
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interfacial activity and Pickering stabilization of kraft lignin particles obtained by solvent fractionation†
Jing
Tian
ab,
Jingqian
Chen
 b,
Peipei
Wang
ab,
Jiaqi
Guo
a,
Wenyuan
Zhu
a,
Mohammad Rizwan
Khan
c,
Yongcan
Jin
a,
Junlong
Song
*a and
Orlando J.
Rojas
*bd
b,
Peipei
Wang
ab,
Jiaqi
Guo
a,
Wenyuan
Zhu
a,
Mohammad Rizwan
Khan
c,
Yongcan
Jin
a,
Junlong
Song
*a and
Orlando J.
Rojas
*bd
aJiangsu Co-Innovation Center for Efficient Processing and Utilization of Forest Resources and International Innovation Center for Forest Chemicals and Materials, Nanjing Forestry University, Nanjing, 210037, China. E-mail: junlong.song@njfu.edu.cn
bBioproducts Institute, Department of Chemical and Biological Engineering, Department of Chemistry and Department of Wood Science, University of British Columbia, 2360 East Mall, Vancouver, BC V6T 1Z3, Canada. E-mail: orlando.rojas@ubc.ca
cDepartment of Chemistry, College of Science, King Saud University, Riyadh 11451, Saudi Arabia
dDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, Vuorimiehentie 1, FI-00076, Espoo, Finland
First published on 6th April 2023
Abstract
This study aims to overcome the challenges of high-value utilization of technical lignins that encounters chemical complexity, heterogeneity and broad molecular mass distribution. Kraft lignin was fractionated through a practical strategy involving sequential dissolution in solvents of different polarities (water, tetrahydrofuran, and water/tetrahydrofuran v/v = 3/7). Lignin self-assembled drawing on the anti-solvent method, generating particles of controllable surface energy. Considering the governing factors in multiphase stabilization, the morphology, wetting characteristics and chemical composition of the kraft lignin particles (KLPs) were examined as a means to achieve controllable Pickering stabilization of oil-in-water (O/W) emulsions. The most anionic and hydrophilic KLPs formed aggregated networks (∼185 nm), which acted as a surfactant-type emulsifier following reversible adsorption at the O/W interfaces. Notably, the particles of the least hydrophilicity (∼146 nm) displayed a characteristic hollow structure. Such particles adsorbed slowly and weakly at the O/W interface, forming a viscoelastic layer around the oil droplets. The smallest lignin particles (∼39 nm) presented an intermediate hydrophilic character. They were uniform in size and formed rigid interfacial layers on the oil droplets. Accordingly, the devised relationship between particle morphogenesis and O/W stabilization capacity enabled the customization of lignin (size and wetting characteristics), allowing broader utilization in multiphase systems.
1 Introduction
Emulsions stabilized by solid particles (Pickering emulsions) are possible due to the balanced affinity with the oil and aqueous phases, paving the way for the formulation of foodstuff, cosmetics, and household and pharmaceutical products, among others.1 The interfacial layer formed by biomass-derived solid particles prevents coalescence, increasing the kinetic stability of the emulsions.2,3 In this context, amphiphilic lignin particles have emerged as a suitable stabilizer.4,5 Particularly, Pickering systems that integrate kraft lignin particles (KLPs) have been demonstrated to template macroporous foams, micro- and nanocapsules, multilayer composites, and other materials.6–8 Expectedly, the valorization of lignin for the above purposes is hindered by the KLP complexity and heterogeneity (chemical, surface, and morphological).9 For instance, lignin's functional groups affect the hydrophobic/hydrophilic character of KLP, while the molecular weight defines its solubility. Both parameters contribute to nonuniform KLP, especially produced by the solvent-shift method.10 Notably, a complex chemical makeup and broad molecular mass significantly impact the morphology, adsorption behavior, and interfacial activity of KLP.11To address the issue of lignin heterogeneity, acid precipitation, membrane separation, and solvent fractionation have been proposed.12 Both membrane fractionation and acid precipitation of lignin suffer from low efficiency.13,14 Solvent fractionation, on the other hand, has relied on single-component solvents (acetone, ethanol and methanol)15–17 and mixed solvents (ethanol/glycerol, acetone/ethyl acetate, ethanol/acetone/tetrahydrofuran, dichloromethane/acetic ether/n-butyl alcohol, and methyl t-butyl ether/ethyl acetate/ethanol/methanol/acetone/dioxane).12,18,19 Recently, aqueous solvents have been considered for their lignin dissolution capability, e.g., by varying the water-to-organic ratio.16 Using a gradient strategy based on water/ethanol, acetone, and γ-valerolactone, lignin fractions presented a gradually reduced molecular mass and polydispersity.20,21 The observed outcomes included lignin particles of different morphologies and lignin–organic solvent interactions.22 The fractionation methods reported in the literature have focused on the role of the water fraction in the co-solvent (such as 20–60% in ethanol/water and 40–80% in acetone/water). The yield of lignin was observed to largely depend on the water concentration and resulted in a low fractionation efficiency.12
Tetrahydrofuran (THF) has been used to solubilize lignin and to form particles by anti-solvent precipitation. The latter process depends strongly on the lignin concentration, temperature, water ratio, THF purity, and other factors, which inevitably lead to heterogeneous particles. Previous attempts to remediate this challenge have relied on the adjustment of lignin concentration and filtration strategies.23 THF/water cosolvent was used to solubilize all lignin fractions, leading to an up-scalable technology.24 However, the obtained particles were relatively large (200–710 nm) and displayed structural heterogeneity. Recently, fractionation prior to anti-solvent precipitation was proposed to reduce lignin nanoparticle heterogeneity.17 Despite the reported progress achieved so far, most methodologies still demand a large volume of organic solvents. Here, the THF solvent was useful for both lignin fractionation and particle formation. In addition, the cosolvent system (water/THF) facilitated the utilization of all lignin fractions, realizing the promise of a sustainable production and application of lignin nanoparticles. However, we acknowledge the relatively toxic nature of THF, which should be considered carefully in any large-scale application and with regard to the precautions and safety measures needed for laboratory work.
This study presents a new route to generate lignin particles by using THF and water/THF as fractionation media. The in-depth characterization of these particles was correlated with the morphology, homogeneity, and yield of each fraction. Specifically, kraft lignin was sequentially dissolved in water, THF and water/THF cosolvent (v/v = 3/7) to yield three fractions which were used for particle formation by using water as anti-solvent. As a proof of concept, different kraft lignin particles were used as Pickering stabilizers. We investigated KLP adsorption behavior at the oil/water interfaces according to affinity factors that relate to particle morphogenesis and heterogeneity.
2 Experimental
2.1 Materials
Softwood kraft lignin (KL) was extracted from an industrial black liquor using acid precipitation at pH 2, sourced from Zhejiang Jiefa Corp. (Shaoxing, China). Sunflower oil was purchased from a local market. Tetrahydrofuran (THF), a green, recyclable solvent, was provided by Sigma-Aldrich Corp. (Canada) along with trichloromethane (CHCl3), pyridine, and ethanol.2.2 Kraft lignin fractionation
Kraft lignin fractionation was carried out as illustrated in Fig. 1, using aqueous suspensions of lignin (with a solid-to-liquid ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 wt/v) that were previously subjected to ultrasonication for 30 min and centrifuged at 8000 rpm for 10 min. The first lignin fraction, referred hereafter as F1-KL, was obtained as the supernatant. The precipitate was dried and dissolved in pure THF with the same solid-to-liquid ratio and ultrasonicated for 30 min. Then, the second fraction, F2-KL, was the supernatant collected in the THF suspension after centrifugation at 8000 rpm for 10 min. The precipitate, fraction F3-KL, was dried in air and dispersed in THF/H2O (v/v = 7/3). This third fraction was fully dissolved in the cosolvent. A reference fraction was obtained by direct dissolution of the precursor-extracted lignin in THF/H2O (v/v = 7/3), F0-KL fraction.
:50 wt/v) that were previously subjected to ultrasonication for 30 min and centrifuged at 8000 rpm for 10 min. The first lignin fraction, referred hereafter as F1-KL, was obtained as the supernatant. The precipitate was dried and dissolved in pure THF with the same solid-to-liquid ratio and ultrasonicated for 30 min. Then, the second fraction, F2-KL, was the supernatant collected in the THF suspension after centrifugation at 8000 rpm for 10 min. The precipitate, fraction F3-KL, was dried in air and dispersed in THF/H2O (v/v = 7/3). This third fraction was fully dissolved in the cosolvent. A reference fraction was obtained by direct dissolution of the precursor-extracted lignin in THF/H2O (v/v = 7/3), F0-KL fraction.
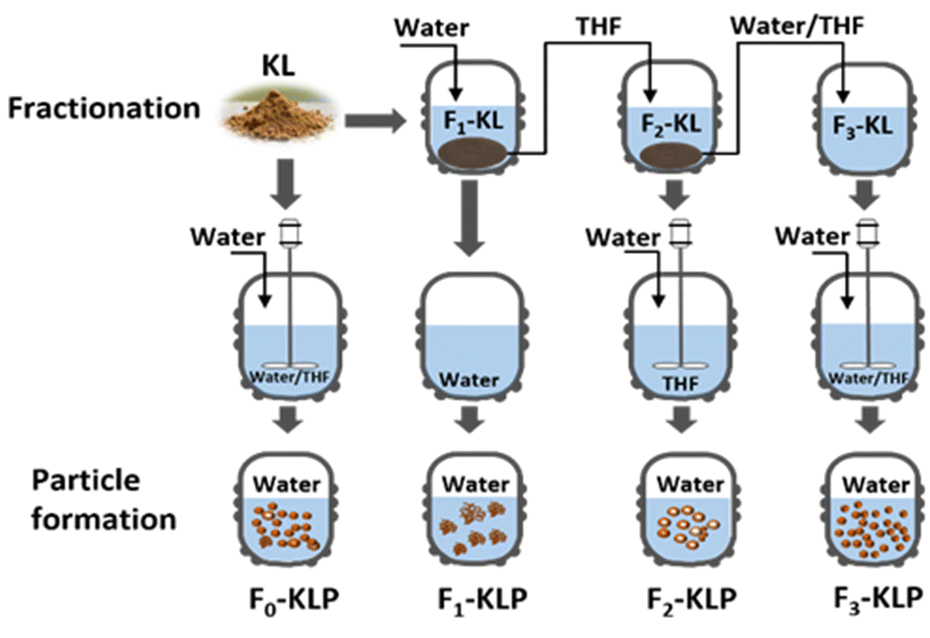 | ||
| Fig. 1 Schematic illustration of kraft lignin fractionation by sequentially dissolving kraft lignin (KL) in water, THF and water/THF (v/v = 3/7). The corresponding fractions are labeled as Fn-KL with n = 1, 2, 3, representing the respective solvent, in the same order listed above. The respective particles (KLPs) were produced by the solvent-shifting method, using water as the anti-solvent. The obtained particles are labeled following the corresponding nomenclature, Fn-KLP (n = 1, 2, 3). | ||
The Kraft lignin particles (KLPs) were obtained from the respective KL fractions using the anti-solvent method. Fn-KL solutions were added separately into a beaker containing water and stirred (magnetic stirring, 600 rpm) for 24 h at room temperature. The lignin concentration was adjusted to 0.5 wt%.
For comparison, the non-fractionated KLP (F0-KLP) was obtained using the same process as that of F3-KLP but using lignin fully dissolved in the cosolvent, F0-KL.
2.3 Chemical composition, surface characterization and emulsion preparation
The molecular weight and distribution of the fractionated kraft lignins (Fn-KL) were assessed by gel permeation chromatography (GPC, KF-806L, Shimadzu). Specifically, lignin samples were acetylated to improve their solubility in THF prior to GPC tests.25 The hydroxy type and content were analyzed by 31P nuclear magnetic resonance (31P-NMR) spectroscopy using a Bruker 600 MHz spectrometer (Bruker 600, Germany), following the sample preparation process reported before.26The morphology of the obtained particles, Fn-KLPs, was observed using a transmission electron microscope (TEM, Tecnai Spirit, FEI, USA). For this purpose, a drop of diluted Fn-KLP dispersion (0.005%) was deposited on a copper grid, dried at room temperature, and observed at an acceleration voltage of 120 kV. The size and ζ-potential of Fn-KLP were measured using a Nano Zetasizer system (Malvern, UK). The interfacial tension (IT) at the oil/water interface was measured by the pendant-drop method using an optical tensiometer (Theta Flex 300, Biolin Scientific, Finland). The three-phase contact angle (TCA) was used to characterize the wettability of Fn-KLP at the oil/water interface using the sessile drop method. Briefly, the given Fn-KLP dispersion (0.5 mL) was deposited on a clean glass slide, dried at 40 °C to form a particulate film, and the system was transferred to a chamber filled with sunflower oil. A water droplet was generated on a syringe tip and moved to the surface of the film. Thereafter, the droplet shape was monitored with a digital camera for 2 min. The TCA is reported as the angle at the three-phase line (film, water and oil).
Pickering emulsions were prepared by mixing the respective kraft lignin particles (Fn-KLP) with oil and water under high-speed homogenization (T-25 Ultra-Turrax, IKA, Germany). In this process, Fn-KLP dispersions (5 mL) were mixed with the same volume of sunflower oil under vigorous stirring (18000 rpm) for 2 min. The concentration of Fn-KLP was maintained at 0.5 wt%.
2.4 Pickering emulsions stabilized with Fn-KLP
The static stability of emulsions was evaluated using optical microscopy. The emulsion stability was measured using a LUMISizer analyzer (GmbH, Berlin, Germany). The instrumental parameters included a total emulsion volume of 0.4 mL, a wavelength of 710 nm, a centrifugal speed of 2000 rpm, a measurement time of 52 min with 10 s intervals between the recordings, and a temperature of 25 °C.The microstructure of the Pickering emulsion droplets stabilized with Fn-KLP was observed by optical microscopy (Leica DM 750, Leica, Germany). A drop of the emulsion was placed onto a microscope slide and covered with a glass coverslip. The droplet size and distribution were determined using a Mastersizer 3000 analyzer (Malvern, UK).
The viscosity and dynamic viscoelasticity of the Pickering emulsions were measured using a rheometer (MCR 302, Anton Paar, Germany) equipped with a plate-and-cone geometry using a gap of 0.1 mm. The shear viscosity was monitored at a shear rate ranging from 0.01 to 1000 s−1. For dynamic viscoelastic measurements, the linear viscoelastic range was determined using a strain sweep (0.01 to 100%) at a fixed frequency of 1 rad s−1. The dynamic frequency sweep measurements (0.1 and 100 rad s−1) were performed by applying a constant strain of 1.0% within the linear region.
2.5 Adsorption of Fn-KLP at the oil/water interface
The interfacial and adsorption behaviors of the systems were determined by using a quartz crystal microbalance with dissipation monitoring (QCM-D) (QCM-D E4 system, Biolin Scientific AB, Sweden). Prior to the experiment, gold sensors were rinsed (10 min at 75 °C) with a solution consisting of Millipore water, 25% ammonia, and 33% hydrogen peroxide at a ratio of 5:1:1. The sensors were then rinsed with hot Millipore water and then dried. An oil film was deposited on the sensors by using a previously reported procedure based on spin-coating.27 Briefly, 0.5% w/v sunflower oil in CHCl3 was prepared, and 100 μL solution was dropped on the sensor for spinning at 2000 rpm (Laurell WS-650Mz-23NPPB, USA). The excess solvent was removed by spinning at 4500 rpm for an additional 45 s. Then the oil-coated sensor was placed in an oven at 40 °C for 12 h. The changes in the sensor were determined before and after oil coating (Attension Theta, Biolin Scientific, Finland).
3. Results and discussion
3.1. Lignin fractions (Fn-KL)
KL was readily fractionated (Fig. 1) into water-soluble (F1-KL, 9.5 ± 6.4%), THF-soluble (F2-KL, 19.8 ± 4.4%) and cosolvent-soluble (F3-KL, 66.5 ± 4.8%) fractions, with the latter showing the highest yield, Table S1.† The 31P-NMR spectra and molecular weight distribution of each lignin fraction are shown in Fig. 2 and a data summary is given in Table 1. The F1-KL and F2-KL fractions had a similar molecular weight (4.8k to 6 kDa), exhibiting significantly reduced heterogeneity compared to the original kraft lignin. Based on its high carboxy content and low aliphatic and phenolic OH concentration, F1-KL was expected to be more hydrophilic than F2-KL.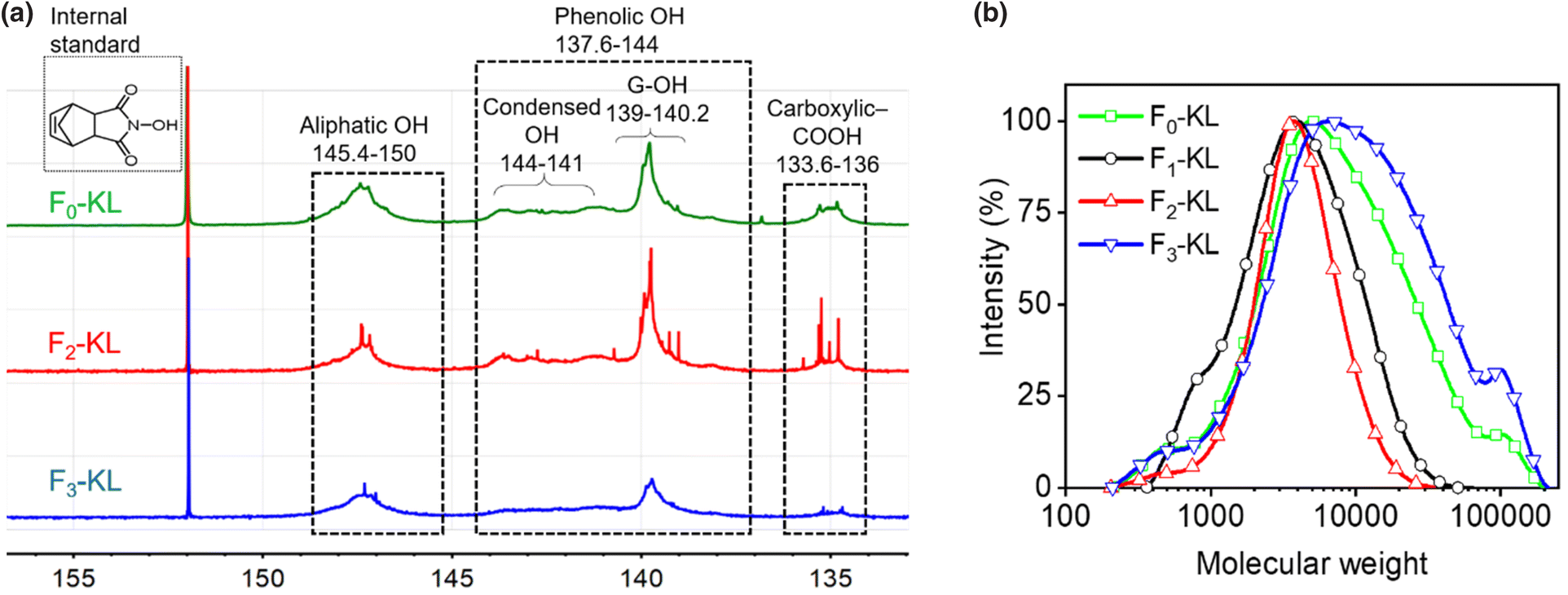 | ||
| Fig. 2 Chemical characterization of Fn-KL: (a) 31P-NMR spectra and (b) molecular weight distribution (GPC). | ||
| Fraction | Yield (%) | Aliphatic OH (*) (mmol g−1) | Phenolic OH (*) (mmol g−1) | COOH (*) (mmol g−1) | M w (**) | M n (**) | PDI |
|---|---|---|---|---|---|---|---|
| (*) determined by 31P-NMR; (**) determined by GPC; and (***) F1-KL could not be fully dissolved in the anhydrous CDCl3/pyridine system. The hydroxy content was assessed by the balance between F0 and its fractions. PDI: Polymer dispersity index. | |||||||
| F0-KL | 100 | 2.36 | 4.69 | 0.68 | 15942 |
4006 | 3.98 |
| F1-KL | 12 ± 2 | 0.7(**) | 0.7(***) | 3.0(**) | 5787 | 2710 | 2.13 |
| F2-KL | 20 ± 4 | 1.92 | 6.01 | 0.61 | 4886 | 2969 | 1.64 |
| F3-KL | 67 ± 4 | 2.85 | 5.14 | 0.31 | 21984 |
4820 | 4.56 |
The total hydroxy group content was almost the same for F2-KL and F3-KL, while a significant difference in molecular weight was noted (F3-KL with the highest molecular weight, ∼22000 and PDI). The results point to the effect of solvency on the chemical composition and molecular weight. Given that the hydroxy and carboxy groups contribute to lignin wettability, such hydrophilic functional groups together with molecular weight affect the colloidal interactions (lignin–lignin, lignin–water, and lignin–THF) which also regulate particle formation in the antisolvent.22,28 Hence, the morphology and surface properties of the particles ultimately depended on the fraction source.
3.2. Particle formation and properties (Fn-KLP)
Colloidally stable suspensions of Fn-KLP were obtained using antisolvent precipitation (Fig. 3a). Interestingly, F2-KLP exhibited a much lighter color than the other two fractions. Similarly, the average particle size was the highest for the same fraction, indicating a non-monotonic trend (the sizes were found to be 60, 185, 146, and 39 nm for F0-, F1-, F2- and F3-KLP, respectively, Fig. 3b). The ζ-potential of Fn-KLP suspended in water was ca. −40 mV, with all fractions being more negatively charged than the particles obtained from the non-fractionated (original) lignin, suggesting that the colloidal stability improved upon fractionation (Fig. 3c). No correlation was found for the ζ-potential with the measured carboxy and phenolic OH content (NMR). This is likely related to lignin self-assembly and surface organization on the particles, which are the leading factors affecting their surface properties.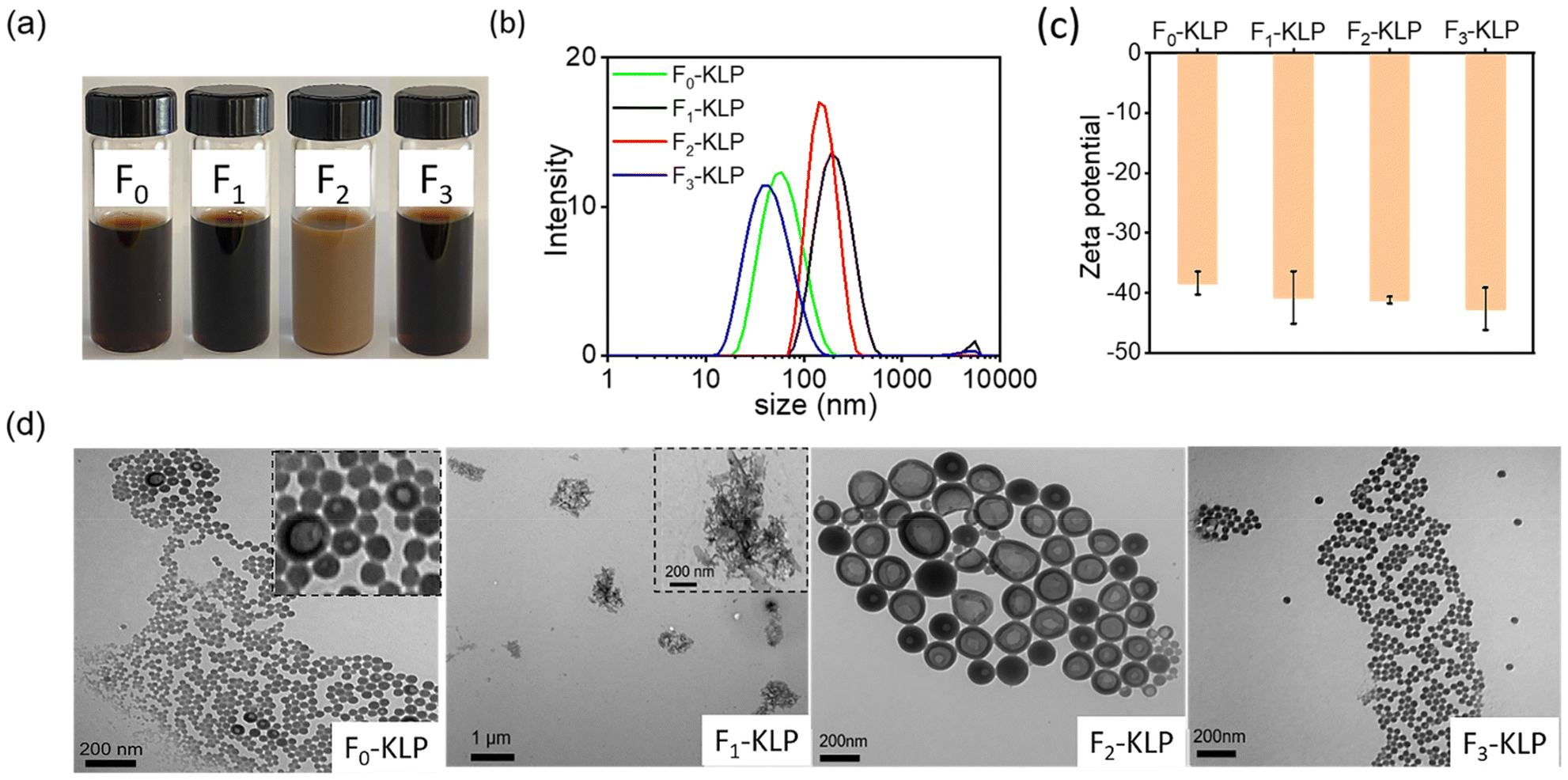 | ||
| Fig. 3 Fn-KLP colloidal characteristics: (a) digital photographs of the colloidal suspensions of Fn-KLP (0.5%), (b) size distribution, (c) ζ-potential and (d) TEM images of Fn-KLP. | ||
The TEM images of Fn-KLP (Fig. 3d) reveal interesting self-assembly processes. First, the observed TEM particle size was close to the values measured by light scattering (Fig. 3b). Pure water (high polarity solvent) is a poor solvent for lignin. The corresponding water-soluble fraction, F1-KL, formed a network-like assembly rather than regular spherical particles. This is likely related to the inherent structure of this lignin fraction and induced structural changes, leading to macromolecular self-assemblies as opposed to nucleation and growth into particles.29 F2-KLP particles were hollow and showed a large particle size. Meanwhile, F3-KLP presented the smallest size and exhibited solid, spherical structures. Interestingly, the particles from non-fractionated lignin, F0-KLP, showed the characteristics of the three fractions, with large agglomerates, hollow and solid (major component) particles (Fig. 3d). F1- and F2-KL were also dissolved in the cosolvent and then precipitated in water. As a result, no changes were observed in the particle size or morphology (Fig. S2†), indicating that the cosolvent was more effective for lignin fractions of high molecular weight.
The F2-KLP morphology showed a clear contrast between the core and the shell, indicating the presence of cavities. Xiong et al.32 and Li et al.30 attributed this hollow morphology to a low initial lignin concentration (<2 mg ml−1). However, in our case, F2-KLP was produced at a much higher lignin concentration (50 mg ml−1 in THF). Qian et al.31 and Pang et al.17 interpreted that the hollow structures were a consequence of the formation of a nanoemulsion system due to the presence of amphiphilic molecules. In our case, F2-KL can be regarded as amphiphilic since it could be completely dissolved in THF, given the relative higher content of hydrophilic groups. Amphiphilic F2-KL molecules tend to accumulate at the interface by self-assembly due to their amphiphilicity, which stabilizes a nanoemulsion (Fig. S1†). Once THF and water were evaporated during drying, hollow lignin particles were obtained. Hence, the organic phase (THF–lignin solution) was dispersed in water. As a result, lignin molecules aggregated at the interface by self-assembly due to their amphiphilicity. It is also worth noting the uneven wall thickness of F2-KLP. This can be explained by the pressure difference between the inside and outside of the shell.30,32 In contrast, the lignin fraction with more affinity to THF is preferentially located in the core (where a higher proportion of THF is present).33 The surface functional groups are expected to play an important role as the hydrophilic moieties tend to orient towards water while the hydrophobic components enriched the THF. In sum, we hypothesize that the hollow morphologies form in the course of THF evaporation during the preparation process (although the contribution of water evaporation during TEM sample preparation cannot be discounted).34
In the process of particle formation, the abundant aromatic rings of lignin would result in strong π–π interactions that drive self-assembly into KLP.28,35 As predicted by the classical nucleation theory, in the binary solvent (THF/water), the diffusion of THF in water promotes uniform supersaturation, leading to small particles.36 The solvent interaction plays a vital role in F3-KLP formation. Water molecules form H-bonds with lignin compared to those between organic solvents and lignin in the cosolvent.21 This explains the higher solubility of lignin in the cosolvent compared to neat water or neat THF. In summary, the larger hollow particles consisted of fractions of lower Mw and higher hydrophilic functional group content. Meanwhile, the fractions of higher Mw and higher hydrophobic group content produced smaller particles. This is consistent with the observations made by Lee et al.37
The polar phenolic and carboxylic groups at the interface form H-bonds with water,34 favoring a hydrophilic character. Hence, lignin molecules can be solvated. This explains the fact that F3-KLP is essentially compact and homogeneous, despite its broad molecular-weight distribution. Nevertheless, since F3-KLP is formed with KL of the highest molecular weight, hydrophobic interactions cannot be neglected. A high molecular weight contributes to a fast nucleation rate due to the lower hydrophilicity of the larger lignin molecules, which translate into small and uniform particles, Fig. 3d.
To summarize the results so far, sequential fractionation based on the dissolution of kraft lignin in water, THF, and the cosolvent (THF/water) effectively reduces the heterogeneity of lignin, and therefore improves the uniformity of the particles produced in an anti-solvent. Compared to other cosolvent systems,10,16,17 F3-KL yielded more spherical, uniform and smaller particles, suggesting that the THF/water cosolvent is most effective in reducing heterogeneity.
The surface tension and water contact angle of Fn-KLP are shown in Fig. 4. Due to the amphiphilic nature of KL molecules, there is an expectation that the hydrophilic moieties would orient toward the aqueous phase (outer layer) and the hydrophobic component to enrich the inner layer (THF) during particle formation. Among the three fractions, the hydrophilicity decreased in the following order: F1-KLP > F3-KLP > F2-KLP. The F1-KLP particles significantly reduced the surface and interfacial tension (Fig. 4a and c), indicating a high surface activity. Meanwhile, F2- and F3-KLP showed weak surface activity. Generally, KL has weak surface activity due to the very limited water solubility at normal pH.38
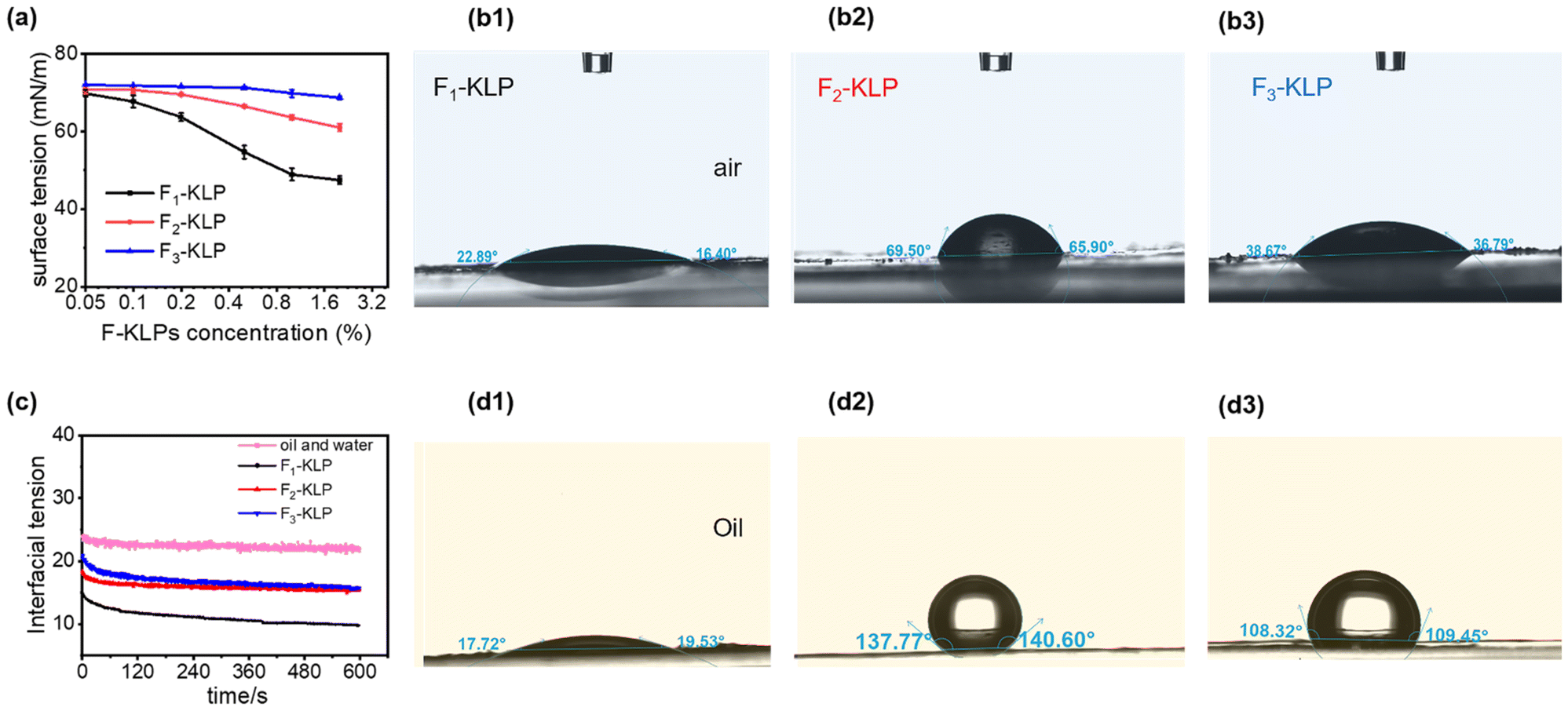 | ||
| Fig. 4 Wettability of Fn-KLP at the air/water and oil/water interfaces. (a) Surface tension as a function of Fn-KLP concentration. (b1–b3) Water contact angles of Fn-KLP. (c) Changes in interfacial tension as a function of time, and (d1–d3) three-phase contact angles for Fn-KLP between sunflower oil and water. | ||
Lignin particles reduced the interfacial tension between sunflower oil and water, a factor that enables Pickering stabilization.39 For this purpose, the three-phase contact angle (θow) is the most important parameter determining the stability of the Pickering emulsion. F1-, F2- and F3-KLP produced θow values (measured after 120 s) of ca. 18°, 138° and 108° (Fig. 4d1–d3). On the basis of theory40 and assuming a single interfacial layer, solid particles with a three-phase contact angle (θow) between 15° and 90° and between 90° and 165° are effective in stabilizing O/W and W/O Pickering emulsions, respectively. In the case of the emulsions stabilized by solid particles with a 3D network, θow shifts to 15°–129° and 51°–165° for O/W and W/O emulsions, respectively. The 3D network gives extra physical stability to the emulsion.41 In our case, all Fn-KLP-based Pickering emulsions were of the oil-in-water (O/W) type, which formed following a mechanism which is explained in more detail in the following sections.
3.3. Fn-KLP-stabilized Pickering emulsions
The physical stability of the emulsions (Fig. 5a) indicated a fast creaming rate that reached equilibrium in 2 days; all emulsions presented excellent long-term stability. The creaming layer was almost the same for the different Fn-KLP fractions. The emulsion stabilized with F2-KLP had a larger droplet size and showed irregular morphologies that coalesced into larger droplets (Fig. 5b and S3†). The F2-KLP stabilized Pickering emulsions were the least stable as also indicated by the accelerated instability index (Fig. 5c): the instability index of emulsions prepared with F2-KLP was ∼0.8, implying a severe phase separation of oil and water after centrifugation. Hence, F2-KLP easily desorbed from the oil/water interface, while F1- and F3-KLP stabilized the emulsions, which remained stable. Particle bridging between droplets in Fn-KLP-stabilized Pickering systems is an additional consideration. Therein, the 3D network of lignin favors the bridging effect, which makes the emulsion more stable.42 Finally, the aggregation of excess lignin nanoparticles in the continuous phase did not affect the emulsion's stability (Fig. 5d).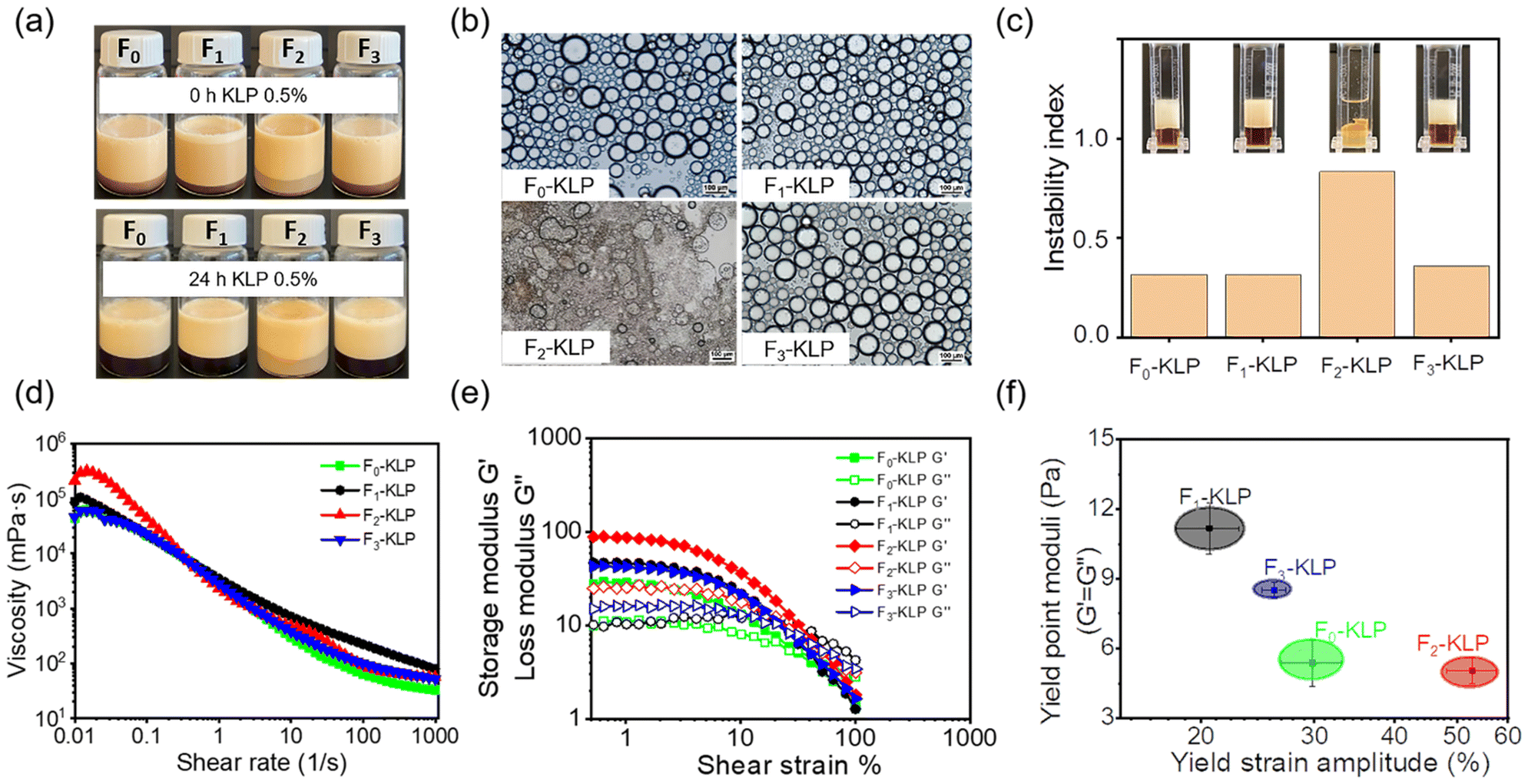 | ||
| Fig. 5 Characterization of Pickering emulsions stabilized with Fn-KLP. (a) Photographs of Pickering emulsions 0 h and 24 h after preparation. (b) Optical micrographs of freshly prepared Pickering emulsions stabilized by Fn-KLP. (c) The instability index of Fn-KLP-based emulsions measured at 2000 rpm. (d) The shear viscosity and (e) strain sweep of Fn-KLP-based Pickering emulsions; and (f) the yield strain amplitude of Fn-KLP-based emulsions. The concentration of Fn-KLP was fixed at 0.5%. | ||
The resistance to the external shear of emulsions stabilized with Fn-KLP was examined by rheology tests (Fig. 5e and S4†). Generally, all emulsions exhibited a shear-thinning behavior, reflecting a reduced apparent viscosity with the shear rate (Fig. 5d). All the emulsions exhibited a gel-like behavior (G′ > G′′) at small amplitudes under oscillatory shear and turned into liquid-like systems (G′ < G′′) at a critical strain amplitude, by entering the large amplitude oscillatory shear region, wherein the critical transition (G′ = G′′) corresponds to the yield point.43 The strain amplitude value at the yield point is here taken as an indicator of the slightest deformation required for the emulsion to flow. As shown in Fig. 5f, F2-KLP-based emulsion showed the largest amplitude and the lowest moduli at the yield point, while the F1-KLP-based emulsion had the lowest amplitude and moduli. These results indicate that particles with a hollow morphology and large size imparted viscoelasticity to the emulsion.
3.4. Fn-KLP adsorption at the oil/water interface
A uniform and fairly smooth oil layer was spin-coated on QCM gold sensors. The contact angles of deionized water on the bare sensor and the oil-coated sensor were determined to be ∼18° and ∼97°, respectively (Fig. 6a1 and a2). The QCM-D reveals the real-time frequency shift and energy dissipation of the quartz crystal oscillator, which can be used to reveal the adsorption behavior of the particles at the oil/water interface. Such information helps us understand the Pickering emulsification mechanism.27 The adsorption of Fn-KLP on the sunflower oil film produced a negative shift in frequency, Δf, and positive ΔD shifts, Fig. 6b and c. For F1-KLP, the adsorption and desorption were very fast. The process was largely reversible since most particles were easily rinsed off. This is a typical characteristic of thermodynamically unstable adsorption.44 F2-KLP experienced a very slow adsorption and equilibrium was reached after a long time. The drop of Δf for F2-KLP adsorbing on sunflower oil was pronounced, and the particles were desorbed quickly upon rinsing. For F3-KLP, the Δf initially increased quickly and then slowly, until reaching a plateau. Most of the particles remained at the interface after rinsing with water and therefore F3-KLP could be considered to adsorb irreversibly. The corresponding, stable adsorbed layer points to a stable interface. The ΔD values indicated that the particle layer formed by F2-KLP was the most viscoelastic, while that formed by F3-KLP was the most rigid, which is in agreement with the bulk rheological results (Fig. 5e and f). The drop of ΔD upon rinsing implies an irreversible layer that becomes stiffer and more compact (the loose particles are rinsed off).27,45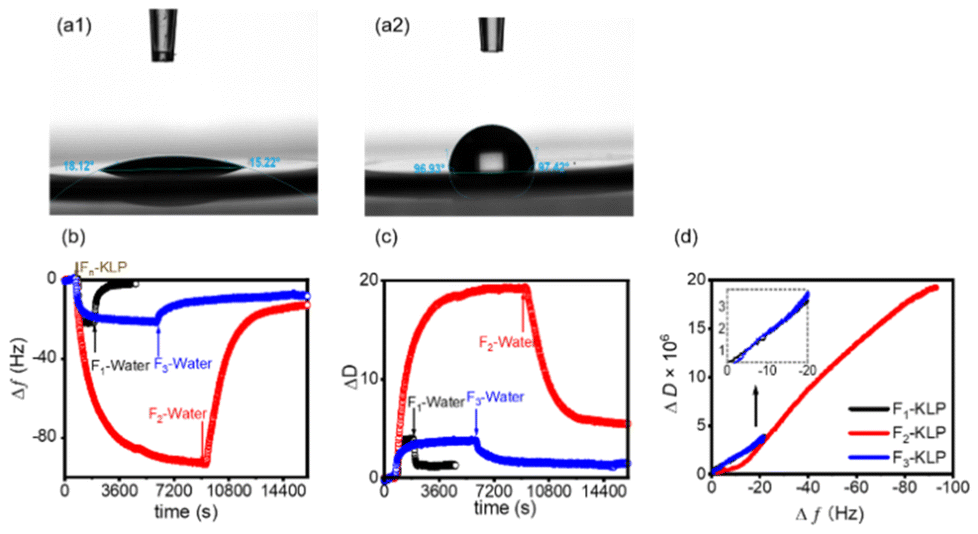 | ||
| Fig. 6 Contact angles of water on (a1) a gold-coated QCM sensor and (a2) a sensor coated with sunflower oil. Changes of Δf (b) and ΔD (c) with time during the adsorption and desorption of Fn-KLP on a quartz crystal sensor coated with sunflower oil. (d) ΔD–Δf plots describing the particle adsorption process. | ||
In order to gain deeper insights into the conformation of the adsorption layer, ΔD–Δf plots were generated. The slope of the ΔD–Δf plots relates to the dynamics of the adsorption process.46 The profiles were essentially straight for F1- and F3-KLP, indicating the buildup of an adsorbed layer with no significant conformational changes; the relatively low ΔD values indicated a layer with low viscoelasticity. In the case of F2-KLP, two distinct stages occurred, suggesting a change in conformation from a more rigid to a less compact adsorbed structure (Fig. 6d).
The stability of a solid particle at the oil/water interface is often described by the desorption energy (Edesorption), the energy required to remove the particle from its equilibrium position at the interface to the bulk phase, eqn (1):40
| Edesorption = πR2γO/W(1 ± cosθ)2 | (1) |
The interfacial adsorption of the particles and the emulsion stability scales with Edesorption, Table 2. The Edesorption value in units of thermal energy was rather low for F1-KLP (2.5kBT), indicating that particles were weakly attached at the oil/water interface. This result is consistent with the QCM data, which indicated that most of the particles were rinsed off with water. It is worth noting that the Edesorption value of F2-KLP was very high, implying that the particles were strongly adsorbed (particles remained at the interface, as verified by the Δf data in the QCM test). The Edesorption value of F3-KLP was much higher than that of most molecular surfactants.47 Therefore, from the energy perspective, the adsorption of F2- and F3-KLP was irreversible. The F2-KLP particles were less homogeneous in size and the average diameter was used to calculate the energy. However, the population with size extremes (largest and smallest sizes) may not be stably attached at the interface. Hence, the uniformity and homogeneity of the particle are important parameters affecting the stability of KLP-based Pickering emulsions.41 We show the advantage of using particles of uniform size and morphology for Pickering emulsion stabilization. We further demonstrate the irreversible adsorption of lignin fractions and their role in Pickering stabilization by using quartz crystal microgravimetry. Finally, the electrostatic interactions are expected to affect the adsorption and packing of KLP at the oil/water interface,8 a factor that was not accounted for in eqn (1).
| Particle | Time/s | Particle size/nm | Θ o/w | E desorption (kBT) |
|---|---|---|---|---|
| F0-KLP | 120 | 54 | 72° ± 2° | 102 ± 8 |
| F1-KLP | 120 | 185 | 13° ± 1° | 2.5 ± 0.6 |
| F2-KLP | 120 | 146 | 136° ± 9° | 4555 ± 618 |
| F3-KLP | 120 | 39 | 109° ± 1° | 196 ± 7 |
4. Conclusions
Upgrading kraft lignin (KL) by selective dissolution with single or binary solvents facilitates the fractionation of black liquors into lignins of different molecular masses and interfacial properties. These fractions lead to particles whose properties change with their solubility and precipitation in water, used as an anti-solvent. For instance, the particles obtained from water-soluble KL (F1-KLP) exhibited the highest surface activity and stabilization ability, offering promise in applications requiring interfacial stabilization.48 Hollow nanoparticles (F2-KLP) were obtained from lignin fractions solubilized in THF and led to systems suited for encapsulation, core–shell materials, templates, and other high-value materials.49 A formation mechanism leading to hollow morphologies was put forward but gaining control on the thickness of the hollow particle remains a subject that deserves future attention. The particles that were more uniform in size and presented higher molecular weight (F3-KLP) were obtained using the cosolvent, THF/water. By reducing the kraft lignin heterogeneity, the derived particles can be designed for tunable three-phase contact angles, making them options for Pickering stabilization. Though this work revealed the Pickering stabilization with Fn-KLP, the in-depth process that leads to size and morphology control along with the adsorption behavior deserves future investigation. The reasons explaining how the hollow lignin particles feature high desorption energy and low emulsion stability also need elucidation. Overall, the proposed strategy for upgrading lignin by sequential dissolution, e.g., to reduce the heterogeneity, paves the way for the more integral valorization of lignin fractions, a subject that can be translated to other lignin resources.Author contributions
The manuscript was written with contributions from all of the authors. All of the authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the support from the National Natural Science Foundation of China (Grant no. 31770623 and 31730106), the Chinese Scholar Committee, the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX20_0856), the Priority Academic Program Development of the Jiangsu Higher Education Institutions of China, the Canada Excellence Research Chair Program (CERC-2018-00006), the Canada Foundation for Innovation (Project number 38623), and the Researchers Supporting Project number (RSP2023R138), King Saud University, Riyadh, Saudi Arabia.References
- F. Wang, J. Tang, H. Liu, G. Yu and Y. Zou, Mater. Chem. Front., 2019, 3, 356–364 RSC.
- M. R. V. Bertolo, L. B. B. de Paiva, V. M. Nascimento, C. A. Gandin, M. O. Neto, C. E. Driemeier and S. C. Rabelo, Ind. Crops Prod., 2019, 140, 111591 CrossRef CAS.
- W. Liu, K. Liu, Y. Wang, Q. Lin, J. Liu, H. Du, B. Pang and C. Si, Ind. Crops Prod., 2022, 185, 115123 CrossRef CAS.
- M. H. Sipponen, M. Smyth, T. Leskinen, L. S. Johansson and M. Osterberg, Green Chem., 2017, 19, 5831–5840 RSC.
- M. Ago, S. Huan, M. Borghei, J. Raula, E. I. Kauppinen and O. J. Rojas, ACS Appl. Mater. Interfaces, 2016, 8, 23302–23310 CrossRef CAS PubMed.
- H. Yi, Y. Yang, X. Gu, J. Huang and C. Wang, J. Mater. Chem. A, 2015, 3, 13749–13757 RSC.
- Y. X. Pang, X. Li, S. W. Wang, X. Q. Qiu, D. J. Yang and H. M. Lou, React. Funct. Polym., 2018, 123, 115–121 CrossRef CAS.
- M. Österberg, M. H. Sipponen, B. D. Mattos and O. J. Rojas, Green Chem., 2020, 22, 2712–2733 RSC.
- W. Zhao, B. Simmons, S. Singh, A. Ragauskas and G. Cheng, Green Chem., 2016, 18, 5693–5700 RSC.
- T. Pang, G. Wang, H. Sun, L. Wang, Q. Liu, W. Sui, A. M. Parvez and C. Si, ACS Sustainable Chem. Eng., 2020, 8, 9174–9183 CrossRef CAS.
- M. Zembyla, B. S. Murray and A. Sarkar, Trends Food Sci. Technol., 2020, 104, 49–59 CrossRef CAS.
- T. Pang, G. Wang, H. Sun, W. Sui and C. Si, Ind. Crops Prod., 2021, 165, 113442 CrossRef CAS.
- G. Wang and H. Chen, Sep. Purif. Technol., 2013, 105, 98–105 CrossRef CAS.
- C. Allegretti, S. Fontanay, K. Rischka, A. Strini, J. Troquet, S. Turri, G. Griffini and P. D'Arrigo, ACS Omega, 2019, 4, 4615–4626 CrossRef CAS PubMed.
- H. Li and A. G. McDonald, Ind. Crops Prod., 2014, 62, 67–76 CrossRef CAS.
- X. Li, J. Shen, B. Wang, X. Feng, Z. Mao and X. Sui, ACS Sustainable Chem. Eng., 2021, 9, 5470–5480 CrossRef CAS.
- T. Pang, G. Wang, H. Sun, L. Wang, Q. Liu, W. Sui, A. M. Parvez and C. Si, ACS Sustainable Chem. Eng., 2020, 8, 9174–9183 CrossRef CAS.
- R. Liu, A. Smeds, L. Wang, A. Pranovich, J. Hemming, S. Willför, H. Zhang and C. Xu, ACS Sustainable Chem. Eng., 2021, 9, 13862–13873 CrossRef CAS.
- Z. Zhang, Y. Zhang, Z. Lin, A. Mulyadi, W. Mu and Y. Deng, Chem. Eng. Sci., 2017, 165, 55–64 CrossRef CAS.
- X. Liu, M. Xie, Y. Hu, S. Li, S. Nie, A. Zhang, H. Wu, C. Li, Z. Xiao and C. Hu, Ind. Crops Prod., 2022, 183, 114943 CrossRef CAS.
- T. Zou, N. Nonappa, M. Khavani, M. Vuorte, P. Penttila, A. Zitting, J. J. Valle-Delgado, A. M. Elert, D. Silbernagl, M. Balakshin, M. Sammalkorpi and M. Osterberg, J. Phys. Chem. B, 2021, 125, 12315–12328 CrossRef CAS PubMed.
- M. D. Smith, B. Mostofian, X. Cheng, L. Petridis, C. M. Cai, C. E. Wyman and J. C. Smith, Green Chem., 2016, 18, 1268–1277 RSC.
- M. Lievonen, J. J. V. Delgado, M. L. Mattinen, E. L. Hult, K. Lintinen, M. A. Kostiainen, A. Paananen, G. R. Szilvay, H. Setälä and M. Österberg, Green Chem., 2016, 18, 1416–1422 RSC.
- T. Leskinen, M. Smyth, Y. Xiao, K. Lintinen, M. L. Mattinen, M. A. Kostiainen, P. Oinas and M. Österberg, Nord. Pulp Pap. Res. J., 2017, 32, 586–596 CrossRef CAS.
- K. Shayesteh, G. Mohammadzadeh and M. Zamanloo, Int. J. Biol. Macromol., 2020, 163, 1810–1820 CrossRef CAS PubMed.
- J. Tian, Y. Yang and J. Song, Int. J. Biol. Macromol., 2019, 141, 919–926 CrossRef CAS PubMed.
- Y. Wei, Y. Xie, Z. Cai, Y. Guo, M. Wu, P. Wang, R. Li and H. Zhang, J. Colloid Interface Sci., 2020, 580, 480–492 CrossRef CAS PubMed.
- S. Salentinig and M. Schubert, Biomacromolecules, 2017, 18, 2649–2653 CrossRef CAS PubMed.
- S. V. Pingali, M. D. Smith, S. H. Liu, T. B. Rawal, Y. Pu, R. Shah, B. R. Evans, V. S. Urban, B. H. Davison, C. M. Cai, A. J. Ragauskas, H. M. O'Neill, J. C. Smith and L. Petridis, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16776–16781 CrossRef CAS PubMed.
- H. Li, Y. Deng, B. Liu, Y. Ren, J. Liang, Y. Qian, X. Qiu, C. Li and D. Zheng, ACS Sustainable Chem. Eng., 2016, 4, 1946–1953 CrossRef CAS.
- Y. Qian, Y. Deng, X. Qiu, H. Li and D. Yang, Green Chem., 2014, 16, 2156 RSC.
- F. Xiong, Y. Han, S. Wang, G. Li, T. Qin, Y. Chen and F. Chu, ACS Sustainable Chem. Eng., 2017, 5, 2273–2281 CrossRef CAS.
- M. D. Smith, L. Petridis, X. Cheng, B. Mostofian and J. C. Smith, Phys. Chem. Chem. Phys., 2016, 18, 6394–6398 RSC.
- H. Trevisan and C. A. Rezende, Ind. Crops Prod., 2020, 145, 112105 CrossRef CAS.
- Y. Qian, X. Zhong, Y. Li and X. Qiu, Ind. Crops Prod., 2017, 101, 54–60 CrossRef CAS.
- E. Lepeltier, C. Bourgaux and P. Couvreur, Adv. Drug Delivery Rev., 2014, 71, 86–97 CrossRef CAS PubMed.
- J. H. Lee, K. Kim, X. Jin, T. M. Kim, I. G. Choi and J. W. Choi, Int. J. Biol. Macromol., 2021, 183, 660–667 CrossRef CAS PubMed.
- S. B. Selyanina, M. V. Trufanova, N. I. Afanas'ev and N. V. Selivanova, Russ. J. Appl. Chem., 2007, 80, 1832–1835 CrossRef CAS.
- T. E. Nypelo, C. A. Carrillo and O. J. Rojas, Soft Matter, 2015, 11, 2046–2054 RSC.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21–41 CrossRef CAS.
- G. Kaptay, Colloids Surf., A, 2006, 282–283, 387–401 CrossRef.
- J. Wu and G. H. Ma, Small, 2016, 12, 4633–4648 CrossRef CAS PubMed.
- M. Kamkar, R. Salehiyan, T. B. Goudoulas, M. Abbasi, C. Saengow, E. Erfanian, S. Sadeghi, G. Natale, S. A. Rogers, A. J. Giacomin and U. Sundararaj, Prog. Polym. Sci., 2022, 132, 101580 CrossRef CAS.
- N. Ghavidel and P. Fatehi, Langmuir, 2021, 37, 3346–3358 CrossRef CAS PubMed.
- B. Xiang, Q. Liu and J. Long, Energy Fuels, 2018, 32, 7451–7457 CrossRef CAS.
- J. Kou and S. Xu, Colloids Surf., A, 2016, 490, 110–120 CrossRef CAS.
- N. Ghavidel and P. Fatehi, ChemSusChem, 2020, 13, 4169–4770 CrossRef PubMed.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- J. Becker and C. Wittmann, Biotechnol. Adv., 2019, 37, 107360 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00692a |
| This journal is © The Royal Society of Chemistry 2023 |