrolyte, the percentage of P element in SEI is considerably high when using the 1 M KFSI–TEP electrolyte, indicating a severe decomposition of TEP solvent than that of FTEP solvent. Besides, SEI's P and S elements stem from the decomposition of solvent (TEP or FTEP) and anion (FSI−), respectively. Thus, the atomic ratio of S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P could be used to evaluate the solvent- or anion-dominated electrolyte decomposition (Fig. S17b, ESI†). For 1 M KFSI–FTEP electrolyte, the atomic ratio of S:P fluctuated slightly around 3.12 (±0.08) at different etching times, demonstrating the formation of an anion-dominated and homogeneous SEI at different depths. While for 1 M KFSI–TEP electrolyte, the atomic ratio of S:P decreased to around 1.20 (±0.20), indicating the solvent decomposition is more severe in this electrolyte. These results could be further confirmed by the atomic contents of P and S elements obtained from the SEM energy spectrum of Cu (Table S3, ESI†) and graphite (Table S4, ESI†) electrodes in different electrolytes.
:P could be used to evaluate the solvent- or anion-dominated electrolyte decomposition (Fig. S17b, ESI†). For 1 M KFSI–FTEP electrolyte, the atomic ratio of S:P fluctuated slightly around 3.12 (±0.08) at different etching times, demonstrating the formation of an anion-dominated and homogeneous SEI at different depths. While for 1 M KFSI–TEP electrolyte, the atomic ratio of S:P decreased to around 1.20 (±0.20), indicating the solvent decomposition is more severe in this electrolyte. These results could be further confirmed by the atomic contents of P and S elements obtained from the SEM energy spectrum of Cu (Table S3, ESI†) and graphite (Table S4, ESI†) electrodes in different electrolytes.
In addition, the F1s XPS depth profiling with two different electrolytes reveal three main peaks, representing the KF and the bond of S–F/C–F, respectively (Fig. 4k and l). Particularly, the atomic ratio of the F element reaches up to 12.38% on the graphite surface with 1 M KFSI–FTEP electrolyte, much higher than that of 8.23% with 1 M KFSI–TEP electrolyte. This indicates that the SEI surface contains more F-rich compounds with 1 M KFSI–FTEP electrolyte (Fig. S17a, ESI†), which is conducive to achieving superior electrochemical performance. Moreover, the XPS depth profiling of graphite electrodes after 50 cycles also exhibit similar results (Fig. S18, ESI†). Specially, the atomic ratios of the F element on the SEI surface are 10.46% and 7.11%, while the atomic ratios of S:P are around 2.67 and 0.97 for 1 M KFSI–FTEP and 1 M KFSI–TEP electrolytes, respectively. These results indicate that the 50th cycle SEI is nearly the same as the 5th cycle SEI, further verifying the anion-dominated and solvent-dominated SEI formation for 1 M KFSI–FTEP and 1 M KFSI–TEP electrolyte, respectively.
At the same time, TOF-SIMS depth profiling (Fig. 4m and n) and 3D render region (Fig. 4o and p) are further used to uncover the compositions and structures of SEI on the cycled graphite electrode. Typically, the ion fragment of SO2− and F− is mainly derived from the decomposition of FSI− anion, while the PO2− and CP− are stem from the decomposition of TEP or FTEP solvent. Obviously, with 1 M KFSI–FTEP electrolyte (Fig. 4m), the signals of SO2−, PO2− and F− are sharply decreased during the initial 50 seconds, and the signals of F− and SO2− are much higher than the PO2− and CP−, verifying the formation of anion-derived SEI. By contrast, with 1 M KFSI–TEP electrolyte (Fig. 4n), the PO2− signal is continuous decreased within 200 seconds, while the CP− and F− signals are slightly increased during the initial sputtering time. These phenomena demonstrated that the decomposition of TEP solvent is more severe, resulting in the formation of a thick and solvent-derived SEI, agree well with the TEM and XPS results. Meanwhile, compared to the 1 M KFSI–TEP electrolyte, the 3D renders of SO2−, PO2−, and F− are mainly homogeneously distributed on the surface of graphite electrode in 1 M KFSI–FTEP electrolyte (Fig. 4o and p). Such differences in the SEI compositions and structures should be responsible for the disparities in the electrochemical properties of these two electrolytes.
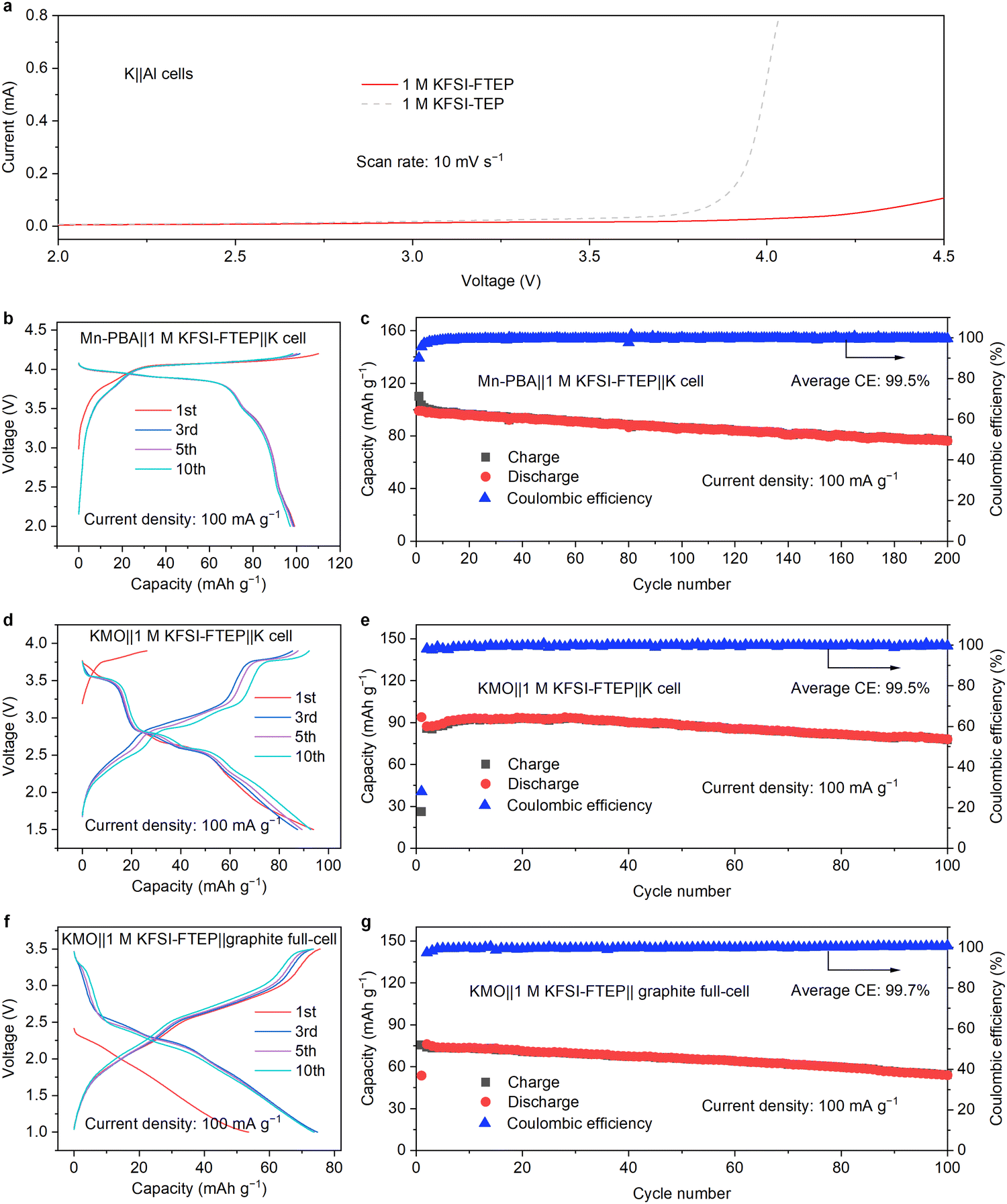 | ||
| Fig. 5 The high voltage properties and full-cell performance. (a) Linear sweep voltammetry (LSV) profiles of the two electrolytes. (b) Charge–discharge curves and (c) cycling performance of Mn-PBA cathode. (d) Charge–discharge curves and (e) cycling performance of KMO cathode. (f) Charge–discharge curves and (g) cycling performance of KMO‖graphite full cell. (c–g) 1 M KFSI–FTEP electrolyte. | ||
Considering the high oxidation stability of 1 M KFSI–FTEP electrolyte, two cathode materials of Mn-based Prussian blue analogue (Mn-PBA) and Mn-based oxides (KMO) are used to evaluate the corresponding electrochemical performances. According to the charge–discharge profiles (Fig. 5b), the Mn-PBA with 1 M KFSI–FTEP exhibits good CE and reversible capacity of 99 mA h g−1, whereas it failed with 1 M KFSI–TEP electrolyte due to the electrolyte decomposition and Al corrosion (Fig. S19, ESI†). Moreover, the Mn-PBA with 1 M KFSI–FTEP exhibits good stability of 200 cycles with a capacity retention of 77.3% and a high average CE of 99.5% (Fig. 5c). Furthermore, the 1 M KFSI–FTEP electrolyte also enables the KMO cathode to operate steadily for 100 cycles with a capacity retention of 83.2% (Fig. 5d and e). The full cell with graphite anode and KMO cathode also delivers stable 100 cycles with capacity retention of 75.2% and a high CE of 99.7% (Fig. 5f and g).
Discussion
We developed a low-concentration, non-flammable, and weakly solvating electrolyte (1 M KFSI–FTEP), which significantly strengthens the cycling stability and security of PIBs. The intrinsically non-flammable nature of FTEP significantly improves the security of the battery. The weakly solvating feature of FTEP strengthens the K+-anion interactions, which is conducive to forming an anion-derived and F-rich SEI on the anode and further reducing the decomposition of the electrolyte. Consequently, this electrolyte enables the durable cycling of K‖graphite (running time for over 2 years) and K‖Cu cells (running time for over 200 days). Furthermore, this electrolyte also enables high voltage stability up to 4.2 V. As a result, with this electrolyte, the K‖Mn-PBA cell delivers a high average Coulombic efficiency of 99.5% for 200 cycles. The KMO‖graphite full cell exhibits 100 cycles with good capacity retention of 75.2% and high average Coulombic efficiency of 99.7%. This study provides a guiding principle in tailoring electrolytes for achieving safe and long-lasting potassium ion batteries.Methods
Materials
Potassium bis(fluorosulfonyl)imide (KFSI), potassium metal, triethyl phosphate (TEP), and tris(2,2,2-trifluoroethyl)phosphate (FTEP) were purchased from Sigma-Aldrich. The TEP and FTEP are pretreated with molecular sieves for 2 days before use. Other materials and chemicals were purchased commercially and used as received.Preparation of cathode materials
The Mn-based Prussian blue analogue (Mn-PBA) was prepared as follows. First, K4Fe(CN)6·3H2O (4 mmol) was dissolved into 100 ml H2O, denoted as solution A. Next, MnSO4 (2.8 mmol) and FeSO4 (1.2 mmol) were dissolved into 100 ml H2O, denoted as solution B. Then solution B was added to solution A under stirring. Then the mixed solution was transferred to Teflon-lined autoclave and maintained at 80 °C for 12 h. Then the Mn-PBA was washed with water and ethanol, then dried at 80 °C for 24 hours under a vacuum. The K0.5MnO2 (KMO) was prepared by annealing the mixtures of Mn2O3 (4 mmol) and K2CO3 (2 mmol) at 800 °C for 12 hours with a heat rate of 2 °C min−1.Preparation of electrolytes and electrodes
The electrolytes were prepared by dissolving the KFSI in corresponding solvents. For instance, the 1 M KFSI–FTEP electrolyte was prepared by dissolving 10 mmol KFSI salt into 10 ml FTEP solvent, then stirred for one hour. All electrolytes were prepared in an Ar-filled glove box. The graphite electrode was prepared by mixing the graphite: Super P:CMC at a ratio of 8:1:1 in H2O:C2H5OH solutions to form a uniform slurry, which was coated onto Cu foil and dried at 80 °C for 24 hours under vacuum. The Mn-PBA or KMO electrodes were prepared by mixing the corresponding cathode materials:ketjen black:CPVDF at a ratio of 7:2:1 in NMP solutions to form a uniform slurry, which was coated onto Al foil and dried at 80 °C for 24 hours under vacuum. The average mass loadings of graphite, Mn-PBA, and KMO are around 1.2, 0.6, and 0.6 mg cm−2, respectively.
Materials characterizations
Transmission electron microscope (Titan G2 60-300) and scanning electron microscope (Jeol, JSM-7610FPlus) were used to analyze the structure and morphology. X-ray photoelectron spectroscopy (ESCALAB 250xi) and time-of-Flight secondary ion mass spectrometry were used to investigate the chemical compositions of SEI. Raman (Renishaw 2000) was used to examine the solvation structure of electrolytes. The flammability and self-extinguishing time of the various electrolytes were measured by burning Whatman glass fibre (soaked by 150 μl different electrolytes).Electrochemical testing
The CR2032 coin cells were prepared by using graphite, Mn-PBA, or KMO as the working electrode, glass fiber (Whatman) as the separator, and potassium foil as the counter electrode. The electrolyte dosage for each cell is 100 μl. Appropriate charge–discharge voltage ranges were used for K‖graphite (0.01–3.0 V), K‖Mn-PBA (2.0–4.2 V), and K‖KMO (1.5–3.9 V) cells. The K‖Cu cells were tested by a repeated step of plating K (0.5 or 2.0 mA h cm−2) on Cu foil, followed by a stripping process (charge to 1.0 V) under a current density of 0.1 or 0.5 mA cm−2. The Aurbach efficiency test was performed as follows, (1) plating 2.5 mA h cm−2 K on Cu foil, followed by charging to 1.0 V; (2) plating 2.5 mA h cm−2 K on Cu foil as a K reservoir; (3) repeatedly stripping/plating of K (0.5 mA h cm−2) for ten cycles; (4) charging to 1.0 V under a current density of 0.25 mA cm−2. The K‖K symmetric cells were cycled with an areal capacity of 0.5 mA h cm−2 under a current density of 0.1 mA cm−2. The electrochemical stabilities of the electrolytes in K‖Al cells were conducted by the linear sweep voltammetry method at a scan rate of 10 mV s−1. For the preparation of KMO‖graphite full cells, the mass loadings of active material in graphite anode and KMO cathode are around 1.1 and 0.63 mg cm−2, respectively. And the capacity in full cell was calculated based on the cathode material. Before the preparation of full cell, the graphite and KMO electrodes were pre-processed as follows: the graphite‖K and KMO‖K half-cells were cycled for 5 cycles, followed by a fully discharge process (0.01 V) for graphite electrode and a fully charge process (3.9 V) for KMO cathode. Then the graphite‖K and KMO‖K half-cells were disassembled and the obtained graphite and KMO electrodes were further used to the full-cell assemble.Theoretical calculations
The density functional theory (DFT) calculations were performed by the Vienna Ab initio Simulation Package (VASP) using the generalized gradient approximation (GGA) of PerdewBurke–Ernzerhof (PBE) functional.53 The energy cutoff of the plane wave was set to 520 eV with an energy precision of 10−5 eV. The atomic positions were fully relaxed until the maximum force on each atom was less than 10−3 eV per Å. The Brillouin zone was sampled with a 1 × 1 × 1 Monkhorst–Pack k-mesh grid for geometry optimizations. According to the experimental results, the 1 M KFSI–TEP electrolyte was simulated with 4 KSFI and 23 TEP molecules in a supercell with 18.33039 × 18.68141 × 18.78886 Å3 dimensions. The 1 M KFSI–FTEP electrolyte was simulated with 5 KSFI and 22 FTEP molecules in a supercell with 20.55906 × 20.181 × 20.51954 Å3 dimensions. Periodic boundary conditions were used in all directions. Gaussian smearing with a width of 0.01 eV was used to aid the convergence of the DFT calculations. The DFT-D3 method of the Grimme van der Waals correction54 was used to account for the long-range interactions. Ab initio molecular dynamics (AIMD) simulations were performed with the NVT ensemble by using the Nosé–Hoover method.55 All electrolyte systems were simulated with a 5 ps pre-equilibrated process followed by a 10 ps production process with a time step of 0.1 fs.Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the ESI.†Author contributions
L. Fan and B. Lu conceived and supervised the project. L. Fan and H. Xie performed the experimental measurements. H. Xie and Y. Hu performed the characterizations. Z. Caixiang synthesized the KMO cathodes. The manuscript draft was written and revised by L. Fan, A. M. Rao, and B. Lu. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
L. Fan acknowledges the National Natural Science Foundation of China (No. 22005093), the Natural Science Foundation of Hunan Province (2022JJ20011), and the Fundamental Research Funds for the Central Universities (No. 531119200156). B. Lu acknowledges the National Natural Science Foundation of China (No. U20A20247, and 51922038). A. M. Rao acknowledges the seed funding provided by the R. A. Bowen Professorship funds at Clemson University.References
- X. Y. Wu, D. P. Leonard and X. L. Ji, Chem. Mater., 2017, 29, 5031–5042 CrossRef
.
- W. Zhang, Y. Liu and Z. Guo, Sci. Adv., 2019, 5, eaav7412 CrossRef PubMed
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120, 6358–6466 CrossRef
- Z. Jian, W. Luo and X. Ji, J. Am. Chem. Soc., 2015, 137, 11566–11569 CrossRef
- S. Komaba, T. Hasegawa, M. Dahbi and K. Kubota, Electrochem. Commun., 2015, 60, 172–175 CrossRef
- J. Zhao, X. X. Zou, Y. J. Zhu, Y. H. Xu and C. S. Wang, Adv. Funct. Mater., 2016, 26, 8103–8110 CrossRef
- L. Fan, Y. Hu, A. M. Rao, J. Zhou, Z. Hou, C. Wang and B. Lu, Small Methods, 2021, 5, e2101131 CrossRef PubMed
- L. Deng, J. Qu, X. Niu, J. Liu, J. Zhang, Y. Hong, M. Feng, J. Wang, M. Hu, L. Zeng, Q. Zhang, L. Guo and Y. Zhu, Nat. Commun., 2021, 12, 2167 CrossRef CAS PubMed
- T. Masese, K. Yoshii, Y. Yamaguchi, T. Okumura, Z. D. Huang, M. Kato, K. Kubota, J. Furutani, Y. Orikasa, H. Senoh, H. Sakaebe and M. Shikano, Nat. Commun., 2018, 9, 3823 CrossRef
- L. Fan, R. Ma, J. Wang, H. Yang and B. Lu, Adv. Mater., 2018, 30, e1805486 CrossRef PubMed
- L. Xue, Y. Li, H. Gao, W. Zhou, X. Lu, W. Kaveevivitchai, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2017, 139, 2164–2167 CrossRef CAS
- J. Ding, H. Zhang, H. Zhou, J. Feng, X. Zheng, C. Zhong, E. Paek, W. Hu and D. Mitlin, Adv. Mater., 2019, 31, e1900429 CrossRef PubMed
- L. Fan, R. Ma, Q. Zhang, X. Jia and B. Lu, Angew. Chem., Int. Ed., 2019, 58, 10500–10505 CrossRef CAS PubMed
- S. Liu, J. Mao, L. Zhang, W. K. Pang, A. Du and Z. Guo, Adv. Mater., 2021, 33, e2006313 CrossRef PubMed
- Y. Hu, L. Fan, A. M. Rao, W. Yu, C. Zhuoma, Y. Feng, Z. Qin, J. Zhou and B. Lu, Natl. Sci. Rev., 2022, 9, nwac134 CrossRef PubMed
- M. Zhou, P. Bai, X. Ji, J. Yang, C. Wang and Y. Xu, Adv. Mater., 2021, 33, e2003741 CrossRef
- S. Liu, J. Mao, Q. Zhang, Z. Wang, W. K. Pang, L. Zhang, A. Du, V. Sencadas, W. Zhang and Z. Guo, Angew. Chem., Int. Ed., 2020, 59, 3638–3644 CrossRef CAS
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef PubMed
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef
- D. A. Rakov, F. Chen, S. A. Ferdousi, H. Li, T. Pathirana, A. N. Simonov, P. C. Howlett, R. Atkin and M. Forsyth, Nat. Mater., 2020, 19, 1096–1101 CrossRef PubMed
- N. Dubouis, T. Marchandier, G. Rousse, F. Marchini, F. Fauth, M. Avdeev, A. Iadecola, B. Porcheron, M. Deschamps, J. M. Tarascon and A. Grimaud, Nat. Mater., 2021, 20, 1545–1550 CrossRef
- T. D. Pham and K.-K. Lee, Small, 2021, 17, 2100133 CrossRef
- Y. Zhao, T. Zhou, T. Ashirov, M. E. Kazzi, C. Cancellieri, L. P. H. Jeurgens, J. W. Choi and A. Coskun, Nat. Commun., 2022, 13, 2575 CrossRef PubMed
- Q. F. Zheng, Y. Yamada, R. Shang, S. Ko, Y. Y. Lee, K. Kim, E. Nakamura and A. Yamada, Nat. Energy, 2020, 5, 291–298 CrossRef
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef
- Z. Yu, P. E. Rudnicki, Z. W. Zhang, Z. J. Huang, H. Celik, S. T. Oyakhire, Y. L. Chen, X. Kong, S. C. Kim, X. Xiao, H. S. Wang, Y. Zheng, G. A. Kamat, M. S. Kim, S. F. Bent, J. Qin, Y. Cui and Z. N. Bao, Nat. Energy, 2022, 7, 94–106 CrossRef
- M. Li, C. Wang, Z. Chen, K. Xu and J. Lu, Chem. Rev., 2020, 120, 6783–6819 CrossRef
- L. Qin, N. Xiao, J. F. Zheng, Y. Lei, D. Y. Zhai and Y. Y. Wu, Adv. Energy Mater., 2019, 9, 1902618 CrossRef
- J. Wang, Y. Yamada, K. Sodeyama, E. Watanabe, K. Takada, Y. Tateyama and A. Yamada, Nat. Energy, 2017, 3, 22–29 CrossRef
- J. Li, Y. Hu, H. Xie, J. Peng, L. Fan, J. Zhou and B. Lu, Angew. Chem., Int. Ed., 2022, 61, e202208291 Search PubMed
- Z. Yu, H. S. Wang, X. Kong, W. Huang, Y. C. Tsao, D. G. Mackanic, K. C. Wang, X. C. Wang, W. X. Huang, S. Choudhury, Y. Zheng, C. V. Amanchukwu, S. T. Hung, Y. T. Ma, E. G. Lomeli, J. Qin, Y. Cui and Z. N. Bao, Nat. Energy, 2020, 5, 526–533 CrossRef
- Y. Q. Li, Q. Zhou, S. T. Weng, F. X. Ding, X. G. Qi, J. Z. Lu, Y. Li, X. Zhang, X. H. Rong, Y. X. Lu, X. F. Wang, R. J. Xiao, H. Li, X. J. Huang, L. Q. Chen and Y. S. Hu, Nat. Energy, 2022, 7, 511–519 CrossRef
- P. Liu, Y. Wang, H. Hao, S. Basu, X. Feng, Y. Xu, J. A. Boscoboinik, J. Nanda, J. Watt and D. Mitlin, Adv. Mater., 2020, 32, e2002908 CrossRef
- N. Xiao, W. D. McCulloch and Y. Wu, J. Am. Chem. Soc., 2017, 139, 9475–9478 CrossRef PubMed
- N. Xiao, G. Gourdin and Y. Wu, Angew. Chem., Int. Ed., 2018, 57, 10864–10867 CrossRef PubMed
- J. Zhang, Z. Cao, L. Zhou, G. T. Park, L. Cavallo, L. M. Wang, H. N. Alshareef, Y. K. Sun and J. Ming, ACS Energy Lett., 2020, 5, 3124–3131 CrossRef CAS
- B. D. Adams, J. Zheng, X. Ren, W. Xu and J. G. Zhang, Adv. Energy Mater., 2017, 8, 1702097 CrossRef
- Z. L. Jian, S. Hwang, Z. F. Li, A. S. Hernandez, X. F. Wang, Z. Y. Xing, D. Su and X. L. Ji, Adv. Funct. Mater., 2017, 27, 1700324 CrossRef
- K. X. Lei, F. J. Li, C. N. Mu, J. B. Wang, Q. Zhao, C. C. Chen and J. Chen, Energy Environ. Sci., 2017, 10, 552–557 RSC
- M. Chen, W. Wang, X. Liang, S. Gong, J. Liu, Q. Wang, S. J. Guo and H. Yang, Adv. Energy Mater., 2018, 8, 1800171 CrossRef
- L. Fan, S. Chen, R. Ma, J. Wang, L. Wang, Q. Zhang, E. Zhang, Z. Liu and B. Lu, Small, 2018, 14, e1801806 CrossRef PubMed
- K. Lei, C. Wang, L. Liu, Y. Luo, C. Mu, F. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 4687–4691 CrossRef PubMed
- Y. Xu, C. Zhang, M. Zhou, Q. Fu, C. Zhao, M. Wu and Y. Lei, Nat. Commun., 2018, 9, 1720 CrossRef PubMed
- W. C. Zhang, W. K. Pang, V. Sencadas and Z. P. Guo, Joule, 2018, 2, 1534–1547 CrossRef
- J. Y. Liao, Q. Hu, B. Che, X. Ding, F. Chen and C. H. Chen, J. Mater. Chem. A, 2019, 7, 15244–15251 RSC
- L. Q. Deng, T. S. Wang, Y. R. Hong, M. Y. Feng, R. T. Wang, J. Zhang, A. F. Zhang, J. W. Wang, L. Zeng, Y. J. Zhu and L. Guo, ACS Energy Lett., 2020, 5, 1916–1922 CrossRef
- Y. Liu, Y. X. Lu, Y. S. Xu, Q. S. Meng, J. C. Gao, Y. G. Sun, Y. S. Hu, B. B. Chang, C. T. Liu and A. M. Cao, Adv. Mater., 2020, 32, e2000505 CrossRef PubMed
- H. Yang, C. Y. Chen, J. Hwang, K. Kubota, K. Matsumoto and R. Hagiwara, ACS Appl. Mater. Interfaces, 2020, 12, 36168–36176 CrossRef PubMed
- M. Gu, L. Fan, J. Zhou, A. M. Rao and B. Lu, ACS Nano, 2021, 15, 9167–9175 CrossRef PubMed
- M. Shimizu, T. Koya, A. Nakahigashi, N. Urakami, T. Yamakami and S. Arai, J. Phys. Chem. C, 2020, 124, 13008–13016 CrossRef CAS
- S. Q. Liu, L. C. Meyer, L. Medenbach and A. Balducci, Energy Storage Mater., 2022, 47, 534–541 CrossRef
- T. Hosaka, K. Kubota, H. Kojima and S. Komaba, Chem. Commun., 2018, 54, 8387–8390 RSC
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee03294e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |