
Sodium dithionite mediated one-pot, tandem chemoselective reduction/cyclization for the synthesis of pyrrole fused N-heterocycles†
Joydev K.
Laha
 *,
Surabhi
Panday
,
Pankaj
Gupta
and
Shiv Raj
Seth
*,
Surabhi
Panday
,
Pankaj
Gupta
and
Shiv Raj
Seth
Department of Pharmaceutical Technology (Process Chemistry), National Institute of Pharmaceutical Education and Research, S. A. S. Nagar, Punjab 160062, India. E-mail: jlaha@niper.ac.in
First published on 2nd December 2022
Abstract
A chemoselective reduction of a nitro group in the presence of an aldehyde or ester group integrated with another synthetic transformation leading to the expedient synthesis of important heterocycles is the subject of this current investigation. The chemoselective reductive cyclization of N-(2-nitrophenyl)pyrrole-2-carboxaldehydes accompanied exclusively by an industrial reagent sodium dithionite (Na2S2O4) yielding diversely substituted pyrrole fused N-heterocycles has been developed for the first time. The amino group generated in situ via chemoselective reduction of the nitro group undergoes condensation with the aldehyde group to form quinoxalines, or undergoes reaction with the ester group to form quinoxalones. The protocol features an efficient one-pot, tandem reductive cyclization performed at room temperature, very short reaction time (1 h), no aqueous work up, purification by crystallization, isolated yields generally >90%, appreciable functional group tolerance, and wide substrate scope. The scalability of the developed protocol is further demonstrated by a gram-scale synthesis.
Introduction
The reduction of a nitro group to the corresponding amino group is a vital organic transformation practised both in academia and industry for the preparation of valuable compounds that are used as, or for developing, pharmaceuticals, agrochemicals, dyes, and polymers.1 As nitro compounds are abundant, cheap and readily available feedstock, this reduction is widely used to prepare amines and derivatives thereof. The classical reduction of aromatic nitro compounds to the corresponding aromatic amines accompanied largely by metal salts in the presence of an acid is a method of choice especially in industry causing production of tremendous waste.2 The practical disadvantages of these reactions include sluggish reactions, expensive steam distillation, tedious work up, long reaction time, and incompatibility of acid-sensitive functional groups. In addition, chemoselective reduction of a nitro group especially in the presence of a carbonyl functionality presented a significant challenge, albeit limited to a few successful reports.3 Moreover, a tandem method involving nitro reduction and subsequent transformation is underexplored.4 Therefore, for the development of a practical method avoiding accompanied limitations (vide supra), mild and greener nitro group reduction is highly desirable.Sodium dithionite (Na2S2O4) is largely used in the textile industry and for the preparation of dyes.5 However, Na2S2O4 has never been employed in the reduction of a nitro group in industry. Despite being a cheap (30$ per kg, ©Thermo Fisher), non-toxic, greener and more sustainable alternative to conventional metal-acid reduction, the use of Na2S2O4 in the reduction of nitro groups remains underexplored and limited to only a few recent reports.6 Na2S2O4 mediated synthesis of benzimidazole via reductive cyclization of ortho-nitroaniline with aldehydes is also reported.7 More importantly, chemoselective reduction of a nitro group especially in the presence of an aldehyde or ester group in the presence of Na2S2O4 remains unexplored, neither the scope of in situ reduction of nitro compounds coupled with a further synthetic transformation to prepare important heterocycles has been demonstrated. The key question is whether a one-pot, tandem chemoselective reductive cyclization could be developed expanding the scope of the use of Na2S2O4 to the preparation of pyrrolo[1,2-a]quinoxalines, 5H-benzo[e]pyrrolo[1,2-a][1,4]diazepines, and pyrrolo[1,2-a]quinoxaline-4(5H)-ones which are present in natural products and pharmaceuticals showing a broad range of biological activities (Fig. 1).8–11
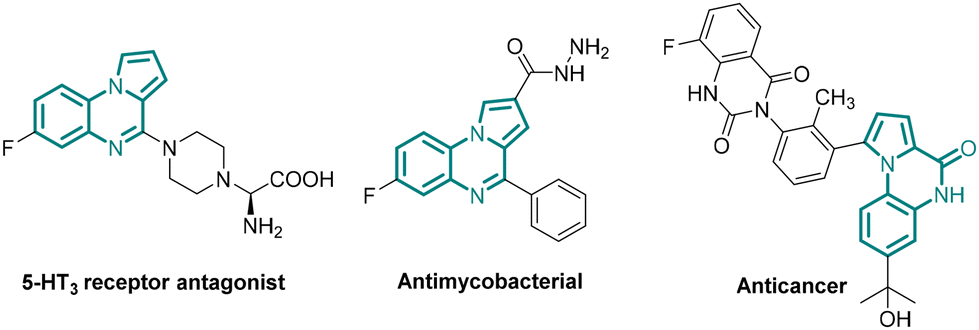 | ||
| Fig. 1 Biologically active pyrrole fused N-heterocycles. | ||
The synthesis of 4-substituted pyrrolo[1,2-a]quinoxalines invariably uses N-(2-aminophenyl)pyrrole and an electrophilic carbon synthon (Scheme 1a)12–20 with an exception of a few intermolecular reactions involving pyrrole-2-carboxaldehyde and iodoanilines performed in the presence of a copper catalyst and sparteine.21,22 On the other hand, the synthesis of pyrrolo[1,2-a]quinoxalin-4(5H)-one is reported to involve a multi-step synthesis,23 reductive cyclization of N-(2-nitrophenyl)pyrrole-2-carboxylate,24 and intramolecular cyclization of pyrrole-2-carboxamides (Scheme 1b).25 To the best of our knowledge, a direct synthetic strategy for the synthesis of pyrrolo[1,2-a]quinoxaline and pyrrolo[1,2-a]quinoxalin-4(5H)-one employing sodium dithionite as a green reductant is currently unknown. This prompted us to develop a new tandem chemoselective reductive cyclization process for the synthesis of pyrrole fused N-heterocycles utilizing sodium dithionite as a green reductant. In this strategy, the nitro group in N-(2-nitrophenyl)-pyrrole-2-carboxaldehyde and carboxylate is reduced to aniline (in situ), which undergoes cyclization to form the corresponding desired product. This approach features mild reaction conditions, high atom-economy, quantitative yields, wide substrate scope, and broad functional group tolerance. The products were obtained simply by a successive process including extraction, concentration, precipitation, and crystallization, without tedious column chromatography.
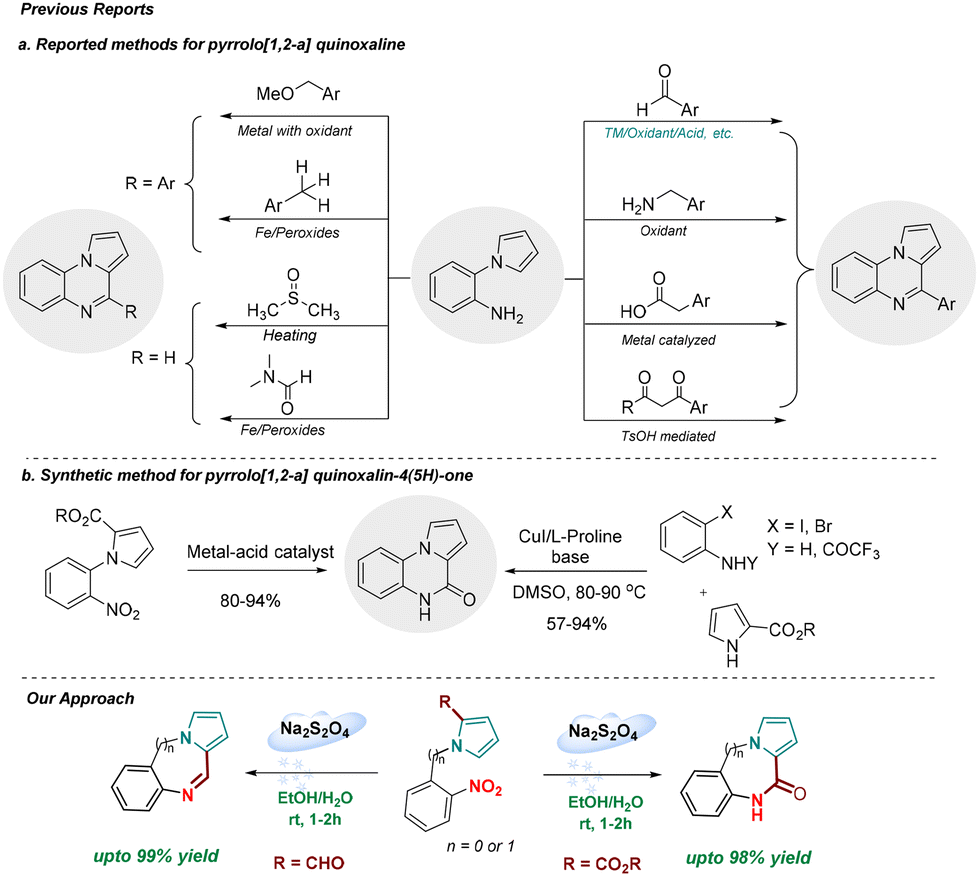 | ||
| Scheme 1 Literature reported methods for the synthesis of pyrrolo[1,2-a]quinoxalines and pyrrolo[1,2-a]quinoxalin-4(5H)-ones. | ||
Results and discussion
We began our investigation with N-(2-nitrophenyl)pyrrole-2-carboxaldehyde 1a as a substrate and sodium dithionite (3 equiv.) as the exclusive reagent. Heating 1a in ethanol at 70 °C for 12 h did not yield the desired pyrroloquinoxaline 2a (entry 1, Table 1). Likewise, replacing ethanol with water also did not produce 2a (entry 2). To our delight, a mixture of solvents (EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O, 1:1) furnished 2a with 40% isolated yield (entry 3). It appeared from our subsequent studies that the ethanol percentage in the solvent mixture played a crucial role in increasing the yield (entries 4 and 5). Next, efforts were made to find the quantity of Na2S2O4 that could be used to obtain the product with a maximum yield. Increasing the stoichiometry of Na2S2O4 from 3 to 4 equiv. significantly increased the yield of 2a to 90% (entry 6). It was found that a comparable yield was obtained when the time was reduced to 6 h (entry 7). While a higher temperature (90 °C) caused a detrimental effect, a reduced temperature (50 °C) did not affect the product formation significantly (entries 8 and 9). Finally, we were pleased to observe that the chemoselective reductive cyclization of 1a to 2a could be achieved at room temperature just in 1 h with a nearly quantitative yield of 2a (entries 10 and 11). Unlike the reported synthesis, the reaction for the synthesis of pyrrolo[1,2-a]quinoxalines in this case was performed at room temperature, and it required only a very short reaction time (1 h), and did not involve any aqueous work up; purification was performed only by crystallization and the yield was >90%.
:H2O, 1:1) furnished 2a with 40% isolated yield (entry 3). It appeared from our subsequent studies that the ethanol percentage in the solvent mixture played a crucial role in increasing the yield (entries 4 and 5). Next, efforts were made to find the quantity of Na2S2O4 that could be used to obtain the product with a maximum yield. Increasing the stoichiometry of Na2S2O4 from 3 to 4 equiv. significantly increased the yield of 2a to 90% (entry 6). It was found that a comparable yield was obtained when the time was reduced to 6 h (entry 7). While a higher temperature (90 °C) caused a detrimental effect, a reduced temperature (50 °C) did not affect the product formation significantly (entries 8 and 9). Finally, we were pleased to observe that the chemoselective reductive cyclization of 1a to 2a could be achieved at room temperature just in 1 h with a nearly quantitative yield of 2a (entries 10 and 11). Unlike the reported synthesis, the reaction for the synthesis of pyrrolo[1,2-a]quinoxalines in this case was performed at room temperature, and it required only a very short reaction time (1 h), and did not involve any aqueous work up; purification was performed only by crystallization and the yield was >90%.
|
|
||||
|---|---|---|---|---|
| Entry | Reductant (equiv.) | Solvent | Temp./time | Yieldb (%) |
| n.d. = not detected.a Reaction conditions: 1 (0.2 mmol) and reductant (3–4 equiv.) in EtOH:H2O (1 mL) under open air.b Isolated yield.c Under N2 conditions. | ||||
| 1 | Na2S2O4 (3) | EtOH | 70 °C/12 h | n.d. |
| 2 | Na2S2O4 (3) | H2O | 70 °C/12 h | n.d. |
| 3 | Na2S2O4 (3) | EtOH:H2O (1:1) |
70 °C/12 h | 40 |
| 4 | Na2S2O4 (3) | EtOH:H2O (2:1) |
70 °C/12 h | 55 |
| 5 | Na2S2O4 (3) | EtOH:H2O (3:1) |
70 °C/12 h | 82 |
| 6 | Na2S2O4 (4) | EtOH:H2O (3:1) |
70 °C/12 h | 90 |
| 7 | Na2S2O4 (4) | EtOH:H2O (3:1) |
70 °C/6 h | 92 |
| 8 | Na2S2O4 (4) | EtOH:H2O (3:1) |
90 °C/6 h | 85 |
| 9 | Na2S2O4 (4) | EtOH:H2O (3:1) |
50 °C/6 h | 95 |
| 10 | Na2S2O4 (4) | EtOH:H2O (3:1) |
rt/6 h | 99 |
| 11 | Na2S2O4 (4) | EtOH:H2O (3:1) |
rt/1 h | 99 |
| 12c | Na2S2O4 (4) | EtOH:H2O (3:1) |
rt/1 h | 96 |
To demonstrate the substrate scope, various N-aryl-pyrrole-2-carboxaldehydes were investigated that could potentially participate in the chemoselective reductive cyclization in the presence of Na2S2O4 (Scheme 2). N-Aryl-pyrrole-2-carboxaldehydes having an electron donating or withdrawing group present on the benzene ring were found to be compatible affording pyrrolo[1,2-a]quinoxalines (2a–g) in yields >90%. The presence of the methylsulfone substituent on the benzene ring of 1h also gave the desired product 2h in 94% yield. Likewise, alkyl, aryl and halo substitution on the pyrrole ring also worked well delivering compounds 2i–k in 90–94% yields. To demonstrate further substrate scope, N-heteroaryl-pyrrole-2-carboxaldehydes containing a pyridine ring were investigated in the chemoselective reductive cyclization. Thus, the reaction of N-(3-nitropyridin-2-yl)-1H-pyrrole-2-carboxaldehydes with sodium dithionite, to our satisfaction, afforded pyrido[3,2-e]pyrrolo[1,2-a]pyrazines (2l and 2m) in 92% and 90% yields, respectively. Similarly, N-heteroaryl-2-carboxaldehydes containing an indole or imidazole ring were also found to be compatible for chemoselective reductive cyclization under the optimized conditions affording 2n and 2o in 98% and 96% yields, respectively.
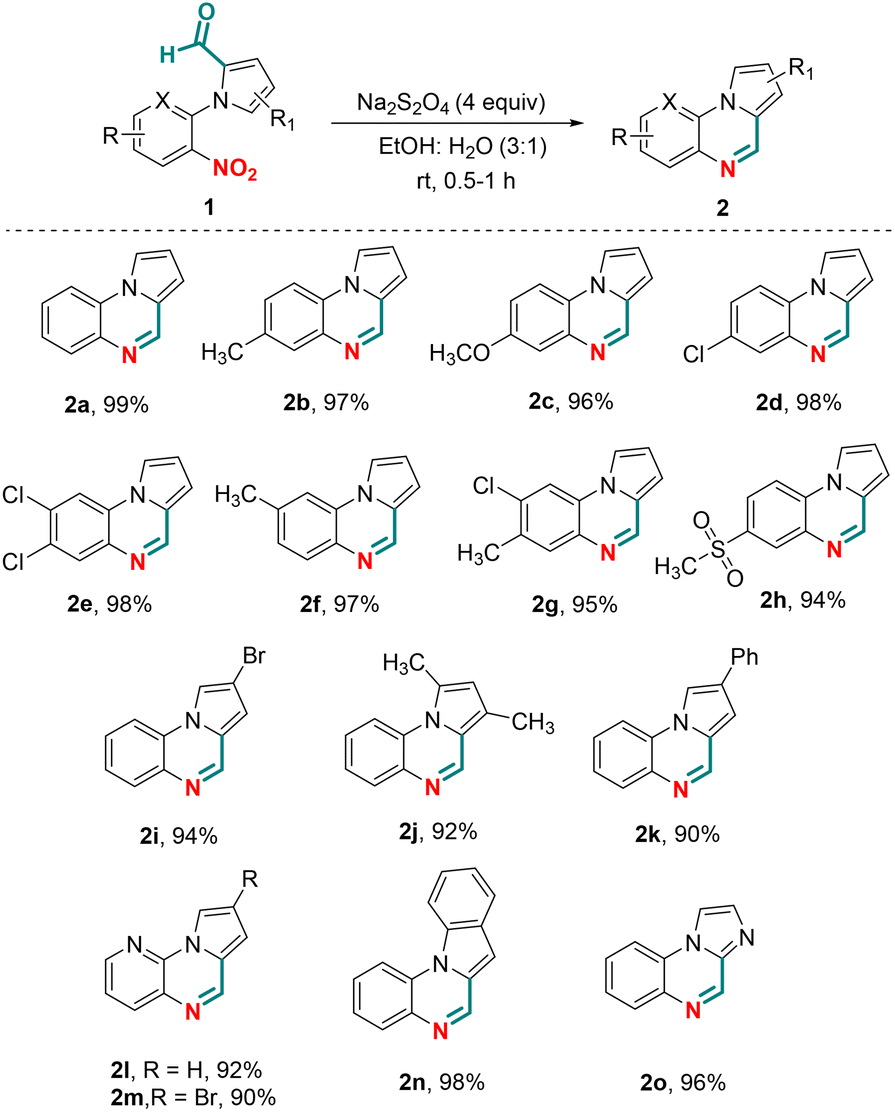 | ||
| Scheme 2 Substrate scope for pyrrolo[1,2-a]-quinoxalines. Reaction conditions: 1 (0.2 mmol) and Na2S2O4 (0.8 mmol) in EtOH:H2O (3:1) (1 mL) were taken in a sealed tube and stirred at room temperature for 0.5–2 h. | ||
Besides using N-(aryl/heteroaryl)-pyrrole-2-carboxaldehydes as substrates, we also explored chemoselective reductive cyclization of N-benzyl-pyrrole-2-carboxaldehydes (Scheme 3). Unlike N-(aryl/heteroaryl)-pyrrole-2-carboxaldehydes, N-benzyl-pyrrole-2-carboxaldehydes gave seven membered cyclized products, pyrrole-fused benzodiazepines.26 Towards this endeavor, when unsubstituted N-benzyl-pyrrole-2-carboxaldehyde 3a was exposed to the optimized reaction conditions, the pyrrolo-fused benzodiazepine 4a was obtained in an isolated yield of 98%. Limited substrate scope was observed under the optimized conditions giving pyrrole fused benzodiazepines 4b–d in 92–96% yields. N-Ts pyrrole-2-carboxaldehyde 3f did not react under the optimized reaction conditions and failed to give the corresponding cyclized product 4f. Perhaps more importantly, chemoselective reduction of a nitro group in the presence of an aldehyde group unveiled for the first time, which upon subsequent cyclization paved the way for the formation of pyrrolo[1,2-a]quinoxalines. Leveraging the practical advantages of the chemistry described herein, the current protocol may be considered as a sustainable alternative to classical methods available for the preparation of this class of compounds.
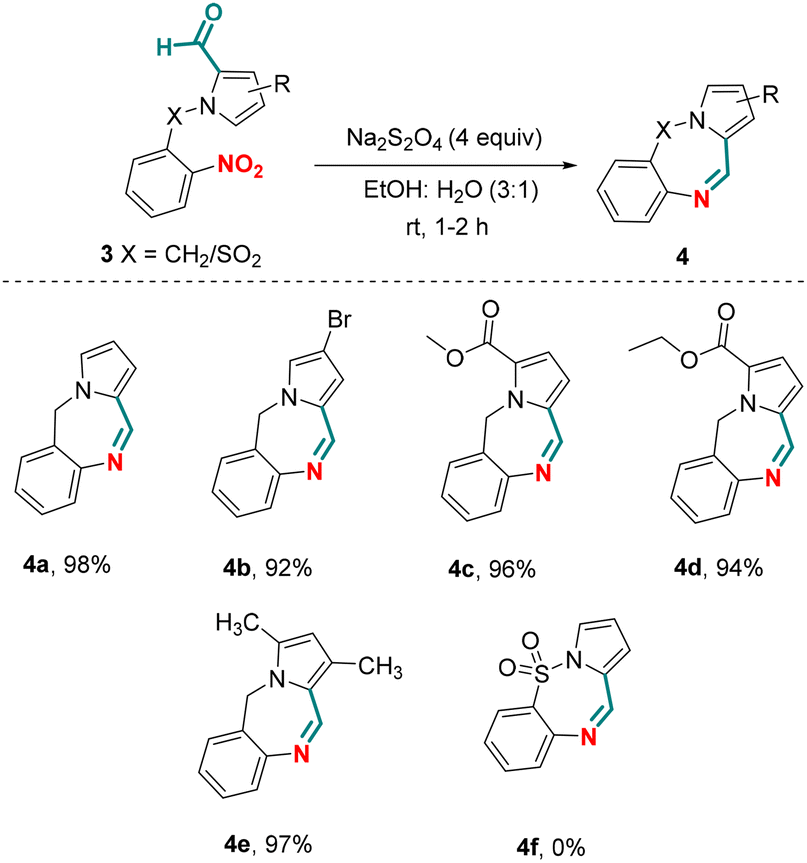 | ||
| Scheme 3 Substrate scope for pyrrole-fused benzodiazepines. Reaction conditions: 3 (0.2 mmol) and Na2S2O4 (0.8 mmol) in EtOH:H2O (3:1) (1 mL) were taken in a sealed tube and stirred at room temperature for 1–2 h. | ||
Subsequent to successful chemoselective nitro reduction in the presence of an aldehyde group, our next effort was directed at realizing a chemoselective nitro reduction in the presence of an ester group, which could further be coupled with amidation reaction involving the in situ generated amine and the ester group. The amidation of an ester with an amine generally requires activation of the ester group.27 However, the reductive amidation of a nitro group has been reported under metal-catalyzed conditions, which also requires in situ activation of the ester group.28 The question remains whether a tandem chemoselective nitro reduction in the presence of an ester group followed by amidation could be achieved possibly without the activation of the ester group. To explore further the scope of our optimized conditions, we investigated the chemoselective reductive cyclization of alkyl N-(2-nitrophenyl)-1H-pyrrole-2-carboxylates 5 for the synthesis of pyrrolo[1,2-a]quinoxaline-4(5H)-ones 6 (Scheme 4). Exposure of methyl 1-(2-nitrophenyl)-1H-pyrrole-2-carboxylate 5a to the optimized conditions gave pyrrolo[1,2-a]quinoxaline-4(5H)-one 6a in 98% yield. Substituted alkyl N-aryl-pyrrole-2-carboxylates also reacted effectively under the optimized conditions furnishing the products 6b–c in excellent yield (92–95%). Notably, similar to alkyl 1-(2-nitrophenyl)-1H-pyrrole-2-carboxylates, alkyl N-benzyl-pyrrole-2-carboxaldehydes did not give the desired product 6d; however a nitro-reduced product of 5d was obtained in 90% yield. The attempted reaction of alkyl N-sulfonyl-pyrrole-2-carboxylate 5e to produce 6e in the presence of Na2S2O4 was futile under the optimized reaction conditions. Unlike previous reports available for the synthesis of pyrrolo[1,2-a]quinoxaline-4(5H)-one 6 suffering from the limitations of using harsh and toxic chemicals, the protocol described herein to synthesize these compounds avoids the use of any such reagents.
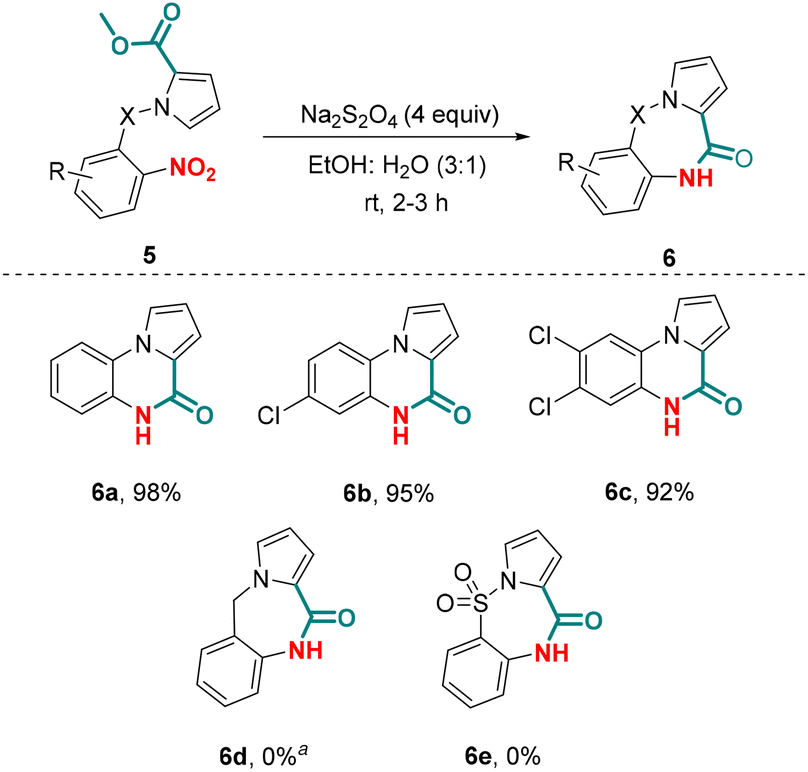 | ||
| Scheme 4 Substrate scope for pyrrolo[1,2-a]-quinoxalin-4(5H)-ones. Reaction conditions: 5 (0.2 mmol) and Na2S2O4 (0.8 mmol) in EtOH:H2O (3:1) (1 mL) were taken in a sealed tube and stirred at rt for 2–3 h. aThe corresponding amine product is obtained. | ||
To demonstrate the scalability of the developed protocol, the synthesis of 2a and 6a was carried out over a gram scale (Scheme 5). Moreover, the simple crystallization yielded pure cyclized products in nearly quantitative yields.
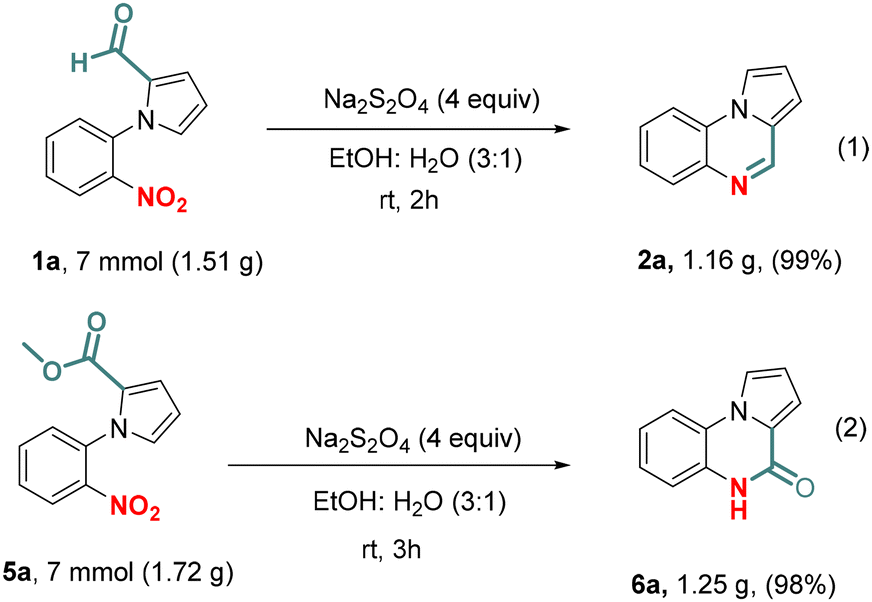 | ||
| Scheme 5 Gram scale synthesis of pyrrole fused N-heterocycles. | ||
Notably, the PMI of these reactions is in the range of 14–16, which is much lower than those of conventional metal-mediated reactions indicating the greener feature of this protocol. Additionally, other quantitative green metrics such as the E-factor and atom economy also comply with green chemistry (see the ESI†).
Next, we sought to understand the mechanism of the tandem chemoselective reductive cyclization. Some control experiments were carried out to understand whether the reaction occurred via a radical pathway. Then, we were interested to know if any radical quenchers (TEMPO and BHT) could play a role in inhibiting the reaction. When the standard reaction was performed in the presence of TEMPO (4 equiv.), the reaction yield significantly reduced to 40% (Scheme 6a). With a higher amount of TEMPO (10 equiv.), only a trace amount of 2a was obtained. Moreover, the reaction was completely halted with BHT (4 equiv.) indicating that a radical pathway is involved (Scheme 6b).
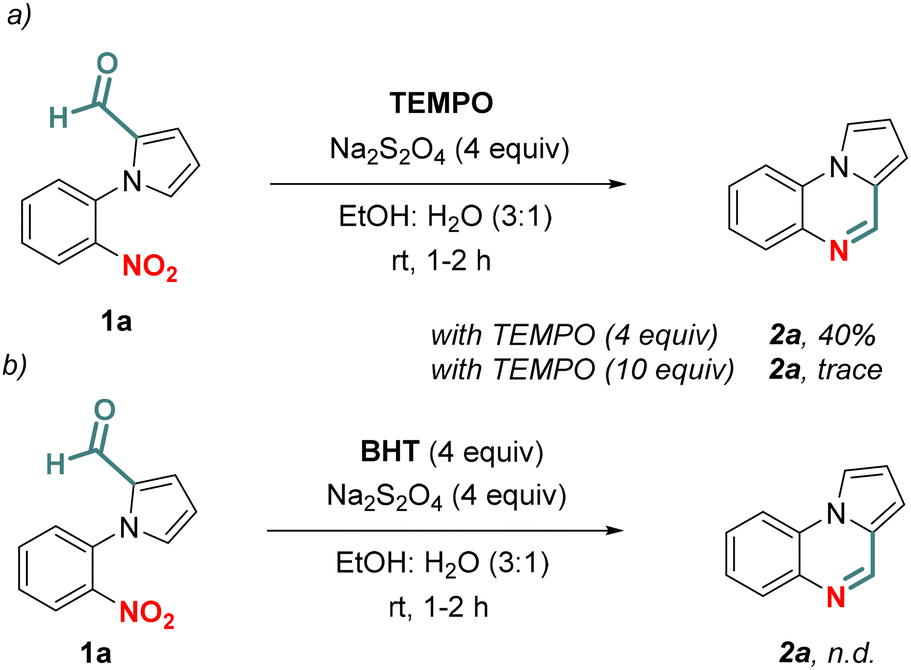 | ||
| Scheme 6 Control experiments. | ||
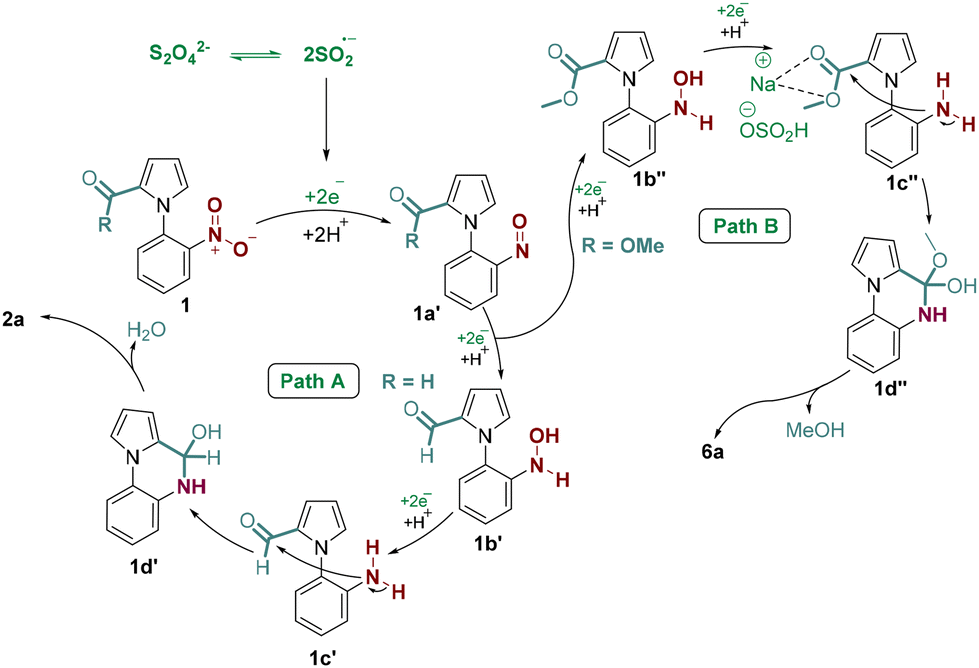
Based on the available literature reports6 and the experimental observation of our reaction, a plausible reaction mechanism is proposed as shown in Scheme 7. Initially, the sulfoxylate radical anion (SO2˙−) generated from the homolytic cleavage of the dithionite anion [S2O42−] reduces the nitro group in 1 to the nitroso intermediate 1a′ by sequential electron transfer. The sulfoxylate radical anion could transfer two more electrons to 1a′ to form a hydroxylamine intermediate 1b′. This intermediate 1b′ then could follow path A or B depending upon whether aldehyde or ester functional groups were present at the C-2 position of pyrrole. In path A, where the aldehyde group is present, the intermediate 1b′ could be reduced to the amine intermediate 1c′ by accepting two electrons from the sulfoxylate anion radical. This amine intermediate 1c′ then undergoes intramolecular nucleophilic addition with the aldehyde group to form intermediate 1d′, which upon aromatization could yield the desired cyclized product 2a. In path B, the intermediate 1b′′ is formed initially as in path A. Following this, the sodium bisulfite (NaHSO3) generated in situ from sodium dithionite and water could activate the ester in 1c′′ by 1,3-chelation. The subsequent intramolecular nucleophilic addition to the activated ester could form the intermediate 1d′′. Finally, the removal of MeOH could give the desired amide 6a.
 | ||
| Scheme 7 Plausible reaction mechanism for chemoselective reductive cyclization. | ||
Conclusions
In conclusion, we have developed a previously unexplored chemoselective reductive cyclization of N-(2-nitrophenyl)-pyrrole-2-carboxaldehydes and esters for the synthesis of diversely substituted pyrrolo[1,2-a]quinoxalines and pyrrolo[1,2-a]quinoxalin-4(5H)-ones, respectively, using Na2S2O4. The method involves a chemoselective reduction of the nitro group in the presence of an aldehyde or ester group followed by intramolecular imine or amide formation. The protocol features a very short reaction time (1 h), reaction at room temperature, no aqueous work up, purification by crystallization, and yields over 90%. The potential scalability of the method is demonstrated by a gram-scale synthesis. The chemistry reported herein is substantially different from the available literature in terms of adopting a different strategy, involving a cheap industrial reagent, and easy purification by simple crystallization. Further scope of the chemoselective reductive cyclization to form other heterocycles that are otherwise difficult to prepare is currently under investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors strongly acknowledge the CSIR, New Delhi (Ref. No. 02(0337)/18/EMR-II) and SERB, New Delhi (award number CRG/2020/000462) for the financial support.Notes and references
- N. Ono, The Nitro Group in Organic Chemistry, Wiley VCH, 1st edn, 2001 Search PubMed.
- M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Org. Process Res. Dev., 2018, 22, 430–445 CrossRef CAS.
- (a) M. Jang, T. Lim, B. Yong Park and M. Sua Hang, J. Org. Chem., 2022, 87, 910–919 CrossRef CAS PubMed; (b) S. Sambher, C. Baskar and R. S. Dhillon, ARKIVOC, 2009, 10, 141–145 Search PubMed.
- (a) B. Paul, S. Shee, K. Chakrabarti and S. Kundu, ChemSusChem, 2017, 10, 2370–2374 CrossRef CAS PubMed.
- C. Yi, X. Tan, B. Bie, H. Ma and H. Yi, Sci. Rep., 2020, 10, 1–8 CrossRef PubMed.
- (a) K. K. Park and C. H. Oh, Tetrahedron Lett., 1993, 34, 7445–7446 CrossRef CAS; (b) W. K. Joung, K. Koh, O. Chang Hun and S. Woo Jeon, J. Org. Chem., 1995, 60, 6202–6204 CrossRef; (c) C. Neyt and D. L. Riley, Beilstein J. Org. Chem., 2018, 14, 1529–1536 CrossRef PubMed; (d) C. T. Redemann and C. E. Redemann, Org. Synth., 1949, 29, 8–10 CrossRef CAS; (e) R. A. Scheuerman and D. Tumelty, Tetrahedron Lett., 2000, 41, 6531–6535 CrossRef CAS.
- (a) S. Oda, H. Shimizu, Y. Aoyama, T. Ueki, S. Shimizu, H. Osato and Y. Takeuchi, Org. Process Res. Dev., 2012, 16, 96–101 CrossRef CAS; (b) V. Kumar, B. Poojary, A. Prathibha and N. Shruthi, Synth. Commun., 2014, 44, 3414–3425 CrossRef CAS.
- B. Parrino, A. Carbone, C. Ciancimino, V. Spano, A. Montalbano, P. Barraja, G. Cirrincione, P. Diana, C. Sissi, M. Palumbo, O. Pinato, M. Pennati, G. Beretta, M. Folini, P. Matyus, B. Balogh and N. Zaffaroni, Eur. J. Med. Chem., 2015, 94, 149–162 CrossRef CAS PubMed.
- J. Guillon, E. Mouray, S. Moreau, C. Mullie, I. Forfar, V. Desplat, S. Belisle-Fabre, N. Pinaud, F. Ravanello, A. Le-Naour, J. M. Leger, G. Gosmann, C. Jarry, G. Deleris, P. Sonnet and P. Grellier, Eur. J. Med. Chem., 2011, 46, 2310–2326 CrossRef CAS PubMed.
- G. Campiani, E. Morelli, S. Gemma, V. Nacci, S. Butini, M. Hamon, E. Novellino, G. Greco, A. Cagnotto, M. Goegan, L. Cervo, F. Dalla Valle, C. Fracasso, S. Caccia and T. Mennini, J. Med. Chem., 1999, 42, 4362–4379 CrossRef CAS PubMed.
- J. Guillon, M. Le Borgne, C. Rimbault, S. Moreau, S. Savrimoutou, N. Pinaud, S. Baratin, M. Marchivie, S. Roche, A. Bollacke, A. Pecci, L. Alvarez, V. Desplat and J. Vose, Eur. J. Med. Chem., 2013, 65, 205–222 CrossRef CAS PubMed.
- (a) M. Ramamohan, R. Sridhar, K. Raghavendrarao, N. Paradesi, K. B. Chandrasekhar and S. Jayaprakash, Synlett, 2015, 1096–1100 CAS; (b) C. Wang, Y. Li, R. Guo, J. Tian, C. Tao, B. Cheng, H. Wang, J. Zhang and H. Zhai, Asian J. Org. Chem., 2015, 4, 866–869 CrossRef CAS.
- (a) G. S. Mani, A. V. S. Rao, Y. Tangella, S. Sunkari, F. Sultana, H. K. Namballa, N. Shankaraiah and A. Kamal, New J. Chem., 2018, 42, 15820–15829 RSC; (b) G. Jiang, S. Wang, J. Zhang, J. Yu, Z. Zhang and F. Ji, Adv. Synth. Catal., 2019, 361, 1798–1802 CrossRef CAS.
- (a) J. J. Lade, B. N. Patil, M. V. Vhatkar, K. S. Vadagaonkar and A. C. Chaskar, Asian J. Org. Chem., 2017, 6, 1579–1583 CrossRef CAS; (b) M. Lecomte, J. M. Lipshultz, S. H. Kim-Lee and G. Li, J. Am. Chem. Soc., 2019, 141, 12507–12512 CrossRef CAS PubMed.
- C. Xie, L. Feng, W. Li, X. Ma, Y. Liu and C. Ma, Org. Biomol. Chem., 2016, 14, 8529–8535 RSC.
- Z. An, L. Zhao, M. Wu, J. Ni, Z. Qi, G. Yu and R. Yan, Chem. Commun., 2017, 53, 11572–11575 RSC.
- J. Ahn, S. Beom, I. Song, S. Chun, D. Chann-Oh and S. Chang, J. Org. Chem., 2021, 86, 7390–7402 CrossRef CAS PubMed.
- H. Liu, F. Zhou, W. Luo, Y. Chen, C. Zhang and C. Ma, Org. Biomol. Chem., 2017, 15, 7157–7164 RSC.
- C. Xie, Z. Zhang, Z. Li, J. Gong, X. Han, X. Liu and C. Ma, J. Org. Chem., 2017, 82, 3491–3499 CrossRef CAS PubMed.
- X. Wang, X. Liu, C. Xie, F. Zhou and C. Ma, New J. Chem., 2020, 44, 2465–2470 RSC.
- J. K. Reeves, D. K. Fangrick, Z. Tan, J. J. Song, H. Lee, N. K. Yee and C. H. Senanayake, J. Org. Chem., 2010, 75, 992–994 CrossRef CAS PubMed.
- Z. Li, N. Yan, J. Xie, P. Liu, J. Zhang and B. Dai, Chin. J. Chem., 2015, 33, 589–593 CrossRef CAS.
- Q. Yuan and D. Ma, J. Org. Chem., 2008, 73, 5159–5162 CrossRef CAS PubMed.
- J. Miyashiro, K. W. Woods, C. H. Park, X. Liu, Y. Shi, E. F. Johnson, J. J. Bouska, A. M. Olson, Y. Luo, E. H. Fry and V. L. Giranda, Bioorg. Med. Chem. Lett., 2009, 19, 4050–4054 CrossRef CAS PubMed.
- G. Rotas, A. Kimbaris and G. Varvounis, Tetrahedron, 2004, 60, 10825–10827 CrossRef CAS.
- (a) M. Artico, G. De Martinor, R. Giuliano, S. Massa and G. C. Porretta, J. Chem. Soc. D, 1969, 671 RSC; (b) A. Mai, R. D. Santo, S. Massa, M. Artico, G. C. Pantaleoni, R. Giorgi, M. F. Coppolino and A. Barrachinni, Eur. J. Med. Chem., 1995, 30, 593–601 CrossRef CAS.
- S. C. Allen, S. Y. Hsieh, O. Gutierezz, J. W. Bode and M. C. Kozlowski, J. Am. Chem. Soc., 2014, 136, 11783–11791 CrossRef CAS PubMed.
- C. W. Cheung, M. L. Ploeger and X. Hu, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Scanned copy of 1H and 13C NMR spectra. See DOI: https://doi.org/10.1039/d2gc03749a |
| This journal is © The Royal Society of Chemistry 2023 |