
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A hydrated deep eutectic electrolyte with finely-tuned solvation chemistry for high-performance zinc-ion batteries†
Ruwei
Chen‡
ab,
Chengyi
Zhang‡
c,
Jianwei
Li‡
a,
Zijuan
Du
a,
Fei
Guo
a,
Wei
Zhang
 a,
Yuhang
Dai
a,
Wei
Zong
a,
Xuan
Gao
a,
Jiexin
Zhu
a,
Yan
Zhao
*c,
Xiaohui
Wang
*b and
Guanjie
He
*a
a,
Yuhang
Dai
a,
Wei
Zong
a,
Xuan
Gao
a,
Jiexin
Zhu
a,
Yan
Zhao
*c,
Xiaohui
Wang
*b and
Guanjie
He
*a
aElectrochemical Innovation Lab, Department of Chemical Engineering, University College London, London WC1E 7JE, UK. E-mail: g.he@ucl.ac.uk
bState Key Laboratory of Pulp and Paper Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: fewangxh@scut.edu.cn
cInstitute of Technological Sciences, Wuhan University, Hubei, Wuhan 430072, China. E-mail: yan2000@whut.edu.cn
First published on 14th April 2023
Abstract
Despite their cost-effectiveness and intrinsic safety, aqueous zinc-ion batteries have faced challenges with poor reversibility originating from various active water-induced side reactions. After systematically scrutinizing the effects of water on the evolution of solvation structures, electrolyte properties, and electrochemical performances through experimental and theoretical approaches, a hydrated deep eutectic electrolyte with a water-deficient solvation structure ([Zn(H2O)2(eg)2(otf)2]) and reduced free water content in the bulk solution is proposed in this work. This electrolyte can dramatically suppress water-induced side reactions and provide high Zn2+ mass transfer kinetics, resulting in highly reversible Zn anodes (∼99.6% Coulombic efficiency over 1000 cycles and stable cycling over 4500 h) and high capacity Zn//NVO full cells (436 mA h g−1). This work will aid the understanding of electrolyte solvation structure–electrolyte property–electrochemical performance relationships of aqueous electrolytes in aqueous zinc-ion batteries.
Broader contextDeveloping a better understanding of the double-edged sword role of water, establishing solvation chemistry–electrolyte property–electrochemical performance relationships, and maintaining the dynamic balance between the advantages and disadvantages of water are particularly important for the industrial application of aqueous zinc-ion batteries. In this work, a new type of hydrated deep eutectic electrolyte composed of hydrogen bond donors and hydrogen bond acceptors was developed for aqueous zinc-ion batteries. After systematically scrutinizing the evolution of solvation chemistry, and studying the effects of water on electrolyte properties and electrochemical performance through experimental and theoretical approaches, a hydrated deep eutectic electrolyte with a water-deficient solvation structure ([Zn(H2O)x(eg)y(otf)z]2−z) and reduced free water in the bulk solution was developed. This electrolyte can achieve a balance between improved reversibility and satisfactory Zn2+ kinetics, resulting in highly reversible Zn anodes and high capacity cathodes. The clarification of these scientific questions in this work will aid the understanding, development, and application of aqueous electrolytes in aqueous zinc-ion batteries. |
Introduction
Lithium-ion batteries have dominated the electrochemical energy storage field in the past four decades. However, the safety issues arising from flammable organic electrolytes and the high cost due to the shortage of lithium resources hinder their grid-scale applications.1–3 Aqueous zinc-ion batteries are regarded as a promising alternative battery technology for grid-scale energy storage applications in the post-lithium era arising from their low cost, inherent safety, and high volumetric energy density of zinc anodes (820 mA h g−1 and 5855 mA h cm−3).4–6 Unfortunately, the implementation of this technology is still restricted by poor reversibility in terms of both anodes and cathodes.In common aqueous electrolytes (AEs), Zn2+ ions are generally solvated by water in the form of [Zn(H2O)6]2+ with free anions and unsolvated water molecules in the bulk electrolytes.7,8 The solvation effect impels electron transfer via Zn–OH2 coordination, strikingly weakens the O–H bonds and accelerates the decomposition of solvated water molecules during the zinc deposition process, which subsequently leads to the notorious hydrogen evolution reaction, surface passivation and dendrite growth at the electrolyte/anode interface.9–12 Moreover, free water molecules with strong polarity can induce irreversible collapse of lattice structures, thereby leading to severe dissolution of active materials at the electrolyte/cathode interface during the discharge process.13–15 Therefore, strategies to solve these bottlenecks are of great research significance and application value.
Electrolyte solvation chemistry, that is, the interactions among cations, anions, and solvent molecules, is regarded as the root cause of these uncontrollable parasitic reactions and plays an essential role in governing ion migration/desolvation processes, electrode/electrolyte interfacial stabilities, and other electrolyte properties.16,17 In this context, the emerging concentrated electrolytes offer great opportunities to re-adjust the coordination environment and the solvation sheath of Zn2+ ions. As the concentration of zinc salts gradually increases, the number of water molecules coordinating with Zn2+ ions decreases dramatically, and the corresponding anion gradually participates in the solvation sheath of Zn2+ ions.18,19 The manipulated solvation sheath with limited active water molecules can suppress the water activity, thus providing a high possibility for reversible Zn plating/stripping and Zn2+ insertion/extraction.20 Nevertheless, most concentrated electrolytes are costly, compromising the economic benefits anticipated for aqueous zinc-ion batteries.21 Hence, the development of cost-effective electrolytes with manipulated solvation chemistry is critical to boost the reversibility of aqueous zinc-ion batteries toward large-scale applications.
Deep eutectic solvents (DESs), mainly composed of hydrogen bond donors and hydrogen bond acceptors, represent a set of intrinsic “designer solvents” because they offer high tunability in terms of their compositions and molecular chemical moieties.22,23 DESs have been attracting increasing research interest in the field of energy storage applications due to their outstanding electrochemical stability, easy preparation, and low cost.24 Different from AEs ([Zn(H2O)6]2+), Zn2+ ions are solvated by hydrogen bond donors, thereby effectively eliminating active water-induced side reactions.25,26 However, DESs generally exhibit high viscosity and low ionic conductivity, stemming from the insufficient solvation and ion clustering, resulting in inferior Zn2+ kinetics.27 The inferior Zn2+ kinetics not only leads to sluggish Zn plating/stripping on the anode, but also severely limits the performance of cathode materials. Very recently, Cui et al. have demonstrated that introducing hydrated salts can improve the ionic conductivity and lower the viscosity of DESs to some extent.28 In fact, the internal coordination and H-bonding species of DESs render them strongly water-miscible, and water molecules can be easily integrated into the eutectic network, thereby changing the solvation chemistry of DESs. However, water is a double-edged sword, and the precise control of the dynamic balance between the merits and demerits of water, the evolution of DES solvation chemistry, and its effect on the electrode/electrolyte interfacial stabilities are rarely studied and unclear. The clarification of these scientific questions will aid the understanding, development, and application of DESs in aqueous zinc-ion batteries.
Here, a new type of hydrated DES electrolyte (HDES) is developed based on ethylene glycol (eg), zinc trifluorosulfonate (Znotf), and H2O for aqueous zinc-ion batteries (Fig. S1, ESI†). Ethylene glycol was chosen as a hydrogen bond donor due to its low cost and liquid state, which is beneficial for obtaining low-cost DESs with relatively low viscosity. Importantly, the evolution of the solvation structure and the effect of water content on DESs’ properties and electrochemical performances were systematically investigated through experimental and theoretical approaches. As the water content increases, water molecules gradually replace eg molecules and anions to participate in the solvation sheath of Zn2+ ions, resulting in an improved ion dissociation degree, ionic conductivity, and Zn2+ kinetics. When the water content is increased further, the DES structure is disrupted. Instead, H2O–H2O and H2O–Zn2+ interactions dominate the solution structure, indicating the transition from DES to aqueous solution properties with dramatically increased water-induced side reactions at electrolyte/electrode interfaces. Therefore, the HDES with precisely controlled water content is an intermediate between a DES and an AE, and by forming a water-deficient solvation structure ([Zn(H2O)x(eg)y(otf)z]2−z) it can achieve a balance between improved reversibility and satisfactory Zn2+ kinetics. As a result, the HDES can realize stable Zn plating/stripping for 1000 cycles with an average Coulombic efficiency of 99.6% and sustain long-term cycling over 4500 h. The Zn//NH4V4O10 (NVO) full cell with HDES exhibits a high capacity of 436 mA h g−1 and excellent rate capability. Meanwhile, all water molecules are bound with the DES network, thereby significantly reducing water activity and ensuring the reversible reactions of NVO cathodes with suppressed dissolution. The Zn-ion batteries with HDES show obviously improved capacity retention compared to those with an AE.
Results and discussion
To understand the solvation structure–electrolyte property–electrochemical performance relationships, theoretical calculations and experimental studies were correlated. We first used DFT to determine the optimized binding configurations of different electrolyte systems (Fig. S2, ESI†). The AE showed a 6 water molecule coordination geometry with the weakest binding strength (−2.8 eV, [Zn(H2O)6]2+), which is consistent with previous reports (Fig. 1a and Fig. S2, ESI†).29 The solvation structure of DES involves both eg molecules and anions (Fig. 1f). This anion-involved solvation structure results from the weaker solvation capacity of eg compared with H2O, leading to low ionic conductivity and low binding energy (−10.0 eV, Fig. S3, ESI†). With the increase of water content, the number of active water molecules gradually increased and anions gradually decreased in the solvation sheath while the binding energy approached that of AE, indicating a gradual transition from the DES to the AE (Fig. 1b–e).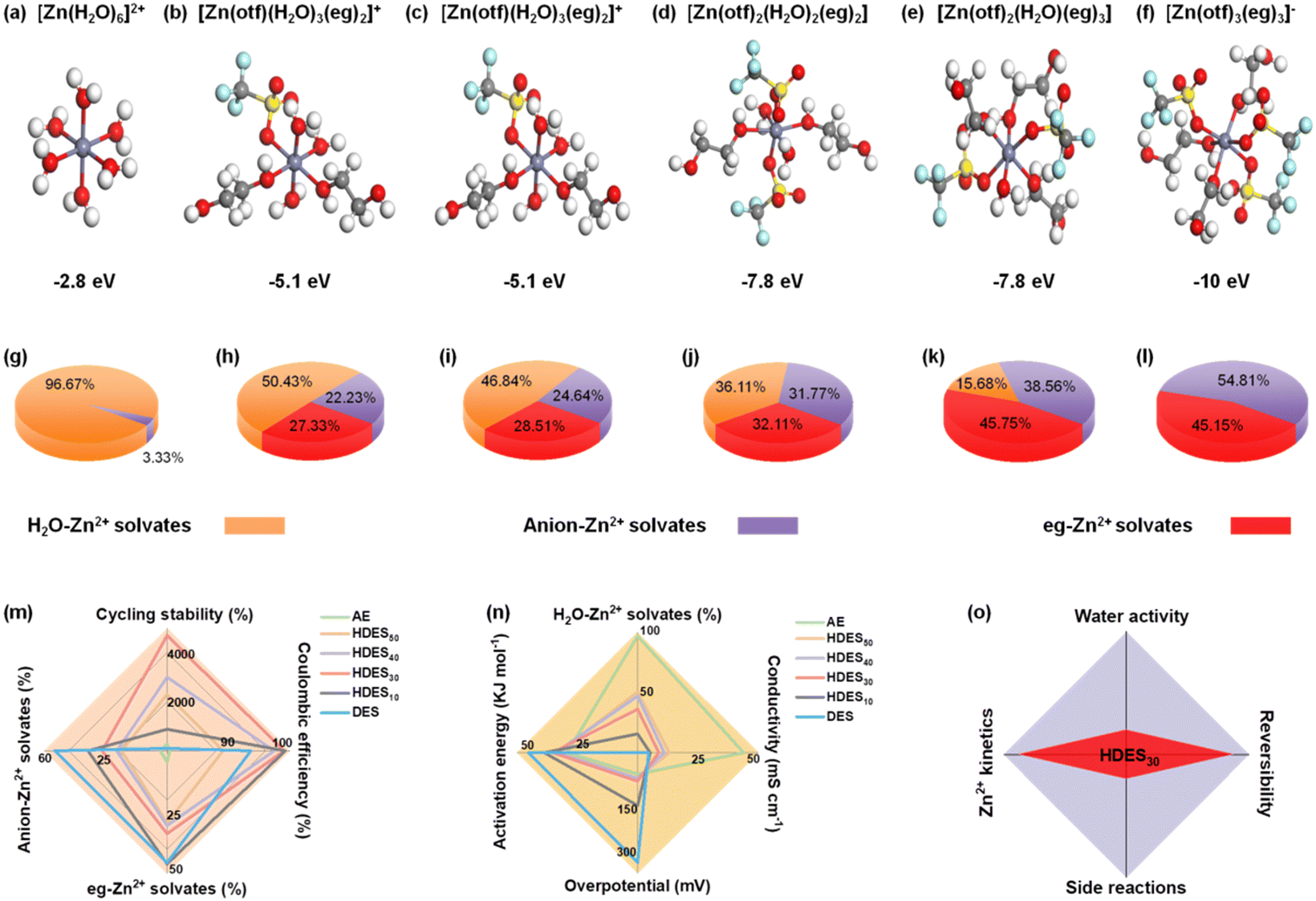 | ||
| Fig. 1 Theoretical and experimental studies on Zn2+ solvation structures and the structure–property correlations of the aqueous electrolyte, DES, and HDESs with different water contents. (a–f) Coordination structures and average binding energies of AE, HDES50, HDES40, HDES30, HDES10, and DES, respectively. The white, gray, red, yellow, light blue and purple balls represent hydrogen, carbon, oxygen, sulfur, fluorine atoms and zinc ions, respectively. (g–l) The most possible solvation structures and the distribution of different Zn2+ solvates calculated from molecular dynamics simulations. Structure–property–performance relationship: (m) reversibility: a radar chart comparing cycling stability, Coulombic efficiency, proportion of eg–Zn2+ solvates, and proportion of anion–Zn2+ solvates. (n) kinetics: a radar chart comparing ionic conductivity, overpotential, activation energy, and proportion of H2O–Zn2+ solvates. (o) The balance between reversibility and kinetics of the optimized HDES30. | ||
Molecular dynamics simulations were conducted to further investigate the Zn2+ solvation sheath and determine the distribution of Zn2+ solvates. The most possible solvation structures of different electrolytes were consistent with those in the aforementioned DFT results. More information was provided by the distribution of Zn2+ solvates, that is, the percentages of H2O-involved Zn2+, eg-involved Zn2+, and anion-involved Zn2+ (loose ion pair). In the AE, H2O-involved Zn2+ solvates dominate the solvation structure (Fig. 1g). In contrast, anion involved-Zn2+ solvates dominate the solvation structure in the DES, indicating an inferior ion dissociation degree (Fig. 1l).17 When a small amount of water is added to the DES, the percentage of anion-involved Zn2+ solvates dramatically decreases, indicating an improved ion dissociation degree (Fig. 1j and k). When the water content is further increased, the percentage of eg-involved Zn2+ solvates gradually decreases while the H2O-involved Zn2+ solvates gradually dominate the solvation structures (Fig. 1h and i), further proving the gradual transition from a DES to an AE.
To better understand structure–property–performance correlations, five parameters related to reversibility and Zn2+ kinetics were evaluated in conjunction with the aforementioned calculation results. First, less anion-involved Zn2+ solvates and more H2O-involved Zn2+ solvates among the electrolyte solvation structures showed a lower percentage of ion clusters and a higher ion dissociation degree, contributing to lower activation energy, lower overpotential, better ionic conductivity and improved Zn2+ kinetics (Fig. 1n and Fig. S6–S8, ESI†). Second, a lower percentage of eg-involved Zn2+ solvates and a higher percentage of H2O-involved Zn2+ solvates, i.e., more active water molecules in the solvation sheath, cause more water-induced side reactions, leading to lower stability and reversibility (Fig. 1m and Fig. S4, S5, ESI†). Therefore, the AE exhibited inferior Coulombic efficiency and cycling stability despite delivering the best ionic conductivity, the lowest activation energy and overpotential. In contrast, HDES30 with a dramatically decreased percentage of anion-involved Zn2+ solvates and moderate percentage of H2O-involved Zn2+ solvates showed the best Coulombic efficiency and cycling stability in terms of better ionic conductivity, lower activation energy, and overpotential. Therefore, HDES30 with a finely-tuned solvation structure with water enables the balance between reversibility and Zn2+ kinetics (Fig. 1o). In addition to excellent electrochemical performance, HDES30 also possesses high thermal stability, low viscosity, low density, and low cost, which are comparable to those of AE, showing great promise for use in large-scale aqueous zinc-ion batteries (Table S1 and Fig. S9–S10, ESI†).
The spectroscopic characterization studies were further utilized to demonstrate the structure–property relationship. The Znotf powder shows Vs(SO3), Vas(SO3) and Vs(CF3), Vas(CF3) bands at 1034, 1256 cm−1 and 1224, 1182 cm−1, respectively, among which the asymmetric bands all disappeared in AE, DES and HDES, illustrating the solvation of the zinc salt and the successful formation of DES (Fig. 2a and Fig. S11a, ESI†).30 Compared with eg, the melting point of DES was significantly depressed, further proving the successful formation of DES (Fig. S12, ESI†). In the Raman spectra, the characteristic peak of eg (C–O) located at ∼865 cm−1 split into two peaks after introducing zinc salt and water, indicating the strong coordination among eg, Zn2+, and water molecules (Fig. 2b and Fig. S11b, ESI†).31 This was further confirmed by the shift of C–H stretching vibrations (Fig. 2c and Fig. S11c–f, ESI†).32 It is worth noting that the intensity of the split peak at ∼892 cm−1 became weaker after adding water to DES, indicating that water molecules replaced some eg molecules to participate in the coordination structure of Zn2+ ions. These results are consistent with the solvation structure of HDES obtained from the aforementioned theoretical study.
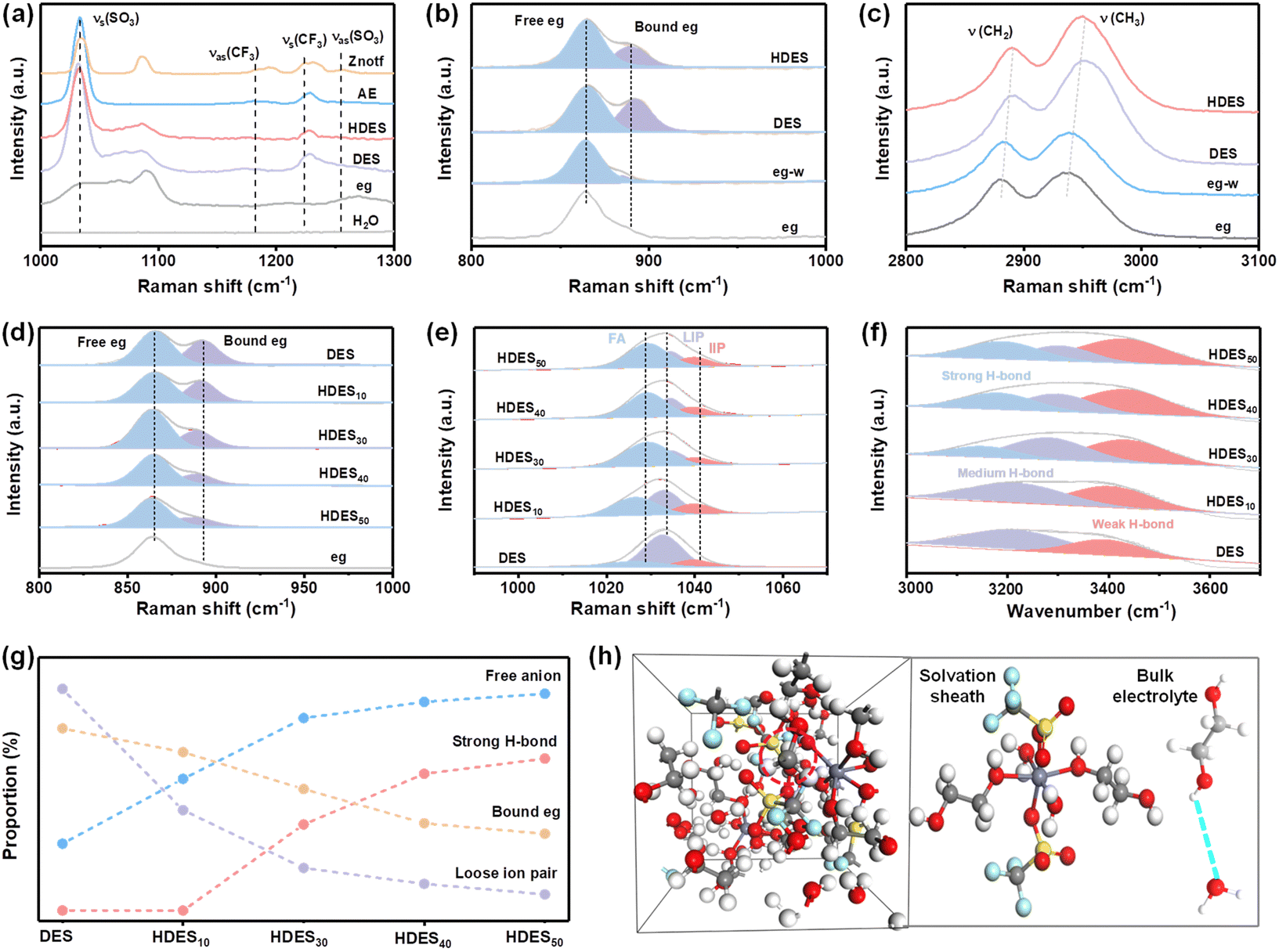 | ||
| Fig. 2 Solvation structure characterization. Comparison of DES and HDES. (a and c) Original Raman spectra. (b) Fitted Raman spectra. Comparison of HDESs with different water contents. (d and e) Fitted Raman spectra. (f) Fitted FT-IR spectra. (g) Summary of the correlations of the bound eg ratio, strong H-bond ratio, free anion ratio, and loose ion pair ratio. (h) The coordination environment of HDES30. The white, gray, red, yellow, light blue and purple balls represent hydrogen, carbon, oxygen, sulfur, fluorine atoms and zinc ions, respectively. | ||
The effect of water content on HDESs was also evaluated. As shown in Fig. 2d, the ratio of bound eg gradually decreased with the increase in water contents, demonstrating reduced eg–Zn2+ coordination and increased H2O–Zn2+ coordination (Fig. S13, ESI†). In Raman spectra, the Vs(SO3) of CF3SO3− is rather susceptible to changes in cation–anion interactions and aggregation behavior of zinc salts.33 It can be deconvoluted into three types at 1028, 1033, and 1041 cm−1, originating from free anions (FA), loose ion pairs (LIP), and intimate ion pairs (IIP), respectively.34 All electrolytes showed limited IIP species, but the amount of FA and LIP varied dramatically from one electrolyte to another (Fig. 2e). In DES, the majority of CF3SO3− exists as LIP, suggesting dominant Zn2+–CF3SO3− species. With the increase in water content, the LIP ratio dramatically decreases while the FA ratio gradually increases, indicating decreased ion clusters and an improved ion dissociation degree. In other words, the Zn2+–CF3SO3− coordination is gradually replaced by Zn2+–H2O coordination, and water molecules gradually participate in the solvation sheath of Zn2+ ions, which is consistent with the above MD results. Meanwhile, the strong H-bond and weak H-bond ratios gradually dominate the O–H peak in FT-IR spectra, suggesting that H2O–H2O and H2O–Zn2+ interactions dominate in HDES3 and HDES4 (Fig. 2f). All the abovementioned parameters are summarized in Fig. 2g. Among them, HDES30 showed a dramatically decreased LIP ratio, increased FA ratio, and moderate strong-H bond ratio, indicating a significantly improved ion dissociation degree and limited free water molecules. As a result, a well-designed hydrated deep eutectic solvent (HDES30) with reduced active water molecules in the solvation sheath and bound-free water molecules in the bulk electrolyte has been prepared (Fig. 2h), which is in good agreement with the calculation results and worth further discussion.
Through theoretical and spectroscopic studies, it has been found that HDES30 with finely-tuned water-deficient solvation structures ([Zn(H2O)2(eg)2(otf)2]) can achieve a high ion dissociation degree while inhibiting the activity of water. Correspondingly, boosted reversibility and Zn2+ kinetics are predictable for aqueous zinc-ion batteries. It is evident that HDES shows a higher oxygen evolution overpotential and lower hydrogen evolution overpotential, providing an expanded electrochemical stable potential window over 2 V (vs. Zn/Zn2+, Fig. 3a). This can be ascribed to the combined contribution of a decrease in the number of active water molecules in the solvation sheath and the presence of bound-free water in the bulk DES network. A lower current density and a higher corrosion potential also prove the suppressed water activity and improved stability of HDES against zinc anodes (Fig. 3b).35 In addition, chronoamperometry curves were recorded to investigate the Zn nucleation and growth. The current response in the AE electrolyte kept increasing, suggesting that a continuous 2D diffusion caused Zn dendrite formation (Fig. S14, ESI†). In contrast, the symmetrical cell with HDES showed a steady current response after the nucleation process, indicating successful suppression of side reactions. As shown in Fig. 3c, strong peaks corresponding to Znxotfy(OH)2x−y·nH2O byproducts can be clearly observed on the zinc anode after 10 cycles in an AE.36,37 In comparison, no obvious peaks of byproducts were detected on the zinc anode after 10 cycles in HDES, suggesting substantially suppressed side reactions. Raman and XPS spectra also show consistent results, further proving that HDES can substantially suppress side reactions (Fig. S15, ESI†).
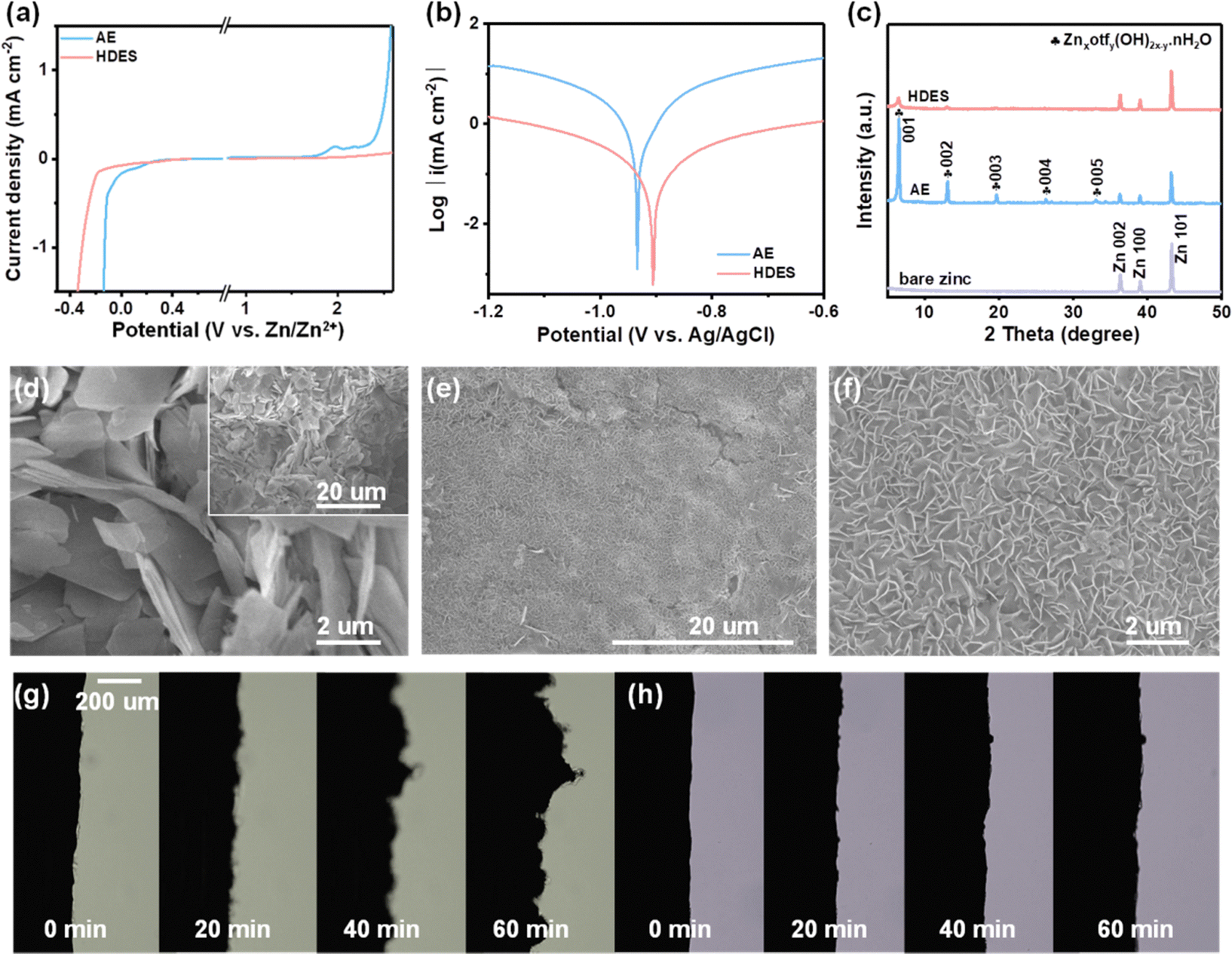 | ||
| Fig. 3 Electrolyte performance characterization. (a) LSV curves to determine the electrochemical stable potential window at a scan rate of 2 mV s−1. (b) Linear polarization curves. (c) XRD patterns. (d) SEM images of the zinc anode surface after cycling in an AE. (e and f) SEM images of the zinc anode surface after cycling in HDES. (g) In situ observation of Zn plating in the Zn//Zn cell with an AE. (h) In situ observation of Zn plating in a Zn//Zn cell with HDES. | ||
Fig. 3d–f shows SEM images of the typical morphology of the zinc growth in the AE and HDES. Severe cracking and irregular protuberances are shown in the AE, which will cause the short-circuit and failure of the cells (Fig. 3d and Fig. S16a, b, ESI†). In contrast, DES and HDES produced a highly uniform surface featured with tightly packed smaller zinc flakes, indicating that Zn dendrites are significantly inhibited due to the reduced water activity (Fig. 3e and f and Fig. S16c, d, ESI†). To understand this result in depth, 3D optical profiles of cycled Zn anodes were obtained. The zinc anode after cycling in AE exhibits an uneven surface with a large number of protuberances and large roughness, indicating severe Zn dendrites (Fig. S17a, ESI†). In contrast, the zinc anode after cycling in HDES possesses a flat surface with significantly reduced roughness, further proving the above result (Fig. S17b, ESI†). To visualize the effect of HDES on restraining side reactions and dendrite growth, the Zn plating process was captured by an in-situ optical microscopy measurement at a current density of 1 mA cm−2. In the AE, random Zn protrusions started forming after 20 min of plating and continuously grew into Zn dendrites after 40 min of plating. Meanwhile, gas bubbles resulting from the H2 evolution reaction were also detected due to the high reactivity of water (Fig. 3g). In contrast, the Zn surface remains flat without any gas evolution or Zn dendrites in the whole plating process in the HDES (Fig. 3h).
As a result, HDES successfully suppressed the growth of Zn dendrites and the side reactions, which are beneficial to the reversibility of the Zn anode. The Coulombic efficiency (CE) is an important index to evaluate the reversibility during the repeated cycling process.38 Hence, Zn//Cu half cells were employed for investigating the CE of different electrolytes at a current density of 1 mA cm−2 and a capacity of 0.5 mA h cm−2. Fig. 4a–c presents the CE and corresponding voltage profiles in different electrolytes. AE exhibited an inferior average CE of 80.4%. Moreover, the CE and voltage profiles show obvious fluctuations in the AE, reflecting the severe concurrent side reactions and poor reversibility (Fig. 4a and b). In HDES, steady CE and voltage profiles of 1000 cycles were obtained with a superior average CE of 99.6%, demonstrating substantially suppressed side reactions and excellent reversibility (Fig. 4c). Excellent cycling performances of Zn//Zn symmetric cells at various current densities were also observed. As displayed in Fig. 4d, symmetric cells with the HDES electrolyte were able to stably cycle for 4500 h at 0.5 mA cm−2–0.5 mA h cm−2 and 1 mA cm−2–1 mA h cm−2. Even at higher current densities and capacities of 3 mA cm−2–3 mA h cm−2 and 5 mA cm−2–5 mA h cm−2, a long cycle life of about 400 h can be maintained (Fig. S18, ESI†). In addition to excellent stability and reversibility, the rate performances of the symmetric cells in different electrolytes were also scrutinized at various current densities from 1 to 10 mA cm−2. A symmetric cell in DES exhibits very large and fluctuating overpotentials even at a small current density of 1 mA cm−2, which is a common problem with DESs due to high viscosity and low ionic conductivity (Fig. S19, ESI†). The overpotentials of a symmetrical cell in AE fluctuated violently at 8 mA cm−2 due to the occurrence of a short circuit (Fig. 4e). In contrast, a symmetric cell in HDES showed steady overpotentials even at a large current density of 10 mA cm−2, proving good rate performances and fast Zn2+ kinetics. As a result, the HDES can achieve integrated excellent reversibility and good Zn2+ kinetics, surpassing most of other reported DESs (Fig. 4e).25,28,30,31,34,35,39–45
 | ||
| Fig. 4 Zn plating/stripping behaviors in AE and HDES. (a) Coulombic efficiency comparison. (b) Corresponding voltage profiles in an AE. (c) Corresponding voltage profiles in HDES. (d) Long-term cycling performances of Zn//Zn symmetric cells at 0.5 mA cm−2–0.5 mA h cm−2 and 1 mA cm−2–1 mA h cm−2. (e) Rate performance of Zn//Zn symmetric cells in AE and HDES electrolytes. (f) Comparison of the cycling stability and current density of the HDES in this work with recently reported state-of-the-art eutectic electrolytes. | ||
To explore the feasibility of HDES electrolyte for practical use, Zn-NVO full cells were assembled. As shown in Fig. 5a and Fig. S20a, ESI,† the NVO cathodes exhibited multiple redox couples in both electrolytes, which were attributed to the Zn2+/H+ co-insertion mechanism.46,47 In particular, the NVO cathode in the HDES electrolyte presents more steady CV profiles compared with those in the AE. The rate performances were evaluated at various current densities. Although the initial capacity of NVO cathodes in HDES is lower than that in AE, their capacities are almost the same after 10 cycles at 0.5 A g−1, which is due to the rapid capacity decay of vanadium-based cathodes at low current densities in AE (Fig. 5b).1 More importantly, after long-term cycling, the rate capability of NVO cathodes in HDES electrolyte surpasses that in AE regardless of current densities, and the capacities in the HDES electrolyte are recoverable when the current density is shifted back, indicating better stability and rate performance (Fig. 5c and Fig. S20b, ESI†).
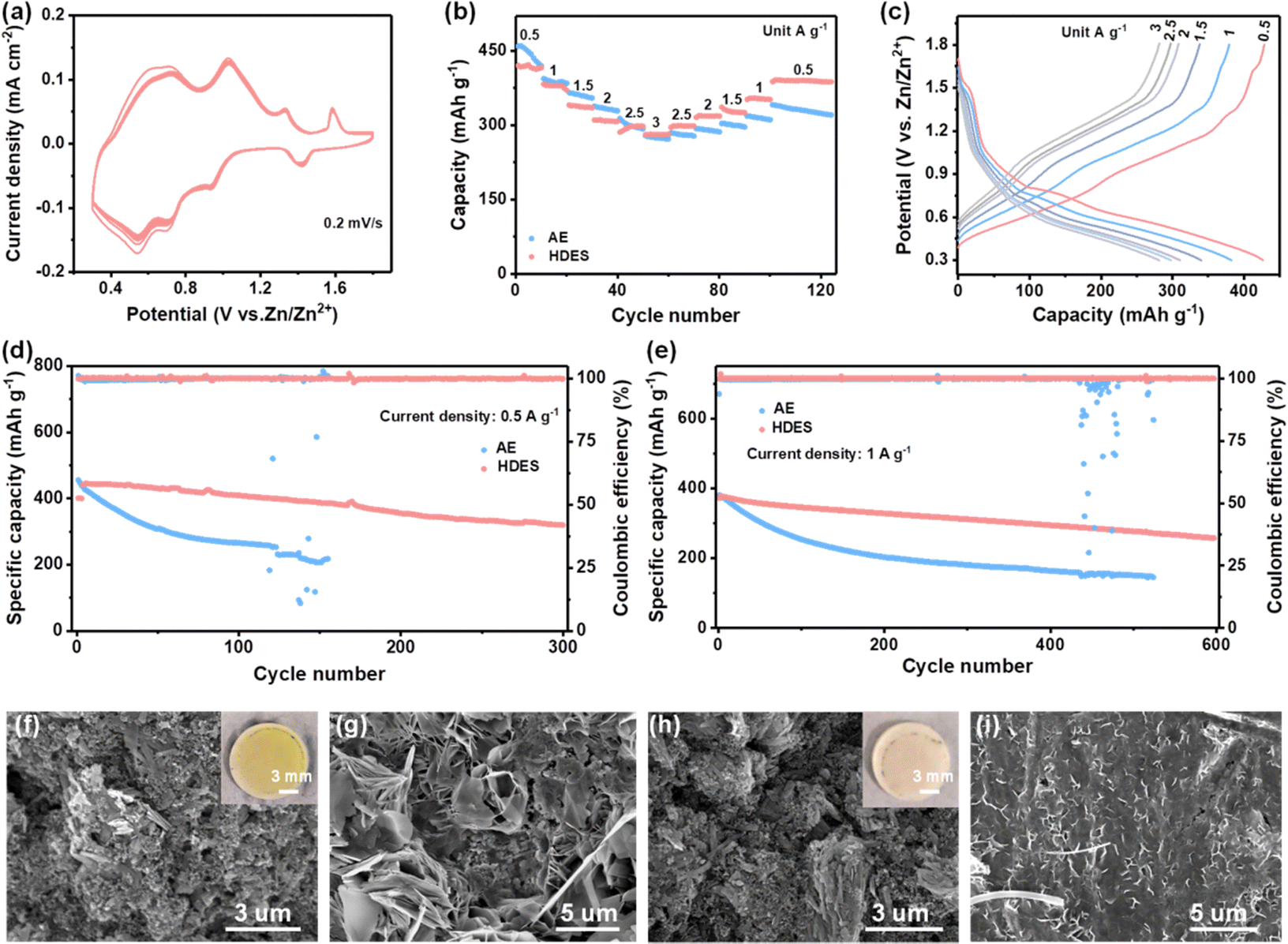 | ||
| Fig. 5 Electrochemical performance of Zn–NVO batteries. (a) CV curves of Zn–NVO batteries in HDES electrolyte for 10 cycles at 0.2 mV s−1. (b) Comparison of rate performances. (c) Corresponding voltage profiles of Zn–NVO in the HDES electrolyte. (d and e) Comparison of the cycling performances of Zn–NVO batteries at 0.5 A g−1 and 1 A g−1, respectively. (f and h) SEM images of cycled NVO cathodes in AE and HDES electrolytes, respectively. Insets show the corresponding optical photographs of the cycled separators. (g and i) SEM images of cycled Zn anodes in AE and HDES electrolytes, respectively. | ||
The long-term cycling performance was further studied. As for the battery with AE, although a slightly higher initial capacity of 455 mA h g−1 is delivered, only 40% of the initial capacity is retained (Fig. 5d). As shown in Fig. 5f, large NVO sheets break into small pieces after cycling in AE, which results from severe V dissolution (Fig. S21a, ESI†).48 This was further confirmed by an optical image observation of the yellow separators after being used in AE (the inset of Fig. 5f). On the other hand, a corroded rough Zn anode surface with obvious dendrites was observed in AE, which may further accelerate capacity fading and cell failure (Fig. 5g and Fig. S21b, ESI†). In contrast, the cathode retained its morphology with large NVO sheets and the Zn anode still exhibited a uniform and flat surface in HDES electrolyte, suggesting greatly inhibited V dissolution at the cathode and suppressed side reactions at the anode (Fig. 5h and I and Fig. S21c, ESI†). Therefore, the Zn-NVO full cells using HDES electrolyte deliver a high capacity retention of 75% after 300 cycles at 0.5 A g−1 and 70% after 600 cycles at 1 A g−1, indicating much better cycling stability (Fig. 5d and e). It is should be noted that, in addition to good cycling stability, the capacities (−436 mA h g−1 at 0.5 A g−1) of full cells in the HDES electrolyte also exceed those of most reported eutectic solvents due to the excellent Zn2+ transfer kinetics from the relatively high ionic conductivity.25,28,31,34,35,39,40,42,43,45
Multiple ex situ and in situ characterizations were carried out to study the Zn2+ storage mechanism behind the electrochemical performance. The X-ray photoelectron spectroscopy (XPS) spectra present changes in the valence state of V and intercalation/deintercalation of Zn2+ during charge/discharge processes in HDES electrolyte. As shown in Fig. 6b, no signal belonging to Zn2+ was detected in the pristine NVO electrode. A substantive Zn 2p peak was observed in the fully discharged state, suggesting the successful insertion of Zn2+. Instead, a pair of inperceptible peaks related to Zn 2p signals were found in the fully charged state, demonstrating the extraction of Zn2+.49,50 In the V 2p region, the peaks assigned to V3+ species increase, accompanied by an increase in V4+ and a decrease in V5+ (Fig. 6a). Subsequently, hybrid V species recovered to their original states in the fully charged state, suggesting that highly reversible redox reactions of V species occur during the intercalation/deintercalation of Zn2+.47,51 Moreover, the peaks related to NH4+ always exist in whole discharge/charge processes, indicating a stable structural support within the VOx polyhedral network (Fig. 6c).
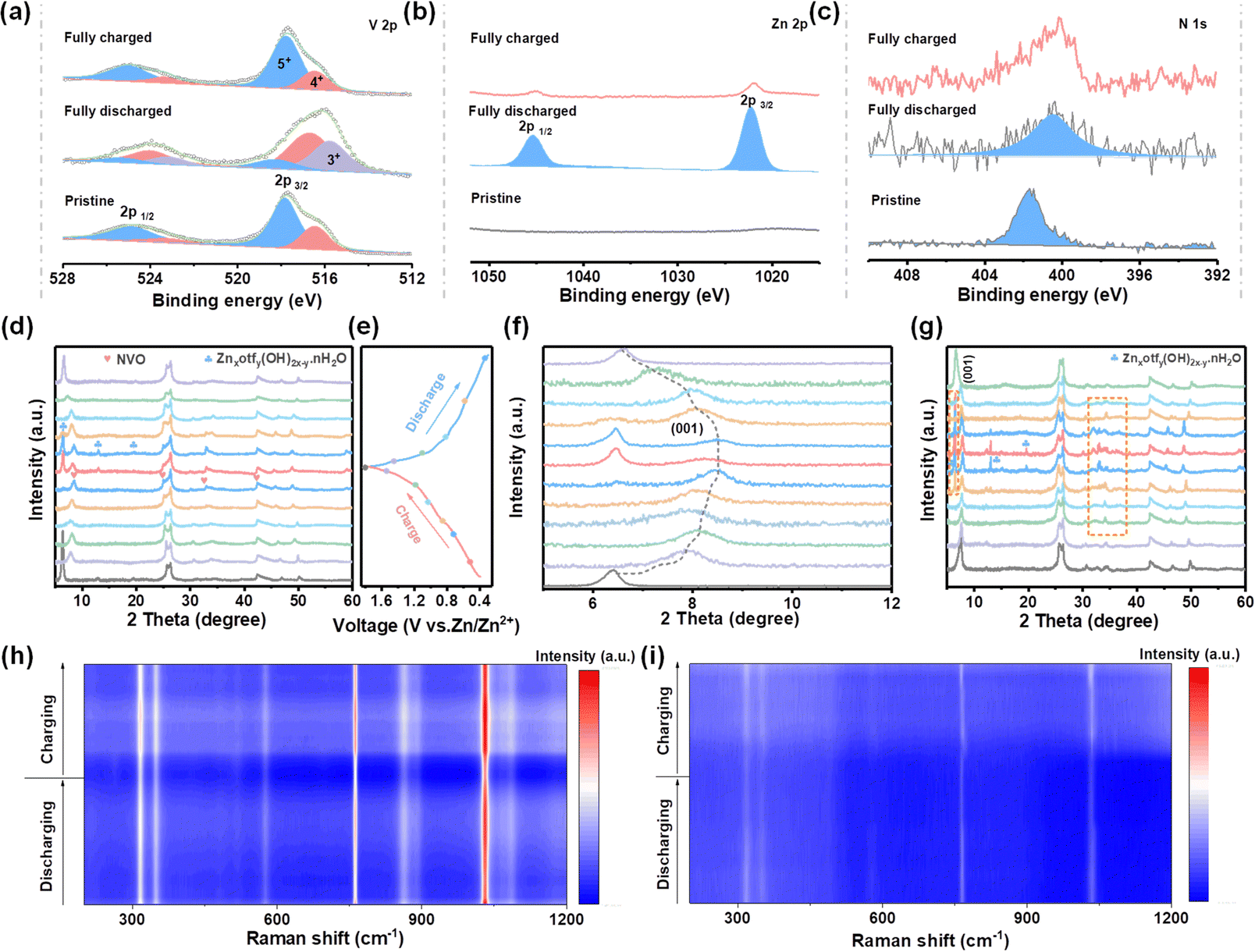 | ||
| Fig. 6 Charge storage mechanism characterizations. (a–c) Ex situ XPS spectra of V 2p, Zn 2p, and N 1s of NVO cathodes in HDES electrolyte, respectively. (d and f) Ex situ XRD patterns of NVO cathodes in HDES electrolyte at different charge/discharge states. (e) Corresponding GCD curves of (d) at 0.3 A g−1. (g) Ex situ XRD patterns of NVO cathodes in AE at different charge/discharge states. (h) In situ Raman spectra of NVO cathodes in HDES electrolyte. (i) In situ Raman spectra of NVO cathodes in AE. | ||
After clarifying the variation of chemical states, ex situ XRD was used to analyze the crystal phase evolution of NVO cathodes at different discharge/charge states. Fig. 6d and f show the ex situ characterized XRD patterns of NVO cathodes in HDES electrolyte at different discharge/charge states according to the GCD curves (Fig. 6e). The peak located at 6.5° representing the (001) crystal plane of NVO shifts towards high 2θ values during the discharge process, and then recovers to its original state during the charge process, which indicates reversible cation intercalation/deintercalation and phase changes.52 Additionally, new peaks emerge at 6.4, 12.9, and 19.5 during discharge to low states, which can be indexed to Znxotfy(OH)2x−y·nH2O, suggesting that OH− ions react with electrolyte and facilitate proton intercalation (Fig. 6d and f).53,54 Moreover, the peaks for Znxotfy(OH)2x−y·nH2O are significantly weaker in the HDES electrolyte compared to those in AE, indicating less water decomposition due to the inhibited water activity, which is consistent with aforementioned results (Fig. 6g and Fig. S22, ESI†). To further gain insight into the storage mechanism, in situ Raman spectra were recorded.55 The Raman peaks at 310–360 cm−1 and 574 cm−1 are related to the bending mode of the V–O–V bonds (Fig. S23, ESI†). The peaks at 760 cm−1 and 1031 cm−1 are derived from the stretching mode of the V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.56,57 These peaks, especially those located at 760 cm−1 and 1031 cm−1, gradually weakened during the discharge process and then slightly strengthened during the charge process, which was due to the intercalation/deintercalation of Zn2+ (Fig. 6h and i).58 Although the Raman intensity is reduced due to the resonance loss caused by cation intercalation, the peak position of the cathode was not obviously shifted, which means that the crystal structure was stable. Notably, the intensity and recovery of these peaks in the HDES electrolyte are better compared to those in AE, proving higher cathode reversibility. Hence, the reversible phase changes and Zn2+/H+ intercalation/deintercalation mechanism further validate a robust cycling performance of NVO cathodes in the HDES electrolyte. According to theoretical calculation, the reduced active water molecules are relatively away from the cathode during the de-solvation process, which may be the reason for the better stability and reversibility of NVO cathodes in the HDES electrolyte (Fig. S24, ESI†).
O bond.56,57 These peaks, especially those located at 760 cm−1 and 1031 cm−1, gradually weakened during the discharge process and then slightly strengthened during the charge process, which was due to the intercalation/deintercalation of Zn2+ (Fig. 6h and i).58 Although the Raman intensity is reduced due to the resonance loss caused by cation intercalation, the peak position of the cathode was not obviously shifted, which means that the crystal structure was stable. Notably, the intensity and recovery of these peaks in the HDES electrolyte are better compared to those in AE, proving higher cathode reversibility. Hence, the reversible phase changes and Zn2+/H+ intercalation/deintercalation mechanism further validate a robust cycling performance of NVO cathodes in the HDES electrolyte. According to theoretical calculation, the reduced active water molecules are relatively away from the cathode during the de-solvation process, which may be the reason for the better stability and reversibility of NVO cathodes in the HDES electrolyte (Fig. S24, ESI†).
Conclusions
In summary, we have developed a new type of hydrated eutectic solvent electrolyte based on ethylene glycol, zinc trifluorosulfonate, and H2O for aqueous zinc-ion batteries with integrated high reversibility and Zn2+ transfer kinetics. The evolution of solvation structures, and the effect of water content on eutectic electrolyte properties and electrochemical performance have been systematically studied through experimental and theoretical approaches. As the water content increased, water molecules gradually replaced ethylene glycol molecules and anions to participate in the solvation sheath of Zn2+ ions, resulting in an improved ion dissociation degree and increased zinc ion kinetics while sacrificing stability and reversibility. The developed electrolyte, HDES30 with a water-deficient solvation structure ([Zn(H2O)2(eg)2(otf)2]) and reduced free water in bulk solution, simultaneously possesses excellent reversibility and high Zn2+ kinetics. Therefore, the Zn anode with the HDES30 electrolyte can realize reversible Zn plating/stripping for 1000 cycles with an average Coulombic efficiency of 99.6% and sustain long-term cycling over 4500 h. The Zn//NVO full cell with the HDES30 electrolyte exhibits a much better cycling stability and a high capacity of 420 mA h g−1. This work will aid in the understanding of electrolyte solvation structure–electrolyte property–electrochemical performance relationships of DESs in aqueous zinc-ion batteries.Author contributions
R. C.: conceptualization, methodology, software, data acquisition, data curation, formal analysis, investigation, validation, visualization, writing–original draft, and writing–review and editing. Y. Z.: methodology, software, data acquisition, and writing–review and editing. J. L.: conceptualization, methodology, and formal analysis. Z. D., F. G., W. Z., Y. D., W. Z., X. G., and J. Z.: data acquisition and writing–review and editing. Y. Z.: methodology, software, data acquisition, and writing–review and editing. G. H.: funding acquisition, supervision, and writing – review and editing. X. W.: funding acquisition, supervision, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2019YFE0114400), the Guangdong Basic and Applied Basic Research Foundation (2021B1515120005), the State Key Laboratory of Pulp & Paper Engineering (2022C01), the National Natural Science Foundation of China (32171721) and the Engineering and Physical Sciences Research Council (EPSRC; EP/V027433/1; EP/V027433/2; EP/Y008707/1).References
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS
.
- M. Li, Z. Li, X. Wang, J. Meng, X. Liu, B. Wu, C. Han and L. Mai, Energy Environ. Sci., 2021, 14, 3796–3839 RSC
- J. Cao, D. Zhang, X. Zhang, Z. Zeng, J. Qin and Y. Huang, Energy Environ. Sci., 2022, 15, 499–528 RSC
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC
- Y. Liu, X. Lu, F. Lai, T. Liu, P. R. Shearing, I. P. Parkin, G. He and D. J. Brett, Joule, 2021, 5, 2845–2903 CrossRef CAS
- P. Ruan, X. Xu, D. Zheng, X. Chen, X. Yin, S. Liang, X. Wu, W. Shi, X. Cao and J. Zhou, ChemSusChem, 2022, 15, e202201118 CrossRef CAS PubMed
- N. Dalleska, K. Honma, L. Sunderlin and P. Armentrout, J. Am. Chem. Soc., 1994, 116, 3519–3528 CrossRef CAS
- S. Liu, J. Mao, W. K. Pang, J. Vongsvivut, X. Zeng, L. Thomsen, Y. Wang, J. Liu, D. Li and Z. Guo, Adv. Funct. Mater., 2021, 31, 2104281 CrossRef CAS
- Z. Cao, P. Zhuang, X. Zhang, M. Ye, J. Shen and P. M. Ajayan, Adv. Energy Mater., 2020, 10, 2001599 CrossRef CAS
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS
- J. Cao, D. Zhang, X. Zhang, Z. Zeng, J. Qin and Y. Huang, Energy Environ. Sci., 2022, 15, 499–528 RSC
- S. Zhu, Y. Dai, J. Li, C. Ye, W. Zhou, R. Yu, X. Liao, J. Li, W. Zhang and W. Zong, Sci. Bull., 2022, 67, 1882–1889 CrossRef CAS PubMed
- C. Li, S. Jin, L. A. Archer and L. F. Nazar, Joule, 2022, 6, 1733–1738 CrossRef
- J. Yue, L. Lin, L. Jiang, Q. Zhang, Y. Tong, L. Suo, Y. S. Hu, H. Li, X. Huang and L. Chen, Adv. Energy Mater., 2020, 10, 2000665 CrossRef CAS
- J. Guo, J. Ming, Y. Lei, W. Zhang, C. Xia, Y. Cui and H. N. Alshareef, ACS Energy Lett., 2019, 4, 2776–2781 CrossRef CAS
- Z. Tian, Y. Zou, G. Liu, Y. Wang, J. Yin, J. Ming and H. N. Alshareef, Adv. Sci., 2022, 9, e2201207 CrossRef PubMed
- Z. Yu, N. P. Balsara, O. Borodin, A. A. Gewirth, N. T. Hahn, E. J. Maginn, K. A. Persson, V. Srinivasan, M. F. Toney, K. Xu, K. R. Zavadil, L. A. Curtiss and L. Cheng, ACS Energy Lett., 2021, 7, 461–470 CrossRef
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed
- C. Li, R. Kingsbury, L. Zhou, A. Shyamsunder, K. A. Persson and L. F. Nazar, ACS Energy Lett., 2022, 7, 533–540 CrossRef CAS
- Y. Zhu, J. Yin, X. Zheng, A.-H. Emwas, Y. Lei, O. F. Mohammed, Y. Cui and H. N. Alshareef, Energy Environ. Sci., 2021, 14, 4463–4473 RSC
- W. Yang, Y. Yang, H. Yang and H. Zhou, ACS Energy Lett., 2022, 7, 2515–2530 CrossRef CAS
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich and B. W. Doherty, Chem. Rev., 2020, 121, 1232–1285 CrossRef PubMed
- S. Azmi, M. F. Koudahi and E. Frackowiak, Energy Environ. Sci., 2022, 15, 1156–1171 RSC
- J. Ma, J. Wang, K. Jia, Z. Liang, G. Ji, Z. Zhuang, G. Zhou and H. M. Cheng, J. Am. Chem. Soc., 2022, 144, 20306–20314 CrossRef CAS PubMed
- L. Geng, J. Meng, X. Wang, C. Han, K. Han, Z. Xiao, M. Huang, P. Xu, L. Zhang and L. Zhou, Angew. Chem., 2022, 134, e202206717 Search PubMed
- J. Wu, Q. Liang, X. Yu, Q. F. Lü, L. Ma, X. Qin, G. Chen and B. Li, Adv. Funct. Mater., 2021, 31, 2011102 CrossRef CAS
- X. Lu, E. J. Hansen, G. He and J. Liu, Small, 2022, 18, e2200550 CrossRef PubMed
- W. Yang, X. Du, J. Zhao, Z. Chen, J. Li, J. Xie, Y. Zhang, Z. Cui, Q. Kong, Z. Zhao, C. Wang, Q. Zhang and G. Cui, Joule, 2020, 4, 1557–1574 CrossRef CAS
- Q. Zhang, K. Xia, Y. Ma, Y. Lu, L. Li, J. Liang, S. Chou and J. Chen, ACS Energy Lett., 2021, 6, 2704–2712 CrossRef CAS
- Y. Yang, S. Liang, B. Lu and J. Zhou, Energy Environ. Sci., 2022, 15, 1192–1200 RSC
- D. Han, C. Cui, K. Zhang, Z. Wang, J. Gao, Y. Guo, Z. Zhang, S. Wu, L. Yin, Z. Weng, F. Kang and Q.-H. Yang, Nat. Sustainability, 2021, 5, 205–213 CrossRef
- H. Du, K. Wang, T. Sun, J. Shi, X. Zhou, W. Cai and Z. Tao, Chem. Eng. J., 2022, 427, 131705 CrossRef CAS
- H. Ao, W. Zhu, M. Liu, W. Zhang, Z. Hou, X. Wu, Y. Zhu and Y. Qian, Small Methods, 2021, 5, e2100418 CrossRef PubMed
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 CrossRef PubMed
- M. Han, J. Huang, X. Xie, T. C. Li, J. Huang, S. Liang, J. Zhou and H. J. Fan, Adv. Funct. Mater., 2022, 32, 2110957 CrossRef CAS
- X. Zeng, J. Mao, J. Hao, J. Liu, S. Liu, Z. Wang, Y. Wang, S. Zhang, T. Zheng and J. Liu, Adv. Mater., 2021, 33, 2007416 CrossRef CAS PubMed
- W. Yuan, G. Ma, X. Nie, Y. Wang, S. Di, L. Wang, J. Wang, S. Shen and N. Zhang, Chem. Eng. J., 2022, 431, 134076 CrossRef CAS
- Y. Yang, C. Liu, Z. Lv, H. Yang, Y. Zhang, M. Ye, L. Chen, J. Zhao and C. C. Li, Adv. Mater., 2021, 33, e2007388 CrossRef PubMed
- D. Han, T. Sun, R. Zhang, W. Zhang, T. Ma, H. Du, Q. Wang, D. He, S. Zheng and Z. Tao, Adv. Funct. Mater., 2022, 2209065 CrossRef CAS
- X. Lin, G. Zhou, M. J. Robson, J. Yu, S. C. T. Kwok and F. Ciucci, Adv. Funct. Mater., 2021, 32, 2109322 CrossRef
- J. Zhao, J. Zhang, W. Yang, B. Chen, Z. Zhao, H. Qiu, S. Dong, X. Zhou, G. Cui and L. Chen, Nano Energy, 2019, 57, 625–634 CrossRef CAS
- J. Shi, T. Sun, J. Bao, S. Zheng, H. Du, L. Li, X. Yuan, T. Ma and Z. Tao, Adv. Funct. Mater., 2021, 31, 2102035 CrossRef CAS
- L. Cao, D. Li, E. Hu, J. Xu, T. Deng, L. Ma, Y. Wang, X. Q. Yang and C. Wang, J. Am. Chem. Soc., 2020, 142, 21404–21409 CrossRef CAS PubMed
- F. Ming, Y. Zhu, G. Huang, A. H. Emwas, H. Liang, Y. Cui and H. N. Alshareef, J. Am. Chem. Soc., 2022, 144, 7160–7170 CrossRef CAS PubMed
- H. Qiu, R. Hu, X. Du, Z. Chen, J. Zhao, G. Lu, M. Jiang, Q. Kong, Y. Yan and J. Du, Angew. Chem., Int. Ed., 2022, 61, e202113086 CAS
- D. Bin, W. Huo, Y. Yuan, J. Huang, Y. Liu, Y. Zhang, F. Dong, Y. Wang and Y. Xia, Chem, 2020, 6, 968–984 CAS
- J. Li, N. Luo, F. Wan, S. Zhao, Z. Li, W. Li, J. Guo, P. R. Shearing, D. J. L. Brett, C.
J. Carmalt, G. Chai, G. He and I. P. Parkin, Nanoscale, 2020, 12, 20638–20648 RSC
- H. Zhang, X. Liu, H. Li, I. Hasa and S. Passerini, Angew. Chem., Int. Ed., 2021, 60, 598–616 CrossRef CAS PubMed
- N. Wang, C. Sun, X. Liao, Y. Yuan, H. Cheng, Q. Sun, B. Wang, X. Pan, K. Zhao, Q. Xu, X. Lu and J. Lu, Adv. Energy Mater., 2020, 10, 2002293 CrossRef CAS
- T. He, S. Weng, Y. Ye, J. Cheng, X. Wang, X. Wang and B. Wang, Energy Storage Mater., 2021, 38, 389–396 CrossRef
- J. Li, K. McColl, X. Lu, S. Sathasivam, H. Dong, L. Kang, Z. Li, S. Zhao, A. G. Kafizas, R. Wang, D. J. L. Brett, P. R. Shearing, F. Corà, G. He, C. J. Carmalt and I. P. Parkin, Adv. Energy Mater., 2020, 10, 2000058 CrossRef CAS
- F. Cui, D. Wang, F. Hu, X. Yu, C. Guan, G. Song, F. Xu and K. Zhu, Energy Storage Mater., 2022, 44, 197–205 CrossRef
- W. Wang, D. He, Y. Fang, S. Wang, Z. Zhang, R. Zhao and W. Xue, Electrochim. Acta, 2022, 416, 140270 CrossRef CAS
- X. Liu, G. Xu, S. Huang, L. Li, Y. Wang and L. Yang, Electrochim. Acta, 2021, 368, 137600 CrossRef CAS
- X. Liu, W. Ni, Y. Wang, Y. Liang, B. Wu, G. Xu, X. Wei and L. Yang, Small, 2022, 18, e2105796 CrossRef PubMed
- Y. Zhao, S. Liang, X. Shi, Y. Yang, Y. Tang, B. Lu and J. Zhou, Adv. Funct. Mater., 2022, 32, 2203819 CrossRef CAS
- F. Ming, H. Liang, Y. Lei, S. Kandambeth, M. Eddaoudi and H. N. Alshareef, ACS Energy Lett., 2018, 3, 2602–2609 CrossRef CAS
- J. Cao, D. Zhang, Y. Yue, X. Wang, T. Pakornchote, T. Bovornratanaraks, X. Zhang, Z.-S. Wu and J. Qin, Nano Energy, 2021, 84, 105876 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: experimental and computational details, and supplementary figures and tables. See DOI: https://doi.org/10.1039/d3ee00462g |
| ‡ R. C., C. Z., and J. L. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |