
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Per and polyfluoroalkylated substances (PFAS) target and EOF analyses in ski wax, snowmelts, and soil from skiing areas†
Viktoria
Müller
 ab,
Larissa Cristine
Andrade Costa
c,
Filipe Soares
Rondan
c,
Eleonora
Matic
b,
Marcia Foster
Mesko
c,
Andrew
Kindness
ad and
Jörg
Feldmann
*b
ab,
Larissa Cristine
Andrade Costa
c,
Filipe Soares
Rondan
c,
Eleonora
Matic
b,
Marcia Foster
Mesko
c,
Andrew
Kindness
ad and
Jörg
Feldmann
*b
aThe James Hutton Institute, Craigiebuckler, Aberdeen AB15 8QH, UK
bTESLA-Analytical Chemistry, Institute of Chemistry, University of Graz, Universitaetsplatz 1, 8010 Graz, Austria. E-mail: joerg.feldmann@uni-graz.at
cCentro de Ciências Químicas, Farmacêuticas e de Alimentos, Universidade Federal de Pelotas, Capão do Leão, RS 96160-000, Brazil
dUniversity of KwaZulu Natal, School of Chemistry & Physics, Private Bag X54001, Westville Campus, ZA-4000 Durban, South Africa
First published on 12th October 2023
Abstract
Per and polyfluoroalkylated substances (PFAS) are common additives in ski waxes for their water repellent characteristic. Abrasion of ski wax leaves PFAS on the snow surface, however, little is known about the distribution and concentration of PFAS in snow and soil due to skiing. In this study we analysed different ski waxes, snowmelts and soil from family skiing areas from Alpine locations using targeted high performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS) to understand more about PFAS distribution in the environment. In general, we found a very diverse PFAS pattern in the analysed media. PFAS level was higher in skiing areas compared to the non-skiing ones that were used as control. ∑target PFAS ranged between <1.7 ng L−1 and 143 ng L−1 in snowmelt, <0.62 ng g−1 and 5.35 ng g−1 in soil and <1.89 and 874 ± 240 ng g−1 in ski wax samples. Snowmelt was dominated by short-chained PFAS, while soil and wax contained both short and long-chained PFAS. Extractable organic fluorine (EOF) was several orders of magnitude higher for waxes (0.5–2 mg g−1) than for soils (up to 0.3 μg g−1), while total fluorine (TF) content of the waxes was even higher, up to 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 210 ± 420 μg g−1. We also showed that the ∑ target PFAS accounts for up to 1.5% in EOF content, showing that targeted LC-MS/MS gives a limited measure of the pollution originated from ski waxes and non-targeted analysis and EOF is necessary for a better overview on PFAS distribution.
210 ± 420 μg g−1. We also showed that the ∑ target PFAS accounts for up to 1.5% in EOF content, showing that targeted LC-MS/MS gives a limited measure of the pollution originated from ski waxes and non-targeted analysis and EOF is necessary for a better overview on PFAS distribution.
Environmental significancePer- and polyfluoroalkyl substances are emerging contaminants that are being more and more frequently reported in the literature. They are common additives to ski waxes to boost performance for racers, however, only a few peer-reviewed publications, mostly from Northern Europe, are available on the distribution of PFAS in snow and soil due to skiing. This study showed that public skiing can produce orders of magnitude higher PFAS concentrations in remote Alpine regions and can be a significant source of PFAS in soils. However, other PFAS sources should be also considered. |
Introduction
The presence of easy to ionize per and polyfluoroalkyl substances (PFAS) such as perfluorinated carboxylic acids (PFCA) has been well studied in the environment due to their ease of analysis using targeted LC-MS/MS. However, not all PFAS are easily ionisable, and therefore can be difficult to measure by LC-MS/MS, thus, may go unreported. Additionally, legacy and precursor compounds (organofluorine substances that undergo degradation to form PFCA1) can be attributed to emission and long-range travel in the atmosphere or with sea currents.2,3 Due to their surface active properties,4,5 PFAS enhance gliding between the snow surface and the ski6 making them a desirable additives to ski wax products that are applied on both cross country and downhill skis. During abrasion PFAS could deposit in the snow creating areas with high concentration of PFAS.3 Later, during the melting of the snow, PFAS could be immobilised in the soil or mobilised into surface – and groundwater. The primary ingredients of fluorinated waxes are semi-fluorinated alkanes (SFAs) (CF3(CF2)n(CH2)mCH3) with varied chain length. The manufacturing process involves fluorotelomeristion of perfluoroalkyl iodide.4,7 Fluorotelomer olefins are produced by the dehalogenation of fluorotelomer iodides, and hydrosilylated to produce silanes leading to SFA as byproducts.4 These PFAS cannot be measured with LC-MS/MS due to their inability to ionise well. However, perfluorocarboxylic acids, that can be measured with targeted LC-MS/MS, have been shown to be present as impurities during manufacturing processes in commercially available fluorinated waxes.3,7–10 Fluorine mass balance analysis combines target PFAS analysis and non-specific organofluorine or total fluorine (TF) measurement.11 It is a useful tool to show how much PFAS can account for with target analysis to what is actually available in the samples.Following the addition of perfluoro octanoic acid (PFOA) and related compounds to the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) and restricted substances by European Chemicals Agency (ECHA) they have been banned in products sold in the European Union (EU). Starting 2023/2024 winter season, the use of fluorocarbon-based ski waxes was prohibited by the international ski federation (FIS).12 There was also a voluntarily phase out of longer chain PFAS and PFAS precursors as they could transform to perfluoroalkyl acids (PFAAs).13–16 However, a recent study8 indicated that PFAS are still present in the waxes.
Few publications are available in the literature on PFAS in ski waxes and snow melts, and most of these are focused on targeted analyses. The aim of this study was not only to determine targeted PFAS which are routinely determined by targeted LC-MS/MS but also to determine extractable organofluorine (EOF) and identify whether the PFAS fingerprint of the ski wax is mirrored in the snowmelts, and in the soil from four skiing areas in Austria. Additionally, our objective was also to quantify total organofluorine (EOF) in all samples which can be easily extracted with methanol using combustion ion chromatography (CIC) in order to identify what was not measurable by routine targeted LC-MS/MS methods, and to compare CIC results of waxes with high-resolution graphite furnace molecular absorption spectrometry (HR-GF-MAS). The second objective was to create a full fluorine mass balance for ski wax to determine the fluorine which was not easily extractable and analysed by LC-MS/MS from the ski wax but potentially end up in the soil by determining the total fluorine content using ion chromatography after microwave-induced combustion.
Materials and methods
Chemicals and consumables
Microwave-induced combustion for total fluorine content
Microwave-induced combustion (MIC) can handle higher amount of sample than CIC, which is beneficial if the homogeneity of the samples is unknown. Hence, it was selected for TF analysis. 0.8 g of solid wax samples were used as described in Mesko et al.20 The detailed process can be found in the ESI.† To assess the repeatability and to check for losses during sample preparation, a sample was spiked with 25 μL of a 64 mg mL−1 F standard and prepared the same way as the rest of the samples with MIC.Instrumentation
Eqn (1): calculation of identified PFAS from target PFAS and EOF.
 | (1) |
Quality control and quality assurance
CIC. A 10-point calibration curve (lower and higher concentration) ranging from 0–20
000 μg F L−1 (from fluoride standard) was prepared in methanol. LOD and LOQ were calculated as 3 times and 10 times the standard deviation of the blank, divided by the slope of the calibration curve. Average method LOQ was calculated from instrumental LOQ the same way as the concentration of the samples was determined. Average method LOQ found to be 0.81 ng F g−1 35 ng F g−1 and 1039 ng F g−1 for snowmelt, soil and wax respectively.
BCR-461 (clay) reference material was included to the run to assess the recovery of the methodology used.
HR-GF-MAS. A 5-point calibration curve was prepared using PFOA in methanol ranging from 0.175 to 5 mg F L−1. LOD and LOQ were determined as 3 times and 10 times the standard deviation of the blank divided by the slope of the corresponding calibration curve. Average method LOQ was calculated from instrumental LOQ the same way as the samples. Average method LOQ found to be 1.4 μg F g−1. Method recoveries (%) (for CIC and HR-GF-MAS) were determined from calculated concentrations based on the analyte area in the spiked samples (same as in targeted analysis) compared the ‘theoretical’ spiked concentrations.
IC after MIC. A 6-point calibration curve ranging from 0 to 3000 μg F L−1 (from NaF standard) was prepared in water. LOD and LOQ were determined as 3 times and 10 times the standard deviation of ten sample blank measurements divided by the slope of the corresponding calibration curve and found to be 30 μg F L−1 and 95 μg F L−1 respectively. Considering the final volume and sample mass the LOD and LOQ of the method were 10 μg g−1 and 30 μg g−1, respectively.
All results concerning quality control can be found in the ESI.†
Results and discussion
Ski wax
A limited range of target PFAS, mostly PFCA, were found in the ski waxes (Fig. 1 and Table S5†), apart from wax 3 and 4. Similar pattern was observed by Freberg and co-workers.6 PFAS profile differed between the different wax types. Mostly smaller carbon number (C4–C7, C9 PFCA, FOEA) PFAS were found in the all the waxes but one (Table S5†). Wax 4 contained PFAS above C10 chain length (C4–C7, C9–C11, C13–C14 PFCA, PFHxS and PFOS) confirming previous studies.3,8,10 Previous publications also show the presence of PFOA and/or PFOS in the waxes,3,6,8,23 indicating that no change has happened in the formulations until that point. Here, however, C8 chemistries were not observed in any of samples above the MLOD (13 ng g−1), suggesting a shift in formulations. However, the MLOQ (38 ng g−1) is higher than the limit of PFOA for waxes (25 ng g−1, ref. 8), thus it cannot be definitively concluded that manufacturers have moved from C8 chemistry to other PFAS due to the global ban on PFOA (ECHA). The asked manufacturers could not provide manufacturing date and composition; thus, it remains only an assumption. PFAS profiles observed in the waxes show different results to previously reported PFAS profiles in which long chain PFAA (>C10) were the dominant species.3,6–8,10 This could suggest that different countries might have different commercially available ski waxes, with different formulations and starting products or the production of fluorinated ski waxes changed during the last few years.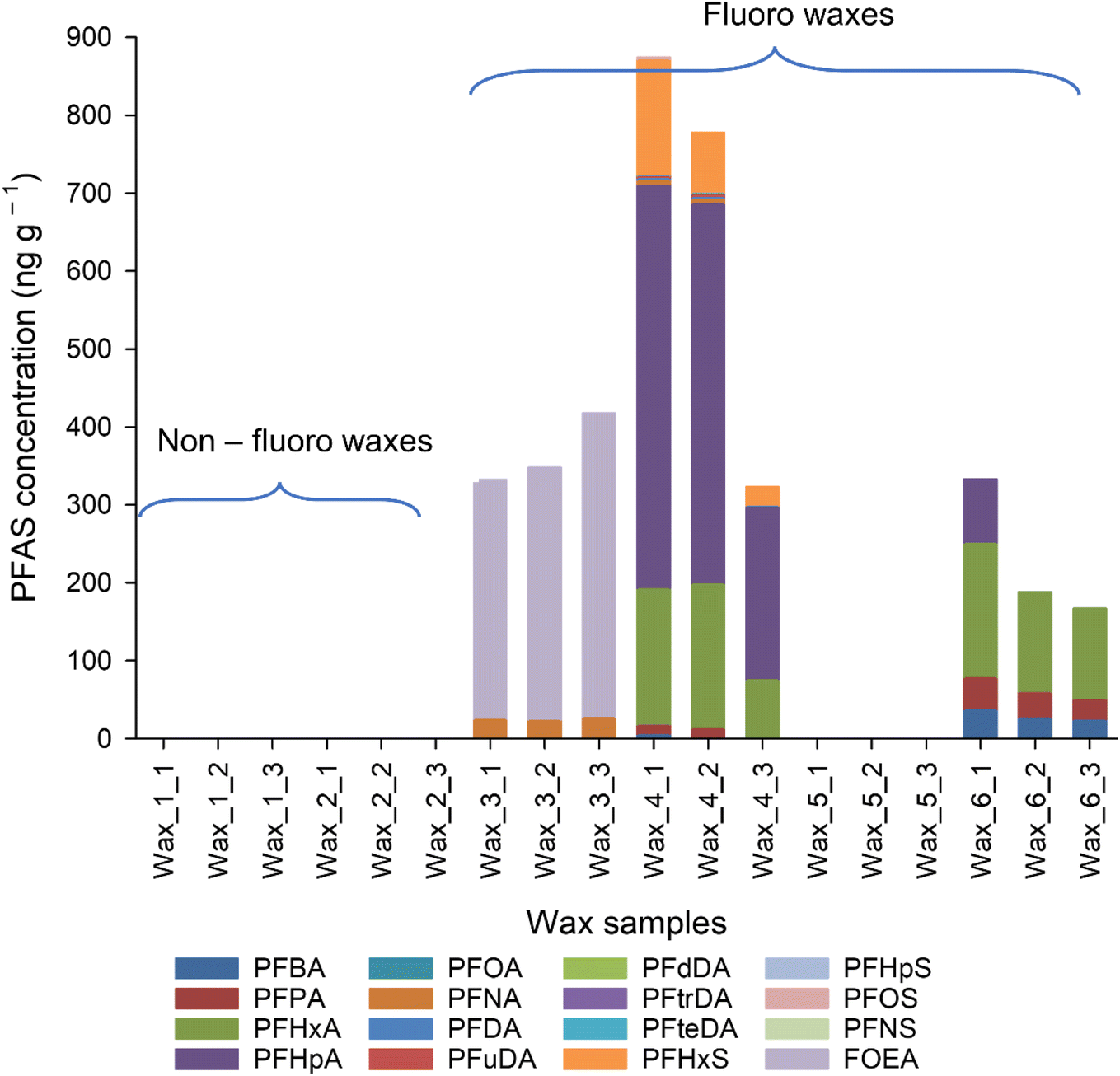 | ||
| Fig. 1 Target PFAS distribution in fluoro and non-fluoro waxes analysed by HPLC-MS/MS. Where there are no results shown, the analytes were either not detected, or below the LOQ. | ||
∑PFAS in ski waxes from previous studies ranged from low ng g−1 to in μg g−1 levels in block waxes,3,6–8 which is in line with our results, ranging from <LOD (non-fluoro waxes) to 874 ± 240 ng g−1 (wax 4) (Fig. 1 and Table S5†). The large quantities of PFAS found in ski waxes in this study as well as in previous publications indicates that snow and soil from skiing areas could also contain high concentration of PFAS.7
CIC analysis showed that non-fluoro waxes have no extractable organic fluorine content, while fluorinated waxes have EOF concentration between 0.52 mg F g−1–2.2 mg F g−1 depending on the manufacturer (Fig. 2a and Table S5†). Block waxes contain long chained paraffins,6 and it is suspected that fluorinated paraffins are present in fluoro waxes. This could explain the high EOF concentrations observed in the waxes, however, the formulation used by the manufacturers is a trade secret, thus no evidence supporting this.
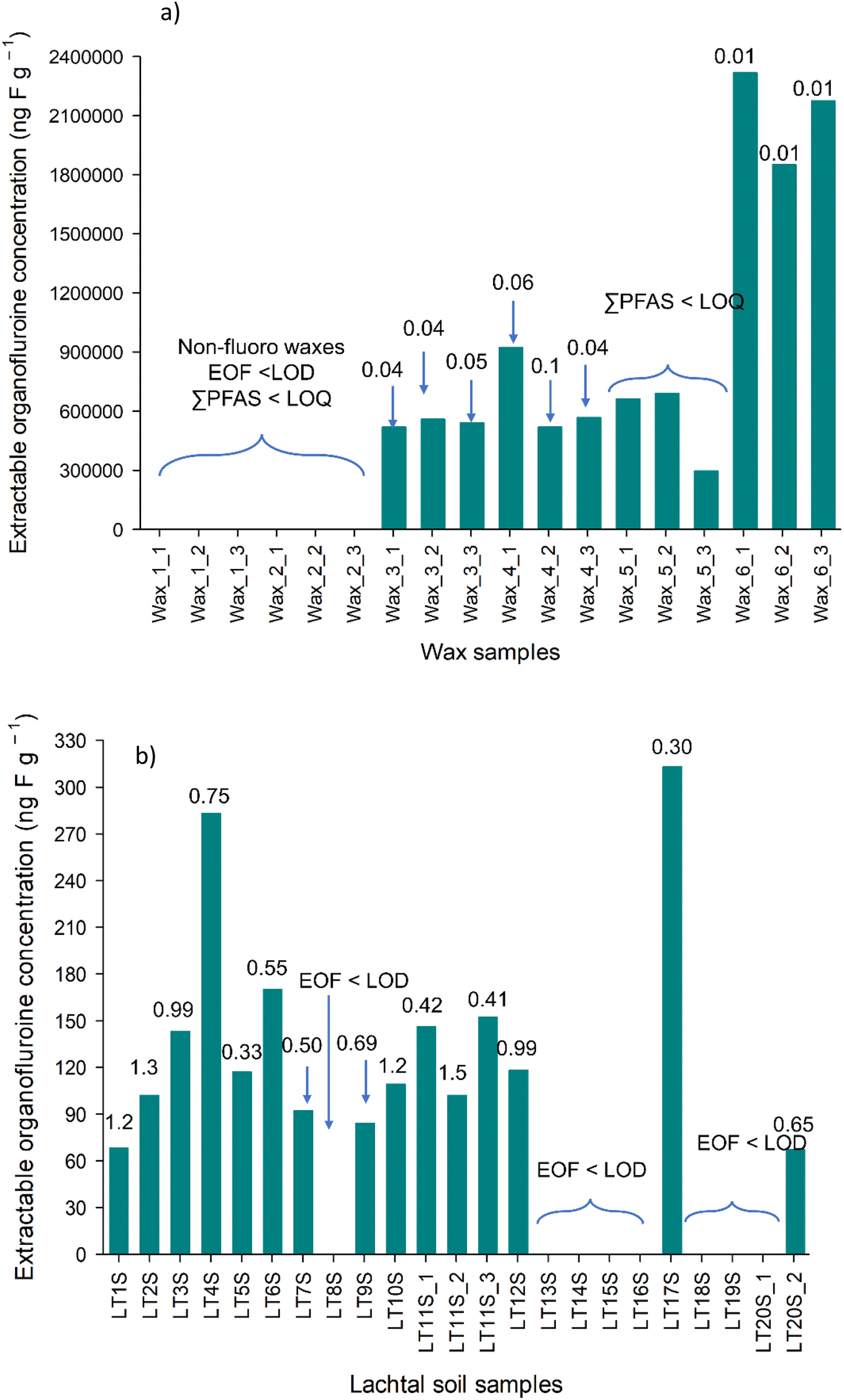 | ||
| Fig. 2 EOF concentrations measured with combustion ion chromatography in wax samples extracted with methanol (a) and in Lachtal soil samples (b). The numbers above the bars represent the ∑ target PFAS in % compared to the EOF. | ||
The identified PFAS fraction was rather low (between 0.001 and 0.1%, Fig. 2a), confirming the presence of target PFAS only as impurities from the manufacturing process. Even including the analytes above LOD but below LOQ would not significantly increase the fraction of identified PFAS in wax samples. No comparison with other studies is possible due to the lack of EOF data for ski wax. PFAS that can be extracted with methanol, due to its polarity, are generally more polar, more water soluble, which in turn more amenable to be determined with LC-MS/MS. Thus, the methanol extractable fraction of PFAS is rather mobile in the environment and can be transported into the groundwater and may form a reservoir of precursor PFAS. It is important to note that SFAs have been shown in ski waxes previously,7 however cyclohexane was used as the extraction solvent. Although the solubility of the SFAs is greater in hexane than in methanol, it is possible to extract these compounds in methanol although in much lower quantity. Nonetheless, SFAs could make up a significant portion of the EOF.
We have observed a difference in EOF using hexane (Fig. 3 and Table S9†) as the extraction solvent instead of methanol, suggesting that the choice of extraction solvent matters. Roesch et al.24 conducted an EOF experiment on soil, extracting them with methanol and with a hexane/acetone mixture. They found that majority of PFAS (since the polar species were the ones that can be analysed using ESI-MS), have a higher mobility in methanol than in hexane/acetone, which is less polar. As waxes contain non-polar components, hexane, being a less polar solvent than methanol, could extract more of these non-polar components, which are suspected to be fluorinated. For those not-easy-to-ionise PFAS, no standard analytical procedure for EOF extraction exists, thus every laboratory have their own developed method, which make the comparison of EOF more challenging.25 Total F concentration reaches up to the % region (0.4–3.1%, Table S13†) in the fluoro waxes. Wax 6 had the highest TF (31210 ± 420 μg g−1) followed by wax 5 (4770 ± 400 μg g−1), 4 (4446 ± 360 μg g−1), and 3 (4277 ± 140 μg g−1), while wax 1 and 2 were below the LOQ (16 μg g−1). This cannot be confirmed by producers and so far, no data have been reported in the literature on total F in ski wax. Ski waxes extracted with hexane showed higher EOF content than methanol extracts (46–79% and 7–13% respectively), suggesting that most of the organofluorine present in the waxes are non-polar and therefore can be well retained in the soil. However, we did not reach 100% extractability, indicating the presence of more F containing compounds such as non-extractable polymers. The fate of these compounds in the environment is totally unclear, although it is known that the wax is removed due to abrasion. So far, no analytical method is available to determine this fraction of F-containing compounds released into the environment, but those compounds may degrade over the time into more mobile PFAS and will be a legacy for many years to come when soils in ski areas leach out PFAS after a ban of PFAS containing ski waxes. However, no study has looked into the degradability of the non-extractable F-fraction in ski wax.
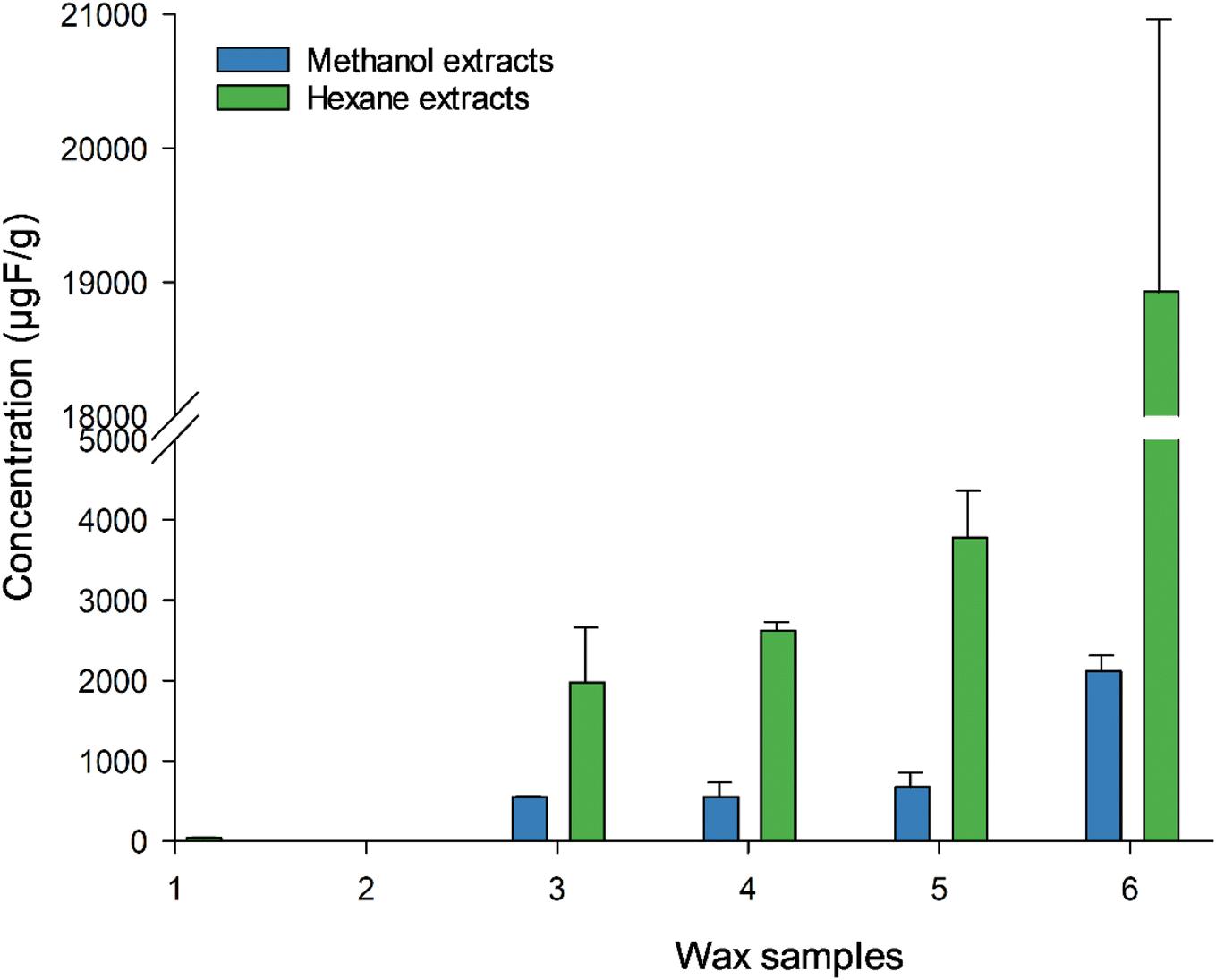 | ||
| Fig. 3 EOF concentrations in wax samples 1–6 using methanol and hexane extraction solvents. Measured with CIC. Error bars represent the standard deviation between the replicates, n = 3. | ||
There is a lack of reported literature that compares EOF concentrations of the same set of samples using CIC and HR-GF-MAS, however, one group reported higher concentrations using HR-GF-MAS than with CIC in river samples,26 we also found higher values using HR GF-MAS. However, they also reported that the concentrations measured with the two different methodologies were in the same order of magnitude in water samples. Our measured relative differences (2-fold at lower concentrations and 7-fold at higher, Fig. 4 and Table S10†) increased with the concentrations in the ski wax. Hence, it might be a concentration effect, since the concentration in the ski wax are multiple order of magnitudes higher than the water samples measured by Gehrenkemper et al.26 may explain also the differences.
 | ||
| Fig. 4 Comparison of EOF concentrations measured with CIC (pink squares) and HR-GF-MAS (blue triangles). The error bars represent the standard deviation between the replicates, n = 3. | ||
Overall, it should be stated that the diversity of PFAS profile of the ski waxes found here makes it difficult to identify a specific ski wax PFAS fingerprint. Hence, the transfer and the contribution of ski wax PFAS to snow and soil is difficult. Therefore, it becomes pertinent to investigate a large market survey of ski wax which are sold worldwide in order to identify the diversity of PFAS profile coming from ski waxes or better to analyse an integral mixture of the used ski wax in one area and that is the snow on the ski slopes.
Snowmelts
The study is conducted on skiing areas which are mainly used for ordinary skiing and not used for ski race competitions. Hence, ski waxes used might, but not necessarily be older and more diverse than those used for competitive races. PFAS profiles in the snow were different then what was found in the ski waxes, possibly due to the different dynamics, for example volatilisation, solubility and mobility of different PFAS, breakdown of precursor compounds. Moreover, the different areas, although all within 100 km range, showed different PFAS profiles. The sampling locations and time for sampling were chosen to ensure maximum contact between the snow and the skiers (e.g., at lift stations), however not all snow samples were collected from lift stations, possibly influencing the different PFAS profiles observed in the different sampling points. Since the few ski waxes already showed a diverse PFAS profile it is most likely that other ski waxes still in use add to the diversity and that makes it difficult to predict the sources of PFAS in the snowmelt and eventual in the soil. Hence, the analysis of the snowmelt directly is useful since, it would give a more comprehensive data on the PFAS release from the skiing activities than analysing the ski waxes directly. In Klippitztörl, Schladming and Teichalm snow, mostly the shorter carbon numbered, more water soluble PFAS were quantified. The species found were C4–C7, C9–C10 and C12 PFCA and PFOS (Fig. 5 and Table S5†) which is in contrast to what was reported the literature, where PFAS found at the highest level were above C10.3,10 Snow from the Teichalm site showed the highest number of quantifiable analytes, which could be attributed to the samples coming from both downhill and cross-country skiing activities.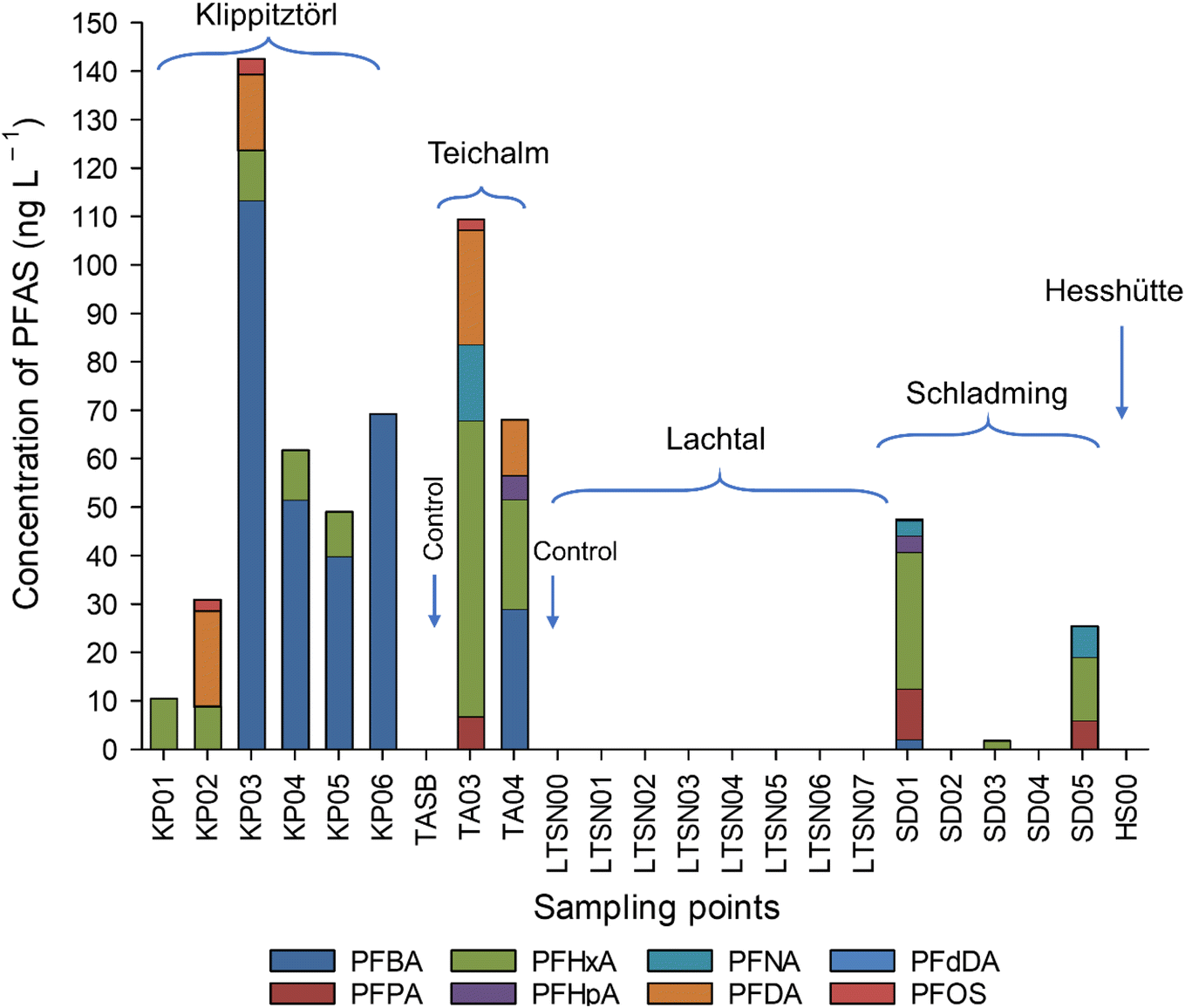 | ||
| Fig. 5 Target PFAS distribution in Teichalm (TA), Klippitztörl (KP), Lachtal (LTSN), Schladming (SD) and Hesshütte (HS) snowmelts analysed with HPLC-MS/MS. Where there are no results shown, the analytes were either not present, or below the LOQ. | ||
In Lachtal snow no PFAS was found above LOQ, however, there were some present above LOD, which was indicated in Table S5.†
In Klippitztörl PFBA was the most dominant species, ranging from <LOQ to 113 ng L−1 followed by PFHxA, PFDA and PFOS. In Teichalm, the most dominant species was PFHxA, ranging between 23 and 61 ng L−1, followed by PFDA > PFBA > PFNA > PFPA > PFHpA and PFOS. In 2 samples from Schladming, no PFAS could be quantified with targeted analysis. In the remaining samples, PFHxA was the most dominant species ranging between 1.7 and 28 ng L−1, followed by PFPA > PFNA > PFHpA > PFBA and PFdDA. Curiously, no precursor PFAS has been observed in any of the snowmelt, which is in line with previous reports on snowmelt from Sweden, Norway and US.3,9,10
∑ target PFAS also varied not just between sampling sites but within each site. In Klippitztörl, ∑ target PFAS was ranging from 10 to 143 ng L−1. In Teichalm, it ranged between 81 and 109 ng L−1, while in Schladming it was between <LOQ and 47 ng L−1. ∑PFAS in previous publications ranged from 38 ng L−1 to 1.4 μg L−1,3 7.6–10.7 μg L−1 (ref. 10) and around 1.25 to 3.8 μg L−1.27 ∑PFAS found in this study (10–143 ng L−1, Fig. 5) is similar to that reported by Plassmann & Berger.3 Due to the low F content in the samples, snowmelts showed no detectable EOF content. The difference seen between the profiles and concentration compared to that study could be attributed to the fact that the above-mentioned studies sampled snow after a professional race took place. Using fluorinated ski wax on professional skis is common, since it improves the performance, thus increasing the PFAS concentration that could be found in the snowmelts after a race.6 Here, we showed results from skiing sites were no skiing competition take place and that is open to the public, who might not use fluorinated ski waxes or waxes at all. However, snow collected from all areas away from ski tracks, and snow collected in the Gesäuse National Park (Styria, Austria), which sits at 1699 m high in the Ennstaler Alps, where no significant skiing takes place contained no detectable amount of PFAS, suggesting that the atmospheric deposition is smaller than the abrasion of ski wax from the skiing during the exposure of the snow sampled. Hence, the analysis of snow for PFAS makes it possible to get a more complete integral value for PFAS coming from the skiing activities in contrast to the atmospheric deposition which potentially contaminate the Alpine soil.
Soil
When we assume relatively low mobility of PFAS we expected to see higher concentrations of PFAS at certain parts of the transect where the tracks used to be. However, the same analytes were present through the soil samples and there is no clear increase or decrease in their concentrations. Mostly smaller carbon numbered PFAS were present with PFNA being the most dominant. Its concentration ranged from 0.23 ng g−1 dry weight (dw) to 0.90 ng g−1 dw followed by PFDA > PFuDA > PFBA > PFHpA > PFOS > PFOA > PFdDA and PFPA (Fig. 6 and Table S5†). Their individual concentrations ranged from <LOQ to 1.55 ng g−1 dw. No PFAS > C12 was present in Lachtal soils. In Klippitztörl site, only 3 soils (control, KPS02 (bottom of hill) and KPS03 (top of hill)) contained quantifiable amounts of PFAS. In contrast to the Lachtal soil, long chained PFAS were the dominating species. PFOS and C9–C10, C13–C14 PFCA were observed in the soils, with PFOS being the most dominant. In the other two soil samples (KPS04, KPS06), no quantifiable PFAS were present, even though they were sampled from similar spots as the snow were sampled from (Fig. 6). PFAS profile seen in Klippitztörl site agrees with other studies.3,10 As the snow melts from the ski slopes, it runs down to the bottom of the hill and the accumulated PFAS found in the snowmelts therefore distributes onto a larger soil area.7,28 PFAS could also move deeper in the soil, can volatilise or be taken up by biota,7,9,10,27,29 therefore, it is difficult to predict their fate. Moreover, PFAS concentration and profile in soil reflect the input from the entire skiing season30 These could be the reason for the concentration difference seen in the samples from these areas as well as the non-quantifiable PFAS from the ski waxes. In the control soil, that was collected away from the skiing slopes in Klippitztörl, PFOS and PFtrDA were observed, although in lower concentration than in the sample from the bottom of the ski slopes. This indicates that PFAS will, indeed, distribute across a larger area due to the snow melting process. | ||
| Fig. 6 Target PFAS distribution in Klippitztörl (KPS) and Lachtal (LT) soil samples analysed with HPLC-MS/MS. Where there are no results shown, the analytes were either not detected, or below the LOQ. | ||
∑PFAS ranged from 0.62 to 5.35 ng g−1 dw, in Lachtal site, while it ranged from <LOQ to 4 ng g−1 dw. with KPS02 having the highest (Fig. 6). ∑PFAS in the collected soils fit into the range previously observed in soils from skiing areas; from 0.059 to 19 ng g−1 dw,3 up to 1.59 ng g−1 dw,9 3.72 and 10.34 3 ng g−1 dw10 and 7.1 to 10.3 ng g−1 dw.31
During EOF analysis of Klippitztörl samples an unknown ion with very close retention time to fluoride appeared (possibly a small organic compound, or something from the column), masking the fluoride peaks (Fig. S1 and S2†), thus Klippitztörl soil samples were excluded from EOF and mass balance analyses. In Lachtal site, EOF concentrations do not show higher concentration in the middle of the transects (Fig. 2b and Table S5†). This suggests two possible scenarios: firstly, the detectable PFAS, which are ionic and reasonably mobile, distribute quickly in the soil, as cross-country skiing areas are relatively flat, the snow melts mostly on top of the soil or the PFAS in the soil is of atmospheric deposition. Although there is no available information about EOF content of soils collected from or around skiing areas, there are information from other parts of the globe. Tan et al.32 reported from Nepali soil (35–200 ng F g−1), where the PFAS contamination comes mostly from long range transport, while Roesch et al.24 found 48–6985 ng F g−1 in PFAS contaminated soil and <LOQ in non-contaminated soil. As the EOF concentrations found in Lachtal soils (up to 313 ng F g−1) are comparable to that observed by Tan et al., atmospheric deposition is the more likely scenario, however further experiments are needed to exclude either scenario. The fraction of identified PFAS found in this study agrees with previous reports of soil; 0.01–1.75% (ref. 24) and 0–1.68%.32 Semi-fluorinated alkanes are showed to end up in soil either due to sorption to particles or due to the melting of last soil,28 possibly contributing to EOF, but going undetected with targeted analysis. Therefore, more efforts have to be made to determine the unknown PFAS by using non-targeted analysis LC-HRMS and other methods, for example gas chromatography for semi-fluorinated alkanes and alkenes7,30 which can determine also the not-easy-to-ionise and particulate PFAS. Direct total oxidisable precursor assay (dTOPA) might also give some idea how the large amount of unknown PFAS would behave in the future and whether this fraction can be a long-term source of PFCA.
Limitations of EOF and the study
Apart from the above mentioned high LOQ for EOF using both analytical systems, needing to have high sample volume for some of the samples and not having a standardised extraction method, EOF analysis has other limitations as well. For example, during sample pre-treatment, water samples are often filtered before sample preparations, which could result in analyte loss. Most of the samples chosen for PFAS analysis (either target or EOF), undergo extraction and SPE for sample concentration and clean-up. The use of SPE selects PFAS that can stick to and can be eluted from the cartridge, lowering the range of analytes, thus lowering the concentration as well. There is no certified reference material for extractable organofluorine analysis, making the comparison between laboratories more difficult.33 Additionally, the organofluorine extracted from the samples could contain not only PFAS but other fluorine containing chemicals as well. Ultrashort chain PFAS, a class of PFAS with ≤C3 could contribute significantly to the unidentified fraction, especially in the case of snow melts, due to atmospheric deposition. Trifluoroacetic acid (TFA) is shown to degrade from hydrofluorocarbons34 which can later enter different water bodies, contributing to EOF concentration. Due to analytical challenges, PFAS below C3 chain length were not targeted in this study, hence lowering the fraction of identified PFAS. Similarly, fluorinated nanoparticles, that can be present in the soil and in the waxes, can contribute to EOF. For F-containing particles at the nanoscale which can be extractable and contribute to the EOF, no analytical method exist so far and is urgently needed to gain a full mass balance of fluorine. The waxes used in this study are only a limited selection of what was available in a single shop. For a more complete overview of waxes, a larger market survey is necessary.Conclusion
This study showed for the first time PFAS in snow and soil from areas where public skiing is taken place in the winter season. The ski wax analysis gave a very diverse picture of the PFAS in the different ski wax products, hence the analysis of snowmelts as an integral average value for the contamination of PFAS from the skiing activity is advisable. The EOF and TF of the ski wax revealed that skiing will produce orders of magnitude higher concentrations of PFAS into remote Alpine regions as well as other places where skiing related activities are taken place, which might need years to break down. The soil analysis revealed that skiing activities can be a significant source of PFAS, but other PFAS sources such as atmospheric deposition cannot be excluded.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Sidney Przygoda, Fiona Reid, Sophia Schlagenhaufen, Walter Goessler, Magdalena Blanz for their help with sampling. Amnah Al Zbedy for her help with the HR-GF-MAS measurement. VM thanks The Macaulay Development Trust (UK) for the financial support.References
- C. A. Mcdonough, W. Li, H. N. Bischel, A. O. De Silva and J. C. Dewitt, Widening the Lens on PFASs: Direct Human Exposure to Perfluoroalkyl Acid Precursors (pre-PFAAs), Environ. Sci. Technol., 2022, 56(10), 6004–6013 CrossRef PubMed.
- UNEP, All POPs Listed in the Stockholm Convention, United Nations Environment Programme, 2020 Search PubMed.
- M. M. Plassmann and U. Berger, Perfluoroalkyl carboxylic acids with up to 22 carbon atoms in snow and soil samples from a ski area, Chemosphere, 2013, 91(6), 832–837 CrossRef PubMed.
- R. C. Buck, J. Franklin, U. Berger, J. M. Conder, I. T. Cousins and P. de Voogt, et al., Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins, Integr. Environ. Assess. Manage., 2011, 7(4), 513–541 CrossRef PubMed.
- E. Kissa, Fluorinated Surfactants and Repellents, Marcel Dekker, New York, 2nd edn, rev. and expanded, 2001 Search PubMed.
- B. I. Freberg, L. S. Haug, R. Olsen, H. L. Daae, M. Hersson and C. Thomsen, et al., Occupational Exposure to Airborne Perfluorinated Compounds during Professional Ski Waxing, Environ. Sci. Technol., 2010, 44(19), 7723–7728 CrossRef PubMed.
- M. M. Plassmann and U. Berger, Trace Analytical Methods for Semifluorinated n -Alkanes in Snow, Soil, and Air, Anal. Chem., 2010, 82(11), 4551–4557 CrossRef PubMed.
- S. Fang, M. M. Plassmann and I. T. Cousins, Levels of per- and polyfluoroalkyl substances (PFAS) in ski wax products on the market in 2019 indicate no changes in formulation, Environ. Sci.: Processes Impacts, 2020, 22(11), 2142–2146 RSC.
- R. Grønnestad, B. P. Vázquez, A. Arukwe, V. L. B. Jaspers, B. M. Jenssen and M. Karimi, et al., Levels, Patterns, and Biomagnification Potential of Perfluoroalkyl Substances in a Terrestrial Food Chain in a Nordic Skiing Area, Environ. Sci. Technol., 2019, 53(22), 13390–13397 CrossRef PubMed.
- G. L. Carlson and S. Tupper, Ski wax use contributes to environmental contamination by per- and polyfluoroalkyl substances, Chemosphere, 2020, 261, 128078 CrossRef PubMed.
- Y. Miyake, N. Yamashita, P. Rostkowski, M. K. So, S. Taniyasu and P. K. S. Lam, et al., Determination of trace levels of total fluorine in water using combustion ion chromatography for fluorine: A mass balance approach to determine individual perfluorinated chemicals in water, J. Chromatogr. A, 2007, 1143(1–2), 98–104 CrossRef PubMed.
- FIS to Fully Implement Fluor Wax Ban at Start of 2023-24 Season, https://www.fis-ski.com/en/international-ski-federation/news-multimedia/news-2022/fis-to-fully-implement-fluor-wax-ban-at-start-of-2023-24-season, last accessed: 14 August 2023.
- A. C. Blaine, C. D. Rich, L. S. Hundal, C. Lau, M. A. Mills and K. M. Harris, et al., Uptake of Perfluoroalkyl Acids into Edible Crops via Land Applied Biosolids: Field and Greenhouse Studies, Environ. Sci. Technol., 2013, 47(24), 14062–14069 CrossRef PubMed.
- S. Brendel, É. Fetter, C. Staude, L. Vierke and A. Biegel-Engler, Short-chain perfluoroalkyl acids: environmental concerns and a regulatory strategy under REACH, Environ. Sci. Eur., 2018, 30(1), 9 CrossRef PubMed.
- M. M. Schultz, C. P. Higgins, C. A. Huset, R. G. Luthy, D. F. Barofsky and J. A. Field, Fluorochemical Mass Flows in a Municipal Wastewater Treatment Facility, Environ. Sci. Technol., 2006, 40(23), 7350–7357 CrossRef PubMed.
- E. Sinclair and K. Kannan, Mass Loading and Fate of Perfluoroalkyl Surfactants in Wastewater Treatment Plants, Environ. Sci. Technol., 2006, 40(5), 1408–1414 CrossRef PubMed.
- S. Taniyasu, K. Kannan, M. K. So, A. Gulkowska, E. Sinclair and T. Okazawa, et al., Analysis of fluorotelomer alcohols, fluorotelomer acids, and short- and long-chain perfluorinated acids in water and biota, J. Chromatogr. A, 2005, 1093(1–2), 89–97 CrossRef PubMed.
- C. P. Higgins, J. A. Field, C. S. Criddle and R. G. Luthy, Quantitative Determination of Perfluorochemicals in Sediments and Domestic Sludge, Environ. Sci. Technol., 2005, 39(11), 3946–3956 CrossRef PubMed.
- S. Taniyasu, K. Kannan, L. W. Y. Yeung, K. Y. Kwok, P. K. S. Lam and N. Yamashita, Analysis of trifluoroacetic acid and other short-chain perfluorinated acids (C2-C4) in precipitation by liquid chromatography-tandem mass spectrometry: comparison to patterns of long-chain perfluorinated acids (C5-C18), Anal. Chim. Acta, 2008, 619(2), 221–230 CrossRef PubMed.
- M. F. Mesko, V. C. Costa, R. M. Pereira and C. A. Hartwig, Chlorine and Fluorine Determination in Eye-Pencil: Development of an Eco-Friendly Sample Preparation Method for Ion Chromatography Analysis, J. Braz. Chem. Soc., 2019, 30(10), 2191–2198 Search PubMed.
- L. Schultes, G. F. Peaslee, J. D. Brockman, A. Majumdar, S. R. McGuinness and J. T. Wilkinson, et al., Total Fluorine Measurements in Food Packaging: How Do Current Methods Perform?, Environ. Sci. Technol. Lett., 2019, 6(2), 73–78 CrossRef.
- A. Akhdhar, M. Schneider, A. Orme, L. Schultes, A. Raab and E. M. Krupp, et al., The use of high resolution graphite furnace molecular absorption spectrometry (HR -MAS) for total fluorine determination in extractable organofluorines (EOF), Talanta, 2020, 209, 120466 CrossRef PubMed.
- M. Kotthoff, J. Müller, H. Jürling, M. Schlummer and D. Fiedler, Perfluoroalkyl and polyfluoroalkyl substances in consumer products, Environ. Sci. Pollut. Res., 2015, 22(19), 14546–14559 CrossRef PubMed.
- P. Roesch, C. Vogel, T. Huthwelker, P. Wittwer and F. G. Simon, Investigation of per- and polyfluoroalkyl substances (PFAS) in soils and sewage sludges by fluorine K-edge XANES spectroscopy and combustion ion chromatography, Environ. Sci. Pollut. Res., 2022, 29, 26889–26899 CrossRef PubMed.
- R. Aro, U. Eriksson, A. Kärrman, K. Jakobsson and L. W. Y. Yeung, Extractable organofluorine analysis: A way to screen for elevated per- and polyfluoroalkyl substance contamination in humans?, Environ. Int., 2022, 159, 107035 CrossRef PubMed.
- L. Gehrenkemper, F. Simon, P. Roesch, E. Fischer, M. von der Au and J. Pfeifer, et al., Determination of organically bound fluorine sum parameters in river water samples—comparison of combustion ion chromatography (CIC) and high resolution-continuum source-graphite furnace molecular absorption spectrometry (HR-CS-GFMAS), Anal. Bioanal. Chem., 2021, 413(1), 103–115 CrossRef PubMed.
- L. Hanssen, D. Herzke, V. Nikiforov, B. Moe, T. Nygård, J. Van Dijk, G. Wing Gabrielsen, F. Fuglei, L. Yeung, C. Vogelsang and P. M. Carlsson, Screening New PFAS Compounds, Norwegian Environment Agency report, O-118084 (NILU report, 23/2019), NILU, Kjeller, 2018 Search PubMed.
- M. M. Plassmann, T. Meyer, Y. D. Lei, F. Wania, M. S. McLachlan and U. Berger, Theoretical and experimental simulation of the fate of semifluorinated n-alkanes during snowmelt, Environ. Sci. Technol., 2010, 44(17), 6692–6697 CrossRef PubMed.
- M. Chropeňová, P. Karásková, R. Kallenborn, E. K. Gregušková and P. Čupr, Pine needles for the screening of perfluorinated alkylated substances (PFASs) along ski tracks, Environ. Sci. Technol., 2016, 50(17), 9487–9496 CrossRef PubMed.
- M. M. Plassmann, A. Denninger and U. Berger, Environmental occurrence and fate of semifluorinated n-alkanes in snow and soil samples from a ski area, Chemosphere, 2011, 85(9), 1458–1463 CrossRef PubMed.
- E. S. Heimstad, T. Nygård, D. Hezke and P. Bohlin-Nizzetto, Environmental pollutants in the terrestrial and urban environment, (Norwegian Environment Agency report, M-1076|2018) (NILU report, 20/2018), NILU, Kjeller, 2018 Search PubMed.
- B. Tan, T. Wang, P. Wang, W. Luo, Y. Lu and K. Y. Romesh, et al., Perfluoroalkyl substances in soils around the Nepali Koshi River: Levels, distribution, and mass balance, Environ. Sci. Pollut. Res., 2014, 21(15), 9201–9211 CrossRef PubMed.
- A. Kärrman, L. W. Y. Yeung, K. M. Spaan, F. T. Lange, M. A. Nguyen and M. Plassmann, et al., Can determination of extractable organofluorine (EOF) be standardized? First interlaboratory comparisons of EOF and fluorine mass balance in sludge and water matrices, Environ. Sci.: Processes Impacts, 2021, 23(10), 1458–1465 RSC.
- T. J. Wallington, W. F. Schneider, D. R. Worsnop, O. J. Nielsen, J. Sehested and J. Warren, The Environmental Impact of CFC Replacements-HFCs and HCFCs, Environ. Sci. Technol., 1994, 28(7), 320A–326A Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3em00375b |
| This journal is © The Royal Society of Chemistry 2023 |