
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous production of amines directly from alkenes via cyclodextrin-mediated hydroaminomethylation using only water as the solvent†
Thomas
Roth
a,
Rebecca
Evertz
a,
Niklas
Kopplin
a,
Sébastien
Tilloy
 b,
Eric
Monflier
b,
Dieter
Vogt
a and
Thomas
Seidensticker
*a
b,
Eric
Monflier
b,
Dieter
Vogt
a and
Thomas
Seidensticker
*a
aTU Dortmund University, Department for Biochemical and Chemical Engineering, Laboratory of Industrial Chemistry, Emil-Figge-Straße 66, 44227 Dortmund, Germany. E-mail: thomas.seidensticker@tu-dortmund.de
bUniversity Artois, CNRS, Centrale Lille, Univ. Lille, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, F-62300 Lens, France
First published on 28th March 2023
Abstract
Aqueous hydroaminomethylation (HAM) is an atom economical route for the efficient production of amines in one reaction step, starting from basic chemicals like alkenes. Herein we present the first successful establishment of a continuous process for HAM in an aqueous multiphase system. The green mass transfer agents randomly methylated-β-cyclodextrins (CD) enabled the catalytic system consisting of rhodium/sulfoXantphos to achieve high yields of up to 70% with selectivities of up to 80% in several continuous experiments with a total run time of more than 220 h. The key here is that water and products have large polarity differences, but the reaction still proceeds effectively due to the addition of cyclodextrin, which made the application of solvents obsolete. The main achievements in this way were the investigation of the influence of the randomly methylated-β-cyclodextrin concentration on the reaction rate and the selectivity in batch studies and finding promising operating points in the first continuous experiments. In a final experiment, the separation temperature was investigated. It was shown that the catalyst loss in the product phase is enormously small at 0.003% h−1 of the initial mass (0.24% in total), which is the lowest ever reported value for the HAM on this scale. Within a run time of 78 hours, 2.87 kg of tertiary amine were produced using only 0.2 g (>14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000:1) of transition metal, while the loss of rhodium per kg of product produced was mostly around 0.15 mg kg−1, suggesting possible economical applicability.
000:1) of transition metal, while the loss of rhodium per kg of product produced was mostly around 0.15 mg kg−1, suggesting possible economical applicability.
Introduction
One of the most urgent challenges facing humanity is anthropogenic climate change. To cope with this burden, we must perform a comprehensive structural change in our value creation system to reduce environmental damage and resource consumption to a permanently sustainable level.1,2 However, a competitive industry with a high level of economic efficiency must be maintained. Regarding the chemical industry, this implies the development of highly selective and efficient reactions and processes. In this context, the use of specific catalysts is imperative as they enable atom economical reactions, thus minimizing waste and reducing the overall downstream effort. This is further highlighted by the fact that catalysis is one of the 12 principles of green chemistry, and the production of the majority of chemical intermediates and end products incorporates at least one catalytic step.3,4Despite their advantages, one major challenge remains: their efficient recycling and reuse are indispensable for the ecological feasibility and economic success of industrial processes, especially when utilizing expensive transition metals such as rhodium. An essential requirement for recycling is that the catalyst and reaction products are separated into two distinct phases, regardless of whether this segregation occurs before, during, or after the reaction. In large-scale processes, the generation of a new phase from a homogeneous mixture causes an enormous demand for energy and equipment, which leads to costly processes, leading to costly procedures. This results in the majority of industrial processes being heterogeneously catalyzed. However, the described requirement for highly efficient reactions makes the use of homogeneous transition metal catalysis particularly advantageous.5 To combine their high activities and selectivity with the beneficial separability of heterogeneous catalysts, the concept of liquid multiphase systems was developed.6 Here, a common homogeneous catalyst is immobilized in one liquid phase while the reaction product resides in another. Since most industrially relevant products are nonpolar, using water as a solvent for the catalyst is especially practical. The high polarity and density of water lead to a fast and reliable separation even with products of medium polarity while being non-hazardous and non-flammable as well as odorless. In addition, the great accessibility at a comparably low cost emphasizes the beneficial properties from an economical and environmental viewpoint.7 Established water-soluble ligands, which are already commercially available, can be used to reduce the loss of the catalyst to the nonpolar product phase. Without the need for an organic solvent, the possibility of generating a pure product phase exists depending on the polarity of the products. This has many advantages for application in technical processes, such as lower liquid holdups, resulting in higher space–time yields and a lower cost downstream. Therefore, combining highly efficient and selective reactions by utilizing common homogeneous transition metal catalysts with the good separation performance of aqueous biphasic reaction systems offers excellent potential for the design of sustainable processes (Fig. 1).
 | ||
| Fig. 1 Basic process design for the recycling of homogeneous catalysts using aqueous biphasic reaction systems. | ||
While products having low water solubility offer good separation with an expected low catalyst leaching, they often arise from low water-soluble substrates, leading to a biphasic system under reaction conditions and, therefore, low substrate concentration in the catalyst phase. This results in a low overall reaction rate since the catalyst cannot reach its intrinsic activity like it would in homogeneous reaction media. Hence, intensification techniques are required to exploit the usually high activities of homogeneous catalysts. A variety of different strategies have been explored in the literature in the context of batch experiments. However, establishing a stable continuous operation is essential to emphasize the advantages of intensified aqueous biphasic reaction systems and demonstrate their competitiveness among large-scale processes. An in-depth literature review on intensification strategies in continuous processes was given by Vorholt, Leitner, and coworkers in 2019, highlighting the feasibility and importance of this research field.8 Intensified mixing, e.g., by utilizing innovative reactor concepts, aims to increase the contact area between the substrate and catalyst molecules due to an increased surface area between the two liquid phases.9–11 Another approach is the application of co-solvents to alter the miscibility gap and increase the substrate solubility in the catalyst phase. This also affects the solubility of the catalyst in the product, leading to potentially increased catalyst loss.
An especially promising approach in this respect that increases the substrate solubility in the polar phase without significantly changing the catalyst's solubility in the substrate phase is the addition of cyclodextrins.12 These cyclic oligosaccharide molecules consist of α-D-glucopyranose units and form conical cylinders with a hydrophilic outer surface and a hydrophobic cavity. Many different hydrophobic substrates exhibit strong binding affinity to the latter, leading to a wide variety of applications in practice, e.g., in analytical chemistry, agriculture, and the pharmaceutical industry.13,14 These and other properties of cyclodextrins, such as different solubilities, can be created by different substituents on the hydroxyl groups. The beneficial impact of cyclodextrins in aqueous, biphasic and homogeneously catalyzed reaction systems has been investigated and pointed out for many years (Fig. 2).15–21 It is generally accepted in the literature that the mechanism is best described by the formation of inclusion complexes at the aqueous/organic interface.
 | ||
| Fig. 2 Proposed mechanism for cyclodextrin-mediated reaction systems.22 | ||
As amines are essential building blocks for modern society, now the next step is to enlarge the toolbox of biphasic aqueous reaction systems to amination reactions. One attractive alternative in this regard is hydroaminomethylation, discovered initially by Reppe in 1949.23 Hydroaminomethylation is an auto-tandem catalytic reaction in which amines are accessible from alkenes as basic chemicals in a single preparative step. However, it thus has a rather complex reaction network, with the first reaction being the hydroformylation to an aldehyde. It is followed by reductive amination, which itself involves a condensation reaction between an aldehyde and an amine, may it be ammonia, primary or secondary, to an enamine which is then catalytically hydrogenated to the corresponding amine (Fig. 3). Interestingly, both catalyzed reactions are carried out with two different catalyst species, which are in equilibrium with each other.24,25 This auto-tandem reaction is a potential alternative to industrial processes for amine synthesis starting from unsaturated substrates, being an atom-efficient reaction with no need for isolation and purification of intermediates.25 Although water is formed as a co-product in an equilibrium reaction, which makes aqueous solvent systems appear unsuitable, here, one also benefits from simple separation.
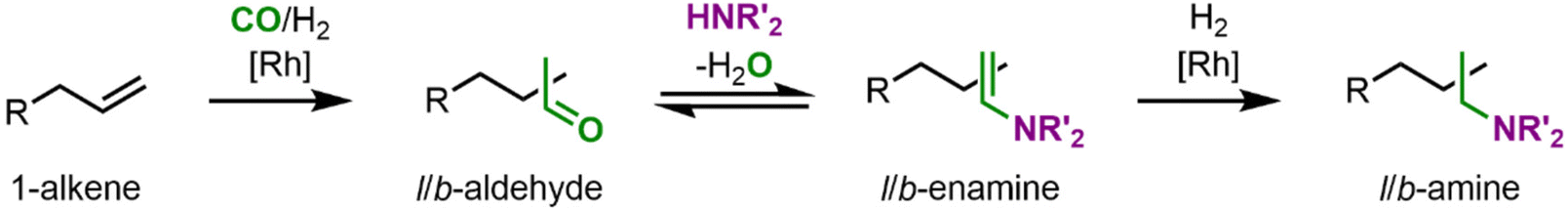 | ||
| Fig. 3 General scheme of the hydroaminomethylation based on a 1-alkene and a secondary amine. | ||
As a result, aqueous hydroaminomethylation has gained more and more attention in recent years. However, to the best of our knowledge, there are no examples of continuous processes to produce amines via hydroaminomethylation in an aqueous reaction system. Several intensification techniques have been presented and others have been investigated in batch reactions in the literature.26–31 Here, cyclodextrin-mediated systems showed the greatest potential due to low leaching but at the expense of a comparably low reaction rate and the formation of solid matter.32 Although this is not a hurdle in batch reactions, it leads to tremendous challenges in continuously operated reactions. The hydroaminomethylation demands long residence times, resulting in small volume flows, which is opposed by the necessity of a minimum flow velocity in the pipes to prevent segregation of the phases. A similar challenge arises, e.g., when selecting the right stirrer speed: while the biphasic reaction system demands a high volumetric power input to present a high gas–liquid–liquid surface area, foam formation must not occur to maintain a stable operation. The combination of exploring these operation windows and the well-known challenges of studying complex reaction networks might be the reason why no continuous process for aqueous hydroaminomethylation has been reported yet. Herein, we aim to fill this gap by demonstrating the upscalability of this reaction system to a continuously operated miniplant. This might allow continuous synthesis with potentially the lowest leaching in a green solvent system without the addition of conventional solvents.
Results and discussion
For the first continuous operation in an aqueous multiphasic reaction system mediated by cyclodextrins, the hydroaminomethylation of 1-octene l-1 and diethylamine with syngas was chosen as a model reaction (Fig. 5). The triphasic system consists of a gas phase, a pure (i.e., solvent-free) substrate/product phase, and an aqueous phase containing the precursor Rh(acac)(CO)2, the ligand sulfoXantphos and randomly-methylated-β-cyclodextrins (CD) as phase transfer agents (Fig. 4). | ||
| Fig. 4 Chemical structures of randomly methylated-β-cyclodextrin (phase transfer agent) and sulfoXantphos (ligand). | ||
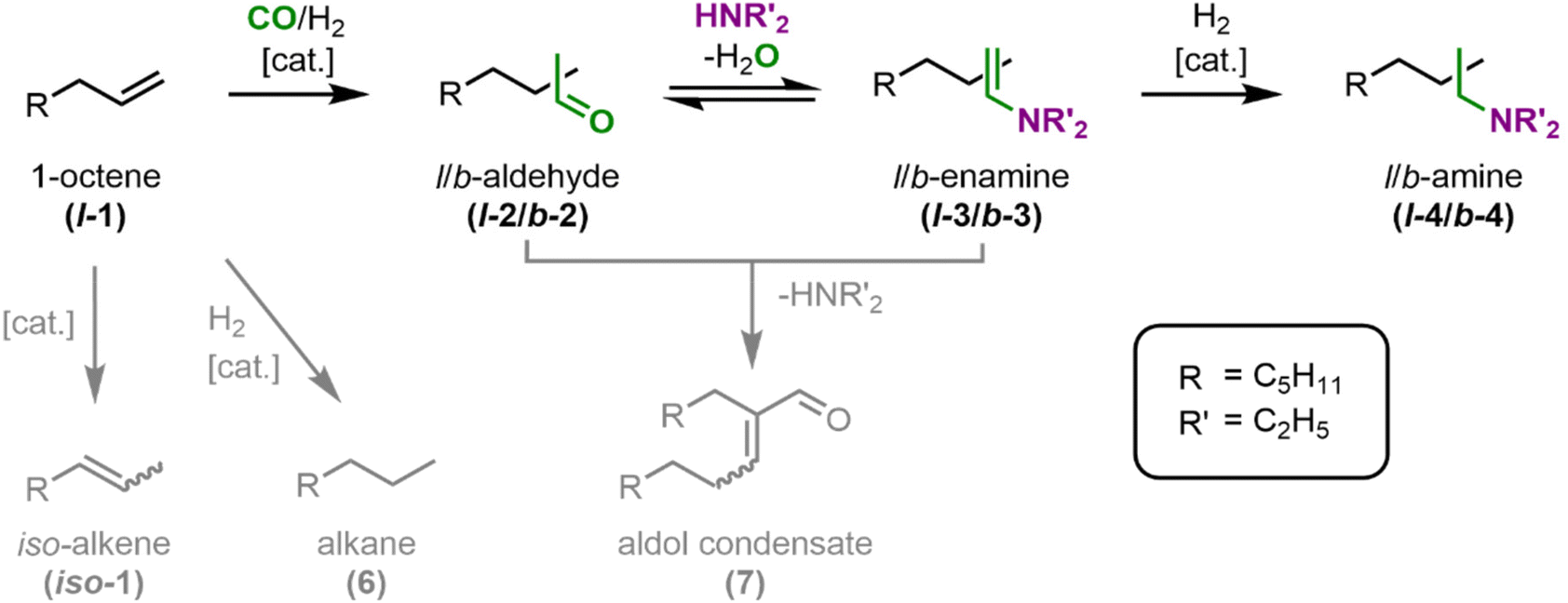 | ||
| Fig. 5 Reaction network for the hydroaminomethylation of 1-octene (l-1) with diethylamine (DEA) to the desired linear tertiary diethyl octylamine (l-4). | ||
The first step in the main reaction path is the hydroformylation of l-1 to the linear aldehyde l-2. In the subsequent reductive amination, l-2 first condenses to the linear enamine l-3 before the linear amine l-4 is formed by catalytic hydrogenation. In case the branched aldehyde b-2 is formed in the hydroformylation, it follows the same reaction path. Other byproducts arising from side reactions are the alkene iso-1 from isomerization, the alkane 6 from hydrogenation, and the aldol condensate 7. Assuming that the solubility of the substrates in the catalyst phase without coordination in the CD can be neglected, the formation of an inclusion complex between the CD and the substrates is necessary for the hydroformylation and hydrogenation to occur. This might lead to a kinetic limitation of the main reaction path, and thus a decrease in selectivity as an aldol side reaction might also occur in the organic phase.
Batch experiments
For realizing a continuous process of cyclodextrin-mediated hydroaminomethylation for the first time, two major aspects from the previous investigations are still limiting: the recyclability of the chosen catalytic system has not been shown in HAM, with one problem being the formation of solid matter that cannot be tolerated during continuous experiments; secondly, a typical reaction time of 24 h for full conversion is not reasonably realized in a continuous experiment. Hence, we decided to first investigate the recycling of the homogeneous Rh-catalyst and, secondly, to decrease reaction time by detailed analysis of the cyclodextrin amount.In three successive recycle batch experiments in a 300 ml autoclave at 120 °C and 35 bar of syngas pressure with a substrate-to-catalyst ratio of 500, which corresponds to standard conditions based on our recent publication,32 almost complete conversion of the substrate was observed (see the ESI for details†). The yield of the desired amine l-4 exceeds 68% in all runs, whereas the byproducts iso-1 and 6 constantly show a combined yield of 13% during the recycling. The yield of the aldol condensate 7 was ca. 9% in all runs. As expected, regioselectivity is constant at a high level of >97%, which is in the range for a rhodium/sulfoXantphos system.33 To our delight, in contrast to our previous investigation, in which 1-decene was used as a substrate, no precipitation of solid matter was observed. The leaching of both Rh and sulfoXantphos is also nearly constant at a low level of around 0.2% of the initial mass per run, proving the recyclability of the system.
In the next step, we investigated an acceleration of the overall reaction rate by increasing the CD amount in the reaction system. An increased CD concentration can work in two ways: it reduces the surface tension of the aqueous phase with air, as shown by Monflier and coworkers.34 This fact allows the assumption that the interfacial tension between water and an organic phase is also reduced, which would result in an increased phase interface at an equal volume-specific energy input. Furthermore, the CD concentration influences the formation of the inclusion complexes A (Fig. 2), which influences the contact of the substrates with the catalyst at the liquid–liquid interface area.22 Thus we suspect that an increased CD concentration may accelerate the conversion of l-1 to achieve higher catalytic activity and therefore contribute to the aim of a reduced reaction time for full conversion. Since, surprisingly, CD-to-catalyst ratios higher than 15 have never been used in hydroaminomethylation or even hydroformylation, we examined this parameter in more detail. So far, it is unknown whether the alkene l-1 and the enamine l-3, respectively, have unequal association constants with CD and, hence, how this affects the complex reaction network of HAM. Hereby, it is possible that one complex is formed more prominently, and thus the reactions are accelerated unequally. This can lead to an accumulation of intermediates and enhanced side reactions such as aldol formation. Accordingly, various CD-to-Rh ratios of up to 250 have been studied (Fig. 6) to find optimized reaction conditions to ensure both high catalytic activity and selectivity for the targeted continuous experiments.
 | ||
| Fig. 6 Conversion (X) and yield (Y) of l-1 over time (left) as well as product distribution (right) at full conversion of the HAM depending on the CD to catalyst ratio (reaction time from left to right: 24 h, 12 h, 6 h, 6 h, 4 h). Conditions: T = 120 °C; p = 35 bar; nH2/nCO = 2:1; u = 800 rpm; cRh,aq. = 3.2 mmol L−1; nP:nRh = 7; φ = 0.2; n1-alkene:nRh = 500; nDEA:n1-alkene = 1.5; nCD:nRh = 15 to 250; t = 6 h to 24 h; preforming: T = 120 °C; p = 35 bar; nH2/nCO = 1:1; u = 800 rpm; t > 12 h. | ||
It is evident that increasing the CD-to-catalyst ratio increases the amount of product produced per time significantly. While it takes 24 and 12 hours to achieve a conversion of 98% with the lower ratios of 15 and 25, respectively, conversions of 98.5% and >99% can be achieved in 6 hours with the medium ratios of 50 and 100. Although the reaction system with a ratio of 250 is even faster with a conversion of >99% after only 4 hours, a reduced selectivity to the main product l-4 can be observed. Apparently, the hydroformylation rate is enhanced more with increasing CD-to-catalyst ratio than the rate of the enamine hydrogenation, which is visible from an increased yield of the intermediates l-2 and l-3 when comparing the reaction profiles (see the ESI†). While this, on the one hand, reduces the selectivity to iso-1 and 6, on the other hand, the resulting accumulation of intermediates l-2 and l-3 leads to an amplified aldol condensation to 7. Nevertheless, it should be noted that the conversion of 90% after 0.5 h in an aqueous HAM is evidence of a remarkably active system. However, the most promising compromise between activity and selectivity is observed at a ratio of 100 with a selectivity of 78% and a turnover frequency of 531 h−1 at 50% conversion. In further investigations, it was found that a further increase of the TOF was possible by reducing the catalyst concentration at high cyclodextrin concentrations. In this case, too, there was a clear decline in selectivity, which is why this approach was not considered further (see the ESI for further information†).
Continuous operation
The findings from the batch tests regarding activity, stability, and recyclability now allow us to carry out continuous experiments. Here the long-term stability and the optimization of various parameters, such as reaction temperature, are carried out under continuous process conditions. As we have recognized with our vast experience in various reaction systems, this strategy is considerably more target-oriented than further optimizing reaction conditions in additional, tedious batch experiments.However, one challenge here is that the process conditions must first be found under which the miniplant (Fig. 7) used can be operated reliably. Here, various effects that are irrelevant in batch reactions come into play. One important parameter for gas–liquid–liquid reaction systems like CD-mediated hydroaminomethylation is the provided surface area.18 High energy input and, thus, high stirring speed are expected to benefit the reaction but can be detrimental due to foam formation. While this is commonly not a problem in a batch operation, in continuous processes, this can be a hurdle.
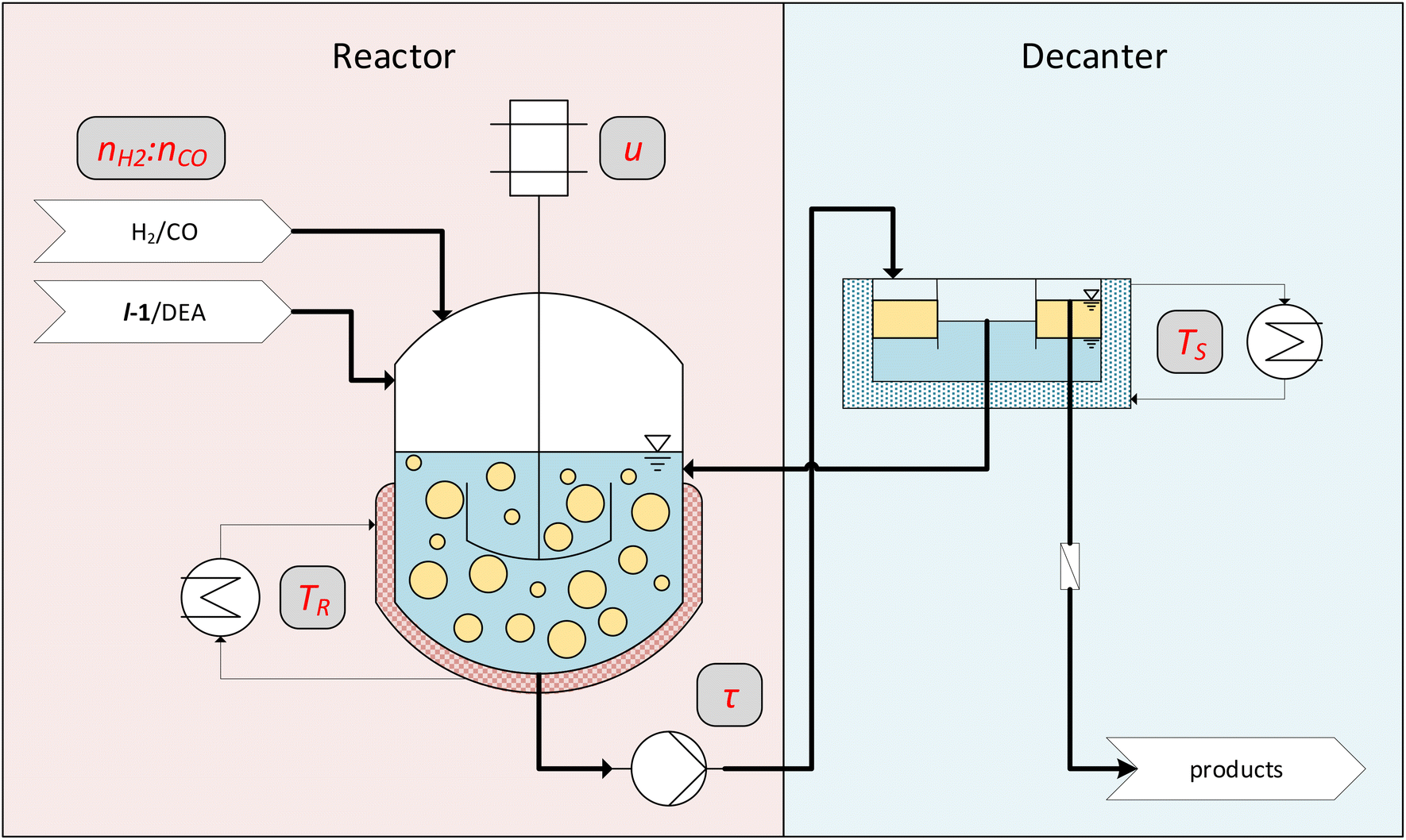 | ||
| Fig. 7 Conceptual flowsheet of the operated miniplant. Parameters that are varied in the continuous experiments are noted in gray boxes. | ||
Another challenge in the investigated biphasic reaction system is the separation of phases in vertical pipes when the flow velocity is too low. This might lead to the accumulation of one phase in the reactor and thus change the organic volume fraction φ, which has a critical influence on the selectivity.32 Residence time is correlated with flow velocity in pipes, which also heavily influences the reaction. The first continuous experiment was carried out (Fig. 8) to determine a suitable operation point for these process conditions.
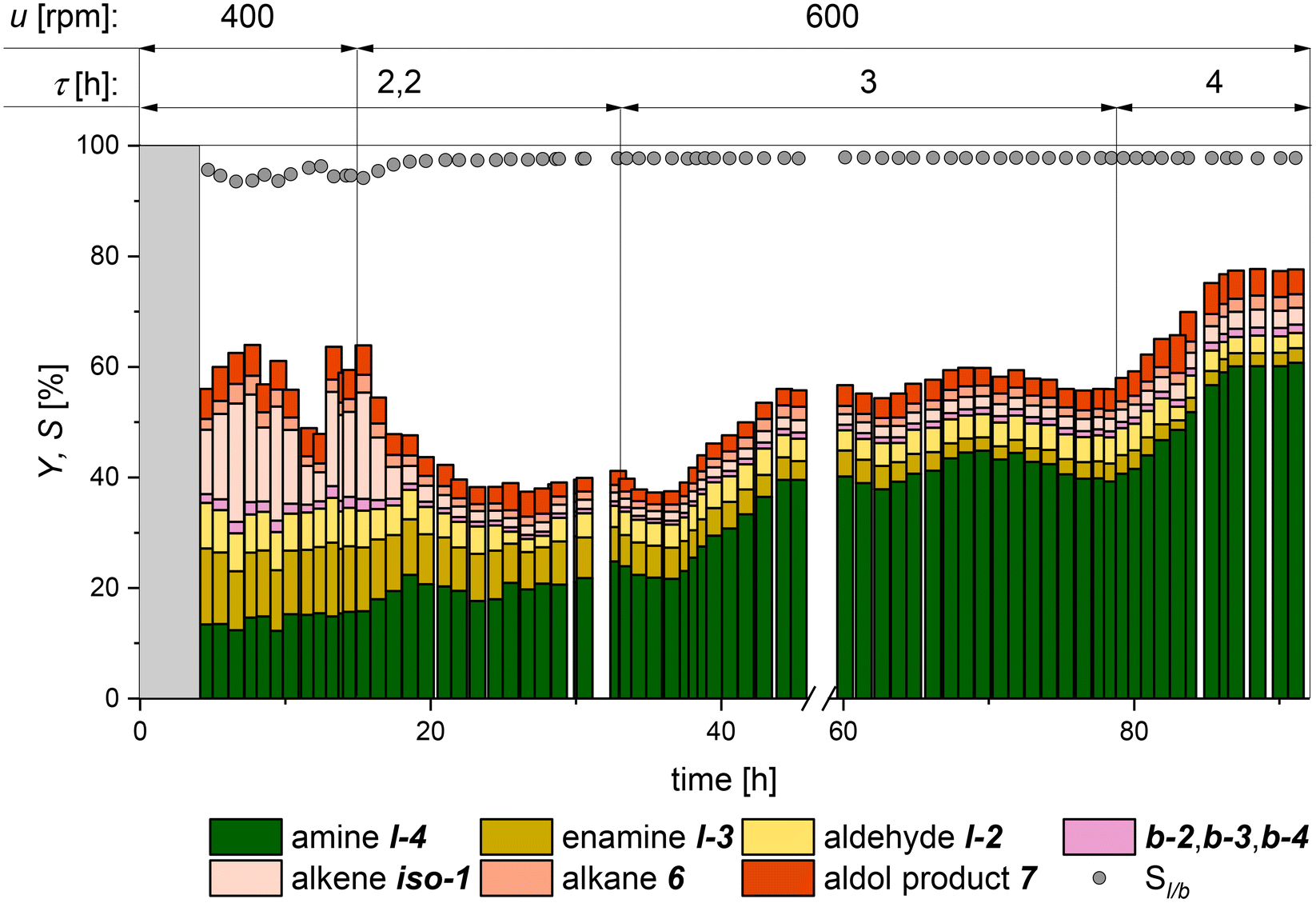 | ||
| Fig. 8 Reaction performance for the continuously operated hydroaminomethylation of 1-octene with diethylamine. Conversion equals the sum of all yields. Yields are calculated based on GC-FID analysis of the product stream with dibutyl ether as the internal standard. Conditions: T = 120 °C; p = 35 bar; u = 400 to 600 rpm; VReactor = 2100 ml, Vliq. = 770 ml; nH2/nCO = 1:1; cRh,aq.,0 = 3.2 mmol L−1; n1-alkene:nRh = 500; nCD/nCat. = 100; nP/nRh = 7; nDEA/n1-alkene = 1.5; φ = 0.2; τ = 2.2 h to 4 h; preforming: T = 120 °C; p = 35 bar; nH2/nCO = 1:1; u = 800 rpm; t > 12 h. | ||
Although HAM requires a molar ratio of 2:1 (H2:CO), the optimum reaction conditions regarding product yield and selectivity can differ from that, for example, due to different activities of solute gasses in the reaction phase and depending on the operation mode. Based on our earlier publication, a ratio of 1:1 in the reactor's gas phase was chosen.26 The gas feed was adjusted depending on the measured gas composition in the reactor to maintain this ratio during the continuous operation. Samples of that gas phase were analyzed at regular intervals via GC (further information is provided in the ESI†). Based on our batch investigation, a CD-to-catalyst ratio of 100 was chosen. In this first continuous experiment, the residence time τ and the stirrer speed are varied, starting with τ = 2.2 h and a stirrer speed of 400 rpm. Furthermore, it will be examined to what extent the selectivity of the side products can be reduced by the changed concentration profiles in a continuously stirred tank reactor (CSTR) compared to that with a conventional batch-stirred tank reactor (STR).
The first 4 hours are the start-up time of the miniplant (marked in grey). In the first 15 hours of continuous operation, a strong fluctuation of the conversion and iso-1 yield can be seen, indicating insufficient mixing in the reactor. Hence, the speed of the pitched 4-blade stirrer was increased from 400 rpm to 600 rpm to increase the availability of the gases in the reaction phase. Consequently, the selectivity to the main product l-4 increased, and an almost stable yield of 20% was achieved. After increasing the residence time to 3 h, a significant rise in the main product yield to above 40% was observed. Also, a slight increase in chemoselectivity could be observed as the reaction intermediates l-2 and l-3 reacted further to give the main product l-4. The same observation could be made for the second increase of residence time to 4 h after 78 h. Here, a stable yield for l-4 of 60% could be maintained for 5 hours before the experiment was shut down. Throughout the whole experiment, a stable miniplant operation was guaranteed, with no need for continuous feed of catalyst, ligand, or CD. A suitable operating window of reaction and process conditions of the reaction system could be identified to ensure stable plant operation. A significantly lower conversion than that in the batch experiments (Fig. 6, right) was observed throughout the experiment, as well as the formation of the side product 7. This can be attributed to the fact that the reaction rate and the accumulation of intermediate products remain at a considerably lower level in a CSTR compared with the reaction profile in a batch reactor. Furthermore, it can be observed that a longer reaction time mainly favors the conversion of the intermediate l-3, which seems to be the rate-limiting step in this reaction network.
In the next step, different reaction conditions were tested on-stream (Fig. 9), while the residence time was kept at 2.2 h to achieve a faster transition into the pseudo-stationary operation points. Since the various partial reactions exhibit different temperature dependencies, the focus here was placed on the variation of the reaction temperature. An increase in activity and possibly a change in chemoselectivity should be observed as the reaction temperature increases. An increased yield is accompanied by an increase in the formation of the co-product water. To verify the estimations made for the ratio of the reaction gases hydrogen and carbon monoxide, a molar ratio of 2:1 was applied at the start of the experiment. As expected, high yields of the side products iso-1 (20%) and 6 (14%) are observed after the start-up phase of the miniplant (marked in gray). With a gas ratio of 1:1, the same yield as in the previous experiments is achieved, highlighting the reliability of this system.
 | ||
| Fig. 9 Reaction profiles for the continuously operated hydroaminomethylation of 1-octene with diethylamine at varying reaction temperatures and gas ratios. Conversion equals the sum of all yields. Yields are calculated based on GC-FID analysis of the product stream with dibutyl ether as the internal standard. Conditions: τ = 2.2 h; p = 35 bar; n = 600 rpm; cRh,aq.,0 = 3.2 mmol L−1; n1-alkene:nRh = 500; nCD:nRh = 100; nP:nRh = 7; nDEA:n1-alkene = 1.5; φ = 0.2; preforming: T = 120 °C; p = 35 bar; nH2/nCO = 1:1; u = 800 rpm; t > 12 h. | ||
At 20 h, the reaction temperature was increased to 130 °C. Unfortunately, due to a technical error, the gas space was simultaneously significantly enriched with hydrogen, which was corrected in the following step. All of this led to a stable increase of the main product amine l-4 yield to nearly 38%, which was maintained in a pseudo-stationary operation point for 6 hours. Here a lower yield of the intermediate product l-3 compared with the last operation point (at 120 °C between 14 h and 20 h) is observed. At 38 h, the reaction temperature was increased to 140 °C. Again, a 10% increase in the yield of l-4 is observed. Note: the residence time was kept constant at the lowest level of 2.2 h only. In order to keep the miniplant at an operable state, the formed co-product water had to be removed in a purge step after 45 h in order to keep the phase ratio within the desired range. The resulting reduction in catalyst concentration led to a slight decrease in selectivity towards the end of the experiment, which was expected from the former results with reduced catalyst concentration. Nevertheless, the positive influence of the increased reaction temperature is evident. Since the literature has shown rhodium catalyst deactivation at 150 °C, no further temperature increase was applied.35
In the final continuous experiment, the optimized conditions from the previous experiments were applied to ensure stable operability and the highest yields. Therefore, a residence time of 4 h was chosen at a reaction temperature of 140 °C and a pressure of 35 bar with a 1:1 H2:CO mixture (Fig. 10). During the experiment, only the separation temperature in the decanter was varied. Although this might influence the leaching of the catalyst into the products, a separation temperature close to the reaction temperature also prevents the need for large energy flows to temper the aqueous catalyst phase. Carrying out hydroaminomethylation in an aqueous multiphase system is a compromise between optimal reaction control and catalyst recycling. In order to be able to evaluate this compromise in terms of its target accuracy, the rhodium loss per product produced is considered in addition to the leaching.
 | ||
| Fig. 10 Yields over time for the continuously operated hydroaminomethylation of 1-octene with diethylamine. Conversion equals the sum of all yields. Yields are calculated based on GC-FID analysis of the product stream with dibutyl ether as the internal standard. Conditions: τ = 4 h; T = 140 °C, p = 35 bar; nH2/nCO = 1:1; u = 600 rpm; cRh,aq.,0 = 3.2 mmol L−1; n1-alkene:nRh = 500; nCD:nRh = 100; nP:nRh = 7; nDEA:n1-alkene = 1.5; φ = 0.2; preforming: T = 120 °C; p = 35 bar; nH2/nCO = 1:1; u = 800 rpm; t > 12 h. | ||
From 18 h on, the experiment was run pseudo-stationary for about 28 hours, which is equivalent to 7 residence times. Here, a yield of the amine l-4 of up to 70% with a chemoselectivity of about 81% was achieved, showcasing the efficiency of this reaction system. After 34 h, the formation of the co-product water made regular purging necessary, as described above. The subsequent deterioration in product yield to 60% with a selectivity of 72% at 70 h was due to the reduced catalyst concentration, which is also consistent with the former results. The separation temperature TS in the decanter was raised from 30 °C to 60 °C from 40 h onwards to investigate the phase separation under real process conditions further. As can be seen from the leaching results (Fig. 11), this results in an immediate increase of the ligand leaching while the leaching of rhodium into the product remained at its previous low level of around 0.003% h−1. An increase in rhodium leaching can be observed, resulting in increased rhodium loss per product beginning after 60 h. However, it can be stated that long-term operation of the reaction system was possible without the need for a ligand or catalyst makeup. This was made possible by the enormously small proportion of rhodium in the product phase of mostly less than 100 ppb. In addition, no loss of cyclodextrins to the product phase was detected (further information is provided in the ESI†). It is noteworthy that the limitation of the reaction carried out here is not its catalyst loss into the product phase but solely the formation of the co-product water. This leaves no doubt that the value of 2.87 kg tertiary amine produced by 0.19 mg of rhodium (>14000:1) used could have been significantly increased with a longer campaign period. In future work, the co-product water could be separated using organophilic nanofiltration membranes, as has already been successfully demonstrated with research work in cooperation with our group on similar reaction systems.36 Here, HAM was performed in a continuously operated thermomorphic multiphase system consisting of the organic solvents dodecane and methanol. In this system, a maximum yield of 66% with a rhodium loss of 11 mg kg−1 product could be achieved at the same residence times. From this comparison, it becomes clear that the system presented in this work not only shines through its enormously low leaching values but also through its competitive yields.
 | ||
| Fig. 11 Leaching results of catalysts and the ligand over the reaction time of the continuous experiment presented in Fig. 10. Leaching corresponds to % of the initial rhodium and sulfoXanphos mass. | ||
Conclusions
The aqueous biphasic hydroaminomethylation (HAM) was performed in a continuous operation mode for the first time. Of the various intensification strategies discussed in the literature, the use of RAME-β-cyclodextrins was selected as the most promising approach, and the transferability to HAM of 1-octene with diethylamines was first verified in recycling experiments. Subsequently, batch experiments were carried out in order to find the experimental conditions for the first time that would allow a reasonable transition to continuous operation. It was found that the overall conversion rate was mainly dependent on the cyclodextrin concentration. However, an unequal dependence of the individual reaction rates in the investigated tandem catalytic reaction network of HAM on the cyclodextrin and catalyst concentration can lead to an unfavorable shift in the product distribution. The determined suitable reaction conditions were then transferred for the first time to a continuously operated miniplant. Once an operating window for the miniplant had been found, various reaction conditions were further investigated on-stream. It was found that an increased reaction temperature not only enhances the conversion but also leads to an increase in the main product selectivity. The characteristic reaction profiles in the CSTR also allowed the formation of undesirable byproducts to be distinctly reduced compared to the batch experiments. Conclusively, the previously obtained findings were combined in a successful long-term experiment without changes in reaction conditions. Over 80 hours, high yields of up to 70% and the main product selectivity of over 80% with linear to branched selectivity of above 96% were achieved without the need for a catalyst- or ligand makeup. During this last experiment, the catalyst leaching was at enormously small values of less than 0.003% h−1 at most times. In particular, the low rhodium loss per produced product of under 0.15 mg kg−1 emphasizes the possible application of this reaction system in large-scale processes.Experimental section
General methods and reagents
The chemicals and reagents used in this work are listed in Table 1. All liquid chemicals used for the reactions were degassed with argon dispersed by a frit for at least one hour in an ultrasonic bath. Process monitoring of liquid phases was performed with a gas chromatography system by Agilent Technologies (7890A) with a flame ionization detector and a capillary column type HP-5 (30 m × 0.32 mm × 0.25 μm). For validating those results, another gas chromatography system by Agilent Technologies (7890A) equipped with a thermal conductivity detector and a capillary column type HP- 5 (30 m × 0.32 mm × 0.25 μm) was used. For this purpose, 0.6 g of tetrahydrofuran as the solvent, 0.05 g of di-n-butyl ether as the internal standard, and 0.35 g of sample were used. For measuring gas phase samples, another gas chromatography system by Agilent Technologies (7890A) equipped with a thermal conductivity detector was used. Separation of gaseous species took place in an HP-Plot Molesieve column (30 m × 0.53 mm × 0.5 μm). Determination of rhodium and phosphor traces took place on an inductively coupled plasma optical emission spectrometer by Analytic Jena (Plasma Quant PQ 9000). If necessary, samples were concentrated in vacuo at elevated temperature to measure values below the limit of quantification.| Chemical | Manufacture | Purity [%] |
|---|---|---|
| 1-Octene | Acros | >99 |
| Diethylamine | Carl Roth | >99.5 |
| Ultrapure water | Generated with PURELAB flex 3 | — |
| Rh(acac)(CO)2 | Umicore | 99.9 |
| SulfoXantphos | MOLISA | — |
| RAME-β-cyclodextrin | Sigma-Aldrich | >98 |
| Di-n-butyl ether | Acros | >99 |
| Tetrahydrofuran | Acros | 99.9 |
| CO | Messer | 98 |
| H2 | Messer | 99999 |
| Argon | Messer | 99996 |
| N2 | Messer | 99.5 |
Procedure for batch experiments
All batch experiments were carried out in a 300 ml overhead stirred autoclave with a pitched blade stirrer at a speed of 800 rpm and continuous gassing during the reaction. For reaction experiments, ultrapure water and cyclodextrin are first dissolved in an ultrasonic bath at 40 °C for 30 minutes. Then the weighed precursor and ligand are added and dissolved for 30 minutes at 40 °C in the ultrasonic bath. The reactor is sealed pressure tight and is inertized by repeatedly drawing a vacuum and applying 5 bar argon. The prepared catalyst solution and diethylamine are drawn into the reactor via vacuo before applying 20 bar of H2/CO pressure and a temperature of 120 °C. Preforming took place overnight for at least 12 hours in most cases. After that, a reaction pressure of 35 bar in the targeted ratio of H2/CO is applied, and 1-octene is added to the reactor via a substrate bomb.Once the reaction time expires, continuous gassing is stopped, and the reactor is cooled in an ice bath to at least 20 °C before stopping the stirrer and degassing. The reactor is then flushed with argon several times. After opening the reactor and separating the phases in a separation funnel, the corresponding analyses were carried out. After all of that, the reactor was cleaned with isopropanol.
In the case of recycling experiments, the aqueous phase was separated and used for the next run as described above but without additional preforming. Samples were taken via a capillary line and a needle valve to determine reaction profiles. The capillary tube was first flushed with reaction media before taking the sample.
Procedure for continuous experiments in the miniplant
The commissioned miniplant (Fig. 12) consists of a 2100 mL overhead stirred tank reactor equipped with a pitched blade stirrer and baffle, providing the same volume-specific power input as in the batch reactions at a speed of 800 rpm. The reaction mixture is conveyed through a riser tube via a gear pump into a decanter. The mixture is separated into the lighter product phase and the heavier aqueous phase. While the latter can flow freely into the reactor via a riser pipe, the product phase leaves the plant through a flash. | ||
| Fig. 12 Flowchart of the commissioned and continuously operated miniplant. | ||
The aqueous phase, consisting of cyclodextrins, precursor, ligand, and ultrapure water, is prepared similarly to the batch reactions described above. The miniplant was leak tested beforehand and then inertized by repeatedly drawing a vacuum and applying 5 bar of nitrogen pressure. The prepared aqueous phase is added to the reactor via a capillary line and vacuum. Preforming takes place at an H2/CO pressure of 20 bar and a temperature of 120 °C for at least 12 hours. The reaction pressure of 35 bar is set, and the aqueous phase is distributed to the miniplant sections by switching on the gear pump. The pump is switched off, and all the liquid-carrying valves are closed. A batch reaction is then started by adding 1-octene to the reactor and enabling continuous gassing with H2 and CO. After 2 hours, continuous plant operation is established by opening the valves and starting the gear and substrate pumps.
During miniplant operation, product phase samples were analyzed periodically with the above-described gas chromatography method. Furthermore, samples for ICP-OES analysis were taken. As the content of rhodium and phosphor is below the determination limit of the available ICP-OES, the samples were concentrated to one-tenth of their original volume before analysis, as described above.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank Umicore AG &Co. KG for donating the rhodium precursor Rh(acac)(CO2).Additionally, the authors thank the networking program ‘Sustainable Chemical Synthesis 2.0′ (SusChemSys 2.0) for the support and fruitful discussions across disciplines.
Finally, we are very thankful to the German Federal Ministry of Food and Agriculture (Bundesministerium für Ernährung und Landwirtschaft), represented by the FNR (Fachagentur Nachwachsende Rohstoffe) for financial support of the young research group “Renewlysis” (project number 2219NR355).
References
- United Nations, Our Common Future. Report of the World Commission on Environment and Development, 1987 Search PubMed.
- Intergovernmental Panel on Climate Change, WGIII, “Climate Change 2022. Mitigation of Climate Change”. Summary for Policymakers, 2022.
- P. T. Anastas and J. C. Warner, 12 Principles of Green Chemistry – American Chemical Society, Oxford University Press, 1998, p. 30 Search PubMed.
- M. Baerns, A. Behr, A. Brehm, J. Gmehling, K.-O. Hinrichsen, H. Hofmann, R. Palkovits, U. Onken and A. Renken, Technische Chemie, Wiley-VCH, Weinheim, 2013 Search PubMed.
- D. J. Cole-Hamilton, Homogeneous catalysis – new approaches to catalyst separation, recovery, and recycling, Science, 2003, 299, 1702–1706 CrossRef CAS PubMed.
- F. Joó, É. Papp and Á. Kathó, Molecular catalysis in liquid multiphase systems, Top. Catal., 1998, 5, 113–124 CrossRef.
- D. J. Adams, P. J. Dyson and S. J. Tavener, Chemistry in alternative reaction media, Wiley, Chichester, West Sussex, 2005 Search PubMed.
- T. Rösler, T. A. Faßbach, M. Schrimpf, A. J. Vorholt and W. Leitner, Toward Water-Based Recycling Techniques: Methodologies for Homogeneous Catalyst Recycling in Liquid/Liquid Multiphase Media and Their Implementation in Continuous Processes, Ind. Eng. Chem. Res., 2019, 58, 2421–2436 CrossRef.
- H. Warmeling, R. Koske and A. J. Vorholt, Procedural Rate Enhancement of Lean Aqueous Hydroformylation of 1-Octene without Additives, Chem. Eng. Technol., 2017, 40, 186–195 CrossRef CAS.
- H. W. F. Warmeling, T. Hafki, T. von Söhnen and A. J. Vorholt, Kinetic investigation of lean aqueous hydroformylation - an engineer's view on homogeneous catalysis, Chem. Eng. J., 2017, 326, 298–307 CrossRef CAS.
- H. W. F. Warmeling, D. Janz, M. Peters and A. J. Vorholt, Acceleration of lean aqueous hydroformylation in an innovative jet loop reactor concept, Chem. Eng. J., 2017, 330, 585–595 CrossRef CAS.
- L. Leclercq, F. Hapiot, S. Tilloy, K. Ramkisoensing, J. N. H. Reek, P. W. N. M. van Leeuwen and E. Monflier, Sulfonated Xantphos Ligand and Methylated Cyclodextrin: A Winning Combination for Rhodium-Catalyzed Hydroformylation of Higher Olefins in Aqueous Medium, Organometallics, 2005, 24, 2070–2075 CrossRef CAS.
- W. Saenger, Cyclodextrin Inclusion Compounds in Research and Industry, Angew. Chem., Int. Ed. Engl., 1980, 19, 344–362 CrossRef.
- A. R. Hedges, Industrial Applications of Cyclodextrins, Chem. Rev., 1998, 98, 2035–2044 CrossRef CAS PubMed.
- A. Z. Triponov and T. T. Nikiforov, Cyclodextrins as phase-transfer catalysts in a nucleophilic displacement reaction, J. Mol. Catal., 1984, 24, 15–18 CrossRef.
- E. Monflier, S. Tilloy, G. Fremy, Y. Castanet and A. Mortreux, A further breakthrough in biphasic, rhodium-catalyzed hydroformylation: the use of Per(2,6-di-O-methyl)-β-cyclodextrin as inverse phase transfer catalyst, Tetrahedron Lett., 1995, 36, 9481–9484 CrossRef CAS.
- F. Hapiot, S. Tilloy and E. Monflier, Cyclodextrins as supramolecular hosts for organometallic complexes, Chem. Rev., 2006, 106, 767–781 CrossRef CAS PubMed.
- F. Hapiot, L. Leclercq, N. Azaroual, S. Fourmentin, S. Tilloy and E. Monflier, Rhodium-Catalyzed Hydroformylation Promoted by Modified Cyclodextrins: Current Scope and Future Developments, Curr. Org. Synth., 2008, 5, 162–172 CrossRef CAS.
- K. U. Künnemann, L. Schurm, D. Lange, T. Seidensticker, S. Tilloy, E. Monflier, D. Vogt and J. M. Dreimann, Continuous hydroformylation of 1-decene in an aqueous biphasic system enabled by methylated cyclodextrins, Green Chem., 2020, 22, 3809–3819 RSC.
- S. A. Jagtap, E. Monflier, A. Ponchel and B. M. Bhanage, Highly regio-selective hydroformylation of biomass derived eugenol using aqueous biphasic Rh/TPPTS/CDs as a greener and recyclable catalyst, Mol. Catal., 2017, 436, 157–163 CrossRef CAS.
- S. Kohata, K. Jyodoi and A. Ohyoshi, Thermal decomposition of cyclodextrins (α-, β-, γ-, and modified β-CyD) and of metal—(β-CyD) complexes in the solid phase, Thermochim. Acta, 1993, 217, 187–198 CrossRef CAS.
- N. Sieffert and G. Wipff, Importance of interfacial adsorption in the biphasic hydroformylation of higher olefins promoted by cyclodextrins: a molecular dynamics study at the decene/water interface, Chem. – Eur. J., 2007, 13, 1978–1990 CrossRef CAS PubMed.
- W. Reppe and H. Vetter, Carbonylierung VI. Synthesen mit Metallcarbonylwasserstoffen, Liebigs Ann. Chem., 1953, 582, 133–161 CrossRef CAS.
- D. Crozet, M. Urrutigoïty and P. Kalck, Recent Advances in Amine Synthesis by Catalytic Hydroaminomethylation of Alkenes, ChemCatChem, 2011, 3, 1102–1118 CrossRef CAS.
- P. Kalck and M. Urrutigoïty, Tandem Hydroaminomethylation Reaction to Synthesize Amines from Alkenes, Chem. Rev., 2018, 118, 3833–3861 CrossRef CAS PubMed.
- B. Gall, M. Bortenschlager, O. Nuyken and R. Weberskirch, Cascade Reactions in Polymeric Nanoreactors: Mono (Rh)- and Bimetallic (Rh/Ir) Micellar Catalysis in the Hydroaminomethylation of 1-Octene, Macromol. Chem. Phys., 2008, 209, 1152–1159 CrossRef CAS.
- T. A. Faßbach, F. O. Sommer and A. J. Vorholt, Hydroaminomethylation in Aqueous Solvent Systems - An Efficient Pathway to Highly Functionalized Amines, Adv. Synth. Catal., 2018, 360, 1473–1482 CrossRef.
- Y. Wang, C. Zhang, M. Luo, H. Chen and X. Li, Hydroaminomethylation of high alkenes with dual-metal catalysts in aqueous/organic biphasic system, ARKIVOC, 2008, 165 Search PubMed.
- K. Li, Y. Wang, Y. Xu, W. Li, M. Niu, J. Jiang and Z. Jin, Thermoregulated phase-transfer rhodium nanoparticle catalyst for hydroaminomethylation of olefins, Catal. Commun., 2013, 34, 73–77 CrossRef CAS.
- S. A. Jagtap, S. P. Gowalkar, E. Monflier, A. Ponchel and B. M. Bhanage, Rhodium catalyzed selective hydroaminomethylation of biorenewable eugenol under aqueous biphasic condition, Mol. Catal., 2018, 452, 108–116 CrossRef CAS.
- K. Cousin, T. Vanbésien, E. Monflier and F. Hapiot, One pot synthesis of aminohydroxylated triglycerides under aqueous biphasic conditions, Catal. Commun., 2019, 125, 37–42 CrossRef CAS.
- K. U. Künnemann, D. Weber, C. Becquet, S. Tilloy, E. Monflier, T. Seidensticker and D. Vogt, Aqueous Biphasic Hydroaminomethylation Enabled by Methylated Cyclodextrins: Sensitivity Analysis for Transfer into a Continuous Process, ACS Sustainable Chem. Eng., 2021, 9, 273–283 CrossRef.
- J. Bianga, K. U. Künnemann, L. Goclik, L. Schurm, D. Vogt and T. Seidensticker, Tandem Catalytic Amine Synthesis from Alkenes in Continuous Flow Enabled by Integrated Catalyst Recycling, ACS Catal., 2020, 10, 6463–6472 CrossRef CAS.
- L. Leclercq, H. Bricout, S. Tilloy and E. Monflier, Biphasic aqueous organometallic catalysis promoted by cyclodextrins: can surface tension measurements explain the efficiency of chemically modified cyclodextrins?, J. Colloid Interface Sci., 2007, 307, 481–487 CrossRef CAS PubMed.
- N. Herrmann, J. Bianga, T. Gaide, M. Drewing, D. Vogt and T. Seidensticker, Aqueous biphasic hydroformylation of methyl oleate: a green solvent-only strategy for homogeneous catalyst recycling, Green Chem., 2019, 21, 6738–6745 RSC.
- S. Schlüter, K. U. Künnemann, M. Freis, T. Roth, D. Vogt, J. M. Dreimann and M. Skiborowski, Continuous co-product separation by organic solvent nanofiltration for the hydroaminomethylation in a thermomorphic multiphase system, Chem. Eng. J., 2021, 409, 128219 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information about the main product identification, the experimental conditions for the recycling experiments, and the leaching results; all reaction profiles for experiments with increased cyclodextrin concentration and experiments with reduced catalyst concentration. Furthermore, the gas ratios of all continuous experiments are shown, followed by the MS of product mixtures. See DOI: https://doi.org/10.1039/d2gc04847g |
| This journal is © The Royal Society of Chemistry 2023 |