
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-purity lignin from selective biomass fractionation with ternary deep eutectic solvents†
Liang Ying
Ee
 ab,
Yong Kuok
Tan
a,
Jiapei
Miao
a,
Hui Ting
Chu
ac and
Sam Fong Yau
Li
*ad
ab,
Yong Kuok
Tan
a,
Jiapei
Miao
a,
Hui Ting
Chu
ac and
Sam Fong Yau
Li
*ad
aDepartment of Chemistry, College of Humanities and Sciences, National University of Singapore, 3 Science Drive 3, Singapore 117543. E-mail: chmlifys@nus.edu.sg
bDepartment of Chemical and Biomolecular Engineering, College of Design and Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585
cSingapore Institute of Food and Biotechnology Innovation, Agency for Science, Technology and Research, 31 Biopolis Drive, Singapore 138669
dNUS Environmental Research Institute, National University of Singapore, T-lab Building, 5A Engineering Drive 1, Singapore 117411
First published on 20th March 2023
Abstract
Conventional caustic soda kraft pulping is not a sustainable pretreatment as it can degrade useful resources from the biomass. Here we design reusable acidic ternary deep eutectic solvents (TDES) composed of alanine, lactic acid, and ethylene glycol for fractionation of high-purity lignin and augmentation of lignocellulosic biomass. The selective fractionation yielded lignin free of sulfur and silica, while ensuring that lignin condensation was avoided by the desired stabilization with diols. Advanced nuclear magnetic resonance (NMR) spectroscopic analysis provided a mechanistic understanding of the delignification process and lignin structural transformation. Besides the predominant demethoxylation and cleavage of β-O-4 linkage, lactic acid induced both acylation and oxidation of the substructures, resulting in lignin with desirable molecular weight and monomeric yield (S/G ratio = 3.39), which are beneficial for applications in reinforcement materials and bio-oils. Overall, the green TDES extraction method could shed new light on alternative pretreatments for sustainable biomass valorization.
Introduction
The valorization and fractionation of lignocellulosic biomass into useful chemicals and desired fuels has become an attractive option over the past few decades to mitigate non-renewable fossil fuel depletion and global warming. The selective transformation of the biomass can be achieved through various processes including partial combustion, fermentation, pyrolysis, and biochemical reactions into downstream products such as biogas, bio-alcohol and biodiesel.1 Besides, the converted solid products, known as biochar, have found important industrial applications in gasification, bio-composites and activated carbon.2 While “biomass” can mean any organic waste, a large fraction of the lignocellulosic source comes from agro-waste, as agricultural production has ballooned over three times in the last five decades.3 Almost all agricultural waste generates some fundamental value, for example, rice husk as soil fertilizer or lignin as biofuel. Nonetheless, strategic and careful pretreatment of the waste can yield much higher value beyond the application as a fuel with the increased economic appeal.4 However, the conventional pretreatment methods involve the use of organic solvents, which can be harmful to the environment and undermine the advocacy of a sustainable and circular economy. To ensure an efficient biomass pretreatment, there are multiple considerations to be made, including: (1) the ability to process biomass particles without requiring their size reduction, (2) prevention of the formation of harmful by-products, and (3) cost-effectiveness and low energy demands.5,6 In general, the composition of an agricultural waste consists of cellulose, hemicellulose, lignin, ash, pectin, proteins, and extractives, ordered from highest to lowest content. Many advancements in pretreatment methods have been reported, deviating from the use of caustic soda, strong acids and volatiles to ionic liquids, supercritical CO2, or steep low-temperature delignification.7,8 When properly designed, ionic liquids can be biocompatible, with negligible vapour pressure and favourable thermal stability.9,10 In hindsight, ionic liquids are expensive to produce and are highly hygroscopic, resulting in compromised extraction efficiency.11–14 In addition, a major hurdle has been the wide variation in the composition of different agricultural wastes,6 and a universal pretreatment technique that has high selectivity for the extraction of specific components would be desirable.In the past, researchers have mainly focused on the extraction of cellulose and hemicellulose for further processing into functional materials, while seeking a solution to remove the stubborn lignin scaffold that protects the fibres. Fortunately, they have found pivotal uses of lignin to produce liquid and gaseous fuels. The native lignin structure consists primarily of ether bonds between monolignols (synapyl, coniferyl, and p-coumaryl alcohols),15,16 with some of the more common linkages being β-O-4 (β-aryl ether), β-β (resinol), β-5 (phenylcoumaran) and β-β′ (pinoresinol). The corresponding phenylpropanoid monomeric units in the lignin polymer are known as syringyl (S), guaiacyl (G) and p-hydroxyphenyl (H) units, respectively. The aromatic cyclic and linear C–O–C bonds in the lignin structure are generally studied for cleavage during depolymerization to produce useful chemicals.17,18 It has been demonstrated experimentally that a plethora range of useful chemicals and polymers can be derived from lignin, including polyurethanes, curing agents, and even vanillin and vanillic acid through catalytic depolymerization.19,20 Furthermore, functional materials with improved mechanical properties, heat and moisture resistance, UV-absorbing ability and shape-memory materials can be fabricated from lignin through reinforcement composite and chemical modification.21–24 Kai et al. have reviewed and analyzed the structure of lignin and interestingly, illustrated that it exhibits bactericidal properties due to the presence of double bonds in the α, β-positions of the side chain and the methyl group in the γ position of the phenolic components, but also depends on the origin of biomass, from which lignin is extracted. Due to its recalcitrant nature, high degree of polymerization, and rigid structure, lignin is highly resistant to chemical attack and harsh conditions are usually required to depolymerize it.25,26 Different pretreatment methods have been reported and they have varied effects on the structural changes of lignin and may affect the cellulosic fibres too. For example, mechanical pretreatment such as ball milling leads to an increase in carbonyl and phenolic hydroxyls, enzymatic hydrolysis reduces the S/G ratio and demethylates the phenolic component into dihydroxybenzene products, and alkali pretreatment results in cleavage of β-O-4 linkage.27,28 The high energy consumption, high cost of using strong chemicals, possible degradation of cellulosic fibres, and corrosion of equipment hinder the feasibility of employing these pretreatment processes.
In recent years, deep eutectic solvents (DES) have emerged as the new breed of ionic liquids made with much cheaper chemicals and in a more environmentally friendly manner. DES is the chemical combination of two or more hydrogen bond acceptors and hydrogen bond donors that have high melting points, experience a large depression in melting point, and exist as a liquid at room temperature. Natural chemicals such as amino acids and sugars are used to enhance the benefit of biodegradability and biocompatibility, with choline chloride-based DESs being the most commonly reported.29,30 Li et al. (2021) showed that choline chloride/oxalic acid (ChCl/OA) at a solubility of 47.85% exhibits excellent separation of lignin from bagasse, although the strong acid tends to carbonize the separated cellulose. They also found that acidic DES performed better than basic DES in dissolving and extracting lignin.31 Other authors screened several combinations of choline chloride-carboxylic acid DES and reported that the weaker lactic acid provided friendlier separation of lignin without damaging the other valuable fibres.32–34 Furthermore, Mankar et al. (2022) were able to achieve an extremely high lignin yield of 82% if the coconut coir was pretreated with choline chloride/lactic acid (ChCl/LA in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 molar ratio) in combination with microwave heating. Although choline chloride is a ubiquitous chemical, there is little literature examining delignification using DES based on other hydrogen bond acceptors. The type and number of functional groups in both the hydrogen bond acceptor and the donor result in different lignin fractionation,35 and research on the process–structure–property relationships of DES pretreatment to obtain useful lignin is still lacking. Moreover, to the best of our knowledge, the selectivity of most prepared and reported DESs for lignin extraction needs to be further improved. Fernandes et al. (2021) reported a novel DES combination of choline chloride/lactic acid/tartaric acid (ChCl/LA/TA in a 1:4:1 molar ratio) that allowed the recovery of 95% lignin from pine sawdust with purity as high as 89%.31 Such a ternary combination is rarely reported elsewhere and encourages further investigation in our study.
:4 molar ratio) in combination with microwave heating. Although choline chloride is a ubiquitous chemical, there is little literature examining delignification using DES based on other hydrogen bond acceptors. The type and number of functional groups in both the hydrogen bond acceptor and the donor result in different lignin fractionation,35 and research on the process–structure–property relationships of DES pretreatment to obtain useful lignin is still lacking. Moreover, to the best of our knowledge, the selectivity of most prepared and reported DESs for lignin extraction needs to be further improved. Fernandes et al. (2021) reported a novel DES combination of choline chloride/lactic acid/tartaric acid (ChCl/LA/TA in a 1:4:1 molar ratio) that allowed the recovery of 95% lignin from pine sawdust with purity as high as 89%.31 Such a ternary combination is rarely reported elsewhere and encourages further investigation in our study.
Herein the present study, we investigate the efficacy and selectivity of alanine/lactic acid/ethylene glycol-based ternary DES in the extraction of pure lignin from rice husk waste. Analytical characterizations and theoretical calculations were also carried out to study the intermolecular interactions within the DES system. Precipitated lignin that is sulfur-free and has a higher S/G ratio is preferred for conversion into useful biofuels and chemicals. Therefore, the mechanistic elucidation of lignin dissolution mechanisms, structure and properties of the precipitated lignin was further studied using different nuclear magnetic resonance (NMR) spectroscopic techniques.
Materials and methods
Materials
Rice husks were purchased from Far East Flora (Singapore), washed, dried, ground, and sieved through 355 μm mesh size. The ground rice husk was subjected to Soxhlet extraction with ethanol for 24 h under reflux to remove extractives. Sodium hydroxide pellets (99–100%), DL-alanine (HPLC, 99%), lactic acid (ACS, 85%), ethylene glycol (anhydrous, 99.8%), Aquastar® solvent and titrant for volumetric Karl Fischer titration, certified reference material (sodium tartrate dihydrate with 15.66% moisture) for Karl Fischer titration, deuterated dimethyl sulfoxide-d6 (DMSO-d6), pyridine-d5, and chloroform (CDCl3), 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP), cyclohexanol (analytical standard), chromium(III) acetylacetonate (99.99% trace metals basis), L-(+)-tartaric acid (≥99.5%), and choline chloride (≥98%) were purchased from Sigma-Aldrich (St Louis, MO, USA). Ultrapure water used was produced by the Milli-Q® water purification system (EMD Millipore Co., Billerica, MA, USA).Preparation of binary and ternary DES
The solid and anhydrous chemicals were dried overnight in the DZ-1BC vacuum oven at 70 °C. The vials and stir bars were dried in an oven to remove moisture and the vial was flushed with nitrogen gas for 45 s before the addition of precursor components. The binary DES was synthesized by mixing alanine and lactic acid, a hydrogen bond acceptor (HBA) and hydrogen bond donor (HBD), in a 1:6 molar ratio in a dried glass vial at 80 °C while stirring with a magnetic stir bar at 500 rpm on a hot plate for 120 min or until a homogeneous solution was formed, whichever was earlier.
Different molar ratios of the BDES to ethylene glycol, 1:6:1, 1:6:2 and 1:6:3 was added to prepare the ternary DES (TDES) labelled TDES 1-6-1, TDES 1-6-2, and TDES 1-6-3, respectively. The effect of ethylene glycol on the physicochemical properties of DESs as well as the lignin dissolution efficiency was then evaluated. Moreover, literature-reported LA/TA/ChCl TDES in 4:1:1 molar ratio was prepared for comparison of its lignin extraction efficiency. 10 g of each ternary DES was synthesized at 80 °C and stirred at 500 rpm for 120 min.
Characterization techniques of DES
DSC analysis using DSC 1 STARe System (Mettler Toledo International Inc., Columbus, OH, USA) was performed from −70 °C to 120 °C at a heating rate of 3 °C min−1 to evaluate the effect on transition temperatures of DESs upon ethylene glycol addition. Approximately 15 mg to 20 mg of DES sample was placed in the DSC sample pan. The environment of the DSC was continuously purged with nitrogen gas at a flow rate of 50 mL min−1 to ensure homogeneity in heat distribution to remove any volatiles that may have evolved during the heating process.Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) was performed using IR Prestige-21 (Shimadzu Co., Kyoto, Japan) between 4000 cm−1 and 600 cm−1 wavenumbers at 4 cm−1 resolution for 40 scans. The Modular Compact Rheometer MCR 302 with the CP25-2 of 25 mm diameter measuring cone (Anton Paar GmbH, Graz, Austria) was used to measure the viscosity of DES samples over a temperature range of 50 °C to 120 °C at a heating rate of 2.5 °C min−1 and a constant shear rate of 80 s−1. The conductivity of DES samples as a function of temperature was investigated between 30 °C and 60 °C using Malvern Zetasizer Nano (Malvern Panalytical Ltd, Malvern, Worcs, UK). The water stability of DES samples was analyzed by storing them at 60% relative humidity and 24 °C for three days, and their moisture content was measured using DL38 volumetric Karl Fischer titrator (Mettler Toledo International Inc., Columbus, OH, USA), calibrated with sodium tartrate dihydrate, and tested with two-components reagents after each day of storage.
It is also important to study and elucidate the intermolecular interactions, particularly the hydrogen bonding involved in the DES samples, which can explain the great melting point depression. 1H NMR spectra and 1H–1H 2D NOESY NMR spectra were recorded using a 600 MHz Varian NMR spectrometer (Agilent Technologies Inc., Santa Clara, CA, USA). 1 drop of DES sample was added to 600 μL of the DMSO-d6. The spectra were obtained at pre-saturation with a relaxation delay of 2.0 s and a NOE mixing time of 700 ms. There were 64 scans for each increment and a total of 256 increments. The process data matrix consisted of 1024 × 2048 points and were zero-filled to 2048 × 2048 points. The spectra were processed with a sine bell-shaped window function along both dimensions.
The presence of water and its interactions in the TDES system were further investigated using NMR spectroscopy. Different concentrations of water (2, 8, 15, 20 and 30% w/w) were first added to freshly prepared TDES 1-6-3 before it was transferred to 5 mm NMR tubes containing a fixed amount of DMSO-d6. 1H NMR spectra were recorded with 8 scans, while 1H self-diffusion coefficients were measured using diffusion ordered spectroscopy (DOSY) with bipolar pulse pair stimulated echo at 25 °C over a spectral width of 16 ppm and diffusion delay of 50 ms. The spectra were processed in MestReNova software (Mestrelab Research, Santiago de Compostela, Spain) and the self-diffusion coefficients were determined after Bayesian DOSY transform.
Pretreatment of rice husk wastes with prepared DES and extraction of lignin
The pretreatment of rice husk wastes with BDES and TDES samples was carried out by adding 5 g of ground rice husk into 50 g of DES that was preheated to 100 °C for 3 h. The solution was stirred at 400 rpm during the dissolution. Thereafter, 200 mL of ultrapure water was added, and the resulting suspension was vacuum filtered. The solid was washed eight times with 100 mL of water each time. The filtered residue was dried in the air-circulated oven at 80 °C overnight to remove moisture. The popular Van Soest method, with some modifications, was used to determine the fibre compositions of the original rice husk and pre-treated samples.36 The hemicellulose, cellulose and lignin were calculated from the difference of neutral detergent fibre (NDF), acid detergent lignin (ADL), acid detergent fibre (ADF) and ash. The insoluble lignin content was determined after calcination with a muffle furnace and the remaining mass corresponds to ash content.The same DES samples (filtrate from vacuum filtration) were used three times to pre-treat a new batch of ground rice husk waste to investigate whether their recyclability maintained the lignin dissolution efficiencies. Before each pretreatment cycle, the recycled DES samples were rotary evaporated under vacuum to remove the water content. The Van Soest procedure was then repeated to quantify the lignin dissolved in the DES samples at each cycle.
Characterization techniques of extracted lignin
The chemical composition was determined by ATR-FTIR analyzed using IR Prestige-21 with 40 scans per spectrum and a resolution of 2 cm−1, covering a wavenumber range of 4000 cm−1 to 500 cm−1. The degree of crystallinity could be analysed with PXRD analysis using D8 Advance (Bruker Daltonics Inc., Billerica, MA, USA), collected over 2θ angles from 5° to 50°, at a scanning rate of 2° min−1. Thermogravimetric Analysis (TGA) was conducted using Discovery TGA 5500 instrument (TA Instruments, New Castle, DE, USA), heating from room temperature to 650 °C at a heating rate of 5 °C min−1 under a flow of nitrogen gas. Differential scanning calorimetry (DSC) was performed using DSC 1 STARe System (Mettler Toledo International Inc., Columbus, OH, USA) between 25 °C and 110 °C and isothermally for 2 min before heating to 450 °C at a heating rate of 5 °C min−1. Optical microscopic images of cellulose, hemicellulose and lignin were captured using an EVOS M7000 microscope (Thermo Fisher Scientific Inc., Waltham, MA, USA). CHNS/O analysis was performed with FlashSmart CHNS Elemental Analyser (Thermo Fisher Scientific, Waltham, MA, USA). JEOL JSM-6701F Field Emission SEM (FESEM) with a JEOL JED-2300F Energy Dispersive X-ray Spectrometer (EDX) (JEOL Co., Akishima, Japan) was used to study the morphologies and elemental contents of the Pt-coated lignin samples at an accelerating voltage of 5 kV. Gel permeation chromatography (GPC) analysis of lignin samples was performed using Waters e2695 Alliance HPLC coupled with Waters 2412 refractive index detector (Waters Co., Milford, MA, USA), conducted at 1.0 mL min−1 with 95% v/v THF mobile phase, Waters Styragel GPC column, and calibrated with polystyrene standards ranging from 580 to 354000 Da molecular weight. The GPC column and detector temperatures were operated at 40 °C and 35 °C, respectively.
All NMR analyses of lignin samples were recorded on 600 MHz Varian NMR spectrometer at 25 °C and the results were processed in MestReNova software (Mestrelab Research, Santiago de Compostela, Spain). For 1H NMR analysis, approximately 7 mg of lignin sample was dissolved in 500 μL of DMSO-d6 with 1 s relaxation delay for 64 scans. The peak of DMSO-d6 was used as the internal reference during data processing. For 13C quantitative NMR (qNMR), 125 mg of lignin sample was dissolved in 500 μL of DMSO-d6 and 20 μL of 0.01 M chromium(III) acetylacetonate as a relaxation agent. The inverse gated decoupling sequence, which allows quantitative analysis and comparison of signal intensities in 13C qNMR, was used with the following parameters: 30° pulse angle, 1.4 s acquisition time, 5 s relaxation delay and 30000 scans. For 31P qNMR, 40 mg of lignin sample was dissolved and vortexed in 500 μL of anhydrous pyridine-d5 and CDCl3 in a 1.6:1 v/v ratio. Then 100 μL of cyclohexanol, prepared as 10 mg mL−1 in a mixture of pyridine-d5 and CDCl3, was added as an internal standard followed by 100 μL of chromium(III) acetylacetonate, prepared as 5 mg mL−1 in a mixture of pyridine-d5 and CDCl3, were added. Finally, the mixture was phosphitylated with 100 μL of TMDP for about 15 min. The inverse gated decoupling sequence, which allows quantitative analysis and comparison of signal intensities in 31P qNMR, was used with the following parameters: 30° pulse angle, 1.2 s acquisition time, 10 s relaxation delay and 256 scans.
For the 1H–13C 2D gradient HSQC (gHSQCAD) NMR analysis, 80 mg of lignin was dissolved in 600 μL of DMSO-d6. The spectral width of 14.0 ppm and 2048 complex points were collected for the 1H dimension. A spectral width of 200 ppm, 128 transients and 512 increments were acquired for the 13C dimension with an acquisition time of 0.2 s and a recycle delay of 1.5 s. 145 Hz was used as the 1J(C–H) coupling constant. Gaussian and squared sine-bell apodizations were processed for 1H and 13C dimensions, respectively. Data matrices for the 13C dimension were zero-filled to 1024 points before Fourier transformation. For the 1H–13C 2D gradient HMBC (gHMBCAD) NMR analysis, 100 mg of lignin was dissolved in 600 μL of DMSO-d6. A spectral width of 14.0 ppm and 2048 complex points were collected for the 1H dimension. A spectral width of 200 ppm, 128 transients and 512 increments were collected for the 13C dimension with an acquisition time of 0.2 s and recycle delay of 1.2 s. 5 Hz was used as the nJ(C–H) multiple bond coupling constant. Gaussian and squared sine-bell apodizations were processed for 1H and 13C dimensions, respectively. For the 1H–13C 2D gradient HSQC-TOCSY (gHSQC-TOCSY) NMR analysis, 100 mg of lignin was dissolved in 600 μL of DMSO-d6. The spectra were recorded with a pulse delay of 2 s, an acquisition time of 0.2 s and a mixing time of 80 ms for 48 scans with 512 increments. A spectral window of 14.0 ppm in F2 (1H) and 150 ppm in F1 (13C) was acquired, and both were processed with a squared sine-bell window function. The central solvent (DMSO) peak was used as an internal chemical shift reference point (δC/δH = 39.5/2.49).
Computational work
Molecular structures of components in DES were optimized with TURBOMOLE® software (TmoleX, COSMOlogic GmbH & Co. KG, Leverkusen, Germany) by DFT BP86 and with valence polarization def-TZVP-FINE before their interactions were investigated by the Conductor-like Screening Model for Real Solvents (COSMO-RS). It predicts the chemical potentials of the individual components in the DES mixture and calculates their sigma profile in the BIOVIA COSMOtherm software (Dassault Systèmes SE, Vélizy-Villacoublay, France). Four β-O-4 lignin model compounds, illustrated in Fig. S1,† were used for molecular simulations and theoretical calculations. Materials Studio 2017 (version 17.1, Accelrys Software Inc., San Diego, CA, USA) was used to minimize the energies of the DES and lignin model compounds structures before calculating their electrostatic interaction in the DMol3 module with B3LYP exchange–correlation functional (fine quality). To further investigate and illustrate the intermolecular interactions between DES samples with each lignin model compound, their optimized structures were combined and analyzed using Cohesive Energy Density in the Forcite module, and the Blends Mixing module at 378 K with alanine, lactic acid, and ethylene glycol as the base while lignin model compound as the screen. Both calculations were performed with COMPASSII forcefield (fine quality).Results and discussion
High-value adhesives and reinforcement materials can be derived from lignin with substantial aliphatic hydroxyl content. It was previously reported by Liu et al. (2021) that extensive recondensation of lignin can be catalyzed by the presence of acid to produce high contents of aliphatic.37 Moreover, several types of diols can stabilize the carbocation species in an acidic environment, which prevents the breakdown of β-O-4 linkages. Therefore, we have established a natural ternary DES (TDES) system incorporating both lactic acid (LA) and ethylene glycol (EG) with alanine (ALA) at different molar ratios based on the aforementioned information. The performance of TDES pretreatment of lignocellulosic source (i.e., rice husks) was compared to binary DES (BDES), containing alanine and lactic acid, and caustic soda. The optimal molar ratio of ALA and LA (1:6) in BDES was determined based on the formation of a homogeneous liquid mixture at room temperature, and the amounts of EG were varied in molar ratios (ALA:LA:EG) of 1:6:1, 1:6:2, and 1:6:3 (Fig. S2†).
Characteristics of binary and ternary deep eutectic solvents
DES consists of HBA and HBDs, which combine chemically to significantly lower melting points. In our case, ALA, with a melting point of about 300 °C, was observed to form a liquid mixture when mixed with LA and/or EG. COSMO-RS prediction was first performed to theoretically study the hydrogen bond interaction mechanism between the HBDs and HBAs in both BDES and TDES prepared, as shown in Fig. 1a and b. The sigma-profiles show the probability distribution of surface area with a particular charge density (σ) and it is divided into HBD (negative σ), non-polar (electroneutral) and HBA (positive σ) regions. Negative σ values mean positive polarity, and vice versa.38 It is noted that both LA (red dotted line) and EG (blue dotted line) have broad peaks between −0.02 e A−2 and −0.01 e A−2, which correspond to the electropositive hydrogen atoms. They also have narrower peaks between +0.01 e A−2 and +0.02 e A−2 that are assigned to their electronegative oxygen atoms. On the other hand, alanine has sharp intense peaks between +0.015 e A−2 and +0.025 e A−2 in the HBA region that represent the electronegative oxygen and nitrogen atoms. More positive charge density is observed in alanine, implying that it could be the dominant HBA in the DES mixture and is further evidenced by its distinctive surface sigma potential profile in Fig. 1b that it can more readily accept hydrogen bonds than donating hydrogens. The 3D molecular structures of BDES and TDES, with surface charge density mapping (red: electronegative, green: electroneutral, blue: electropositive), show that the alanine portion accepts more hydrogen while the other two components donate hydrogen.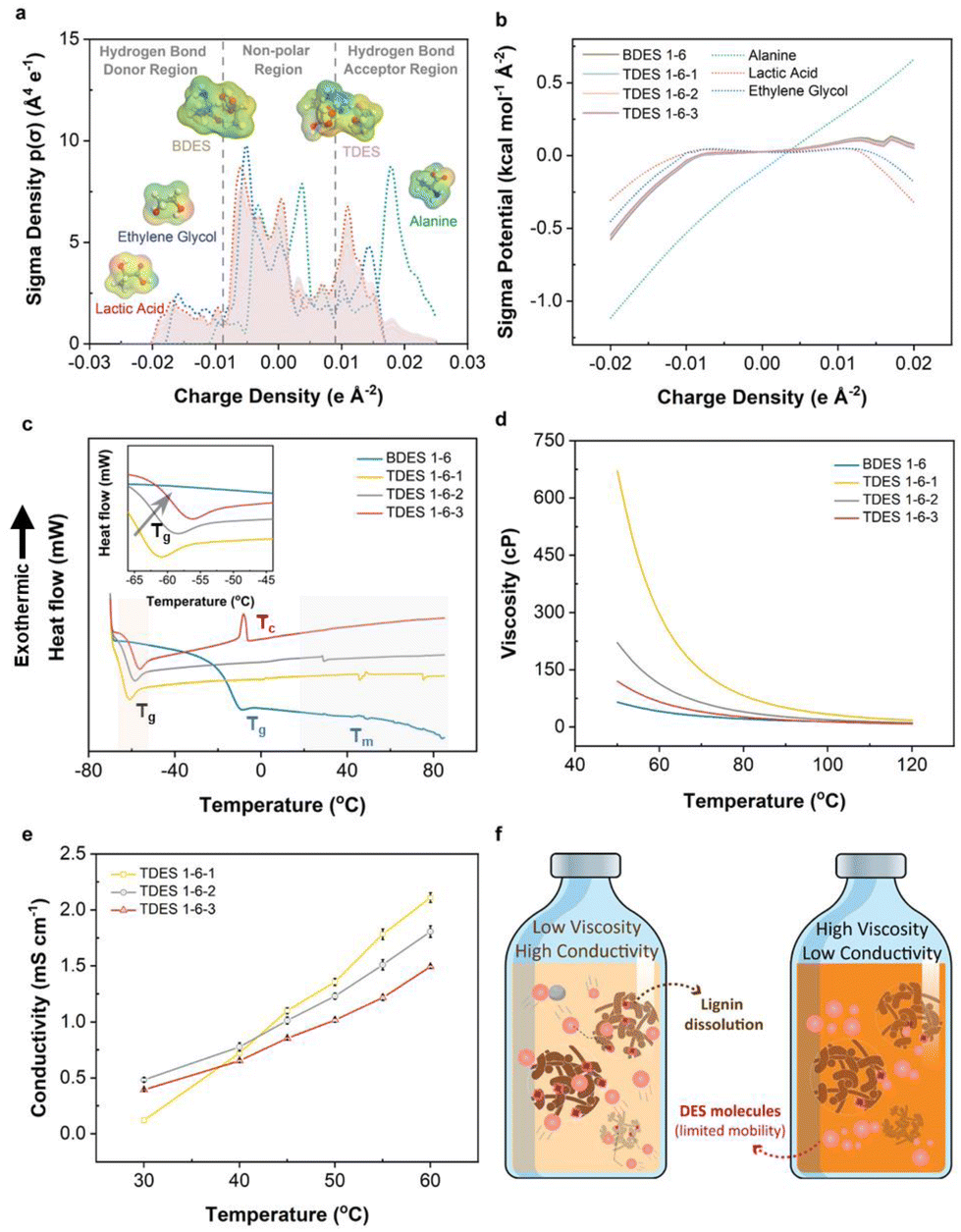 | ||
| Fig. 1 (a) COSMO-RS sigma profile showing sigma density (segmented into hydrogen bond donor, non-polar and hydrogen bond acceptor regions) and (b) sigma potential as a function of charge densities of deep eutectic solvent samples. The sigma potential illustrates the chemical potential of surface polarization charge density in the compound or solvent; (c) glass transition temperatures indicated by a dip in the heat capacity (magnified in inset) from DSC analysis (heating rate = 3 °C min−1), and the effect of temperature on (d) rheology and (e) conductivities of deep eutectic solvent samples. Conductivities of BDES 1–6 as a function of temperature were omitted as the change was insignificant; (f) schematic representation of the effect of bulk viscosity on ion mobility and conductivity. | ||
To confirm the theoretical predictions, 2D 1H–1H NOESY NMR spectroscopy was used to investigate both intra- (grey dotted line) and inter-molecular (red solid line) hydrogen bonding. Results are shown in Fig. S3,† where crosspeaks indicate hydrogen contributions from both HBA and HBDs, with intensity suggesting enhanced contribution. The BDES sample showed intense crosspeaks between the methyl group in LA and the amine and alpha carbon of ALA, while the carboxylic group in LA contributed to the amine group of ALA. In TDES, EG accounted for additional intense crosspeaks between adjacent carbon atoms and carboxylic group in LA, while its hydroxyl groups contributed to the amine of ALA. These findings agreed with theoretical predictions from both σ-profiles and σ potential-profiles. TDES of different molar ratios showed similar 2D NMR spectra, with slight differences due to possible moisture content. The ATR-FTIR spectra of the prepared DES (Fig. S4a†) also show the presence of EG, with a more intense broad band at about 3375 cm−1 corresponding to its hydroxyl groups in TDES samples, and characteristic peak at 1736 cm−1 and 1123 cm−1 assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration mode of LA and the C–N stretching vibration of ALA, respectively.
O stretching vibration mode of LA and the C–N stretching vibration of ALA, respectively.
Water was not desired in the DES systems as it could compete with the lignin fractions for hydrogen bonding with either ALA/LA BDES or ALA/LA/EG TDES, leading to less lignin dissolution.39 To study the hygroscopicity of each DES system, the samples were stored in relative humidity of 60% at room temperature for three days. Results in Fig. S4b† observe an initially slow moisture uptake for all DES samples (≤0.32%), which accelerated after one day of storage with greater change as the EG content increased. It can be deduced that the addition of EG increased the hygroscopicity of the TDES and eventually resulted in 8.11% moisture content in TDES 1-6-3 compared to 2.11% moisture content in BDES 1–6. Therefore, the DES samples were used within 24 h of preparation to avoid significant reduction in the amount of dissolved lignin, as the maximum moisture difference of 2.31% among these samples was considered trivial.
DSC analysis was carried out to determine the phase transition temperatures of BDES and TDES, with their thermograms shown in Fig. 1c. The DSC thermogram of BDES shows a glass transition point (Tg) at −10.1 °C (Table S1†). As expected from the difference in physicochemical properties between BDES and TDES, the addition of EG reduced the Tg to as low as −61.7 °C in TDES 1-6-1. Increased EG content led to higher Tg (rightward shift in magnified inset), which was attributed to the stronger hydrogen bond network formed with the other components. Despite the TDES samples comprising similar components, they exhibit different characteristics of glass transition, vitrification and melting phase in their respective thermograms. TDES 1-6-3, with highest EG content, displays a prominent vitrification temperature (Tc) at −8.1 °C suggesting that the high EG concentration induces crystallization during the slow heating. Generally, the melting points (Tm) of all DES samples are observed to be over a broad range between 35 °C and 75 °C, which are much lower than the melting point of ALA (i.e., ∼300 °C).
Solvent rheology plays a critical role in the delignification process of lignocellulosic materials, as low viscosity solvents increase the probability of intermolecular collisions and permeation into their inter-fibrillated structures. The viscosities of the DES samples as a function of temperature are illustrated in Fig. 1d, wherein all samples show reducing viscosity with increasing temperature due to the gradual weakening of hydrogen bonds. Their viscosities were highly sensitive to a temperature below 110 °C and the addition of EG in TDES leads to a significant increase in viscosity when compared to BDES. Comparatively, TDES 1-6-1 (80.7 cP) is nearly four times more viscous than BDES (21.2 cP) at 80 °C. Again, further addition of EG in TDES 1-6-2 and TDES 1-6-3 made the samples less viscous since more EG molecules could have interrupted the stronger hydrogen bond between ALA and LA. Therefore, TDES 1-6-1 may seem less suitable for our delignification applications, and it is expected that there is slower diffusivity of molecules.
The conductivity of DES is also an important parameter to study the mobility of ions under temperature influence. Fig. 1e shows the conductivities of TDES between 30 °C and 60 °C, which unravels that TDES 1-6-1 possesses lower conductivity (0.12 mS cm−1) than the other two TDES samples (0.48 mS cm−1 for TDES 1-6-2 and 0.39 mS cm−1 for TDES 1-6-3) when the temperature is below 40 °C due to the former's high viscosity. However, at above 40 °C, the effect of viscosity was diminished as the conductivity of TDES 1-6-1 increased to values higher than TDES 1-6-2 and TDES 1-6-3. A close inspection of the results shows that the conductivity values at 60 °C are ranked as follows: TDES 1-6-1 (2.11 mS cm−1) > TDES 1-6-2 (1.81 mS cm−1) > TDES 1-6-3 (1.49 mS cm−1). In theory, a highly viscous DES would normally have poor conductivity, due to limited mobility of its constituent molecules.40 However, in the case of high viscous TDES 1-6-1, it exhibits the highest conductivity among all TDES samples at 60 °C. This phenomenon can be explained by the possible inhibition of LA ionization with a higher concentration of EG in the solution. According to Fig. 1f, it can be visually inferred that DES with low viscosity and high conductivity enables suitable lignin motion within the solvent system for dissolution. Therefore, to promote high mobility of the molecules, the lignin extraction process in this study was conducted at optimally 100 °C, since the viscosities of all DES converge at this temperature to eliminate any disparity that could have arose.
Efficiency of DES pretreatment on rice husk wastes
The DES samples (BDES and TDES) were evaluated for their performance and selectivity in dissolving lignocellulosic materials. The results are summarized in Fig. 2a and Table S2,† assuming that the rice husks contain only hemicellulose, cellulose, lignin, and ash contents. The relative composition (%) of each component remaining in the dried residue after DES pretreatment is shown in Fig. 2a. Initially, the rice husks sample is comprised of 23.4% hemicellulose, 36.5% cellulose, 27.1% lignin, and 13.0% ash. Both BDES and TDES demonstrated high selectivity in solubilizing lignin, with around 80% of the dried residue consisting of hemicellulose and cellulose. The BDES pretreatment resulted in highest cellulose and hemicellulose content of 88.2%, but also a high lignin composition (11.2%) and low ash content (0.6%), indicating more effective removal of ash than lignin. Much lower lignin contents are observed with TDES pretreatment, with the lowest (5.6%) being achieved with TDES 1-6-1 pretreatment. These findings demonstrate that adding a third diol component to the DES system can improve selective fractionation of recalcitrant lignin from cellulose and hemicellulose in lignocellulosic materials.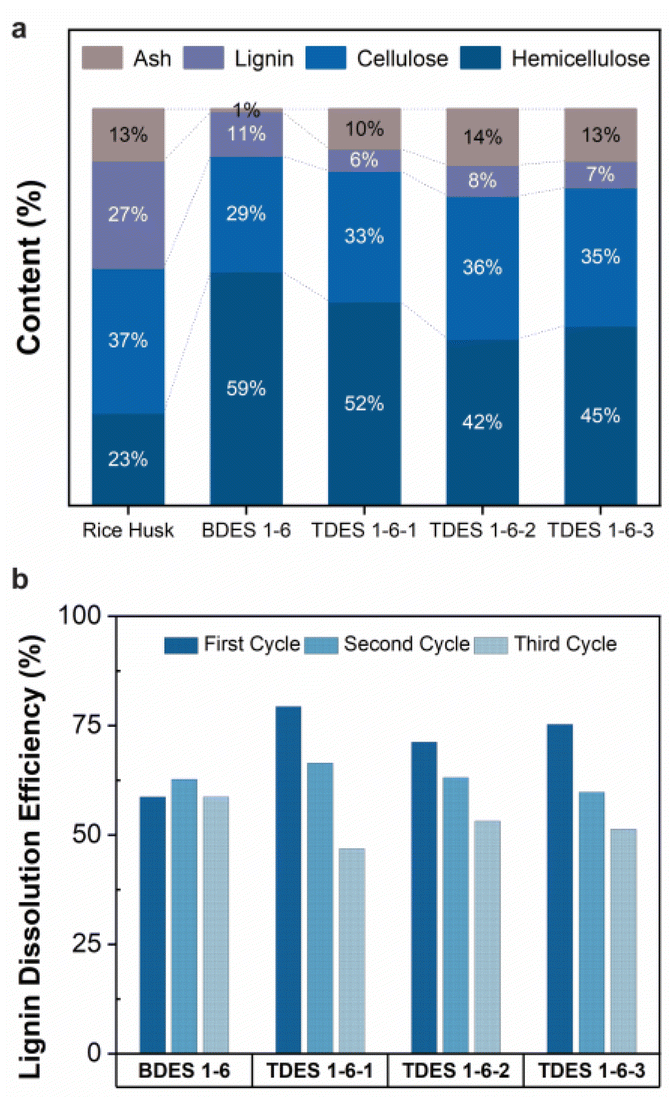 | ||
| Fig. 2 (a) Dissolution selectivity of rice husk contents (assumed to contain only hemicellulose, cellulose, lignin, and ash) in deep eutectic solvent samples, and (b) recyclability of deep eutectic solvent samples over three cycles. A new batch of ground rice husk samples was used every cycle for the recyclability study. | ||
The stability of the DES systems was investigated envisaging their recyclability and reusability over three cycles after the removal of the water used to precipitate the isolated lignin (Fig. 2b). Overall, all DES samples exhibited relatively good stability with lignin dissolution efficiencies of above ∼50% after three cycles. The BDES sample attained a maximum lignin dissolution efficiency of 62.7% and maintained a deviation of 7% in all three cycles. In contrast, all TDES samples exhibit high lignin dissolution efficiency of above 70% during their first cycle but decreased as much as 15.5% in efficiency (TDES 1-6-3) in the second cycle possibly due to their stronger hydrogen bond interactions with water molecules. The upfield or downfield shift of NMR chemical shifts in the 1H NMR of TDES 1-6-3, diluted with varying concentrations of water, reveal the transformation in hydrogen bond network.39 According to Fig. S5a,† the obvious upfield shift (shielding) of Alanine-NH2 suggests a weakened hydrogen bond in the presence of increased water content. Additionally, the slight downfield shift (deshielding) of H2O signal indicates that water formed strong hydrogen bonds with the TDES constituents, disrupting the original network. This disruption is further evidenced by a significant reduction in the self-diffusion coefficients, as observed in the DOSY NMR spectra of all constituents in the diluted TDES 1-6-3 samples (Fig. S5b and Fig. S6†). These results indicate that the water remained tightly bound even at increasing concentrations,41 which can hinder the efficiency of lignin dissolution and thus explain why the recycled TDES underperformed over multiple cycles.
Albeit the huge reduction in performance, the lignin dissolution efficiencies of all TDES samples in the third cycle were very close to that of the BDES sample, with the lowest value of 46.9% in TDES 1-6-1. Available evidence from the literature supports the higher incidence of loss of volatile component, ethylene glycol in this case, in the DES recovery using rotary evaporation, which may account for the significant drop in lignin dissolution efficiency.42,43 However, there is no experimental data to support this claim here and chromatographic separation analysis can be performed as future work. Despite this, the selectivity and dissolution efficiency of the recycled DES remain reasonably high, confirming that they can be reused at least three times.
Characteristics of extracted lignin from DES pretreatment
The chemistry and structure of isolated lignin from alkali, BDES, and TDES 1-6-1 pre-treatments were comprehensively studied using multiple NMR spectroscopic analyses. Fig. 3 presents the 1H, 13C, and 31P quantitative NMR (qNMR) spectra of the precipitated lignin samples, which have distinctive features that differentiate between the pre-treatment methods. 1H NMR spectrum of lignin extracted from alkali pre-treatment show messy signals due to its complex polymeric structure, with intense peaks at 9.3 and 8.2 ppm in the aromatic region (10–6.5 ppm) assigned to the phenolic hydroxyl groups, and lower intensity bands corresponding to the phenylpropane units (S, G, and H-units). Several aliphatic side-chain protons are found between 6.5 and 5.5 ppm, right before the broad band responsible for the protons of the methoxy groups at 5.2 ppm. The huge broad band at around 2.7 ppm corresponds to the acetyl protons of the native esters in the isolated lignin. The absence of peaks between 1.8 and 0.5 ppm implies that there is little to no aliphatic moiety of lignin from alkali pre-treatment.44 In contrast, lignin from BDES and TDES pretreatments show cleaner 1H NMR spectra with higher contents of aliphatic protons and methoxy group. The low-intensity profiles in the aromatic region suggest that the DES pre-treatment resulted in fewer S, G, and H-units compared to alkali pre-treatment.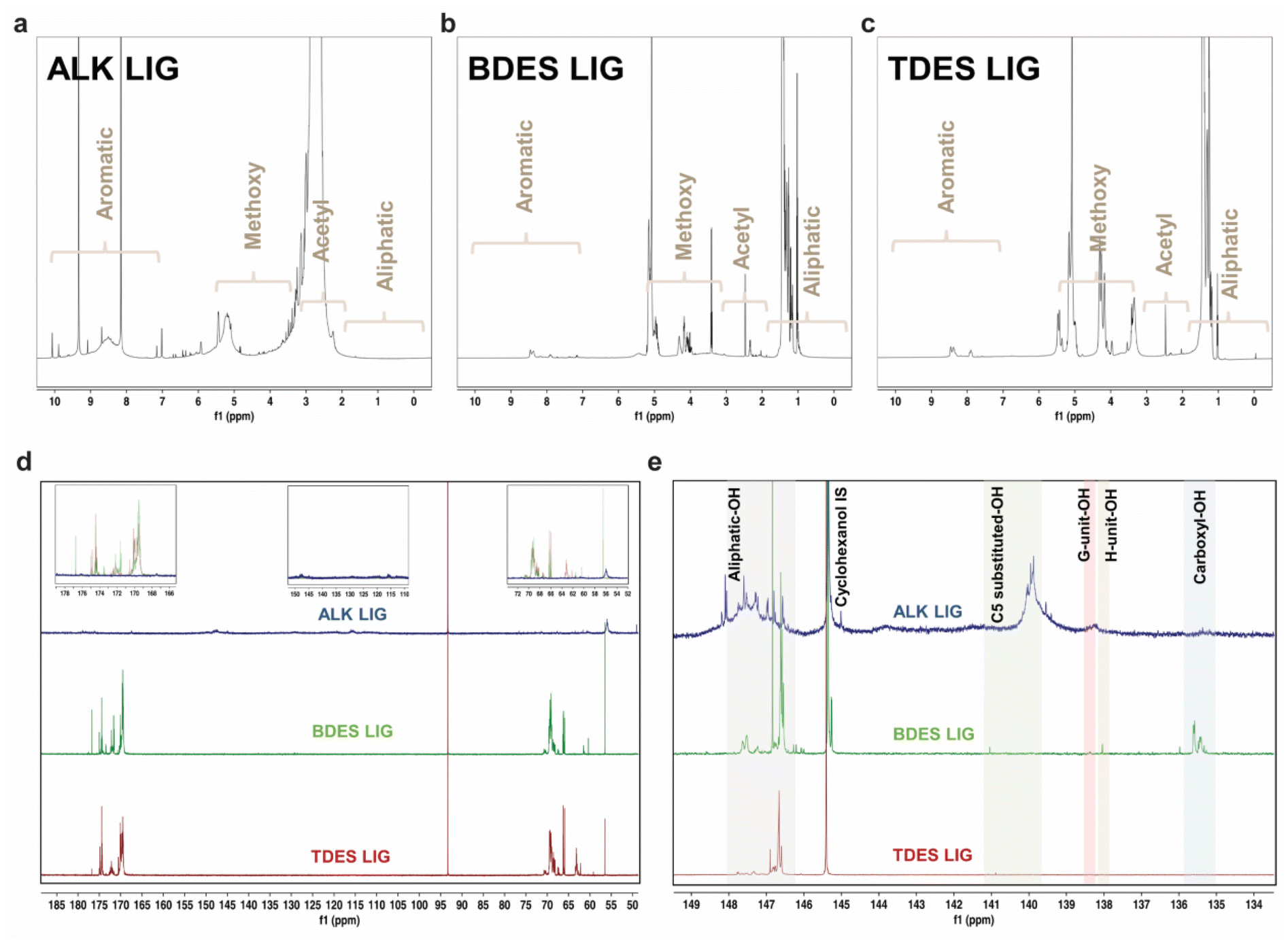 | ||
| Fig. 3 (a)–(c) 1H NMR spectra of alkali, BDES and TDES-precipitated lignin respectively. Regions corresponding to aliphatic, acetyl, methoxy and aromatic content are denoted in the spectra; (d) 13C qNMR and (e) 31P qNMR spectra of precipitated lignin samples. Regions corresponding to aliphatic-OH (grey highlight), C5 substituted-OH (green highlight), G-unit OH (red highlight), H-unit OH (brown highlight) and carboxyl-OH (blue highlight) were identified and denoted in 31P qNMR spectra. Their respective concentrations were calculated based on their peak areas with reference to the cyclohexanol internal standard peak. | ||
13C qNMR allowed us to identify and quantify the lignin-carbohydrate complex linkages, typically the phenyl glycoside, ester, and benzyl ether bonds (Fig. 3d and Table S3†). The absence of carbohydrate peaks between 105–90 ppm indicates a clean fractionation of lignin in high purity.45 Similarly, the absence of ether linkages between 90–72 ppm corresponds to the possible dissolution mechanism through β-O-4 linkage cleavage. The presence of strong peaks in the 13C NMR spectra of alkali isolated lignin at 50.0–48.1 ppm and 117.9–111.5 ppm reveals high phenolic contents, corresponding to the C-β and C-5 in G-units, respectively. Moreover, the high content (0.2 mmol g−1) of combined β-O-4 in S and G phenylpropane units (150.5–146.0 ppm) suggests more rapid β-O-4 cleavage in S-unit since the C-β and C-5 of G-units were detected. Comparatively, the methoxy content in alkali pre-treated lignin is found to be the lowest (0.3 mmol g−1) compared to BDES (9.3 mmol g−1) and TDES (4.1 mmol g−1) pre-treatments, indicating varying degree of demethoxylation among the pre-treatment processes, ranked as follows: alkali pre-treatment > TDES pre-treatment > BDES pre-treatment. Noteworthy, the distinct signals between 178.2 and 167.5 ppm can be attributed to CO in carboxyl group, potentially formed through oxidation by LA in the DES system. The DES isolated lignin samples display various peaks corresponding to the C-α and C-γ of G-units between 72.6 and 58.5 ppm. BDES isolated lignin, for instance, has a high content of 183.3 mmol g−1 of C-α in G type β-O-4 units (erythro) at 72.6–68.0 ppm. These findings unravel the possible protection of the ether linkages from cleavage during DES pre-treatment. Conversely, the absence of these signals in alkali isolated lignin indicates that the predominant cleavage of β-O-4 aryl linkages, resulting in increased phenolic-OH contents.
Hydroxyl group contents in the isolated lignin samples were quantified using 31P qNMR spectroscopy to relate them to respective lignin units (Fig. 3e). The low hydroxyl content in native lignin can only be increased from depolymerization reactions and cleavage of ether linkages. 31P qNMR provides an indirect measurement of possible structural transformation during pre-treatments. The total phenolic-OH in alkali isolated lignin significantly increased to 0.53 mmol g−1, which was much higher than in BDES (0.32 mmol g−1) and TDES (0.09 mmol g−1) isolated lignin samples (Table 1). The extensive cleavage of ether bonds during alkali pretreatment explains this increase. BDES isolated lignin observed higher phenolic-OH content than TDES isolated lignin, implying greater aromatic density and a lower molar mass fraction of lignin being precipitated from the former pre-treatment. Much higher condensed content of C5-substituted-OH is observed in the BDES isolated lignin coming from both S and G lignin units, indicating increased chance of repolymerization. Conversely, TDES isolated lignin possesses higher aliphatic-OH content, corresponding to a higher molar mass fraction dissolved during pretreatment.46 The presence of EG reduces the change of repolymerization by protecting the cleaved units with diols, resulting in slightly less C5-substituted-OH than BDES isolated lignin. TDES precipitated lignin has almost double the aliphatic-OH content of BDES precipitated lignin, likely due to the residual EG, which also prevented dehydration reactions. Besides acylation, possible oxidation with LA in BDES and TDES could have induced the formation of carboxyl-OH, which finds great agreement with the presence of CO groups in the 13C NMR spectra. This was not present in alkali pre-treatment, corroborating the oxidation with LA.47 Conversion of aliphatic-OH to carboxylic groups was more significance in BDES than in TDES pretreatment due to steric hindrance and stronger hydrogen bonds within the solvent system.
| Sample | Content (mmol g−1) | ||||
|---|---|---|---|---|---|
| Phenolics | |||||
| Aliphatic-OH | C5-substituted | Non-condensed | Carboxyl-OH | ||
| Guaiacyl-OH | p-Hydroxyphenyl-OH | ||||
| Precipitated lignin (binary DES) | 17.8 | 0.1 | 0.02 | 0.2 | 6.2 |
| Precipitated lignin (ternary DES) | 31.3 | 0.06 | 0.03 | — | 1.1 |
| Alkali lignin | 0.4 | 0.1 | 0.4 | 0.03 | — |
The molecular weight of the isolated lignin samples was determined using the GPC technique, as shown in Fig. S8a and Table S4.† The alkali isolated lignin showed two major peaks in its GPC chromatogram, corresponding to molecular weights (Mw) of 1890 and 355 g mol−1. These values are approximately half of the molecular weights (3066–3538 g mol−1, and 794 g mol−1) of BDES and TDES isolated lignin samples due to the more extensive cleavage of ether linkages. Despite the higher molecular weights and lower chemical reactivity, the BDES and TDES isolated lignin samples find favourable applications in the synthesis of adhesives, mechanical reinforcement, and energy production.48 Additionally, the thermal degradation behaviour of the different lignin samples was studied using DSC and TGA (Fig. S8b, c and Table S5†). It is observed that the alkali isolated lignin underwent endothermic transition at 353.5 °C due to dehydration and depolymerization. Yet, both BDES and TDES isolated lignin oil observed highly exothermic peaks at 312.1 °C and 324.2 °C, respectively, due to the char formation. This is well reflected in TGA analysis showing similar peak degradation temperatures, which resulted in at least 96% total mass loss. However, pyrolyzed alkali isolated lignin showed only a 41.7% total mass loss from TGA analysis, which can be attributed to the vast amounts of ash and inorganic residues.
The limited structural information and dissolution mechanisms of the lignin could not be fully understood from the 1D NMR data alone, prompting the need for a qualitative 2D 1H–13C HSQC NMR analysis to examine the short-range C–H correlations, as shown in Fig. 4a and b and crosspeaks identified in Table S6.† The alkali isolated lignin was used as a control for reference to unambiguously identify and label the crosspeaks. The 2D NMR spectra are divided into aromatic (δC/δH = 160–90 ppm/8.0–6.0 ppm) and side-chain (δC/δH = 95–45 ppm/6.5–2.0 ppm) regions, where S, G and H units with other individual substructures such as ferulate (FA), p-coumarate (pCA) and p-hydroxybenzoate (pBA) are identified in the former region for BDES isolated lignin. FA, pCA, and pBA are prominent products from the acylation of lignin.49 A slight difference in the chemical shift of the phenylpropane units in BDES isolated lignin from alkali isolated lignin could be the result of new C–C bonds formed during repolymerization, which lead to denser electron cloud around their aromatic rings.50 The missing G5 signal implies that condensation of the lignin could have occurred at the C-5 position of the G type unit during BDES pre-treatment. Whereas from the side-chain region, methoxy group, β-O-4 linkages to free (A) and γ-acetylated-OH (A′), phenyl coumaran (B), resinol (C) and Hibbert's ketone are found to be present. Acetylated-OH was observed, attributing to the acylation of the γ-OH group by LA during the pre-treatment process, which coincides with previous studies in the literature. Meanwhile, only G and S units are observed in the aromatic region of TDES isolated lignin, agreeing well with the 31P qNMR results. Moreover, condensation reactions were mainly observed at the C-2, and C-6 positions of the H-units, hence their signals disappeared. pCA and pBA substructures are also found. As for the side-chain region, it is typically similar to BDES isolated lignin with the additional cinnamyl alcohol end-group due to the etherification of β-O-4 units. It was found that resinol structures formed specifically by the β-β linkages under condensation reactions were present in both BDES and TDES isolated lignin samples, indicating the simultaneous cleavage of β-O-4 aryl ether linkages during DES pre-treatment. Similarly, the existence of uncondensed Hibbert's ketone moieties, useful for the production of aromatic monomers, confirms some extent of ether bond breakage in both isolated lignin samples.
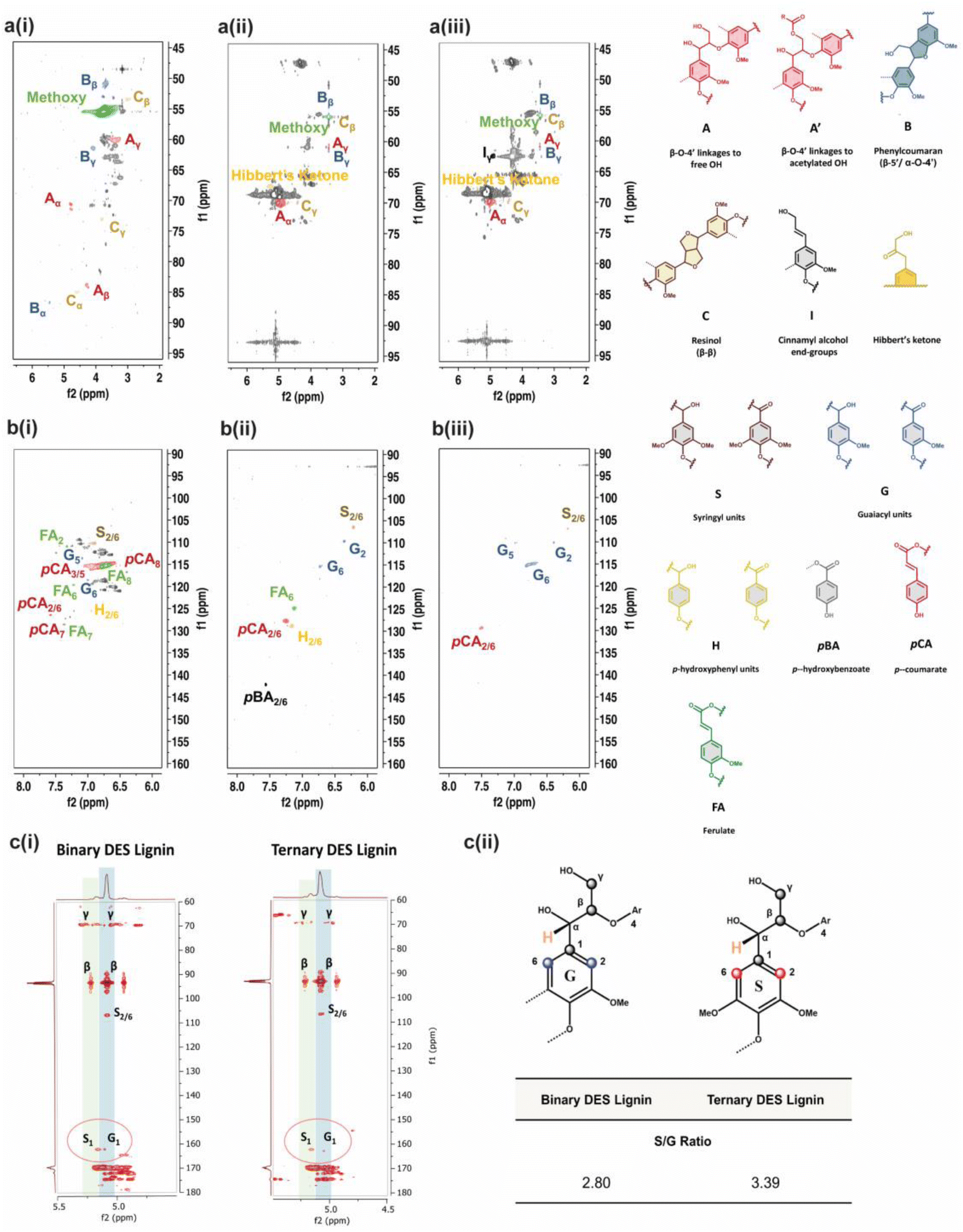 | ||
| Fig. 4 1H–13C 2D HSQC NMR spectra of alkali, BDES and TDES-precipitated lignin samples in a(i)–a(iii) side-chain regions and b(i)–b(iii) aromatic regions. Identified compounds with their molecular structures illustrated were colour-coded similarly as in the 1H–13C 2D HSQC NMR spectra; c(i) Semi-quantitative 1H–13C 2D HMBC NMR spectra and c(ii). calculated S/G ratios of BDES and TDES-precipitated lignin samples based on the cross-peak areas. Carbon positions of lignin G-unit and S-unit were identified and labelled in 1H–13C 2D HMBC NMR spectra, with reference to drawn molecular structures. | ||
Finally, 2D 1H–13C HMBC NMR analysis was performed to identify the long-range C–H correlations and any compositional transformation of the lignin units upon the different DES pre-treatments. The 2D 1H–13C HMBC NMR spectra in Fig. 4c(i) show the presence of S and G-units with assigned positions. The semi-quantitative analysis allows the quantification of the S/G ratio, which translates to the maximum monomeric yield from lignin in the pre-treatment.51 BDES isolated lignin observes a lower S/G ratio of 2.80 compared to TDES isolated lignin (S/G ratio = 3.39), implying that the former has a lower maximum monomeric yield from the degradation of G-units than the latter (Fig. 4c(ii)). This further indicates that more energy and chemical are consumed to effectively release the total aromatic units from BDES in consequence of the higher tendency of BDES lignin to form cross-linkages and condensed bonds from repolymerization leading to increased viscosity. In contrast, the higher S/G ratio in TDES precipitated lignin generally means that there was less extensive condensation of G-units leading to less intense G1 signals. Therefore, it is conclusive that TDES pre-treatment not only improves the selectivity of lignin dissolution but also precipitates higher quality lignin that can be potentially reused as useful resources such as bio-derived fuels.
Both SEM-EDX and CHNS/O analyses were performed to determine the elemental composition of the isolated lignin samples from different pre-treatment methods (Fig. 5 and Table 2). Intriguingly, no sulfur content was detected in either BDES or TDES isolated lignin samples, whilst the alkali isolated lignin contains 1.8% w/w sulfur. Sulfur is reported to exist mainly as its inorganic elemental state with some organically bound components,52 but its presence is found by several authors to promote the cleavage of ether bonds in the non-condensed phenylpropane units.53,54 This phenomenon explains the greater extent of β-O-4 cleavage observed in the NMR analyses. However, sulfur-free lignin is more highly sought after in real-life applications since it can result in the inhibition of depolymerization and has detrimental effects on the production of high-quality bio-oil. It is widely reported that sulfur can participate in hydrogen bonds, with itself being a good HBA while its organic form as HBD.55,56 Therefore, the tuneable hydrogen bonding network in the DES system plays a vital role in ensuring that the sulfur content in isolated lignin is minimized during pre-treatment, which was successfully achieved with either BDES or TDES 1-6-1 blend in this study. In addition, TDES isolated lignin observed no silica content (i.e., ash) unlike BDES isolated lignin, agreeing well with the results from the evaluation of their selectivity in dissolution.
 | ||
| Fig. 5 SEM-EDX mapping of (a) alkali-precipitated, (b) BDES-precipitated and (c) TDES-precipitated lignin samples. Silicon elemental content from ash was not identified in both alkali-precipitated and TDES-precipitated lignin samples. | ||
Theoretical study of interactions between lignin and DES
Several β-O-4 lignin model compounds were used for the molecular simulation and theoretical calculations of interactions with the BDES and TDES. The substitution of acyl and hydroxyl functional groups at different positions of the model compounds is highlighted in grey in their molecular structures (1a, 1b, 1c, and 1d). Calculated intermolecular electrostatic interactions (i.e., hydrogen bonding) between BDES and model compound 1a show the highest value of −45.8 kcal mol−1 and solubility of 18.5 (J cm−3)1/2. The theoretical results in Fig. 6 are essentially in line with the experimental results reported by Liu et al. (2021) that its β-O-4 linkage could be efficiently cleaved in choline chloride/oxalic acid DES without the presence of strong acid. Surprisingly, the electrostatic solubilities of model compounds 1a, 1c, and 1d reduced to 17.3, 13.8, and 12.6 (J cm−3)1/2, respectively, after the addition of EG in the TDES 1-6-1 blend. This can again reiterate the potential protective pathway rendered by EG to minimize the acidolysis and ether bond cleavage. However, the high intermolecular electrostatic interactions (−56.0 kcal mol−1) and solubility (19.8 (J cm−3)1/2) between TDES and model compound 1b can be ascribed to the additional hydroxyl group at C-4 aromatic position that is analogous to the guaiacyl unit. Several authors also reported that the G-type units are more susceptible to degradation and β-O-4 cleavage,51,57 which is supported by the larger S/G ratio determined from 2D 1H–13C HMBC NMR analysis earlier. Therefore, the model compound 1b was used to propose the lignin dissolution mechanisms by TDES 1-6-1 in Fig. 7. | ||
| Fig. 6 a(i)–a(viii) Calculated intermolecular interactions between deep eutectic solvents and lignin model compounds (labelled 1a, 1b, 1c and 1d with molecular structures shown in Fig. S1†). | ||
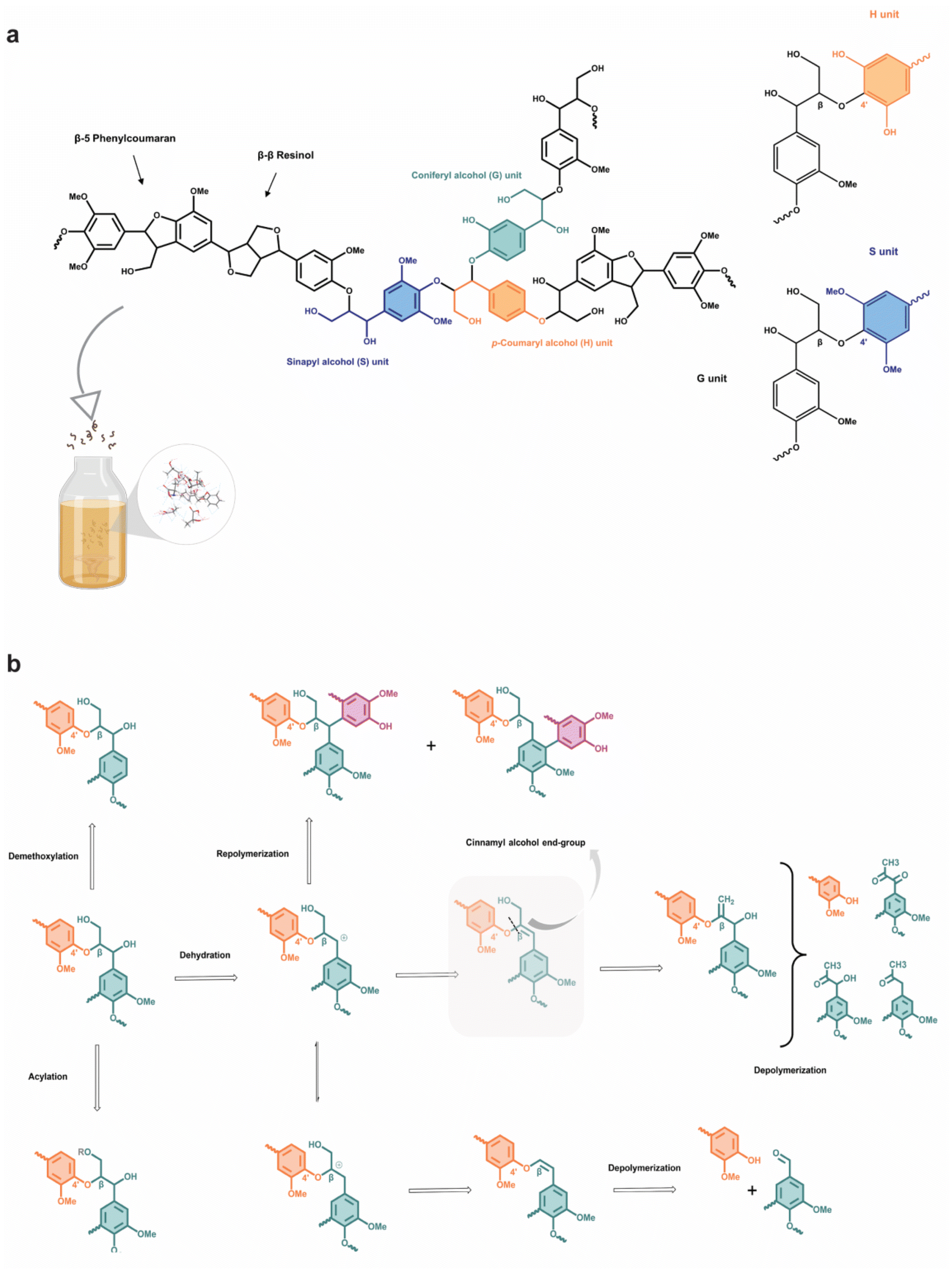 | ||
| Fig. 7 Schematic illustration of (a) lignin and sub-units (G-unit, S-unit, and H-unit), and (b) proposed lignin dissolution mechanism in the ternary deep eutectic solvent system. The proposed lignin dissolution mechanism is based on the identified species and compounds from 1H, 13C, 31P, 1H–13C 2D HSQC and 1H–13C 2D HMBC NMR analyses. | ||
It has been recognized that the ALA/LC/EG TDES in molar ratio 1:6:1 exhibit high efficacies in the selective fractionation of lignin from other biopolymers in lignocellulosic material and inducing structural transformation on the isolated lignin. Better mechanistic insights on lignin dissolution from rice husks in the TDES were gained from the different advanced instrumentation techniques. Based on the theoretical calculations, model compound 1b corresponding to the G-type lignin units was used to elucidate the different mechanisms of preferential depolymerization, dehydration, and demethoxylation. Recondensation or repolymerization could also happen as observed from the 2D 1H–13C HSQC NMR spectra, and the regenerated lignin structure is far more complex than the simplified proposed mechanism. Moreover, it can be envisaged that oxidation and acylation by residual LA in the DES system exist although not shown in the schematic illustration. Overall, the G-type units were found to be the most vulnerable to ether bond cleavage and degradation at 100 °C, catalyzed by the acidic TDES pre-treatment. With all useful findings combined, the possible chemical transformations of the lignin from rice husks during TDES pre-treatment have been proposed, which aims to include all substructures identified from NMR analyses.
Compared to other DES pretreatments of lignocellulosic biomasses reported in the literature (Table 3), the ternary DES mixture formulated in this work achieved excellent lignin extraction efficiency (79.3%) under much milder conditions. Currently, binary DES mixtures are still more commonly explored for the pretreatment of biomasses to obtain lignin. However, most reported binary DES often lack dissolution selectivity and also result in sugars loss, albeit satisfactory lignin extraction efficiency.34 More recently, Fernandes et al. (2021) synthesized an acidic ternary DES based on lactic acid, tartaric acid, and choline chloride, which up to 89.4% lignin extraction efficiency from maritime pine.31 Nonetheless, the high temperature (i.e. 175 °C) used in the pretreatment process poses demanding challenges on both engineering and energy aspects. In contrast, this work not only reports a novel blend of ternary DES mixture, but also extracts lignin with high efficiency under comparatively milder conditions. The favourable results underscored the importance of the interplay between the hydrogen bonding network and the viscosity of DES, which provided essential guidelines for the design of effective DES.
| DES system (molar ratio) | Biomass | Extraction condition | Lignin extraction (%) | Ref. |
|---|---|---|---|---|
| LA: lactic acid; ChCl: choline chloride; TA: tartaric acid; GLY: glycerol. | ||||
| LA/ChCl (2:1) |
Wheat straw | 120 °C; 12 h | 78.7 | 34 |
| LA/TA/ChCl (4:1:1) |
Maritime pine | 175 °C; 1 h | 89.4 | 31 |
| LA/ChCl (5:1) |
Poplar | 150 °C; 2 h | 68.3 | 58 |
| LA/ChCl (2:1) |
145 °C; 6 h | 66.0 | ||
| ChCl/Urea (1:2) |
Oil palm empty fruit bunch | 120 °C; 8 h | 34.0 | 59 |
| K2CO3/GLY (1:6) |
120 °C; 8 h | 51.0 | ||
| LA/TA/ChCl (4:1:1) |
Rice husk | 100 °C; 3 h | 71.6 | This work |
| ALA/LA/EG | Rice husk | 100 °C; 3 h | 79.3 | This work |
Conclusions
In this study, a novel ternary DES mixture based on ALA/LA/EG (1:6:1 molar ratio) was designed and developed to tailor to high lignin dissolution selectivity and recyclability. Over 85% of hemicellulose, cellulose and ash contents were preserved in the residue after TDES pre-treatment, invigorating the remarkable lignin dissolution capacity (79.3%) achieved due to its high ion mobility and complex hydrogen bonding network. The addition of protective diols (i.e., EG) endowed great controllability over the degree of ether bond cleavage in addition to minimizing repolymerization and dehydration. Meanwhile, the presence of LA induced both acylation and oxidation of the substructures as evidenced by the NMR analyses. Altogether, it was found that the sulfur-free lignin was successfully isolated with high purity, molecular weight, and monomeric yield (S/G ratio = 3.39), which are important properties for applications in reinforcing materials and bio-oils. Therefore, the need to optimize the pre-treatment process and structural transformation of lignin underscore the importance of elucidating a mechanistic understanding of the interactions between lignin chemistry and DES through comprehensive analytical investigations.
Author contributions
Liang Ying Ee: conceptualization; methodology; investigation; validation; writing – original draft; writing – review & editing; resources. Yong Kuok Tan: validation; investigation. Jiapei Miao: validation; investigation. Hui Ting Chu: validation; writing – review & editing. Sam Fong Yau Li: conceptualization, supervision; writing – review & editing; funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was financially supported by a grant (R-143-000-B24-592) co-funded by the Singapore National Additive Manufacturing – Innovation Cluster (NAMIC) and MIPS Innovations Pte. Ltd. The authors acknowledge the support from PUB, Singapore's National Water Agency, and the facility support from NUS Chemical, Molecular and Materials Analysis Centre (CMMAC). The first author would like to acknowledge the Singapore Ministry of Education (MOE) for providing a scholarship for his Ph.D. research programme in Chemistry at the National University of Singapore. This work was partially shared as a poster presentation at the Singapore International Water Week 2021, which attained the “Best Student Poster Award”.References
- C. Briens, J. Piskorz and F. Berruti, Int. J. Chem. React. Eng., 2008, 6(1) DOI:10.2202/1542-6580.1674.
- J. A. Okolie, E. I. Epelle, M. E. Tabat, U. Orivri, A. N. Amenaghawon, P. U. Okoye and B. Gunes, Process Saf. Environ. Prot., 2022, 159, 323–344 CrossRef CAS.
- M. Duque-Acevedo, L. J. Belmonte-Ureña, F. J. Cortés-García and F. Camacho-Ferre, Global Ecol. Conserv., 2020, 22, e00902 CrossRef.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- A. R. Mankar, A. Pandey, A. Modak and K. K. Pant, Bioresour. Technol., 2021, 334, 125235 CrossRef CAS PubMed.
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee, M. Holtzapple and M. Ladisch, Bioresour. Technol., 2005, 96, 673–686 CrossRef CAS PubMed.
- S. K. Bhatia, S. S. Jagtap, A. A. Bedekar, R. K. Bhatia, A. K. Patel, D. Pant, J. R. Banu, C. V. Rao, Y. G. Kim and Y. H. Yang, Bioresour. Technol., 2020, 300, 122724 CrossRef CAS PubMed.
- S. S. Hassan, G. A. Williams and A. K. Jaiswal, Bioresour. Technol., 2018, 262, 310–318 CrossRef CAS PubMed.
- E. S. Morais, A. Lopes, M. G. Freire, C. S. R. Freire, J. A. P. Coutinho and A. J. D. Silvestre, Molecules, 2020, 25(16), 3652 CrossRef CAS PubMed.
- L. Wils, S. Hilali and L. Boudesocque-Delaye, Molecules, 2021, 26(21), 6556 CrossRef CAS PubMed.
- D. Fu, G. Mazza and Y. Tamaki, J. Agric. Food Chem., 2010, 58, 2915–2922 CrossRef CAS PubMed.
- Z. Jiang and C. Hu, J. Energy Chem., 2016, 25, 947–956 CrossRef.
- M. Kienberger, S. Maitz, T. Pichler and P. Demmelmayer, Processes, 2021, 9, 804 CrossRef.
- R. Prado, X. Erdocia and J. Labidi, J. Chem. Technol. Biotechnol., 2013, 88, 1248–1257 CrossRef CAS.
- S. A. Arni, Ind. Crops Prod., 2018, 115, 330–339 CrossRef CAS.
- J. C. Carvajal, A. Gomez and C. A. Cardona, Bioresour. Technol., 2016, 214, 468–476 CrossRef CAS PubMed.
- J. A. Poveda-Giraldo, J. C. Solarte-Toro and C. A. C. Alzate, Renewable Sustainable Energy Rev., 2021, 138, 110688 CrossRef CAS.
- S. Suresh, V. Viswanathan, M. Angamuthu, G. P. Dhakshinamoorthy, K. P. Gopinath and A. Bhatnagar, Biomass Convers. Biorefin., 2021 DOI:10.1007/s13399-021-01497-8.
- M. S. Rishikesh, S. Harish, S. Mahendran Prasanth and D. Gnana Prakash, Biomass Convers. Biorefin., 2021, DOI:10.1007/s13399-021-01637-0.
- J. Sternberg, O. Sequerth and S. Pilla, Prog. Polym. Sci., 2021, 113, 101344 CrossRef CAS.
- F. H. M. Graichen, W. J. Grigsby, S. J. Hill, L. G. Raymond, M. Sanglard, D. A. Smith, G. J. Thorlby, K. M. Torr and J. M. Warnes, Ind. Crops Prod., 2017, 106, 74–85 CrossRef CAS.
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC.
- H. Y. Lim, S. Yusup, A. C. M. Loy, S. Samsuri, S. S. K. Ho, A. S. A. Manaf, S. S. Lam, B. L. F. Chin, M. N. Acda, P. Unrean and E. Rianawati, Waste Biomass Valorization, 2020, 12, 5285–5302 CrossRef.
- Y. Zhang and M. Naebe, ACS Sustainable Chem. Eng., 2021, 9, 1427–1442 CrossRef CAS.
- I. Spiridon, Environ. Chem. Lett., 2020, 18, 771–785 CrossRef CAS.
- D. Watkins, M. Nuruddin, M. Hosur, A. Tcherbi-Narteh and S. Jeelani, J. Mater. Res. Technol., 2015, 4, 26–32 CrossRef CAS.
- Y. Sheng, S. S. Lam, Y. Wu, S. Ge, J. Wu, L. Cai, Z. Huang, Q. V. Le, C. Sonne and C. Xia, Bioresour. Technol., 2021, 324, 124631 CrossRef CAS PubMed.
- W. Wang and D. J. Lee, Bioresour. Technol., 2021, 339, 125587 CrossRef CAS PubMed.
- A. K. Kumar, B. S. Parikh and M. Pravakar, Environ. Sci. Pollut. Res., 2016, 23, 9265–9275 CrossRef CAS PubMed.
- H. Malaeke, M. R. Housaindokht, H. Monhemi and M. Izadyar, J. Mol. Liq., 2018, 263, 193–199 CrossRef CAS.
- C. Fernandes, E. Melro, S. Magalhães, L. Alves, R. Craveiro, A. Filipe, A. J. M. Valente, G. Martins, F. E. Antunes, A. Romano and B. Medronho, Int. J. Biol. Macromol., 2021, 177, 294–305 CrossRef CAS PubMed.
- A. R. Mankar, A. Pandey and K. K. Pant, Bioresour. Technol., 2022, 345, 126528 CrossRef CAS PubMed.
- E. O. Owhe, N. Kumar and J. G. Lynam, Biocatal. Agric. Biotechnol., 2021, 32, 101949 CrossRef CAS.
- R. Lou and X. Zhang, Bioresour. Technol., 2022, 344, 126174 CrossRef CAS PubMed.
- M. Zhou, O. A. Fakayode, A. E. A. Yagoub, Q. Ji and C. Zhou, Renewable Sustainable Energy Rev., 2022, 156, 111986 CrossRef CAS.
- P. J. V. Soest, J. Assoc. Off. Agric. Chem., 1963, 46, 829–835 CrossRef.
- Y. Liu, N. Deak, Z. Wang, H. Yu, L. Hameleers, E. Jurak, P. J. Deuss and K. Barta, Nat. Commun., 2021, 12, 5424 CrossRef CAS PubMed.
- J. Palomar, J. S. Torrecilla, J. Lemus, V. R. Ferro and F. Rodríguez, Phys. Chem. Chem. Phys., 2010, 12, 1991–2000 RSC.
- M. E. Di Pietro, M. Tortora, C. Bottari, G. C. Dugoni, R. V. Pivato, B. Rossi, M. Paolantoni and A. Mele, ACS Sustainable Chem. Eng., 2021, 9, 12262–12273 CrossRef CAS.
- A. Kovács, E. C. Neyts, I. Cornet, M. Wijnants and P. Billen, ChemSusChem, 2020, 13, 3789–3804 CrossRef PubMed.
- H. Kivela, M. Salomaki, P. Vainikka, E. Makila, F. Poletti, S. Ruggeri, F. Terzi and J. Lukkari, J. Phys. Chem. B, 2022, 126, 513–527 CrossRef CAS PubMed.
- C.-L. Li, G. Huang, Y. Yu, Q. Xiong, J.-M. Yan and X.-b. Zhang, J. Am. Chem. Soc., 2022, 144, 5827–5833 CrossRef CAS PubMed.
- U. Alshana and M. Soylak, in Analytical Sample Preparation With Nano- and Other High-Performance Materials, ed. R. Lucena and S. Cárdenas, Elsevier, 2021, pp. 471–512, DOI:10.1016/B978-0-12-822139-6.00019-5.
- I. V. Pylypchuk, P. A. Lindén, M. E. Lindström and O. Sevastyanova, ACS Sustainable Chem. Eng., 2020, 8, 13805–13812 CrossRef CAS.
- T.-Q. Yuan, S.-N. Sun, F. Xu and R.-C. Sun, J. Agric. Food Chem., 2011, 59, 10604–10614 CrossRef CAS PubMed.
- N. Giummarella, P. A. Lindén, D. Areskogh and M. Lawoko, ACS Sustainable Chem. Eng., 2020, 8, 1112–1120 CrossRef CAS.
- D. Smink, S. R. A. Kersten and B. Schuur, Sep. Purif. Technol., 2020, 235, 116127 CrossRef.
- P. Solt, A.-S. Jääskeläinen, P. Lingenfelter, J. Konnerth and H. Van Herwijnen, For. Prod. J., 2018, 68, 365 CAS.
- Q. Ma, Z. Li, L. Guo, H. Zhai and H. Ren, Ind. Crops Prod., 2021, 161, 113165 CrossRef CAS.
- J.-Y. Liu, S. Wu and R. Lou, BioResources, 2011, 6(2), 1079–1093 CAS.
- E. Anderson, M. Stone, R. Katahira, M. Reed, W. Muchero, K. Ramirez, G. Beckham and Y. Roman-Leshkov, Nat. Commun., 2019, 10, 2033 CrossRef PubMed.
- D. Daniel, L. Khachatryan, C. Astete, R. Asatryan, C. Marculescu and D. Boldor, Bioresour. Technol. Rep., 2019, 8, 100341 CrossRef.
- S. Guadix-Montero and M. Sankar, Top. Catal., 2018, 61, 183–198 CrossRef CAS.
- C. Gong, M. J. Goundalkar, B. M. Bujanovic and T. E. Amidon, J. Wood Chem. Technol., 2012, 32, 93–104 CrossRef CAS.
- H. S. Biswal, in Noncovalent Forces, ed. S. Scheiner, Springer International Publishing, Cham, 2015, pp. 15–45, DOI:10.1007/978-3-319-14163-3_2.
- P. Zhou, F. Tian, F. Lv and Z. Shang, Proteins: Struct., Funct., Bioinf., 2009, 76, 151–163 CrossRef CAS PubMed.
- R. Santos, P. Hart, H. Jameel and H.-M. Chang, BioResources, 2013, 8, 1456 Search PubMed.
- C. Alvarez-Vasco, R. Ma, M. Quintero, M. Guo, S. Geleynse, K. K. Ramasamy, M. Wolcott and X. Zhang, Green Chem., 2016, 18, 5133–5141 RSC.
- Y. T. Tan, G. C. Ngoh and A. S. M. Chua, Ind. Crops Prod., 2018, 123, 271–277 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00080j |
| This journal is © The Royal Society of Chemistry 2023 |