
Electrochemical conversion of lignin to short-chain carboxylic acids†
Shirong
Sun
 ac,
Xueqing
Qiu
*ac,
Shuhua
Hao
a,
Sabarinathan
Ravichandran
a,
Jinliang
Song
a and
Wenli
Zhang
*abc
ac,
Xueqing
Qiu
*ac,
Shuhua
Hao
a,
Sabarinathan
Ravichandran
a,
Jinliang
Song
a and
Wenli
Zhang
*abc
aSchool of Chemical Engineering and Light Industry, Guangdong University of Technology (GDUT), 100 Waihuan Xi Road, Panyu District, Guangzhou 510006, China. E-mail: cexqqiu@scut.edu.cn; wlzhang@gdut.edu.cn
bGuangdong Provincial Key Laboratory of Plant Resources Biorefinery, Guangdong University of Technology (GDUT), 100 Waihuan Xi Road, Panyu District, Guangzhou 510006, China
cJieyang Branch of Chemistry and Chemical Engineering Guangdong Laboratory (Rongjiang Laboratory), Jieyang 515200, China
First published on 9th March 2023
Abstract
Lignin valorization is a challenge for chemical and materials scientists due to its complex molecular structure and intra- and inter-molecular assembly. Although catalytic conversion of lignin into small aromatic organic molecules has been widely investigated, it faces the problems of low selectivity and complex products. Herein, we report a direct electrochemical conversion method for the selective oxidation of industrial lignin to produce short-chain carboxylic acids (SCCAs) with a wide range of concentrations. In an electrolytic cell, lignin is chemically converted to SCCAs by H2O2 with titanium silicate-1 (TS-1) as a catalyst. H2O2 is generated in situ by the recombination of H+ and HO2− in a three-compartment cell by separately introducing hydrogen (H2) and oxygen (O2) as the sources of protons and HO2− into the anode and cathode compartments. We have achieved a record-high concentration of SCCAs up to 109.8 mg mL−1 through the catalytic depolymerization of lignin. A high concentration of SCCAs of 80.1 mg mL−1 could also be achieved at a cell voltage of 2.13 V by coupling the 2e−-ORR and water oxidation using air and 1 M aqueous H2SO4 solution as feedstocks. Our approach could eliminate the additional economic consumption and environmental hazards caused by the transportation and storage of commercially concentrated and stabilized H2O2. Aside from the SCCAs produced in this process, the formation of new carboxyl groups in the lignin residue during the oxidation reaction greatly broadens the application potential of industrial lignin.
Introduction
Carboxylic acids are a class of essential chemicals widely used in materials, pharmaceuticals, food preservation, cosmetics, pesticides, and other manufactured chemicals, due to the special properties of their carboxylic group (acidity, polarity, surface properties, etc.).1,2 Most of the carboxylic acids are produced through tandem oxidation processes of petrochemical feedstocks or biological fermentation of food resources.3 The use of waste or renewable biomass as feedstock can effectively reduce our dependence on non-renewable carbon resources.4–6 Lignin, the second-most abundant renewable polymer, is currently mostly produced as waste in pulping and bioethanol industries and is commonly burned to produce energy. Consequently, the use of some of the lignin resources as raw materials to convert them into high-value-added products can effectively enhance the value of industrial waste without affecting the energy balance of the pulping industry.7In plant cell walls, lignin is biosynthesized by coupling reactions between three primary precursors (Fig. S1†), i.e., p-coumaryl (4-hydroxycinnamyl), coniferyl (4-hydroxy-3-methoxycinnamyl), and sinapyl (4-hydroxy-3,5-dimethoxycinnamyl) alcohols.8,9 The heterogeneous and complex structural characteristics of lignin, including C–O bonds (β-O-4, α-O-4, α-O-γ, 4-O-5), C–C bonds (β-5, β–β, β-1, 5–5), a complex three-dimensional configuration and intra- and inter-molecular π–π interactions, make it difficult to generate products with regular structures.10–12 The highly selective conversion of lignin becomes a recognized challenge, although various processes were developed, including acid/base catalysis,13 enzymatic hydrolysis,14 pyrolysis (thermolysis),15,16 oxidation,17 reduction (hydrogenolysis),18etc. Among these widely investigated methods, oxidation has been the most commonly used lignin depolymerization method, which is technically employed in the modern pulp bleaching processes.19 Simultaneously, carboxylic acids, phenolic compounds, and furan and benzene compounds are produced in different bleaching stages of pulp treatment. These generated compounds are usually considered to be industrial waste, bringing a series of environmental problems, although in-depth basic research on lignin oxidation was carried out and convincing research results were obtained.7,20,21 Therefore, the development and application of new technological processes for lignin conversion to achieve industrial-scale production of lignin-derived chemicals have stimulated the interest of researchers.22,23
Oxidation reactions can not only weaken the stacking force of π–π interactions but also form hydroxyl and carboxyl groups by inserting oxygen functional groups into lignin side-chains and/or aromatic rings, becoming some of the most potential methods for the preparation of lignin-derived chemicals,24–26 especially chemicals containing carboxyl groups. In this context, the most studied oxidants in lignin oxidation are molecular oxygen, KMnO4, nitrobenzene, metal oxides, and hydrogen peroxide (H2O2). Among these oxidants, the H2O2 oxidation of lignin is highly promising since water is the only product of H2O2 after the reaction, and no heteroatoms are introduced into the low molecular-weight organics.25–27 This avoids the problems of complex post-treatment in industrial applications. In the current pulp bleaching industry, H2O2 is a commonly used bleaching agent that can remove lignin from unbleached pulp with high selectivity.28 The significant decrease in pH during H2O2 bleaching proves the formation of a large amount of carboxyl-containing products.29 In addition, industrial lignin resources including alkaline lignin and enzymatic hydrolysis lignin have been utilized for the preparation of short-chain carboxyl acids (SCCAs) by H2O2 oxidation,30,31 confirming that lignin is an excellent source candidate for the preparation of SCCAs. However, H2O2 is currently produced through the anthraquinone process with high energy consumption and waste production.32–34 The H2O2 concentration produced by this conventional method is only 1–2% by weight, and energy-intensive concentration and complex purification processes are necessary to produce a marketable commodity.34 In addition, harsh storage and transportation conditions increase application costs due to the unstable and hazardous characteristics of highly concentrated H2O2 solutions.
The direct catalytic synthesis process of H2O2 using hydrogen (H2) and oxygen (O2) provides a new route in small-scale H2O2 applications (Fig. 1A). Catalysts such as gold–palladium (AuPd) and palladium–tin (PdSn) have been developed for such reactions with high selectivity (>95%) for in situ H2O2 generation.35,36 Nevertheless, the flammability of high-pressure H2 and O2 mixtures is a disadvantage that necessitates that the mixture must be diluted with N2 or CO2, which limits the synthesis of highly concentrated H2O2. In addition, methanol or t-butanol needs to be added to improve the solubility of H2 to increase the rate of H2O2 synthesis.37 In the present work, an electrochemical process for the direct synthesis of H2O2 is proposed to avoid the problems caused by the strategy of high-pressure production of H2O2. Pure H2 and O2 can be safely introduced separately into anode and cathode compartments to achieve the in situ synthesis of H2O2 at various concentrations by coupling the 2e− O2 reduction reaction (2e−-ORR) and the H2 oxidation reaction (HOR) (Fig. 1B). The direct electrochemical formation of H2O2 in such a three-compartment cell reduces the cost of transportation and storage of concentrated H2O2, as well as the risk associated with high-pressure equipment,36,38 and becomes a potential process for industrial application including lignin conversion.
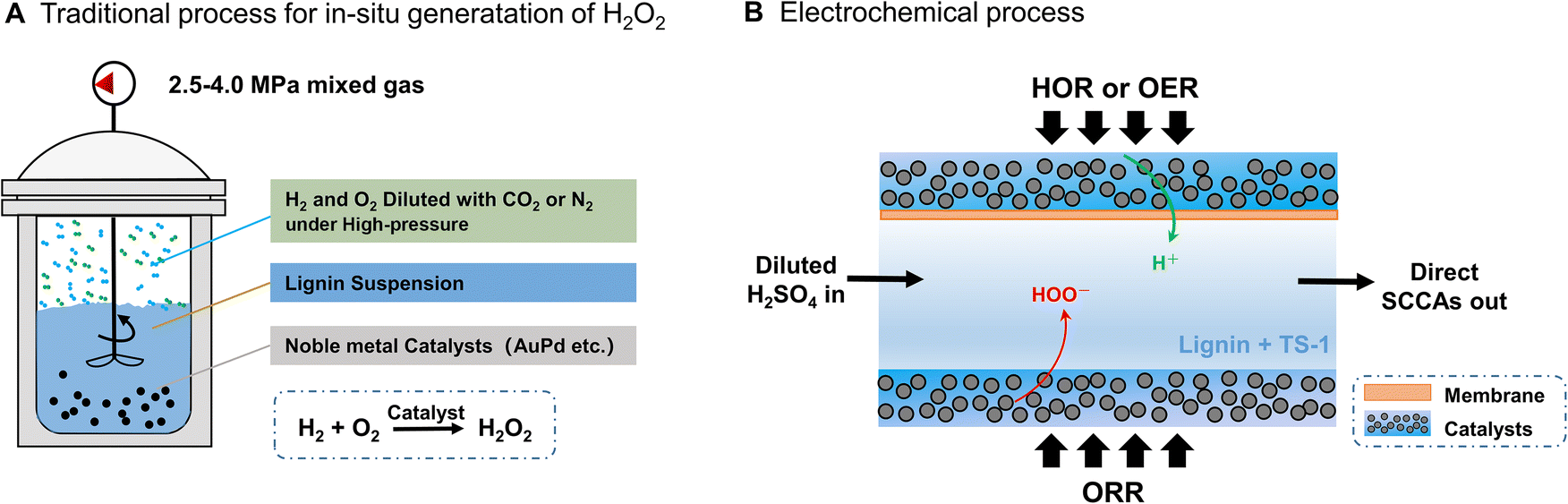 | ||
| Fig. 1 Schematic illustration of the two different methods for producing short-chain carboxylic acids (SCCAs) from lignin using in situ generated H2O2. (A) Traditional direct synthesis of H2O2 with diluted H2 and O2 gases and noble-metal catalysts (e.g., AuPd alloy) for lignin conversion. Alcohols, such as methanol and t-BuOH, were chosen as co-solvents for enhancing the solubility of H2. Sophisticated catalyst structural design was considered to prevent the degradation of in situ generated H2O2. (B) Direct electrosynthesis of H2O2 with pure H2 and O2 introduced into the anode and cathode compartments separately. Diluted H2SO4 was chosen as the lignin dispersion solvent and product carrier. A cation exchange membrane was used at the anode side, and a PTFE membrane was used as a liquid barrier at the cathode side. The catalysts were dispersed on a gas diffusion layer (GDL). The produced short-chain carboxylic acids (SCCAs) were collected directly from the three-compartment cell. | ||
Results and discussion
Reaction procedures for electrochemical production of short-chain carboxylic acids
In our designed direct-electrochemical lignin conversion device, a state-of-the-art platinum–carbon (Pt/C) catalyst was chosen for the HOR to convert H2 to H+ with low overpotentials on the anode side. For the cathode, the research on electrocatalysts with high selectivity for the 2e−-ORR to H2O2 is much less explored than that for the 4e−-ORR to water.39 To meet the needs of industrial applications, surface-oxidized carbon black (O-CB) was applied as the cathode catalyst for the selective 2e−-ORR, because of its low cost, high selectivity, and high faradaic efficiencies (FEs) as described in the ESI (Fig. S2–S4†).40,41 This single-step process could overcome the challenges of the traditional H2O2 application in lignin oxidation, using a three-compartment cell, as a mature lignin conversion method. Fig. 2A shows the current–voltage (I–V) curve of the three-compartment cell with O-CB and Pt/C as electrocatalysts at the cathode and anode compartments, respectively. Pure H2 and O2 gases were directly introduced without any dilution treatments, and a 1 M aqueous H2SO4 solution with a fixed flow rate of 60 mL h−1 continuously flowed through the electrolytic cell with an electrode geometric size of 1 cm−1. At a current of 10 mA cm−2, H2O2 can be synthesized in situ at a rate of 0.20 mmol cm−2 h−1 at −0.01 V without external energy input and even output electricity. The selectivity for H2O2 generation was always higher than 90% over the entire voltage range (Fig. 2B). Different concentrations of H2O2 could be obtained by adjusting the flow rate of the H2SO4 solution (Fig. 2C). For efficient catalytic conversion of lignin with in situ-generated H2O2, titanium silicate-1 (TS-1) was chosen as the catalyst to enhance the oxidation activity by promoting the formation of hydroxyl and superoxide radicals.42 Although many titanium-based catalysts have been developed for the catalytic oxidation process with H2O2, TS-1 is still considered the industrial standard catalyst for reactions of H2O2 applications.43,44 The morphological characterization of TS-1 zeolites was observed with SEM (Fig. S5†), and the powder XRD pattern indicates the presence of an ordered orthorhombic, MFI topology TS-1 framework (Fig. S6†).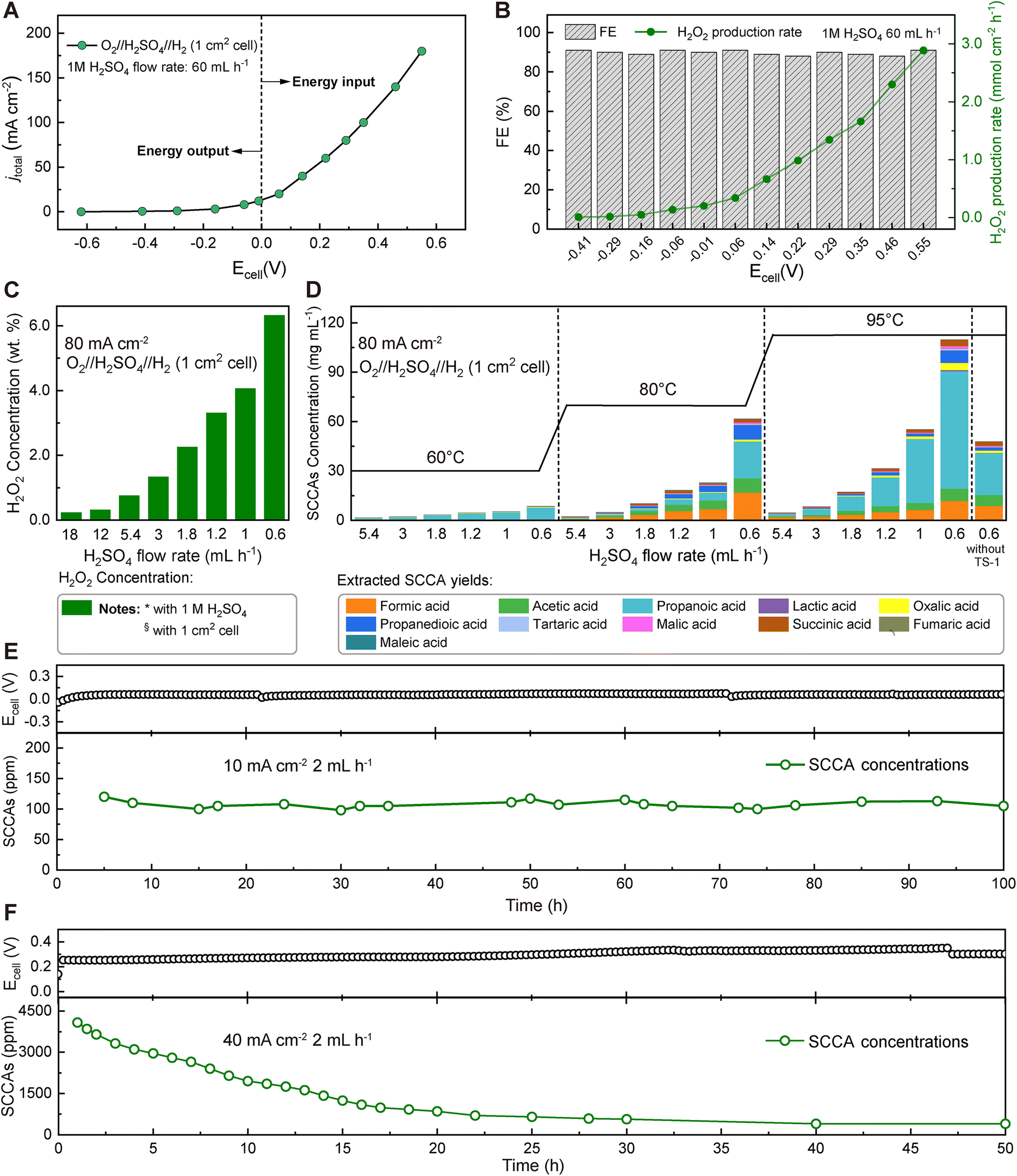 | ||
| Fig. 2 Direct electrochemical production of short-chain carboxylic acids (SCCAs) from lignin using pure H2 and O2. (A) I–V curve for an O2//H2SO4//H2 flow cell (1 cm2) in which Pt/C was chosen as the catalyst at the anode side, and oxygen-doped carbon black (O-CB) was selected as the catalyst at the cathode side. (B) The FEs and H2O2 production rates at different voltage potentials. (C) H2O2 production concentrations at an overall current density of 80 mA cm−2 and different diluted H2SO4 flow rates. (D) SCCA concentrations at different H2SO4 flow rates and temperatures at a fixed current density of 80 mA cm−2. The SCCAs were collected in the first hour under different conditions. (E) The long-term operation test of SCCA production with concentrations of around 100 ppm at a fixed current density of 10 mA cm−2 with negligible energy input. (F) Stability test for the continuous production of SCCAs at a fixed current density of 40 mA cm−2. The concentration of SCCAs gradually decreased with reaction time, indicating that lignin continued to degrade in this long-term test. The structures of the residues at 10 h and 50 h are elucidated in detail in subsequent texts. The 1 M diluted H2SO4 flow rate was 2 mL h−1 in the long-term tests shown in (E) and (F) at the reaction temperature of 80 °C. | ||
Through this industrially available route, enzymatic hydrolysis lignin and commercial TS-1 (10 wt% based on lignin) were mixed and loaded into an O-CB//H2SO4//Pt/C cell for in situ H2O2 oxidation treatment. We observed a correlation between the reaction conditions (i.e., temperature and H2SO4 flow rates) and catalytic performance toward the direct conversion of lignin to SCCAs (Fig. 2D). Concerning the temperature conditions, the concentration of SCCAs significantly increased from 60 °C to 95 °C under different H2SO4 flow rates. Likewise, concentrations of SCCAs significantly increased with decreasing H2SO4 flow rates due to the increased concentration of the in situ generated H2O2, with the highest concentration of SCCAs being up to 109.8 mg mL−1 (32.9 wt%, based on the weight of the initial lignin). The results indicate that the reactivity of the lignin conversion reaction was extremely sensitive to temperature and the concentration of H2O2. Moreover, catalyzed and non-catalyzed reactions were compared at an H2SO4 flow rate of 0.6 mL h−1 and 95 °C, confirming that the TS-1 catalyst improved the conversion efficiency of lignin to SCCAs with almost twice the concentration obtained without the addition of TS-1. The microporosity and hydrophobicity of the TS-1 catalyst, as well as the presence of titanium atoms, can enhance the H2O2 reaction performance by reducing the electron density of the O–O bond, making it susceptible to nucleophilic attack on lignin by H2O2-derived radicals.30,45 The main short-chain carboxylic acid products include four monocarboxylic acids (MCAs), such as formic acid, acetic acid, propionic acid, and lactic acid, and seven dicarboxylic acids (DCAs), such as oxalic acid, propanedioic acid, tartaric acid, malic acid, succinic acid, fumaric acid and maleic acid. Among all products of SCCAs, the contents of formic acid, acetic acid, and propionic acid accounted for more than 80% of all products, especially propionic acid exceeded 60% of the total product content. Our device could continuously output ∼100 ppm SCCA solution in a 100 hour test at a fixed flow rate of 2 mL h−1 and a cell voltage of ∼0 V without external energy input (Fig. 2E). Compared with traditionally used commercial H2O2 produced by the anthraquinone process, the electrochemical in situ H2O2 production method for biomass refinery is safe for human and environmental health. The method eliminates the need for transporting and storing hazardous bulk H2O2. A long-term operation test was also conducted for direct conversion of lignin at a cell current density and H2SO4 flow rate of 40 mA cm−1 and 2 mL h−1, and the device operated stably and continuously output SCCAs with the concentration decreasing over time from ∼4200 ppm to ∼500 ppm during 50 hours of operation until the lignin could not be further oxidized (Fig. 2F).
Simulation of industrial production of short-chain carboxylic acids
To establish the industrial viability, we also used the reaction of water oxidation to O2 to release H+ instead of the HOR at the anode side using a commercial IrO2 electrode to simulate the conditions of no H2 supply in the industrial production process. 1 M H2SO4 solution was circulated at a flow rate of 30 mL h−1 on the anode side to reduce ionic resistance, and no SCCAs entered the H2SO4 solution circulated at the anode side due to the blocking of the CEM membrane. A high H2O2 production rate of 2.79 mmol cm−2 h−1 could be obtained at a cell voltage of 2.19 V in an O-CB//H2SO4//IrO2 cell (Fig. 3A and B). The selectivity for H2O2 production was always higher than 80% (Fig. 3B), which was just slightly lower than that achieved with the O-CB//H2SO4//Pt/C cell with pure H2 and O2. Considering the more stringent conditions, the use of air instead of O2 also enabled in situ synthesis of H2O2, albeit at a higher voltage than when pure O2 was introduced (Fig. 3C). As the flow rate of the H2SO4 solution that flowed through the cell decreased, the concentration of the output SCCAs increased significantly (Fig. 3D). A high concentration of SCCAs of 80.1 mg mL−1 could be observed at an H2SO4 flow rate of 0.6 mL h−1. A 50 hour stability test for continuous generation of SCCAs confirmed that lignin conversion could also be achieved when only air and H2SO4 solutions were applied (Fig. 3E). The concentration of SCCAs decreased from ∼3300 ppm to ∼600 ppm in this long-term test, which could also enable the direct conversion of lignin to SCCAs.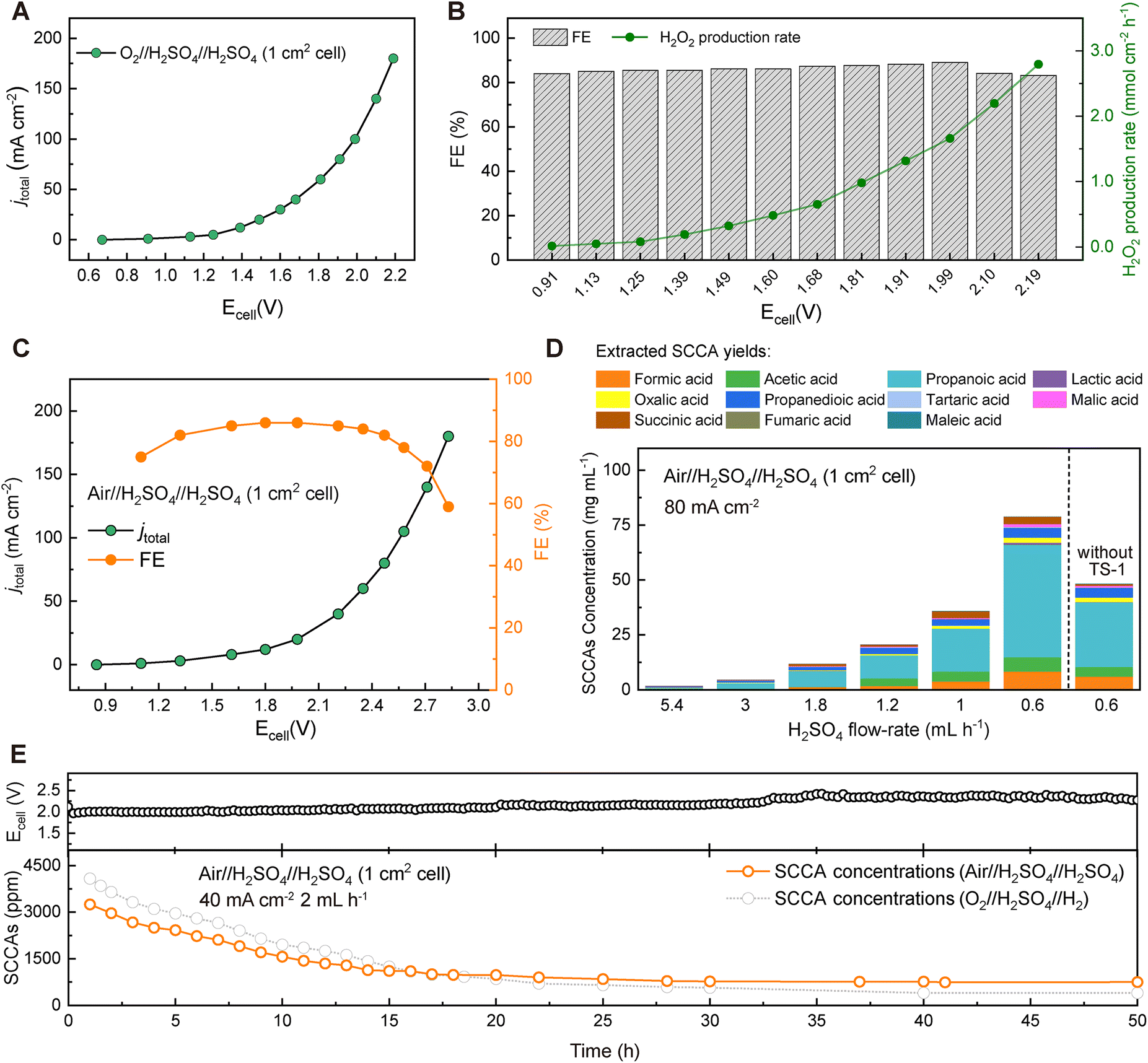 | ||
| Fig. 3 Direct electrochemical production of short-chain carboxylic acids (SCCAs) from lignin by coupling the 2e−-ORR and water oxidation. (A) I–V curve of the O-CB//H2SO4//IrO2 cell in which 1 M H2SO4 solution was circulated at the anode side (30 mL h−1) for improving the ionic conductivity, and H2O was oxidized to form O2 and protons. The flow rate of 1 M H2SO4 solution flowing through the cell was 60 mL h−1. (B) The FEs and H2O2 production rates at different voltage potentials of the O2//H2SO4//H2SO4 cell. (C) I–V curve and FEs of the O-CB//H2SO4//IrO2 cell when O2 was replaced with air. (D) SCCA concentrations at different H2SO4 flow rates flowing through the cell. A fixed current density of 80 mA cm−2 and temperature of 95 °C were chosen with air and 1 M H2SO4 solution as cathode and anode feedstock, respectively, in the O-CB//H2SO4//IrO2 cell. The SCCAs were collected at different H2SO4 flow rates flowing through the cell in the first hour. (E) Stability test for continuous production of SCCAs at a fixed current density of 40 mA cm−2 using the O-CB//H2SO4//IrO2 cell by the 2e−-ORR and water oxidation at the reaction temperature of 80 °C. | ||
Mechanism studies
The potential mechanism was investigated using guaiacol and 1,2-dimethoxy benzene as monomeric models, and several arylglycerol-β-aryl ether (β-O-4 structure) lignin models with guaiacyl (G) and/or syringyl (S) nuclei, erythro or threo stereostructures, with or without terminal hydroxyl groups as dimeric models, respectively. The synthetic route to the dimeric model compounds, the quantitative method of the erythro and threo stereoisomers, and the structures of model compounds used for mechanism studies are listed in the ESI (Fig. S7–S9†). TS-1-catalyzed H2O2 oxidation of monomeric models guaiacol and 1,2-dimethoxy benzene was found to generate different DCAs, mainly including malic acid, succinic acid, tartaric acid, etc., with total yields of 8.7 and 8.2 wt% at the conversion yields of 32 and 25 wt%, respectively. In the oxidation of the non-phenolic dimeric lignin model compounds, not only different MCAs (7.1 wt%) containing formic acid, acetic acid, and lactic acid but also different DCAs (20.5 wt%) containing propanedioic acid, tartaric acid, malic acid, succinic acid, fumaric acid and maleic acid were detected at the conversion yield of 80 wt%. In the oxidation of the phenolic dimeric lignin model compounds, different MCAs (11.1 wt%) and DCAs (18.5 wt%) were detected at the conversion yield of 95 wt%. A relatively high amount of propionic acid was detected only when the lignin dimeric model contained terminal phenolic hydroxyl groups (2.5 wt%). These results pointed out that MCAs were mainly derived from the side-chain oxidation of lignin, while DCAs were primarily derived from the oxidative ring-opening reaction of the aromatic rings, although oxalic acid, lactic acid, malonic acid, etc., may also be derived from the oxidation of aromatic rings. The structure of the lignin models with or without terminal hydroxyl groups affected the distribution of SCCA product yields, possibly due to the easy nucleophilic attack of the hydroxyl group by hydroxyl and superoxide radicals followed by the electron rearrangement on aromatic rings.Fig. 4 shows the 2D HSQC NMR spectra of industrial enzymatic hydrolysis lignin (typical industrial lignin isolated from corncob with S, G and H nuclei and C–O and C–C side-chain structures) before and after the direct conversion treatment of the long-term test in an O-CB//H2SO4//Pt/C cell. The aliphatic regions (top panels of Fig. 4A–C) revealed that 32.9% of the β-ether linkages were cleaved at 10 h, and all were cleaved during the 50 hour treatment. The phenomenon was also observed in phenylcoumaran (β-5 linkage) and spirodienone (β-1 linkage) structures. The aromatic regions (middle panels of Fig. 4A–C) indicated that the assignment of syringyl nuclei (S) decreased with the reaction of 10 h and disappeared at 50 h. The aromatic regions also revealed that the S unit was oxidized to the benzylic ketone analogue S’ unit, although the content of the S′ unit also gradually decreased during the continuous processing. Overall, this preliminary study demonstrates that the electrochemical in situ generation of H2O2 in combination with the TS-1 catalyzed direct conversion of lignin generates free radicals that can simultaneously nucleophilically attack the side chain and aromatic ring of lignin to achieve efficient conversion of lignin to SCCAs. At the same time, the polymerization of lignin occurred, which was manifested by the disappearance of G2 and G6 signals and the presence of the G5 signal. The content of p-coumaric acid (pCA), a typical structure in herbaceous plants, decreased significantly and became one of the main source structures of lignin oxidation to produce SCCAs, especially propionic acid.46,47 The carboxyl group content in the lignin residues was significantly increased to 2.52 mmol g−1 (10 h) and 2.74 mmol g−1 (50 h), from the 0.46 mmol g−1 of the raw corncob lignin measured by aqueous potentiometric titration using an automatic potentiometric titrator (809 Titrando, Metrohm Corp., Switzerland), which greatly broadened the feasibility of lignin applications.48,49
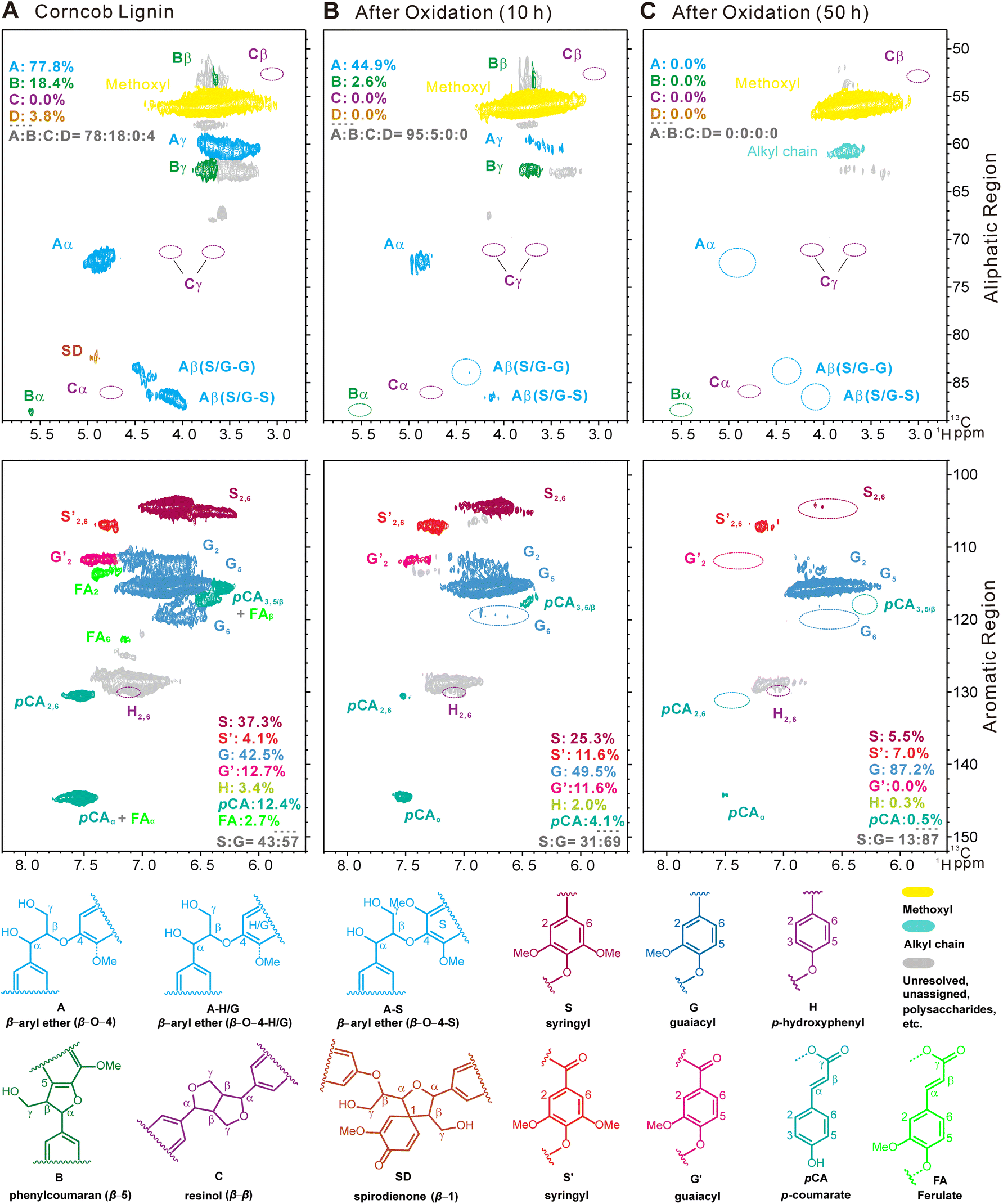 | ||
| Fig. 4 Partial 2D HSQC NMR spectra of an industrial enzymatic hydrolysis corncob lignin (in DMSO-d6) before and after electrochemical conversion. (A) Original enzymatic hydrolysis corncob lignin. (B) The residual lignin resulting from the electrochemical treatment for 10 h and (C) 50 h in an O2//H2SO4//H2 flow cell (1 cm2). The contour areas are color-coded to the corresponding structures and integrated to calculate the interunit linkage (aliphatic regions) and aromatic unit (aromatic regions) distributions. | ||
Conclusions
In summary, we have identified a highly effective electrochemical method for direct conversion of lignin to high concentration SCCAs by the in situ generated H2O2. Radicals derived from H2O2 can selectively attack the lignin side chain to break the conjugated C3-side structures and the lignin aromatic ring part to achieve a ring-opening reaction of the phenylpropane structure to generate the corresponding SCCAs (Fig. 5). The optimized catalyst system that simulated industrial conditions could perform using only air and H2SO4 solution as feedstocks and was potentially suitable for large-scale industrial applications. The prepared SCCAs could be utilized in organic reagents, esterification agents, plasticizers, preservatives, pharmaceutical intermediates, and other high-value applications. This approach could form a basic method for extended chemical transformations that depends on in situ generated H2O2 and TS-1.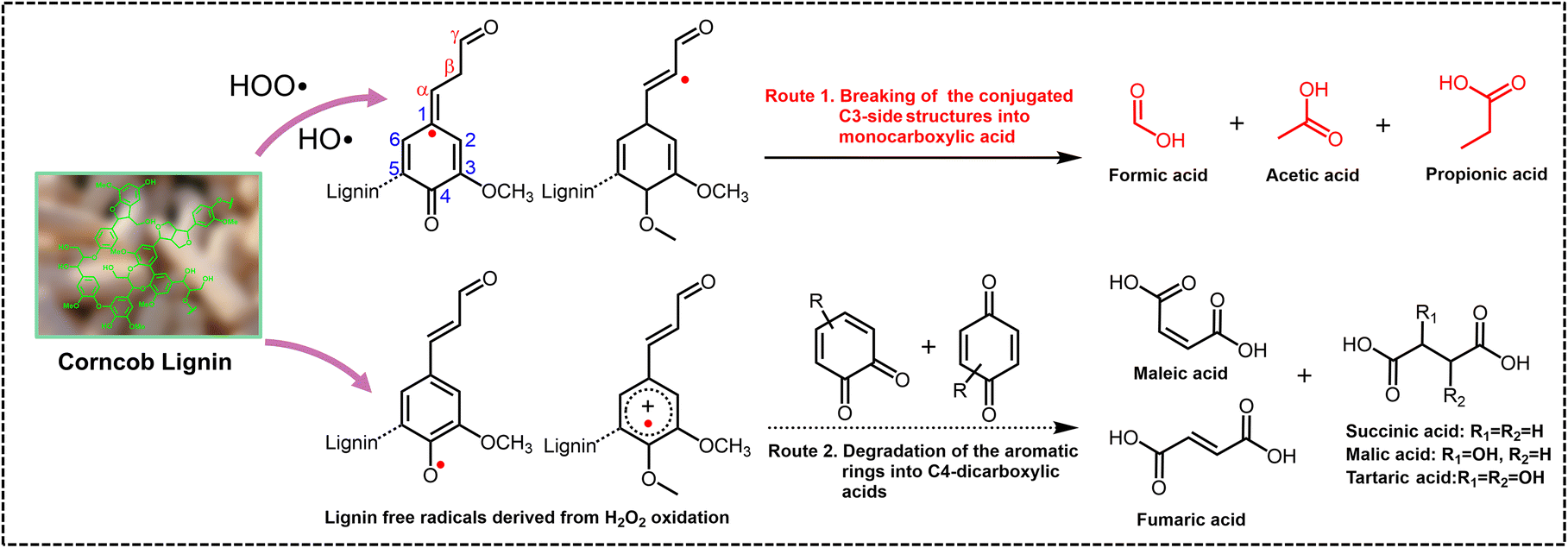 | ||
| Fig. 5 A schematic diagram of the proposed lignin oxidation mechanism by H2O2 to directly produce SCCAs. | ||
Experimental
Materials
The enzymatic hydrolysis lignin (EHL) derived from corncob was provided by Shandong Longlive Biotechnology Co., Ltd. The EHL was purified by sequential alkali dissolution and acid precipitation treatment before use. TS-1 was purchased from Rhawn Corporation. All commercially available chemicals and solvents except those mentioned were purchased from Macklin and Aladdin Corporation and used as received unless stated otherwise. Deionized H2O was used in all the experiments.Synthesis of oxygen-doped carbon black (O-CB) catalysts
Oxygen-doped carbon black (O-CB) catalysts were prepared as described in a previous report.50 In brief, 1.0 g of commercial carbon black (XC-72R, Cabot, USA) was added to 600 mL of 12 mol L−1 HNO3 and refluxed at 85 °C for 1 h, 3 h, 6 h, 9 h, and 12 h, respectively. After the slurries were cooled down to room temperature, they were centrifuged and washed with water until neutral pH values. Finally, the samples were dried at 70 °C in a vacuum oven. The functionalized carbon blacks were obtained with different surface oxygen doping levels.Rotation ring disk electrode (RRDE) test
The ORR activities of O-CB were measured using linear sweep voltammetry (LSV) with a rotation ring disk electrode (RRDE) in a typical three-electrode system (PINE Instrument, USA). To prepare the O-CB working electrode, 3.3 mg of O-CB was mixed with 10 μL of Nafion 117 (dissolved in 1 mL of ethanol) to obtain a homogeneous catalyst slurry by ultrasonication. 6 μL of the as-prepared catalyst slurry was pipetted on the rotating disk glassy carbon electrode with a 0.1 mg cm−2 mass loading. The O-CB cast electrode was vacuum dried before use. 0.1 M Na2SO4 was saturated with high-purity O2 as an electrolyte. A continuous flow of O2 was used as a shielding gas for the electrolyte. A platinum wire and Ag/AgCl electrode were used as the counter and reference electrodes, respectively. All the potentials against Ag/AgCl were calculated by converting to the reversible hydrogen electrode (RHE) using the equation E (vs. RHE) = E (vs. Ag/AgCl) + 0.197 V + 0.0591 × pH. The H2O2 selectivity was calculated using the following equation: H2O2 (%) = 200 × (Iring/N)/(Idisk + Iring/N), where Iring and Idisk are the ring and disk currents, respectively, and N is the ring collection efficiency, determined to be 0.32 in a solution of 2 mM K3Fe(CN)6 and 0.1 M KNO3.Electrochemical cell performance test
For the two-electrode cell for direct electrochemical production of H2O2, commercial Pt/C on a gas diffusion layer (GDL) (1 mg cm−2) was used as the anode, and around 0.5 mg cm−2 O-CB loaded on a GDL was prepared as the cathode. Typically, 10 mg of O-CB was mixed with 0.5 mL of Nafion 117 solution (5% diluted in ethanol), then sonicated and pipetted onto the GDL and vacuum dried before use. For the O2//H2SO4//H2 cell, 50 sccm of humidified O2 and H2 were introduced into the cathode and anode, respectively. A commercial IrO2 electrode was applied when the anode side was replaced with 1 M H2SO4 solution circulating at a flow rate of 30 mL h−1. The O2 gas was also replaced with air when pure O2 was not supplied for mimicking more severe H2O2 production conditions. All electrochemical tests were performed with Admiral Instruments Squidstat™ Plus and Donghua DH7000 electrochemical workstations. Industrial lignin (200 mg) and TS-1 (10 wt% based on lignin) were mixed and loaded on the three-compartment cell for the direct electrochemical conversion of lignin. In the experiments to prepare SCCAs by direct electrochemical oxidation of lignin at different temperatures and H2SO4 flow rates, the output SCCAs in the first hour were collected for quantification.Synthesis, characterization, and reaction of lignin model compounds
The dimeric compounds with β-aryl ether bonds were synthesized according to the method of Adler et al.51 The detailed synthesis route is shown in Fig. S7.† The structure of compound (2-(2-methoxyphenoxy)-1-(3,4,5-trimethoxyphenyl)propane-1,3-diol) was identified by NMR, and the content of each isomer was quantified by the method described in the previous paper.52,53 Degradation experiments of each model compound were performed with sealed bottles. The reaction conditions were as follows: 10 mg of the model compound, 30 mL of solvent containing 5 mL of 1,4-dioxane, 25 mL of 1 mol L−1 H2SO4 solution, and 1 mL of commercial H2O2 (30 wt%) at a reaction temperature of 95 °C.H2O2 and SCCA concentration quantification and faradaic efficiency (FE) calculations
In a two-electrode cell, the produced concentration of H2O2 was determined by redox titration with KI aqueous solution, according to the reported procedures.54,55 The faradaic efficiency (FE) for H2O2 production is calculated according to the following equation: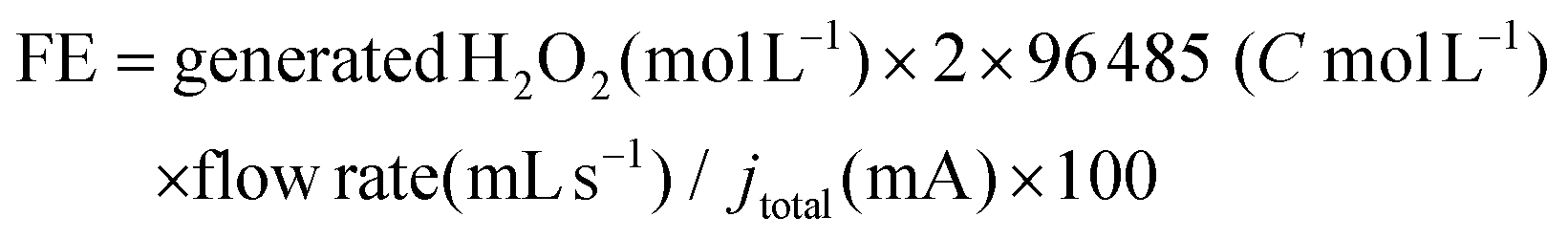
The quantification of SCCAs was conducted with a high-performance liquid chromatography instrument (HPLC, Agilent 1260 Infinity II) equipped with an ultraviolet-visible (UV-Vis, 1260 VWD) absorption detector at 210 nm. In HPLC analyses, an HPLC column, Ultimate AQ-C18 (length: 250 mm, inner diameter: 4.6 mm, and particle size: 5.0 μm, Welch, Shanghai, China), was used at an oven temperature of 30 °C with a solvent flow rate of 0.7 mL min−1. 20 mM dibasic sodium phosphate solvent with a pH of 2.60 adjusted using phosphoric acid containing 1% methanol was used as mobile phase A. Methanol was used as mobile phase B. The solvent system used for quantification of SCCAs was run at a gradient A/B (v/v) of 100/0 for 9 min, from 100/0 to 50/50 within 11 min, and held for 5 min and equilibrated for 5 min.
2D heteronuclear single quantum coherence (HSQC) NMR analysis
For 2D heteronuclear single quantum coherence (HSQC) NMR analysis, 80–90 mg lignin in 0.6 mL of dimethyl sulfoxide-d6 and 40 μL of chromium(III) acetylacetonate (0.01 M) were added to an NMR tube. The 2D HSQC NMR characterization of each lignin sample was performed on a Bruker AVANCE III HD 600 MHz spectrometer equipped with a 5 mm BB probe. Sufficient amounts of scans consisting of 4096 by 256 points were collected.Author contributions
S. Sun: conceptualization, formal analysis, methodology, writing – original draft, writing – review & editing. X. Qiu: funding acquisition, writing – review & editing. S. Hao: data curation, formal analysis, investigation, writing – review & editing. S. Ravichandran: data curation, investigation. W. Zhang: conceptualization, funding acquisition, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (22208061, 22108044), the China Postdoctoral Science Foundation (2022M710828), the Research and Development Program in Key Fields of Guangdong Province (2020B1111380002), the Basic Research and Applicable Basic Research in Guangzhou City (202201010290), and the Guangdong Provincial Key Laboratory of Plant Resources Biorefinery (2021GDKLPRB07).References
- S. H. Hoseinifar, Y.-Z. Sun and C. M. Caipang, Aquacult. Res., 2017, 48, 1380–1391 CrossRef CAS.
- G. V. Polycarpo, I. Andretta, M. Kipper, V. C. Cruz-Polycarpo, J. C. Dadalt, P. H. M. Rodrigues and R. Albuquerque, Poult. Sci., 2017, 96, 3645–3653 CrossRef CAS PubMed.
- M. Zhou, J. Zhou, M. Tan, J. Du, B. Yan, J. W. C. Wong and Y. Zhang, Bioresour. Technol., 2017, 245, 44–51 CrossRef CAS PubMed.
- D. R. Dodds and R. A. Gross, Science, 2007, 318, 1250–1251 CrossRef CAS PubMed.
- K. Zhang, Z. Zhan, M. Zhu, H. Lai, X. He, W. Deng, Q. Zhang and Y. Wang, J. Energy Chem., 2023, 80, 58–67 CrossRef CAS.
- B. Liu, Z. Zheng, Y. Liu, M. Zhang, Y. Wang, Y. Wan and K. Yan, J. Energy Chem., 2023, 78, 412–421 CrossRef CAS.
- E. Subbotina, T. Rukkijakan, M. D. Marquez-Medina, X. Yu, M. Johnsson and J. S. M. Samec, Nat. Chem., 2021, 13, 1118–1125 CrossRef CAS PubMed.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- R. Lou, S. Wu and G. Lyu, J. Anal. Appl. Pyrolysis, 2015, 111, 27–32 CrossRef CAS.
- S. Kang, X. Li, J. Fan and J. Chang, Renewable Sustainable Energy Rev., 2013, 27, 546–558 CrossRef CAS.
- S. Shimizu, T. Yokoyama, T. Akiyama and Y. Matsumoto, J. Agric. Food Chem., 2012, 60, 6471–6476 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- A. Imai, T. Yokoyama, Y. Matsumoto and G. Meshitsuka, J. Agric. Food Chem., 2007, 55, 9043–9046 CrossRef CAS PubMed.
- S.-I. Kwon and A. J. Anderson, Curr. Microbiol., 2001, 42, 8–11 CrossRef CAS PubMed.
- G. Bensidhom, M. Arabiourrutia, A. Ben Hassen Trabelsi, M. Cortazar, S. Ceylan and M. Olazar, J. Energy Inst., 2021, 99, 229–239 CrossRef CAS.
- J.-M. Yuan, H. Li, L.-P. Xiao, T.-P. Wang, W.-F. Ren, Q. Lu and R.-C. Sun, Fuel, 2022, 319, 123758 CrossRef CAS.
- T. Yokoyama, Y. Matsumoto and G. Meshitsuka, J. Wood Chem. Technol., 1999, 19, 187–202 CrossRef CAS.
- W. Lan, M. T. Amiri, C. M. Hunston and J. S. Luterbacher, Angew. Chem., Int. Ed., 2018, 57, 1356–1360 CrossRef CAS PubMed.
- T. Yokoyama, Y. Matsumoto and G. Meshitsuka, Holzforschung, 2005, 59, 269–275 CAS.
- C. Yang, S. Maldonado and C. R. J. Stephenson, ACS Catal., 2021, 11, 10104–10114 CrossRef CAS.
- W. Lan, J. B. de Bueren and J. S. Luterbacher, Angew. Chem., Int. Ed., 2019, 58, 2649–2654 CrossRef CAS PubMed.
- Z. Cai, J. Long, Y. Li, L. Ye, B. Yin, L. J. France, J. Dong, L. Zheng, H. He, S. Liu, S. C. E. Tsang and X. Li, Chem, 2019, 5, 2365–2377 CAS.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- Z. Bi, Z. Li and L. Yan, Green Process. Synth., 2018, 7, 306–315 CAS.
- C. A. Vega-Aguilar, C. Costa, M. F. Barreiro and A. E. Rodrigues, Ind. Eng. Chem. Res., 2022, 61, 3570–3581 CrossRef CAS PubMed.
- C. A. Vega-Aguilar, M. F. Barreiro and A. E. Rodrigues, Ind. Eng. Chem. Res., 2021, 60, 3543–3553 CrossRef CAS.
- X. Li and Y. Zhang, ChemSusChem, 2020, 13, 1740–1745 CrossRef CAS PubMed.
- J. Gierer, Holzforschung, 1997, 51, 34–46 CrossRef CAS.
- J. Gierer and F. Imsgard, Sven. Papperstidn., 1977, 80, 510–518 CAS.
- C. A. Vega-Aguilar, M. F. Barreiro and A. E. Rodrigues, Chem. Eng. Res. Des., 2020, 159, 115–124 CrossRef CAS.
- D. Zhang, B. Sun, L. Duan, Y. Tao, A. Xu and X. Li, Chem. Eng. Technol., 2016, 39, 97–101 CrossRef.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- Y. Yi, L. Wang, G. Li and H. Guo, Catal. Sci. Technol., 2016, 6, 1593–1610 RSC.
- R. J. Lewis and G. J. Hutchings, ChemCatChem, 2019, 11, 298–308 CrossRef CAS.
- J. K. Edwards and G. J. Hutchings, Angew. Chem., Int. Ed., 2008, 47, 9192–9198 CrossRef CAS PubMed.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley, A. Y. Borisevich, C. J. Kiely and G. J. Hutchings, Science, 2016, 351, 965–968 CrossRef CAS PubMed.
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. E. Davies, D. J. Morgan, L. Chen, J. Qi, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, Y. Yamamoto and G. J. Hutchings, Science, 2022, 376, 615–620 CrossRef CAS PubMed.
- J. Su, L. Yang, R. N. Liu and H. Lin, Chin. J. Catal., 2014, 35, 622–630 CrossRef CAS.
- Y. Xia, X. Zhao, C. Xia, Z.-Y. Wu, P. Zhu, J. Y. Kim, X. Bai, G. Gao, Y. Hu, J. Zhong, Y. Liu and H. Wang, Nat. Commun., 2021, 12, 4225 CrossRef CAS PubMed.
- A. J. Plomp, D. S. Su, K. P. d. Jong and J. H. Bitter, J. Phys. Chem. C, 2009, 113, 9865–9869 CrossRef CAS.
- K. A. Wepasnick, B. A. Smith, J. L. Bitter and D. Howard Fairbrother, Anal. Bioanal. Chem., 2010, 396, 1003–1014 CrossRef CAS PubMed.
- R. J. Lewis, K. Ueura, Y. Fukuta, T. E. Davies, D. J. Morgan, C. B. Paris, J. Singleton, J. K. Edwards, S. J. Freakley, Y. Yamamoto and G. J. Hutchings, Green Chem., 2022, 24, 9496–9507 RSC.
- G. Wu, Z. Lin, L. Li, L. Zhang, Y. Hong, W. Wang, C. Chen, Y. Jiang and X. Yan, Chem. Eng. J., 2017, 320, 1–10 CrossRef CAS.
- Q. Chen and E. J. Beckman, Green Chem., 2007, 9, 802–808 RSC.
- B. Wang, X. Peng, W. Zhang, M. Lin, B. Zhu, W. Liao, X. Guo and X. Shu, Catal. Commun., 2017, 101, 26–30 CrossRef CAS.
- C. Lapierre, A. Voxeur, S. D. Karlen, R. F. Helm and J. Ralph, J. Agric. Food Chem., 2018, 66, 5418–5424 CrossRef CAS PubMed.
- V. I. Timokhin, M. Regner, A. H. Motagamwala, C. Sener, S. D. Karlen, J. A. Dumesic and J. Ralph, ACS Sustainable Chem. Eng., 2020, 8, 17427–17438 CrossRef CAS.
- J. Wang, Y. Deng, Y. Qian, X. Qiu, Y. Ren and D. Yang, Green Chem., 2016, 18, 695–699 RSC.
- W. He, W. Gao and P. Fatehi, ACS Sustainable Chem. Eng., 2017, 5, 10597–10605 CrossRef CAS.
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 226–231 CrossRef CAS PubMed.
- E. Adler, B. O. Lindgren and U. Saeden, Sven. Papperstidn., 1952, 55, 245–254 CAS.
- T. Akiyama, T. Sugimoto, Y. Matsumoto and G. Meshitsuka, J. Wood Sci., 2002, 48, 210–215 CrossRef CAS.
- T. Akiyama, H. Goto, D. S. Nawawi, W. Syafii, Y. Matsumoto and G. Meshitsuka, Holzforschung, 2005, 59, 276–281 CAS.
- X. Zhou, F. Yan, A. Lyubartsev, B. Shen, J. Zhai, J. C. Conesa and N. Hedin, Adv. Sci., 2022, 9, 2105792 CrossRef CAS PubMed.
- X. Zhou, B. Shen, J. Zhai and N. Hedin, Adv. Funct. Mater., 2021, 31, 2009594 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00324h |
| This journal is © The Royal Society of Chemistry 2023 |