
Environmental chemistry response of beryllium to diverse soil-solution conditions at a waste disposal site†
Md. Rashidul
Islam
 *ab,
Peter
Sanderson
ab,
Mathew P.
Johansen
c,
Timothy E.
Payne
c and
Ravi
Naidu
*ab
*ab,
Peter
Sanderson
ab,
Mathew P.
Johansen
c,
Timothy E.
Payne
c and
Ravi
Naidu
*ab
aGlobal Centre for Environmental Remediation (GCER), College of Engineering, Science and Environment, The University of Newcastle, University Drive, Callaghan Campus, NSW 2308, Australia. E-mail: ravi.naidu@newcastle.edu.au; md.rashidul.islam@uon.edu.au; Tel: +61470219676 Tel: +61249138705
bCRC for Contamination Assessment and Remediation of the Environment (CARE), The University of Newcastle, University Drive, Callaghan Campus, NSW 2308, Australia
cAustralian Nuclear Science and Technology Organisation (ANSTO), Lucas Heights, NSW 2234, Australia
First published on 29th November 2022
Abstract
This study evaluated how the variation in different sorption conditions of beryllium (Be) in soil–water systems (electrolytes; ionic strengths; competing, counter, and co-existing ions; concentrations of Be and soil; and temperature) affected Be's environmental behaviour. For this reason, potentially contaminated soil was collected from a legacy waste site near Sydney, Australia. The sorption–desorption plateau for Be was found at >12.5 g L−1 (soil/solution), considering higher sorption and limited desorption. Variable surface charges developed by different added ions (competing ions, counter ions, and co-existence of all ions) were not always correlated with Be sorption. However, effects of added ions in Be sorption (increased by counter ions and decreased by competing ions) primarily occurred at low pH, with no noticeable changes at pH > 6 due to the hydration and precipitation behaviour of Be at higher pH. Both laboratory data and modelling indicated the substantial effect of counter ions on increased sorption of Be. Relatively higher amounts of sorption under the co-existence of all added ions were suggested from synergistic actions. Sorption was favourable (KL > 0, and 0 < RL < 1) across all concentrations and temperatures at pH 5.5, and high retention (84–97%) occurred after four desorption cycles indicated specific sorption. The sorption process was exothermic (ΔH > −43 kJ mole−1), while desorption was endothermic (ΔH > +78.4 kJ mole−1). All sorption–desorption reactions were spontaneous (ΔG = −Ve), and executed without any structural deformation (ΔS = nearly zero) of soil particles. However, the effect of temperature on desorption was influenced by the concentrations of Be. Higher retention and different sorption–desorption parameters (Kd-desorption > Kd-sorption; Kf-desorption > Kf-sorption; ndesorption/nsorption < 1) indicate limited mobility of Be and the presence of desorption hysteresis in the studied soil under the experimental conditions.
Environmental significanceThe utilisation of beryllium and beryllium alloys in different scientific and technological applications has risen in recent decades. The waste arising from mining, research, manufacturing, handling, corrosion, infiltration, etc., are emitted into the environment, and potentially impacts plants, animals and humans. Environmental research of Be is limited in comparison to other metals. It is, however, important to understand the environmental behaviour of Be in various soil–water conditions. Knowledge of these is essential for proper management of Be contamination. Therefore, this study evaluates the role of different soil–water conditions (e.g. electrolytes; ionic strengths; competing, counter, and co-existing ions; soil loading; Be loading; and temperature) on the environmental chemistry of Be in sorption–desorption phenomena to understand its management strategy. |
1 Introduction
Due to its wide uses and subsequent emissions, beryllium (Be) has been referred to as a modern industrial hazard as a result of its toxicity.1,2 It is classified as group one category human carcinogen,3 and in the environment, it is very toxic to plants and animals, even at low concentrations,4,5 which reflects the urgency for conducting thorough explorations of the environmental behaviour (e.g. sorption–desorption, retention, etc.) of Be so that its contamination can be effectively managed.Beryllium in the subsurface soils may experience different hydrothermal and geochemical alterations through a series of chemical reactions to form its minerals (most common minerals are beryl, bertrandite, chrysoberyl, etc.) over time.6 However, waste containing Be buried in the ground may also be exposed to hydrothermal and chemical weathering, and the weathered Be (mobile) may enter into surface soil, subsurface soil or groundwater. The behaviours of Be in the surface soils are influenced by different physicochemical factors of soils/environment.7,8 The most important factors are pH, time and types of contact or reaction between soils and metals (quantity of metals–soil interaction), types of organic–inorganic ions or ligands present, index ions effects (e.g. cations, anions, electrolytes, ionic strength) and temperature, etc. These factors significantly control the sorption or transport of any metals from contaminated soil,9,10 and this may also be relevant for Be, which received limited study. In one study, Kaplan et al.11 reported that different geochemical factors affected metal transport at a low-level waste disposal site, and Islam et al.12 reported various physicochemical properties of soil (from low-level radioactive waste disposal site) regulate sorption–desorption of Be under field soil pH. Previous studies13–15 reported pH is the most powerful factor for controlling aqueous solution chemistry or sorption behaviour of Be (soluble at pH < 3 and pH > 11–12; insoluble 6–11), but this role of pH can be influenced by other factors in the real soil–water (complex chemical system) environment. For example, El-Soad et al.16 reported maximum sorption at pH 4, whereas Boschi and Willenbring4 reported it at pH 6 using different sorbent materials, indicating their effects over pH. However, at a constant pH, sorption of metal primarily depends on four factors,9 namely: number of available sites to bind the metals; potential for metal contact with the available sorption sites; the amount of metal present in the solution; and the displacement of ions already occupied at sorption sites by target metal ions (e.g. Be). These phenomena are greatly influenced by the amounts of sorption sites (depend on the amounts of soils in the system) and metal ions (e.g. Be2+) present, including the presence of different organic–inorganic ligands and influenced by other physicochemical factors, which is important to be determined for Be.
When soils are immersed in solutions, different index ions develop the surface charge and regulate the sorption–desorption of metals.9 Surface charges and point of zero charges (pHzpc) are important to identify specific (chemisorption, less desorption) and non-specific sorption (physical or electrostatic interaction) of metals.9,17 However, surface charge can be modified (variable charges) by different electrolytes or ionic strengths, in which the valency of ions (competing and counter) and types of counter ions or ligands significantly control the surface charge and metal sorption.9,17–19 In addition, temperature is the most fluctuating environmental parameter, and Harter and Naidu9 reported each 10 °C temperature variation could significantly alter the sorption–desorption phenomena of metals, which must be determined for Be.
In this communication, we have investigated the environmental behaviour of Be under diverse conditions of the soil-solution environment. Soil samples were collected from the Little Forest Legacy Site (LFLS), Sydney (Australia), where a substantial amount of Be (∼1070 kg) was disposed of in unlined trenches during the 1960s.12,20 Numerous similar sites around the world were reported in the literature.12 In the presence of intense rainfall events, the continuous infiltration and saturation of the waste trenches (bathtub effect) could influence the exposure of Be in the surface soil as a similar mechanism was reported for Plutonium mobility at this study site.21 Exposed Be can be affected by the diverse physicochemical conditions of soils in their (e.g. Be) sorption–desorption phenomena. In a recent study,12 the sorption–desorption behaviour of Be was investigated under a fixed condition of pH, temperature, added electrolytes, and ionic strength, which may vary in the future at the study sites. Alternatively, variations of these parameters may be relevant for other similar sites around the world. Nevertheless, it is crucial to evaluate how different added ions (competing and counter ions/ligand) in the soil-solution interface can govern the sorption of Be in the wider ranges of pH.
Therefore, the specific objective of this study is to explore the environmental behaviour of Be in the LFLS soil under diverse soil-solution conditions. For this reason, we used different batch sorption–desorption techniques to determine how sorption–desorption, retardation and hysteresis are regulated by different environmental parameters of soil. Taken into account here are the variation of sorption–desorption conditions by the addition of electrolytes, ionic strengths, competing ions, counter ions, co-existence of all ions as a function of pH, soil loading, Be loading and temperature at a fixed pH. All these variables are essential for expanding our knowledge of Be chemistry in the LFLS site or any other similar Be contaminated site around the world.
2 Methods and materials
2.1 Study area and sampling information
The studied site (e.g. LFLS) is a historical waste disposal site which was operational during the 1960s and has been extensively monitored and studied during subsequent decades.20 This site is located at the buffer zone boundary of the Australian Nuclear Science and Technology Organisation (ANSTO), on the southern periphery of Sydney, Australia. Beryllium (approximately ∼1070 kg) and other low-level radioactive and non-radioactive, organic and inorganic elements in the waste (liquid, solid, semi-solid etc.) were co-disposed in the unlined trenches following standard practice at the time (1960–1968).20 There are 79 waste trenches in the clay/shale lens covered by 1 m of the surrounding surface soil.22 Moreover, a separate legacy waste disposal site for industrial chemicals and eluents adjacent to the studied site can influence the LFLS site, with potential mixing of contaminants plumes.23 The “bathtub effect” may influence the exposure of Be in the surface soil, as was reported by Payne et al.21 for radionuclides at the same site.For this reason, representative surface soils (0–10 cm depth) were collected from the LFLS, dried in an oven, homogenised by grinding, and sieved (<2 mm) for further analysis. Details of the study site, sample collection, preparation and analysis of physicochemical properties are discussed in the previous study12 and also in the ESI, Table S1.†
2.2 Effect of ionic strengths on the pHzpc of LFLS soil
The pHzpc of an adsorbent (e.g. soil) is changed by the presence of solutions containing different ionic strengths, which was evaluated for LFLS soil using the pH difference method as done elsewhere.24 Briefly, soil samples (<2 mm sieve) were added to solutions (Milli-Q water; 0.01 M NaNO3; 0.1 M NaNO3) at 20 g L−1, adjusted pH to 2, 4, 5, 6, 7, and 9 (desired pHi ± 0.1), shaken for 24 h, recorded final pH (pHf), and the pHpzc was obtained by plotting pHivs. ΔpH (= pHf − pHi).2.3 Surface charge of soil under various added ions as a function of pH
The surface charge and isoelectric point (IEP) were measured by electrophoretic mobility of ions at different pH levels ranging from 1 to 9 (0.01% soil in Milli-Q water), using zeta potential analyser (NanoPlus-HD).12 The effect of cations, including sodium (Na+), potassium (K+), ammonium (NH4+), calcium (Ca2+), magnesium (Mg2+), aluminium (Al3+) (taking their 0.1 M chloride salt); anions are chloride (Cl−), nitrate (NO3−), sulphate (SO42−), phosphate (PO43−), acetate (CH3COO−) (using their 0.1 M sodium salt); and the co-existence of all cations and anions (0.01 M of all competing and counter ions as mentioned above) on the surface charge of soil were also evaluated according to the literature.18,19 Briefly, 20 g of soil per litre electrolyte solutions were adjusted to pH 3, 5, 7, and 9; shaken for 24 h (pH was checked and adjusted if needed); centrifuged (4500g for 10 minutes, 10 °C); recorded final pH and immediately measured surface charge by zeta potential analyser as above. The surface charge was measured up to pH ∼6 for Al due to the gelatinous precipitation of Al at higher pH.252.4 Effect of soil–water conditions and experimental parameters on the environmental chemistry of Be
| Equations | No | References |
|---|---|---|
| a C i, and Ce are the initial and equilibrium concentrations of Be (mg L−1); qe is the amount of sorption (mg g−1); QmL, KL, θ, and RL are the Langmuir maximum sorption capacity (mg g−1), Langmuir constant (L mg−1), surface coverage, and dimensionless constant, respectively; QmF, KF, and n are the Freundlich maximum sorption capacity (calculated), sorption constant, and sorption intensity, respectively; Rf denotes retardation factor; α0, α1, and α2 are the regression parameters calculated using polynomial equation; ΔG, ΔH, and ΔS represent the change of Gibbs free energy, enthalpy and entropy, respectively. | ||
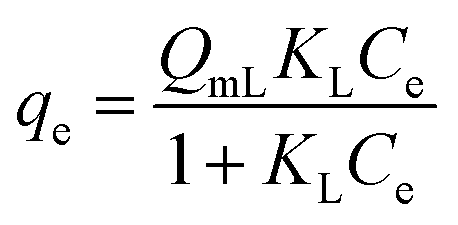
|
(1) | 31,32 |
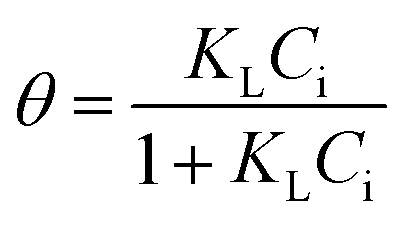
|
(2) | 16 |
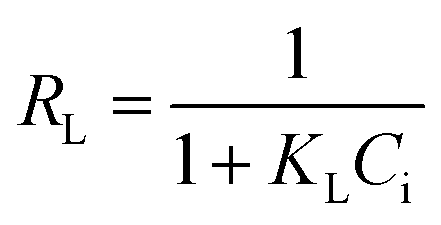
|
(3) | 16 |
| q e = KFCe1/n | (4) | 31,32 |
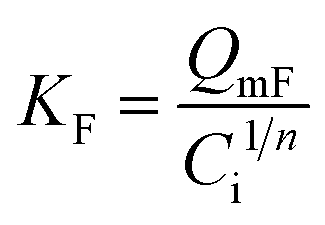
|
(5) | 67 |
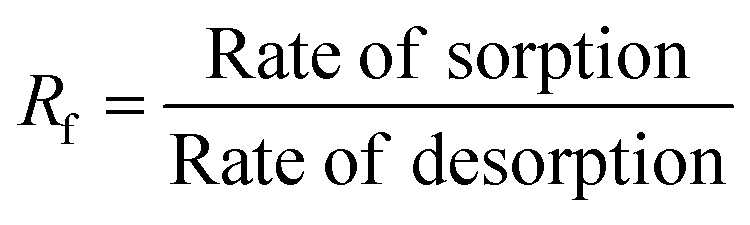
|
(6) | 33,34 |
| ΔG = −RTLnKL | (7) | 58 |
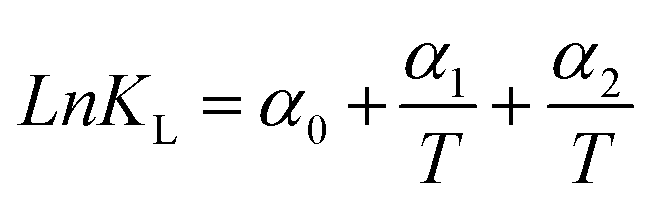
|
(8) | 35–37 |
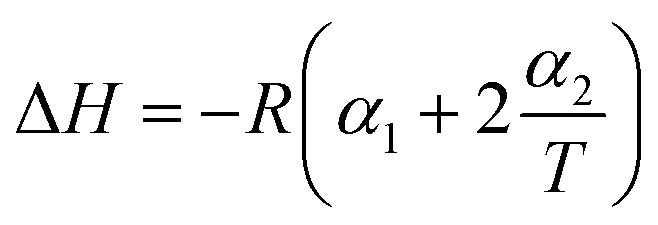
|
(9) | 36 |
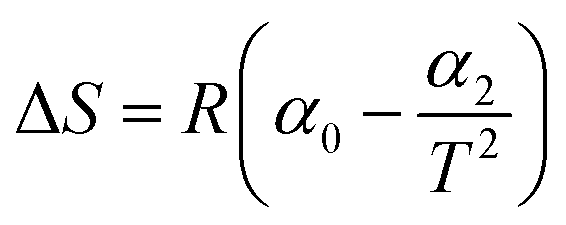
|
(10) | 36 |
Solution speciation chemical modelling using visual MINTEQ 3.1 was run at different pH to interpret the laboratory data; taken into consideration here were the interaction of Be with different electrolytes, competing ions and counter ions in solution under similar experimental conditions of temperature, electrolytes, concentrations of added ions, and concentration Be as run in the laboratory experiments.
The non-linear sorption and desorption isotherms of Be were plotted [i.e. amounts of Be (mg g−1) in the solid-phase (sorbed) vs. amounts of Be (mg L−1) in the liquid phase at equilibrium] and fitted using the Langmuir and Freundlich models.31,32 The different parameters in the sorption–desorption phenomena were calculated as shown in Table 1. The amount of retention (mostly specific sorption) was estimated after each desorption cycle (Retention = total sorption − desorption in each step).32 Dimensionless distribution coefficient is often referred to as the retardation factor (Rf) (eqn 6), which is an effective tool for describing the interaction and transport of contaminants (e.g. Be) in the environmental matrix (soil, sediments, rock, and geological fraction).33,34 If the temperature does not exhibit a linear effect on the sorption–desorption of metal, then it may make better sense to use non-linear Van't Hoff equation to calculate ΔH, and ΔS (eqn (8)–(10)).35–37
3 Results and discussion
3.1. Physicochemical properties of studied soil
The studied soils are acidic in nature (pHCaCl2 = 4.60 ± 0.17; pHwater = 6.06 ± 0.13), sandy loamy classification, high organic matter contents (6.37 ± 0.75%) and water holding capacity (56%) (Table S1†). Quartz is the predominant mineral in the soil, including small amounts of other clay minerals (Fig. S1†). Porous structure (meso and micropores) of soils were confirmed by BET surface area analysis (Fig. S2†), as well as SEM and TEM analysis (Fig. S3†). Details are discussed in the previous study12 and also in the ESI.†3.2 Effect of ionic strength and different added ions on the surface charge of soil
The pHzpc of LFLS soil was 5.24, 4.95, 4.65 in the presence of Milli-Q water, 0.01 M NaNO3 and 0.1 M NaNO3, respectively, representing the diminishment of pHzpc with increasing ionic strength (Fig. 1A). The release of protons increases (exchange with cations) when electrolytes are added to the soil suspension, resulting in falling pH, which is linked with Hzpc. Naidu et al.38 reported that with increasing ionic strength, the positive charge fell below pHzpc, and negative charges rose above pHzpc, which resulted in shifting pHzpc toward the lower value. A similar result was also reported by Appel et al.39 and is consistent with this study.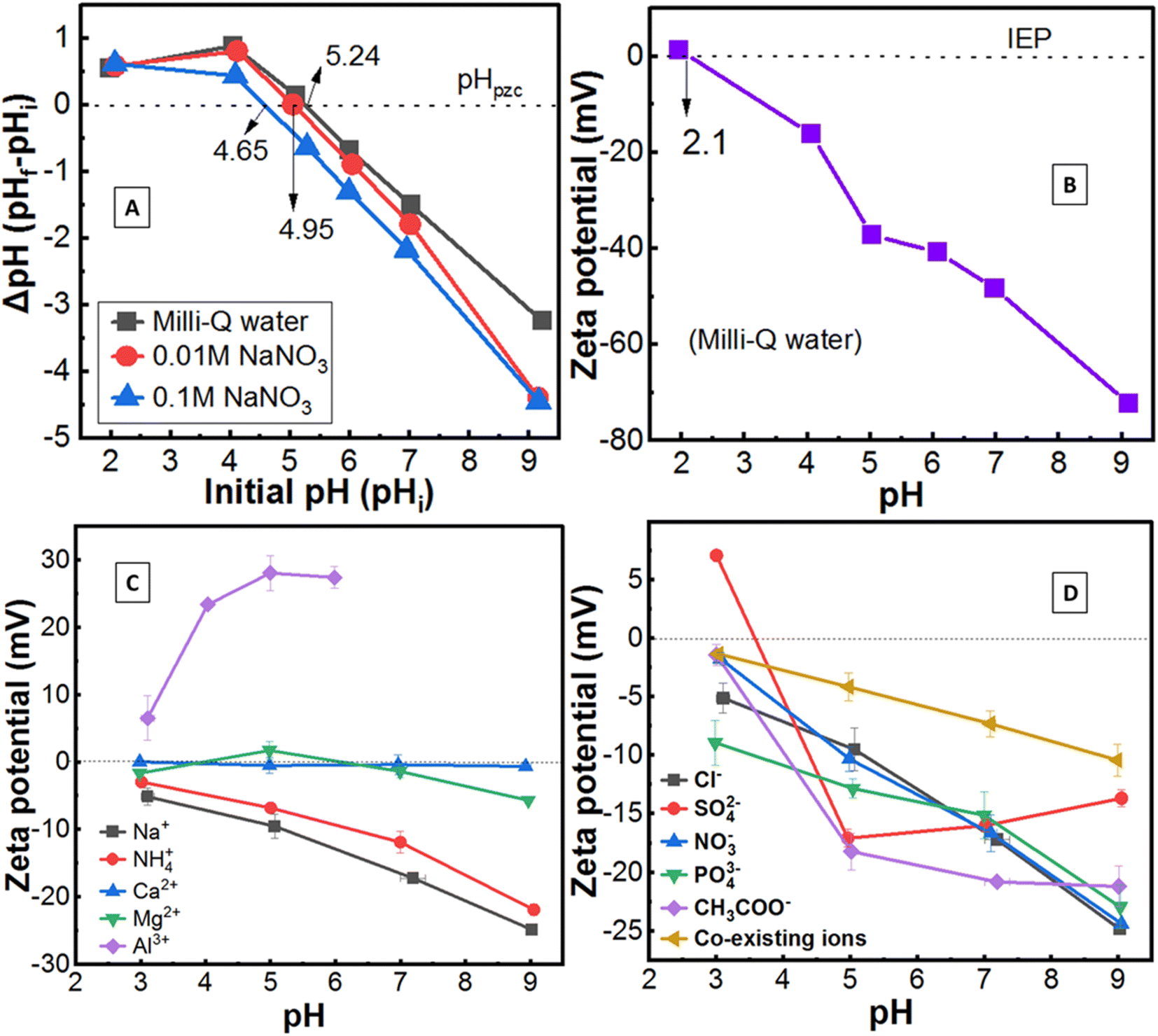 | ||
| Fig. 1 Surface charge of LFLS soil. (A) Effect of ionic strength on the pHzpc; (B) surface charge of soil in Milli-Q water; (C) effect of competing ions (0.1 M of each chloride salt); (D) effect of counter ions (0.1 M of each sodium salt) and co-existing ions (0.01 M of each of the above salts as nominated by competing ions and counter ions) on the surface charge of soil. | ||
On the other hand, the zeta potential method (Fig. 1B) revealed the net zero surface charge of soil at pH 2.1 (referred to as IEP), but pH difference methods showed pHzpc at pH 5.24 under a similar experimental condition using Milli-Q water. The pH difference method (pHzpc) measured only H+ and OH− ions, whereas zeta potential method measured all kinds of charges on the soil surface and found pHzpc > IEP. Suggested here is that the soil has very high permanent negative surface charges (probably specific sorption site, originated from organic–inorganic elements and edges of minerals), along with some free OH− ions at the soil field pH ∼5.5. Liu et al.40 reported specific sorption would be the predominant mechanism if pHpzc and IEP are not identical, which may be applied in this study.
The addition of monovalent cations (Na+, NH4+) increased net negative surface charge from −2.9 mV to −25 mV with increasing pH from 3 to 9; divalent cations (Ca2+, Mg2+) showed limited effect [zeta potential is nearly zero (+0.06 mV to −5.7 mV) at all pH]; while trivalent cation (Al3+) increased positive zeta potential (+6.5 mV to +27 mV) rather than any negative value (Fig. 1C). This indicates the order of negative zeta potential value as follows: monovalent > divalent > trivalent cations, which is consistent with the other research on minerals.18,25,41 However, Tunç and Duman25 reported negative zeta potential value increased with increasing ionic strength from 1 × 10−5 to 1 × 10−2 M, then declined. In contrast, Saka and Guler18 demonstrated zeta potential diminished (−24 mV to −12 mV) with increasing ionic strength (1 × 10−4 M to 1 × 10−1 M) of monovalent electrolytes due to the compression of the electric double layer thickness, which is also applicable in this study (see Fig. 1B and C). In the case of Al3+, the positive hydrolysis ions increase with rising pH up to 5, then slightly drop due start precipitation,18,25,42 which was related to the value of zeta potential (Fig. 1C).
For monovalent counter ions (Cl−, NO3−), the negative zeta potential value increased sharply with increasing pH (Fig. 1B), which might be due to the adsorption of these ions on the soil surface.18 Zeta potential of CH3COO− did not significantly change at pH > 6, which could be attributed to the loss of CH3COO− by different complex formations.43 Surprisingly, the value increased from +7.0 mV to −17 mV between pH 3 to 5 for divalent anion (SO42−), and then a small decline was recorded. Based on the concentrations of SO42− ion, different types of zeta potential curves may be obtained,44 which suggests that further study for soil is required. Almost similar behaviour of mono and trivalent anion (PO43−) on the surface charge of soil was found, which meant that the PO43− ion was not a potentially determining ion since it was also reported earlier.25 Relatively lower surface charges were noticed while considering co-existing ions, which could be attributed to the presence of a relatively lower concentration of ions (0.01 M) in the solution.18
3.3 Environmental chemistry of Be is regulated by diverse soil–water conditions
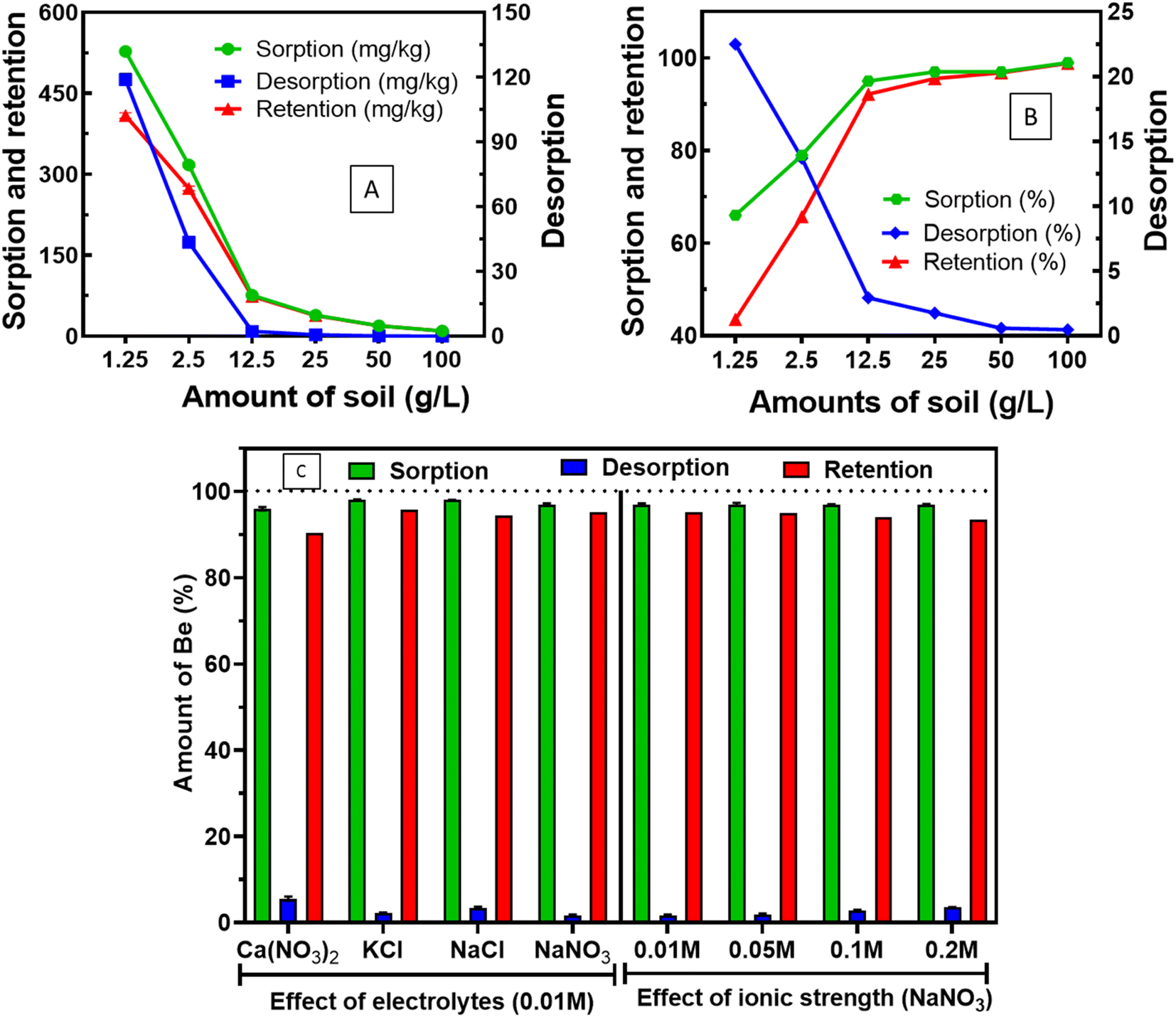 | ||
| Fig. 2 Amounts of soil in the suspension affect sorption, desorption (right axis in A and B) and retention behaviour of Be ion soils at pH 5.5. (A) Sorption per unit mass; and (B) sorption percentage at different soil loading; (C) effect of electrolytes and ionic strengths. | ||
In all ranges of soil loading, we found Kd (desorption) > Kd (sorption), which indicates the presence of hysteresis.45 Moreover, Islam et al.12 investigated the presence of desorption hysteresis at 33.3 g L−1 soil loading. In this study, we confirmed the presence of desorption hysteresis with a wide range of soil loading (1.25–100 g L−1). The interaction of Be with the amounts of soil is important since soil loading modifies reaction equilibrium46 or the aqueous phases chemistry of index metal ions, which also influences the sorption–desorption mechanism of metals.9 We have optimised soil loading at 12.5–25 g L−1 for higher sorption and lower desorption in 24 h at pH 5.5.
Islam et al.12 demonstrated that sorption decreased in the presence of electrolytes (0.01 M NaNO3) compared to the absence of background electrolyte (ultrapure water) at pH 5, whereas this study revealed that the types of electrolytes showed a marginal effect at pH 5.5. In addition, increased ionic strength (0.01 M to 0.2 M) of the same electrolyte (e.g. NaNO3) did not show any significant effect on the sorption (97%), and desorption (1.74–3.58%) of Be (Fig. 2C). Boschi and Willenbring29 reported 8.4–37% Be desorption (from sorbed amount by potentially outer-sphere complexation or electrostatic interaction) from different minerals, and organic ligands under different electrolytes and ionic strengths at pH 3. The limited desorption over a range of electrolytes and ionic strength in this study at pH 5.5 strongly suggests that sorption was specifically caused by chemical complexation, but pH can be an important consideration.
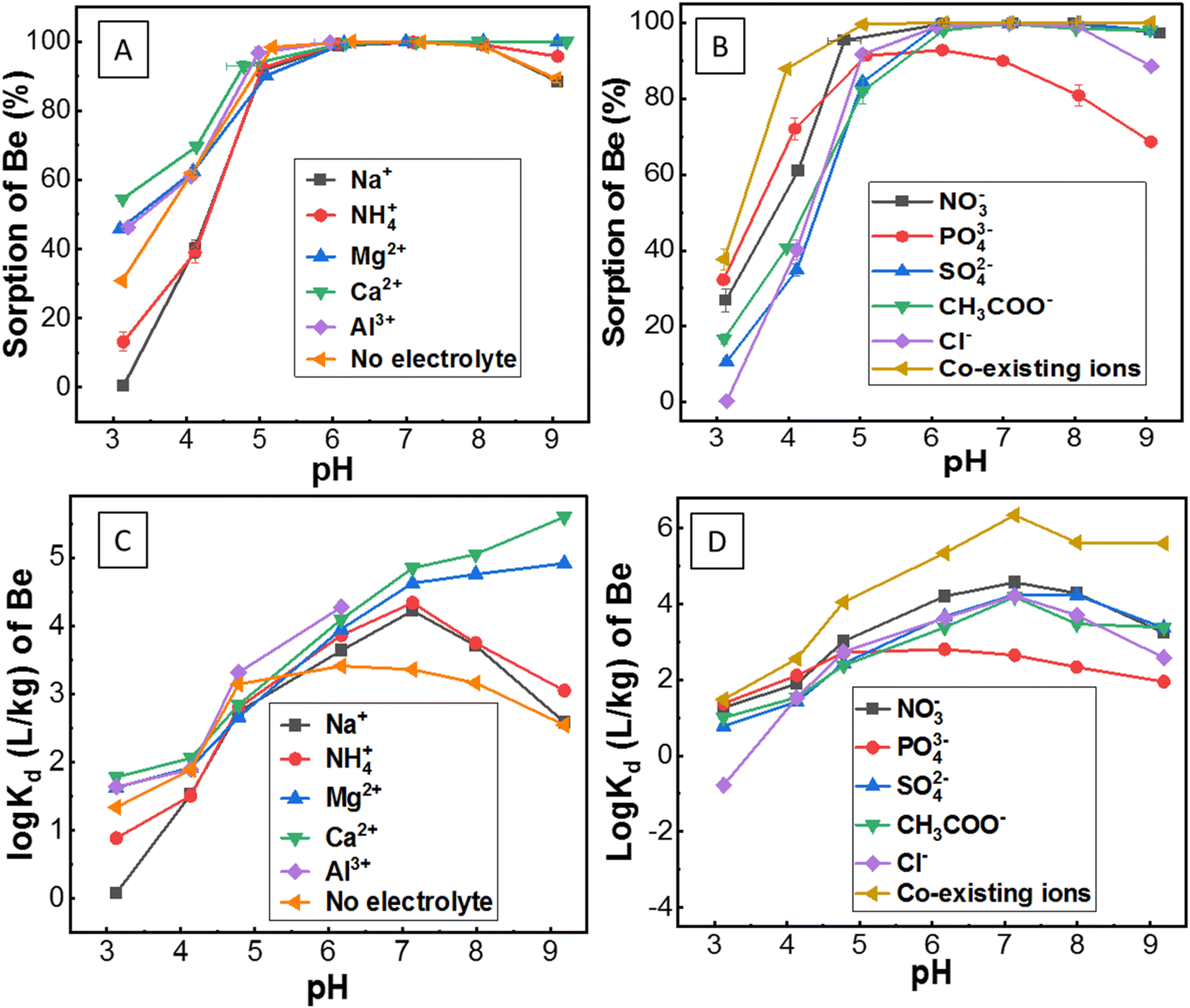 | ||
| Fig. 3 (A) Sorption of Be (initial concentration 1 mg L−1) in the presence of different competing ions; (B) counter ions, and co-existing ions as a function of pH; (C) and (D) represent distribution coefficient (Kd) of Be under similar sorption conditions as stated above [for details of the electrolyte solution, refer to the caption of Fig. 1]. | ||
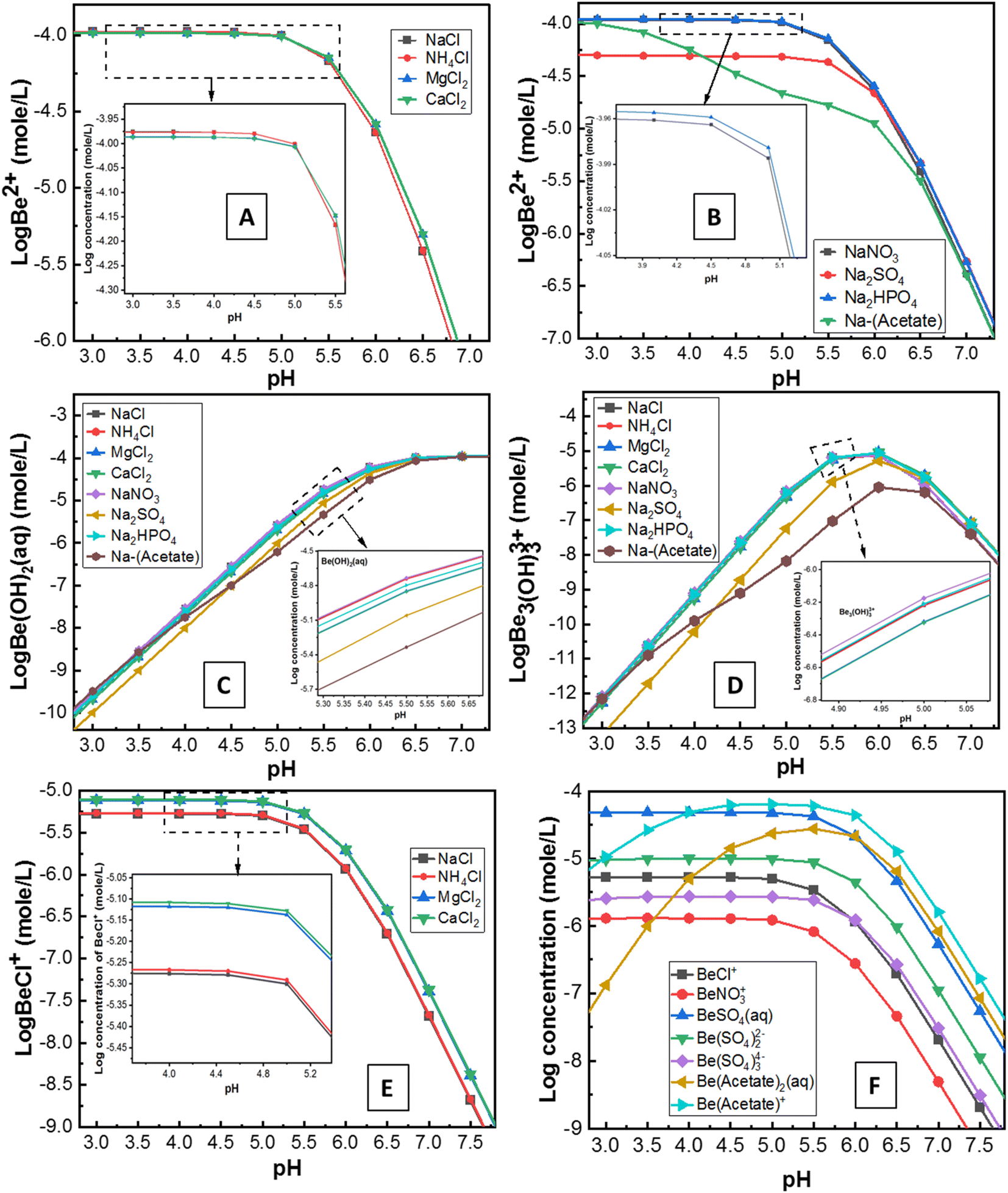 | ||
| Fig. 4 Solution speciation chemical modelling data using visual MINTEQ 3.1 under similar experimental conditions of pH, temperature (25 °C), concentrations of added ions (0.1 M), and Be (1 mg L−1) (see sections 2.4.3 and 3.2). (A) Effect of competing ions and (B) counter ions on Be2+ in the presence Cl− and Na+, respectively. Similarly, effect of competing + counter ions on the formation of (C) Be(OH)aq and (D) Be(OH)33+ (predominant species of Be in solution); (E) effect of chloride salt (electrolytes) on the BeCl+ formation; (F) different Be complexes formation. | ||
Although the surface charge was highly positive in the presence of Al3+, surprisingly, sorption was still evident. The reason for this is unknown, but potentially, it could be attributed to: (1) competition and exchange with Al on the soil surface; or/and (2) hydrolysis and co-precipitation of Be with Al due to the similar electrochemical and aqueous solution behaviour between them.50,51 Al3+ (AlCl3) in solution concentrations ranging from 1 × 10−5 M to 1 × 10−1 M remained in non-hydroxyl form at pH 3.2–6.5, where Al3+ formed different hydroxyl complexes with pumice as reported by Tunç and Duman.25 These mechanisms may also be applied in this study for similar conditions (0.1 M Al3+; pH 3–6) of experiments in the soil suspension. Moreover, Zhang et al.51 reported that if Al ions move into the sorption surface of clay minerals or colloids, the Be ion will be displaced and leach into ground water. However, the reverse of this mechanism (Al3+ ion in solution may accelerate the sorption of Be2+ in soils by exchange or co-precipitation) was apparent in this study. Simulation data (Fig. S4†) showed Al(OH)4− formation increased up to pH 6, which might have a direct relationship with Be sorption (Fig. 3A). However, concentrations of AlCl3 in solutions can be an important consideration since it wields significant effects on the solubility, hydrolysis and sorption–desorption phenomena of both Al and Be in the solution (Fig. S5†).
However, the mechanism of Be complexation with SO42−, and Cl− is still not well understood. Rudolph et al.48 reported SO42− can penetrate the first hydration sphere of Be, and form stable Be-sulphate complexes [e.g. Be(OH2)3OSO3], whereas Lim et al.53 noted ion-pair formation rather than a stable complex. Nevertheless, stable complexes of Be with S-donors ligands were reported previously.4,14 Ion-pair formation (e.g. BeSO4) or anionic complexation [e.g. Be(SO4)22−, Be(SO4)34−] of Be with sulphate was found in this study using MINTEQ simulation (Fig. 4F). Sorption of Be with Cl− was not always higher than the other counter ions. Nonetheless, the amount of Cl− exerts a substantial effect on increasing the sorption of Be, resulting from the decrease of Be2+ (Fig. 4A) and increase of BeCl+ (Fig. 4E and F). Previous studies13,48 reported that Cl− forms a complex with Be, but most of these are often less stable (ion-pair formation) than the other ligands. Rudolph et al.48 demonstrated most of the BeCl2 in the solution were outer-sphere types, with Cl− ion not penetrating the first hydration sphere, which may explain why Cl− showed relatively less sorption than the other counter ions such as NO3− (Fig. 3B).
Sorption was relatively higher (27–95% at pH 3–5) in the presence of NO3−, due to strong complexation by nitrogen ligands using both pi (lone pair) and sigma (bond pair) electrons, as was recently reported by Peplow.54 Furthermore, NO3− in the solution can form [Be(NO3)4]2−, aqueous Be(NO3)2 and eventually, the crystal structure of Be nitrate complexes including (NO)2Be(NO3)4, and Be4O(NO3)6 at different conditions.48,55,56 Consequently, BeNO3+ formation was comparatively lower (Fig. 4F), but sorption of Be was relatively higher in the presence of NO3− (Fig. 3B), which suggests different sorts of Be complexation may occur with nitrogen ligands in the soil suspension, echoing a similar founding in the literature.30
Surprisingly, the sorption pattern of Be in the presence of PO43− ligand (increasing from 32% to 92% from pH 3 to pH 6, then decreasing to 68% up to pH 9) differed from other ligands as described above, which could be related to the competing hydration or precipitation of PO43− ions as reported elsewhere.4,13,57 Earlier studies4,14,15 reported only phosphate/phosphonate ligands could compete with H2O ligands in aqueous solution to form stable Be–phosphate complexes than many other ligands.
Although sorption was almost constant at pH > 6 for all studied ions above, the Kd increased slowly up to pH 7 (majority of the cases) (Fig. 3C and D), which is susceptible to the contribution of the precipitation mechanism of Be at higher pH.13Kd was lower in the presence of monovalent than divalent cations, which is because of higher competition and lower ion-pair formation (due to lower amounts of Cl− ion present in the monovalent electrolytes) as stated above. It is clearly visible that the Kd was relatively higher in the presence of NO3− ion and co-existing ions, which is consistent with the data of larger amounts of Be sorption.
3.3.6.1 Sorption. At constant pH 5.5, soil/solution ratio (20 g L−1), ionic strength and electrolyte (0.01 M NaNO3), the sorption of Be was also controlled by the amount of Be present in solution and different temperatures, as shown in Fig. 5. Results showed that increasing the amount of Be in solution (under a constant mass of soil) resulted in enhancing sorption of Be per unit mass of soil (Fig. 5A), which could be attributed to the displacement of more sorption sites occupied ions by Be. At low Be loading, only specific (high energy) sorption sites were occupied by Be, but at higher loading, high energy sorption sites were occupied first, and then non-specific low energy sites were gradually captured as reported for other metals.9,45,58 However, percentage sorption declined with increasing concentration (Table 2), which indicated the number of Be ions far exceeded the number of available sorption sites at higher concentrations; similar findings have been published on other metals.9
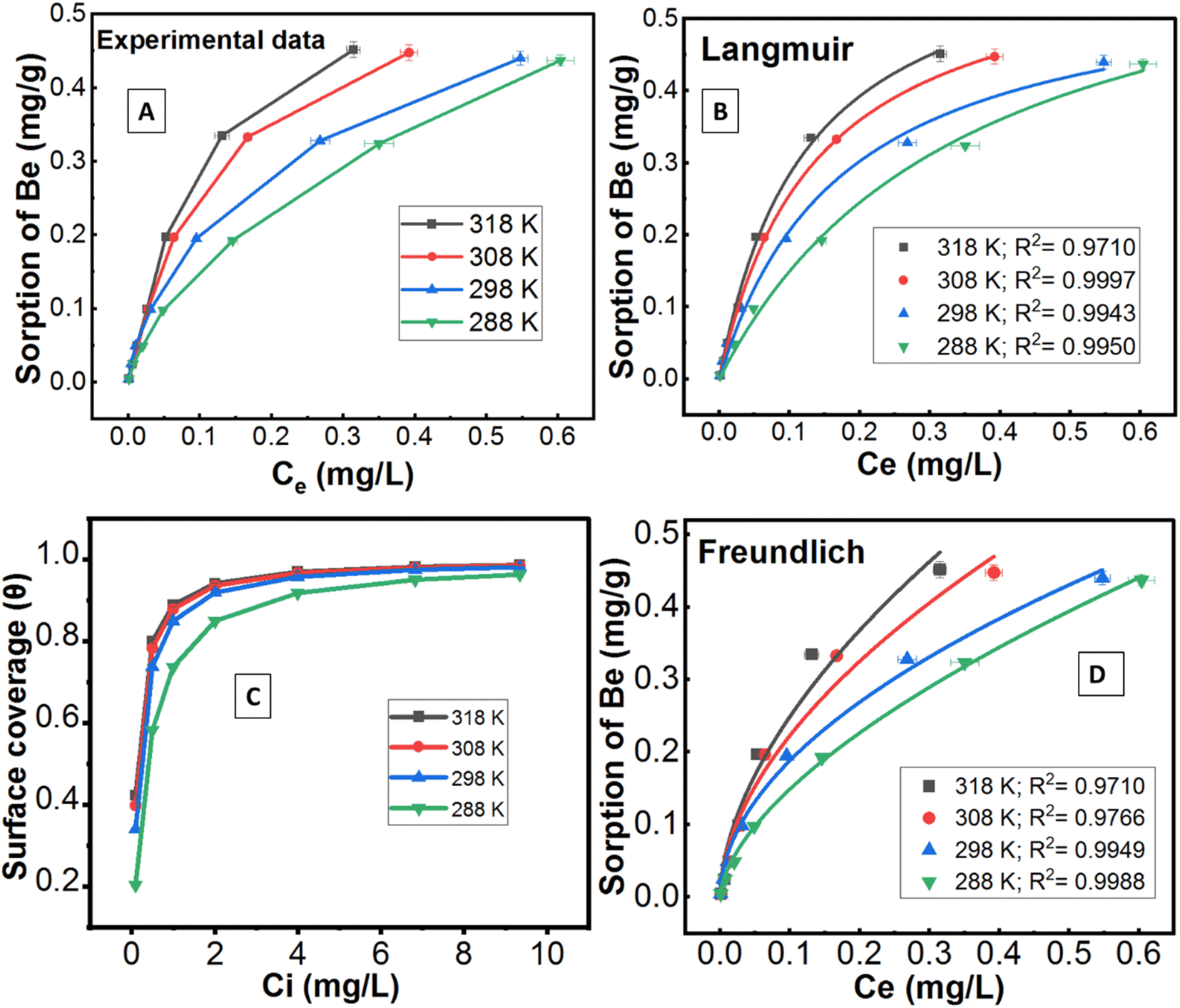 | ||
| Fig. 5 (A) Amount of Be sorption (mg g−1) from experimental data at different temperatures (288–318 K); (B) non-linear Langmuir sorption model of Be; (C) coverage of soil surface (θ) by Be (calculated from Langmuir model) as a function of initial Be concentration; (D) Freundlich sorption model. | ||
| Concentrations (mg L−1) | Sorption (%) | Total desorption (%) | R* (%) | R f | K dr (L kg−1) |
|---|---|---|---|---|---|
| a R* = retention of Be (%) after four cycles; Rf= retardation factor = sorption rate/desorption rate; Kdr = distribution ratio (Kdr = Kd-sorption/Kd-desorption). | |||||
| 0.1 (288 K) | 98.9 ± 0.36 | 3.66 ± 0.90 | 95.2 ± 0.59 | 128 ± 28 | 0.757 |
| 0.5 (′′) | 98.6 ± 0.023 | 4.16 ± 0.56 | 94.4 ± 0.37 | 101 ± 12 | 0.706 |
| 1.0 (′′) | 98.0 ± 0.18 | 5.00 ± 0.40 | 93.0 ± 0.27 | 85.4 ± 8.5 | 0.572 |
| 2.0 (′′) | 97.6 ± 0.17 | 5.46 ± 0.40 | 92.1 ± 0.27 | 77.8 ± 4.9 | 0.523 |
| 4.0 (′′) | 96.3 ± 0.012 | 7.08 ± 0.23 | 89.3 ± 0.07 | 58.7 ± 1.5 | 0.457 |
| 7.0 (′′) | 94.9 ± 0.45 | 8.57 ± 0.64 | 86.3 ± 0.61 | 47.6 ± 3.5 | 0.401 |
| 10 (′′) | 93.5 ± 1.03 | 9.15 ± 0.81 | 84.3 ± 1.24 | 44.5 ± 4.2 | 0.339 |
| 0.1 (298 K) | 99.3 ± 0.04 | 2.63 ± 0.62 | 96.6 ± 0.15 | 177 ± 47 | 0.761 |
| 0.5 (′′) | 99.1 ± 0.13 | 2.89 ± 0.16 | 96.3 ± 0.12 | 141 ± 8.4 | 0.852 |
| 1.0 (′′) | 99.0 ± 0.04 | 3.68 ± 0.15 | 95.3 ± 0.04 | 112 ± 5.2 | 0.860 |
| 2.0 (′′) | 98.4 ± 0.06 | 3.76 ± 0.13 | 94.7 ± 0.09 | 109 ± 3.9 | 0.584 |
| 4.0 (′′) | 97.6 ± 0.06 | 5.21 ± 0.33 | 92.4 ± 0.14 | 78.3 ± 4.6 | 0.530 |
| 7.0 (′′) | 96.1 ± 0.39 | 6.65 ± 0.59 | 89.4 ± 0.54 | 61.2 ± 5.4 | 0.411 |
| 10 (′′) | 94.1 ± 0.39 | 8.12 ± 0.33 | 86.0 ± 0.47 | 49.7 ± 2.0 | 0.330 |
| 0.1 (308 K) | 98.6 ± 0.24 | 2.82 ± 0.51 | 95.8 ± 0.22 | 159 ± 25 | 0.473 |
| 0.5 (′′) | 98.8 ± 0.05 | 3.62 ± 0.28 | 95.1 ± 0.09 | 114 ± 9.9 | 0.709 |
| 1.0 (′′) | 98.6 ± 0.15 | 4.24 ± 0.30 | 94.4 ± 0.23 | 97.6 ± 7.3 | 0.767 |
| 2.0 (′′) | 98.6 ± 0.05 | 4.11 ± 0.16 | 94.5 ± 0.09 | 101 ± 4.3 | 0.715 |
| 4.0 (′′) | 98.4 ± 0.01 | 4.81 ± 0.11 | 93.6 ± 0.02 | 85.6 ± 2.4 | 0.728 |
| 7.0 (′′) | 97.6 ± 0.07 | 5.88 ± 0.18 | 91.7 ± 0.11 | 69.1 ± 1.8 | 0.586 |
| 10 (′′) | 95.8 ± 0.07 | 8.08 ± 0.30 | 87.7 ± 0.14 | 50.6 ± 2.1 | 0.460 |
| 0.1 (318 K) | 98.9 ± 0.15 | 3.87 ± 0.32 | 95.1 ± 0.23 | 116 ± 9.8 | 0.832 |
| 0.5 (′′) | 98.8 ± 0.12 | 3.79 ± 0.35 | 95.0 ± 0.14 | 112 ± 10 | 0.727 |
| 1.0 (′′) | 98.8 ± 0.11 | 4.35 ± 0.25 | 94.4 ± 0.16 | 95.3 ± 5.2 | 0.866 |
| 2.0 (′′) | 98.7 ± <0.01 | 4.92 ± 0.20 | 93.8 ± 0.05 | 83.1 ± 3.2 | 0.917 |
| 4.0 (′′) | 98.7 ± 0.07 | 4.65 ± 0.16 | 94.0 ± 0.10 | 88.4 ± 3.6 | 0.861 |
| 7.0 (′′) | 98.1 ± 0.12 | 6.05 ± 0.11 | 92.0 ± 0.12 | 67.4 ± 1.3 | 0.773 |
| 10 (′′) | 96.6 ± 0.25 | 7.62 ± 0.17 | 89.0 ± 0.45 | 53.9 ± 5.5 | 0.544 |
The sorption data closely fitted both the Langmuir and Freundlich sorption isotherm models (Fig. 5B and D), but the superior fit was noted in the Langmuir model based on the high regression value (R2 = 0.9943 − 0.9997), low chi-square value (X2 = 1.0 × 10−5 to 1.9 × 10−4) and low average percentage error (APE = 2.08–3.96%) between experimental and theoretical data (Table 3). This outcome suggests potential multilayer sorption is possible after complete monolayer coverage.12,16 However, Langmuir's maximum sorption capacity (QmL) was 0.568–0.679 mg g−1 under different temperatures (288–318 K). According to the modelling parameters (Table 3), sorption was favourable (KL > 0, and 0 < RL < 1)12 with a predominant chemisorption mechanism (n > 1, QmF = 1.94–3.22 mg g−1) under experimental conditions.16 Dimensionless constant (RL) and surface coverage (θ) are related to the binding energy and initial concentration of Be, and the result showed surface coverage escalated with rising initial concentration. After a certain point, the plateau curve was noticed as shown in Fig. 5C, which represents complete coverage of the sorption site (present on the soil surface) by Be. Following this, some non-specific sorption may occur, and it would be reversible sorption.
| Parameters | 288 K | 298 K | 308 K | 318 K |
|---|---|---|---|---|
| a R 2, X2, and APE% are the linear regression coefficient, chi-square, and average percentage error, respectively. Subscripts s, and d represent sorption and desorption, respectively. ‘*’ denotes average values of four desorption cycles. | ||||
| Q mL (mg g−1) | 0.679 ± 0.056 | 0.568 ± 0.034 | 0.605 ± 0.008 | 0.635 ± 0.024 |
| K L (L mg−1) | 2.82 ± 0.481 | 5.69 ± 0.873 | 7.31 ± 0.233 | 8.091 ± 0.697 |
| R L | 0.037 | 0.018 | 0.014 | 0.013 |
| θ | 0.963 | 0.982 | 0.986 | 0.987 |
| R 2 | 0.9950 | 0.9943 | 0.9997 | 0.9978 |
| X 2 | 1.7 × 10−4 | 1.9 × 10−4 | 1.0 × 10−5 | 7.6 × 10−5 |
| APE% | 3.96 | 2.08 | 2.51 | 2.90 |
| Q mF (mg g−1) | 2.32 | 1.94 | 2.65 | 3.22 |
| K F (mg g−1).(L mg−1)1/n | 0.599 ± 0.011 | 0.615 ± 0.023 | 0.783 ± 0.074 | 0.913 ± 0.108 |
| n (mg g−1) | 1.65 ± 0.044 | 1.95 ± 0.101 | 1.83 ± 0.186 | 1.77 ± 0.197 |
| R 2 | 0.9988 | 0.9949 | 0.9766 | 0.9710 |
| X 2 | 4.1 × 10−5 | 1.7 × 10−4 | 8.1 × 10−4 | 1.0 × 10−3 |
| APE% | 30.7 | 24.3 | 35.1 | 43.8 |
| ΔGs (kJ mole−1) | −2.48 | −4.31 | −5.09 | −5.53 |
| ΔHs (kJ mole−1) | −43.8 | −63.2 | −81.3 | −98.3 |
| ΔSs (kJ K−1 mole−1) | +0.034 | −0.032 | −0.092 | −0.147 |
| *ΔGd (kJ mole−1) | −2.87 | −5.02 | −5.20 | −4.62 |
| *ΔHd (kJ mole−1) | 206 | 161 | 118 | 78.4 |
| *ΔSd (kJ K−1 mole−1) | 0.495 | 0.340 | 0.199 | 0.072 |
Thermodynamic parameters using the non-linear Van't Hoff equation revealed that the sorption process was exothermic (ΔH = −43.8 kJ mole−1 to −98.3 kJ mole−1), spontaneous (ΔG = −2.48 kJ mole−1 to −5.53 kJ mole−1)) at any temperature and entropically (average ΔS = −0.059 ± 0.067 kJ K−1 mole−1) influenced (Table 3), which was similar to the Cd sorption (Cd and Be are divalent cations) as reported by Rafatullah et al.59 In one study, El-Soad et al.16 demonstrated the exothermic process of Be sorption using sulfonated chitosan, but it may be both exothermic and endothermic48,60–62 depending on the available ligands, types of surface, and chemical reaction. However, previous studies51,58 reported chemisorption would be the predominant sorption mechanism of metals and non-metals if ΔH > 40 kJ mole−1, which may also be applied in this study for Be sorption since we found ΔHs from −44 kJ mole−1 to −98.3 kJ mole−1 considering temperature from 288 K to 318 K. A small negative value of ΔS (near to zero) represents waning randomness, no structural change/deformation of the adsorbent materials (e.g. soil) during sorption, resulting in the irreversibility and stability of the sorption process of Be, as it has been reported for other metals.59,63,64
3.3.6.2 Desorption. Soil showed very low desorption (average 0.42–2.6% from each desorption cycle; total 2.6–9.2% considering four cycles) from the sorbed amount (Fig. 6 A–D), including all concentrations and temperatures studied. It suggested specific sorption of Be with soil since reversible sorption of metal usually occurs from the non-specific sorption site.9 Desorption was relatively higher (0.80–2.6%) in the first cycle (Fig. 6) (Langmuir desorption maximum was also high, see Table S2 and Fig. S6†), which suggests desorption occurred from either outer-sphere complexation or may be from physically sorbed or entrapped Be. Desorption increased with the increasing loaded concentration of Be (Table 2), indicating some non-specific sorption with low energy sorption sites or physical sorption at higher concentrations. Non-specific or physical sorption is susceptible to easy desorption as it was reported previously for other metals.45,58
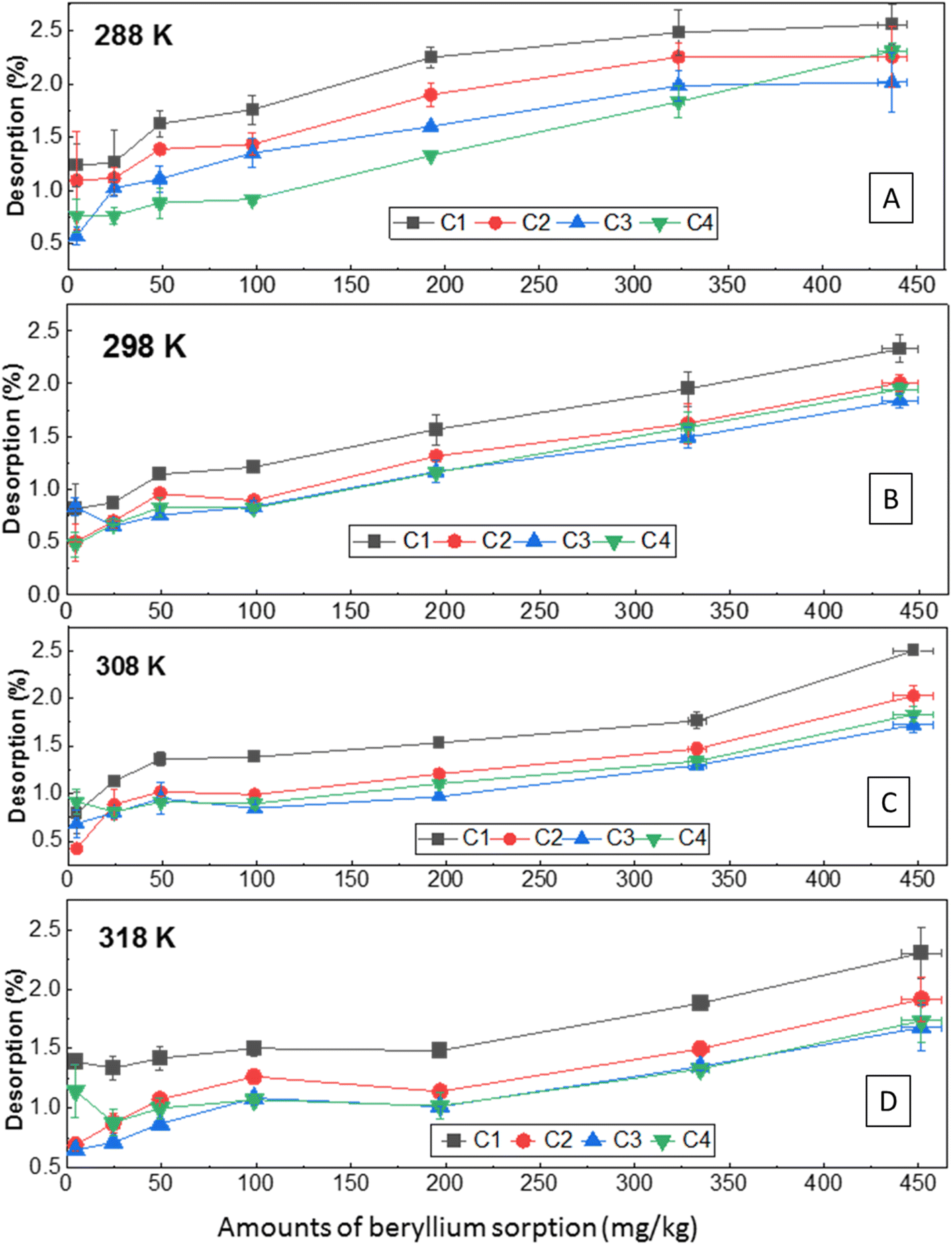 | ||
| Fig. 6 Desorption of Be from the sorbed amount at different initial concentrations and temperatures under four successive desorption cycles (C1, C2, C3, C4). | ||
However, the total amount of desorption (0.16–40 mg kg−1) was comparatively higher at 288 K than at other temperatures considering all concentrations of Be (Fig. S7†). Higher desorption at 288 K was also confirmed by higher desorption maxima using the Langmuir desorption isotherm model (Table S2†). However, non-linear relationships of Be desorption with temperature were revealed (Fig. S7†), which was also reflected by the irregular value of ΔGd (Table 3), and Langmuir constant (KLd) in the desorption phenomena. With rising temperature, metal desorption may either decrease or increase, exhibiting some effect of initial metal loading as reported previously,58,65 but specific data for Be was not found; further study is needed to confirm these observations. In the first three cycles, KLd increased from 288 K to 308 K, and then started to decrease, but no remarkable differences were noted in the fourth cycle (retention of specifically sorbed Be) at all temperatures. Indicated here is that reversible sorption is not affected by temperature, particularly if the sorption occurs with specific sorption sites. However, the desorption reaction was spontaneous (ΔGd = −2.87–5.20 kJ mole−1), endothermic (ΔHd = +78.4 to +206 kJ mole−1), and entropically influenced (ΔSd > 0). The higher positive value of ΔH in the desorption process represents reversible sorption reactions require relatively more energy to desorb from the sorbed Be.
3.4 Retardation/retention and hysteresis
Soil showed very high retention of Be after each desorption cycle, resulting in total retention from 84.3 ± 1.24% to 96.6 ± 0.15%, considering all concentrations (retention decrease with rising Be concentration) and temperatures. Moreover, retardation factor (Rf) decreased from 177 ± 47 to 44.5 ± 4.2 with rising to Be concentrations from 0.1 mg L−1 to 10 mg L−1. This indicates that the mobility or transport of Be in the LFLS soil can be limited if the level of concentration/contamination is comparatively lower since at smaller concentrations of Be, specific sorption is predominantly occurred, which resulted in increasing the degree of Be retention/retardation in the studied soil. However, the temperature did not show any linear relationship (Table 2) with the concentration of Be, which is also consistent with the above data.K d was relatively higher at higher temperatures (Fig. 7A), which is consistent with the sorption data (Fig. 5). This indicates lower mobility of Be in the environment at higher temperatures. In addition, Kd-sorption of soil was 738–6676 L kg−1, while Kd-desorption was 2175–8775, which represents limited transport or higher retardation resulting from the presence of desorption hysteresis (Kd-desorption > Kd-sorption = Kdr < 1) as it was demonstrated by Aşçi et al.45 for heavy metals. However, Vega et al.32 reported that the retention mechanism of metals in the soil is unclearly defined and often referred to as “sorption”, but sorption, surface precipitation and fixation are involved in the contiguous solid-phase during the sorption process. Only the metals with surface precipitation and outer-sphere complexation with non-specific sorption sites are being desorbed, while others are retained.9 Higher retention in this study represents chemical complexations of Be with different organic–inorganic elements in soils.
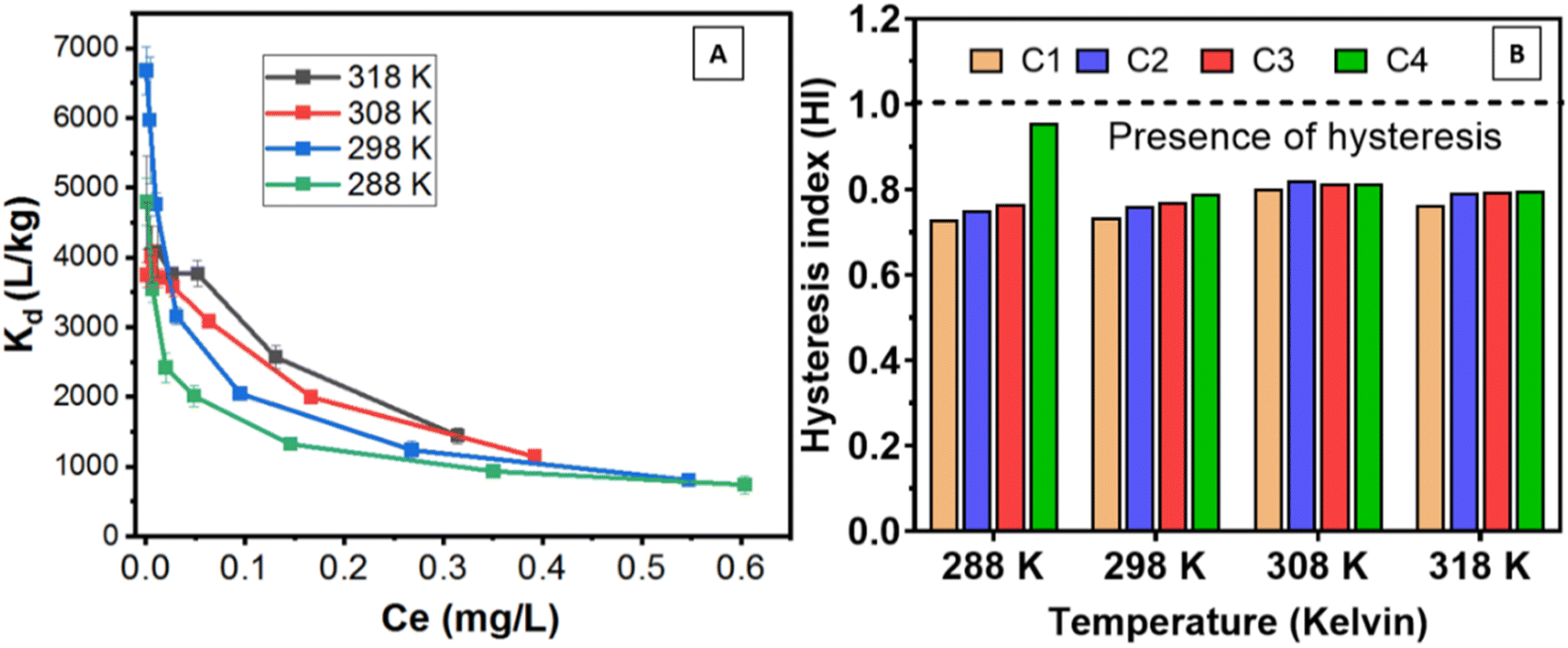 | ||
| Fig. 7 (A) Distribution coefficient (Kd) of Be sorption at different temperatures and Be loading; (B) presence of desorption hysteresis (HI < 1) of Be in all desorption cycles at different temperatures. | ||
However, hysteresis is often best described by Freundlich's sorption–desorption phenomena (e.g. n, Kf). We found desorption hysteresis of Be where both sorption and desorption intensities (n) [HI = ndesorption/nsorption < 1] were considered,12 (Fig. 7B). The sorption–desorption constant (Kf) (Kf-desorption > Kf-sorption)66 of Freundlich model (Tables 3 and S2†), including all temperatures and desorption cycles also confirmed desorption hysteresis.
The recent study by Islam et al.12 confirmed the presence of sorption–desorption hysteresis (HI = 0.68–0.84) of Be at room temperature in the LFLS soil due to the irreversible binding of Be in the clay minerals, oxyhydroxides of metal, SOM, and entrapment in the pores. In this study, we have extensively explained the sorption–desorption mechanism of Be with a wide range of environmentally relevant parameters. This research reported high sorption and limited desorption of Be, reflecting the presence of desorption hysteresis (HI = 0.73–96) irrespective of all temperatures (environmentally feasible), concentrations of Be and other environmentally relevant soil–water conditions during batch sorption–desorption study.
4 Conclusion
This study investigated how physicochemical variations in the soil-solution conditions regulate the environmental behaviour (e.g. sorption–desorption, retention or hysteresis) of Be. Results revealed that negative charges on the soil surface increased, and pHpzc fell in the presence of electrolytes as well as with increasing ionic strengths. However, the developed surface charges were not always correlated with the sorption of Be. But the presence of counter ions (originated from an electrolyte solution) is clearly important to increase Be sorption, which was observed in the laboratory data and the modelling results. Sorption was affected by the addition of different ions/ligands primarily at low pH, but no significant effect was noticed at pH > 6 due to the strong hydration and precipitation behaviour of Be. However, referring to the different ligands, the PO43− ions exerted a unique characteristic for Be sorption (suggesting they effectively compete with hydration of Be to form complexation); relatively higher sorption occurred in the presence of NO3− (utilising both lone pair and bond pair electron); and co-existing ions increased sorption (synergistic ions effect).The sorption process of Be was favourable (KL > 0, and 0 < RL < 1) under the experimental conditions. However, specific sorption was recommended at lower Be concentrations and in the presence of higher amounts of soil. Spontaneous (ΔG = −Ve) chemisorption (ΔH > − 43 kJ mole−1) reaction without affecting soil structure (ΔS = close to zero) was also suggested from the thermodynamic parameters at all temperatures. However, sorption was exothermic and reversible sorption was endothermic, which might require more energy ΔH > +78.4 kJ mole−1 to desorb Be, resulting in limited cumulative desorption (2.6–9.2%) after four desorption cycles. The non-linear relationship of temperature with desorption was noticed, and the effect of Be loading often suppressed the effect of temperature.
Higher sorption and limited desorption were substantiated with higher retention (84.3 ± 1.24% to 96.6 ± 0.15%) after four desorption cycles; higher Rf; Kd-desorption > Kd-sorption; Kf-desorption > Kf-sorption; ndesorption/nsorption < 1. These all represent the presence of desorption hysteresis under all concentrations and temperatures, delineating very specific sorption and limited mobility under diverse soil–water conditions at field pH 5.5. However, pH was the most important parameter controlling the environmental behaviour of Be in which different physicochemical diversities of soil–water conditions were substantially influential in the LFLS soil. This outcome is crucial for understanding the environmental behaviour of Be, which is highly important for effective management of Be contamination in the LFLS soil or any other similar site around the world.
Author contributions
Md. Rashidul Islam: conceptualisation, methodology, investigation, formal analysis, software, writing – original draft, writing – review and editing; Peter Sanderson: conceptualization, writing – review and editing, supervision; Mathew P. Johansen: conceptualization, writing – review and editing, supervision; Tim E. Payne: conceptualisation, writing – review and editing, supervision; Ravi Naidu: conceptualisation, resources, writing – review & editing, supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the University of Newcastle, CRC for Contamination Assessment and Remediation of the Environment, and the Australian Nuclear Science and Technology Organisation for providing financial support and instrumental facilities to complete this work. Special thanks to Global Centre for Environmental Remediation and their expert analytical team, who help with the analysis of the samples.References
- M. E. Kolanz, Introduction to beryllium: uses, regulatory history, and disease, Appl. Occup. Environ. Hyg., 2001, 16, 559–567 Search PubMed.
- K. Kreiss, G. A. Day and C. R. Schuler, Beryllium: a modern industrial hazard, Annu. Rev. Public Health, 2007, 28, 259–277 CrossRef PubMed.
- IARC, International Agency for Research on Cancer (IARC), IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. A Review of Human Carcinogens: Beryllium and Beryllium Compounds. Volume 100 C, Accessed April 2014 at http://monographs.iarc.fr/ENG/Monographs/vol100C/index.php, 2012 Search PubMed.
- V. Boschi and J. K. Willenbring, The effect of pH, organic ligand chemistry and mineralogy on the sorption of beryllium over time, Environ. Chem., 2016, 13, 711–722 CrossRef CAS.
- Y.-G. Gu, L.-G. Wang and Y.-P. Gao, Beryllium in riverine/estuarine sediments from a typical aquaculture wetland, China: Bioavailability and probabilistic ecological risk, Mar. Pollut. Bull., 2018, 137, 549–554 CrossRef CAS.
- D. A. Lindsey, H. Ganow and W. Mountjoy, Hydrothermal Alteration Associated with Beryllium Deposits at Spor Mountain, Report 2330-7102, Utah, 1973 CAS.
- E. S. Grew, Beryllium: Mineralogy, Petrology, and Geochemistry, Walter de Gruyter GmbH & Co KG, 2018 Search PubMed.
- G. Armiento, F. Bellatreccia, C. Cremisini, G. Della Ventura, E. Nardi and R. Pacifico, Beryllium natural background concentration and mobility: a reappraisal examining the case of high Be-bearing pyroclastic rocks, Environ. Monit. Assess., 2013, 185, 559–572 CrossRef CAS.
- R. D. Harter and R. Naidu, An assessment of environmental and solution parameter impact on trace-metal sorption by soils, Soil Sci. Soc. Am. J., 2001, 65, 597–612 CrossRef.
- K. H. Tan, Principles of Soil Chemistry, CRC press, 2010 Search PubMed.
- D. I. Kaplan, R. J. Seme, and M. G. Piepkho, Geochemical Factors Affecting Radionuclide Transport through Near and Far Fields at a Low-Level Waste Disposal Site, United States: N, 1995, p. 1995, Web. DOI:10.2172/28418.
- M. R. Islam, P. Sanderson, M. P. Johansen, T. E. Payne and R. Naidu, The influence of soil properties on sorption-desorption of beryllium at a low level radioactive legacy waste site, Chemosphere, 2021, 268, 129338 CrossRef CAS PubMed.
- L. Alderighi, P. Gans, S. Midollini and A. Vacca, Aqueous solution chemistry of beryllium, Adv. Inorg. Chem., 2000, 50, 109–172 CrossRef CAS.
- M. R. Buchner, Beryllium Chemistry, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, 2017, DOI:10.1016/B978-0-12-409547-2.11024-8.
- L. C. Perera, O. Raymond, W. Henderson, P. J. Brothers and P. G. Plieger, Advances in beryllium coordination chemistry, Coord. Chem. Rev., 2017, 352, 264–290 CrossRef CAS.
- A. M. A. El-Soad, M. O. Abd El-Magied, M. S. Atrees, E. G. Kovaleva and G. Lazzara, Synthesis and characterization of modified sulfonated chitosan for beryllium recovery, Int. J. Biol. Macromol., 2019, 139, 153–160 CrossRef PubMed.
- R. Naidu, R. S. Kookana, M. E. Sumner, R. D. Harter and K. Tiller, Cadmium sorption and transport in variable charge soils: a review, J. Environ. Qual., 1997, 26, 602–617 CrossRef CAS.
- E. Saka and C. Guler, The effects of electrolyte concentration, ion species and pH on the zeta potential and electrokinetic charge density of montmorillonite, Clay Miner., 2006, 41, 853–861 CrossRef CAS.
- A. M. E. Badawy, T. P. Luxton, R. G. Silva, K. G. Scheckel, M. T. Suidan and T. M. Tolaymat, Impact of environmental conditions (pH, ionic strength, and electrolyte type) on the surface charge and aggregation of silver nanoparticles suspensions, Environ. Sci. Technol., 2010, 44, 1260–1266 CrossRef.
- T. Payne, Background Report on the Little Forest Burial Ground Legacy Waste Site (ANSTO/E-780), Australian Nuclear Science and Technology Organisation, Lucas Heights, NSW, 2012 Search PubMed.
- T. E. Payne, J. J. Harrison, C. E. Hughes, M. P. Johansen, S. Thiruvoth, K. L. Wilsher, D. I. Cendón, S. I. Hankin, B. Rowling and A. Zawadzki, Trench ‘bathtubbing’and surface plutonium contamination at a legacy radioactive waste site, Environ. Sci. Technol., 2013, 47, 13284–13293 CrossRef CAS PubMed.
- T. E. Payne, Little Forest Legacy Site – Summary of site history until the commencement of waste disposal in 1960, Report no.ANSTO/E-782, ANSTO Institute for Environmental Research, 2015 Search PubMed.
- D. Cendón, C. Hughes, J. Harrison, S. Hankin, M. Johansen, T. Payne, H. Wong, B. Rowling, M. Vine and K. Wilsher, Identification of sources and processes in a low-level radioactive waste site adjacent to landfills: groundwater hydrogeochemistry and isotopes, Aust. J. Earth Sci., 2015, 62, 123–141 CrossRef.
- E. N. Bakatula, D. Richard, C. M. Neculita and G. J. Zagury, Determination of point of zero charge of natural organic materials, Environ. Sci. Pollut. Res., 2018, 25, 7823–7833 CrossRef CAS PubMed.
- S. Tunç and O. Duman, Effects of electrolytes on the electrokinetic properties of pumice suspensions, J. Dispersion Sci. Technol., 2009, 30, 548–555 CrossRef.
- A. Fotovat, R. Naidu and M. E. Sumner, Water: soil ratio influences aqueous phase chemistry of indigenous copper and zinc in soils, Soil Res., 1997, 35, 687–710 CrossRef CAS.
- T. Townsend, B. Dubey and T. Tolaymat, Interpretation of synthetic precipitation leaching procedure (SPLP) results for assessing risk to groundwater from land-applied granular waste, Environ. Eng. Sci., 2006, 23, 239–251 CrossRef CAS.
- M. R. Islam, P. Sanderson, T. E. Payne, M. P. Johansen and R. Naidu, Desorption and Migration Behavior of Beryllium from Contaminated Soils: Insights for Risk-Based Management, ACS Omega, 2021, 6, 30686–30697 CrossRef PubMed.
- V. Boschi and J. K. Willenbring, Beryllium desorption from minerals and organic ligands over time, Chem. Geol., 2016, 439, 52–58 CrossRef CAS.
- M. R. Islam, P. Sanderson, T. E. Payne, A. K. Deb and R. Naidu, Role of Beryllium in the Environment: Insights from Specific Sorption and Precipitation Studies under Different Conditions, Science of The Total Environment, 2022, 155698 Search PubMed.
- Y. Gao, A. T. Kan and M. B. Tomson, Critical evaluation of desorption phenomena of heavy metals from natural sediments, Environ. Sci. Technol., 2003, 37, 5566–5573 CrossRef CAS PubMed.
- F. A. Vega, E. F. Covelo and M. Andrade, Competitive sorption and desorption of heavy metals in mine soils: influence of mine soil characteristics, J. Colloid Interface Sci., 2006, 298, 582–592 CrossRef CAS.
- S. Krishnaswami, W. C. Graustein, K. K. Turekian and J. F. Dowd, Radium, thorium and radioactive lead isotopes in groundwaters: Application to the in situ determination of adsorption-desorption rate constants and retardation factors, Water Resour. Res., 1982, 18, 1663–1675 CrossRef CAS.
- R. G. Wilhelm, Under standing variation in partition coefficient, Kd value, 2004, https://swap.stanford.edu/20110203095432/http://www.epa.gov/radiation/docs/kdreport/vol3/402-r-04-002c.pdf.
- R. I. Boysen, Y. Wang, H. H. Keah and M. T. Hearn, Observations on the origin of the non-linear van't Hoff behaviour of polypeptides in hydrophobic environments, Biophys. Chem., 1999, 77, 79–97 CrossRef CAS.
- T. Galaon and V. David, Deviation from van't Hoff dependence in RP-LC induced by tautomeric interconversion observed for four compounds, J. Sep. Sci., 2011, 34, 1423–1428 CrossRef CAS PubMed.
- M. Tanase, A. Soare, V. David and S. C. Moldoveanu, Sources of nonlinear van't Hoff temperature dependence in high-performance liquid chromatography, ACS Omega, 2019, 4, 19808–19817 CrossRef CAS PubMed.
- R. Naidu, N. Bolan, R. S. Kookana and K. Tiller, Ionic-strength and pH effects on the sorption of cadmium and the surface charge of soils, Eur. J. Soil Sci., 1994, 45, 419–429 CrossRef CAS.
- C. Appel, L. Q. Ma, R. D. Rhue and E. Kennelley, Point of zero charge determination in soils and minerals via traditional methods and detection of electroacoustic mobility, Geoderma, 2003, 113, 77–93 CrossRef CAS.
- Y. Liu, R. Naidu and H. Ming, Surface electrochemical properties of red mud (bauxite residue): Zeta potential and surface charge density, J. Colloid Interface Sci., 2013, 394, 451–457 CrossRef CAS.
- U. Farooq, M. T. Tweheyo, J. Sjöblom and G. Øye, Surface characterization of model, outcrop, and reservoir samples in low salinity aqueous solutions, J. Dispersion Sci. Technol., 2011, 32, 519–531 CrossRef CAS.
- E. S. Rigobello, A. D. B. Dantas, L. Di Bernardo and E. M. Vieira, Influence of the apparent molecular size of aquatic humic substances on colour removal by coagulation and filtration, Environ. Technol., 2011, 32, 1767–1777 CrossRef CAS.
- P. Fournier, E. H. Oelkers, R. Gout and G. Pokrovski, Experimental determination of aqueous sodium-acetate dissociation constants at temperatures from 20 to 240 C, Chem. Geol., 1998, 151, 69–84 CrossRef CAS.
- W. S. De Lint, N. E. Benes, J. Lyklema, H. J. Bouwmeester, A. J. van der Linde and M. Wessling, Ion adsorption parameters determined from zeta potential and titration data for a γ-alumina nanofiltration membrane, Langmuir, 2003, 19, 5861–5868 CrossRef CAS.
- Y. Aşçi, Ü. Açikel and Y. S. Açikel, Equilibrium, hysteresis and kinetics of cadmium desorption from sodium-feldspar using rhamnolipid biosurfactant, Environ. Technol., 2012, 33, 1857–1868 CrossRef.
- M. Jalali and N. H. Matin, Sorption of phosphorus in calcareous paddy soils of Iran: effects of soil/solution ratio and pH, Environ. Earth Sci., 2015, 73, 2047–2059 CrossRef CAS.
- G. Mattock, The hydrolysis and aggregation of the beryllium ion, J. Am. Chem. Soc., 1954, 76, 4835–4838 CrossRef CAS.
- W. W. Rudolph, D. Fischer, G. Irmer and C. C. Pye, Hydration of beryllium (II) in aqueous solutions of common inorganic salts. A combined vibrational spectroscopic and ab initio molecular orbital study, Dalton Trans., 2009, 6513–6527 RSC.
- M. Mac Naughton and R. James, Adsorption of aqueous mercury (II) complexes at the oxide/water interface, J. Colloid Interface Sci., 1974, 47, 431–440 CrossRef CAS.
- V. Inglezakis, A. Zorpas, M. Loizidou and H. Grigoropoulou, The effect of competitive cations and anions on ion exchange of heavy metals, Sep. Purif. Technol., 2005, 46, 202–207 CrossRef CAS.
- Y. Zhang, J. Sun, J. Liu, G. Huang, X. Chen, J. Wang, J. Jing and X. Xiang, A survey of heavy metals in sediments of Yangzonghai Lake in Yunnan Province: their source and distribution, Environ. Sci. Technol., 2010, 33(12), 171–175 CAS.
- M. Müller and M. R. Buchner, Beryllium Complexes with Bio-Relevant Functional Groups: Coordination Geometries and Binding Affinities, Angew. Chem., Int. Ed., 2018, 57, 9180–9184 CrossRef.
- R. C. Lim, B. De Silva, J. H. Park, V. F. Hodge and R. K. Gary, Aqueous solubility of beryllium (II) at physiological pH: effects of buffer composition and counterions, Prep. Biochem. Biotechnol., 2020, 50, 585–591 CrossRef CAS.
- M. Peplow, Beryllium doubles down on nitrogen bonding, Chem. Eng. News, 2021, 99, 8 Search PubMed.
- H. Noeth and D. Schlosser, Beryllium nitrogen compounds. 1. Monomeric bis (amido) beryllium compounds, Inorg. Chem., 1983, 22, 2700–2703 CrossRef CAS.
- S. Troyanov, G. Tikhomirov, K. Znamenkov and I. Morozov, Crystal structure of beryllium nitrate complexes (NO)2 [Be(NO3)4] and Be4O(NO3)6, J. Inorg. Chem., 2000, 45, 1941–1948 CAS.
- P. Barbaro, F. Cecconi, D. Dakternieks, S. Dominguez, A. Duthie, C. A. Ghilardi, S. Midollini, A. Orlandini and A. Vacca, Beryllium (II) Complexes of the Kläui Tripodal Ligand Cyclopentadienyltris (diethylphosphito-P) cobaltate (−), Inorg. Chem., 2001, 40, 2725–2729 CrossRef CAS PubMed.
- X. Li, Q. Zhou, S. Wei, W. Ren and X. Sun, Adsorption and desorption of carbendazim and cadmium in typical soils in northeastern China as affected by temperature, Geoderma, 2011, 160, 347–354 CrossRef CAS.
- M. Rafatullah, O. Sulaiman, R. Hashim and A. Ahmad, Removal of cadmium (II) from aqueous solutions by adsorption using meranti wood, Wood Sci. Technol., 2012, 46, 221–241 CrossRef CAS.
- S.-i. Ishiguro and H. Ohtaki, A Thermodynamic Study on the Hydrolysis of Beryllium Ion in Dioxane–Water Mixed Solvents, Bull. Chem. Soc. Jpn., 1979, 52, 3198–3203 CrossRef CAS.
- A. Deshmukh, R. Konda, E. Titus and A. Chaudhari, Electronic structure calculations and molecular dynamics simulations of hydrogen adsorption on Beryllium doped complexes, Int. J. Hydrogen Energy, 2017, 42, 23708–23715 CrossRef CAS.
- Y. A. Ermakov, K. Kamaraju, A. Dunina-Barkovskaya, K. S. Vishnyakova, Y. E. Yegorov, A. Anishkin and S. Sukharev, High-affinity interactions of beryllium (2+) with phosphatidylserine result in a cross-linking effect reducing surface recognition of the lipid, Biochemistry, 2017, 56, 5457–5470 CrossRef CAS.
- O. Sulaiman, N. S. Ghani, M. Rafatullah and R. Hashim, Removal of zinc (II) ions from aqueous solutions using surfactant modified bamboo sawdust, Sep. Sci. Technol., 2011, 46, 2275–2282 CrossRef CAS.
- M. N. Sahmoune, Evaluation of thermodynamic parameters for adsorption of heavy metals by green adsorbents, Environ. Chem. Lett., 2019, 17, 697–704 CrossRef CAS.
- G. Mustafa, R. S. Kookana and B. Singh, Desorption of cadmium from goethite: effects of pH, temperature and aging, Chemosphere, 2006, 64, 856–865 CrossRef CAS PubMed.
- M. Hamidpour, M. Kalbasi, M. Afyuni, H. Shariatmadari, P. E. Holm and H. C. B. Hansen, Sorption hysteresis of Cd (II) and Pb (II) on natural zeolite and bentonite, J. Hazard. Mater., 2010, 181, 686–691 CrossRef CAS.
- O. Hamdaoui and E. Naffcrechoux, Modeling of adsorption isotherms of phenol and chlorophenols onto granular activated carbon: Part I. Two-parameter models and equations allowing determination of thermodynamic parameters, J. Hazard. Mater., 2007, 147, 381–394 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2em00313a |
| This journal is © The Royal Society of Chemistry 2023 |