
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A versatile non-fouling multi-step flow reactor platform: demonstration for partial oxidation synthesis of iron oxide nanoparticles†
Maximilian O.
Besenhard
 *a,
Sayan
Pal
a,
Liudmyla
Storozhuk
b,
Simon
Dawes
a,
Nguyen Thi Kim
Thanh
bc,
Laura
Norfolk
d,
Sarah
Staniland
d and
Asterios
Gavriilidis
*a
*a,
Sayan
Pal
a,
Liudmyla
Storozhuk
b,
Simon
Dawes
a,
Nguyen Thi Kim
Thanh
bc,
Laura
Norfolk
d,
Sarah
Staniland
d and
Asterios
Gavriilidis
*a
aDepartment of Chemical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK. E-mail: m.besenhard@ucl.ac.uk; a.gavriilidis@ucl.ac.uk
bBiophysics Group, Department of Physics and Astronomy, University College London, Gower Street, London, WC1E 6BT, UK
cUCL Healthcare Biomagnetics and Nanomaterials Laboratories, University College London, 21 Albemarle Street, London W1S 4BS, UK
dDepartment of Chemistry, The University of Sheffield, Dainton Building, Brook Hill, Sheffield, S3 7HF, UK
First published on 24th November 2022
Abstract
In the last decade flow reactors for material synthesis were firmly established, demonstrating advantageous operating conditions, reproducible and scalable production via continuous operation, as well as high-throughput screening of synthetic conditions. Reactor fouling, however, often restricts flow chemistry and the common fouling prevention via segmented flow comes at the cost of inflexibility. Often, the difficulty of feeding reagents into liquid segments (droplets or slugs) constrains flow syntheses using segmented flow to simple synthetic protocols with a single reagent addition step prior or during segmentation. Hence, the translation of fouling prone syntheses requiring multiple reagent addition steps into flow remains challenging. This work presents a modular flow reactor platform overcoming this bottleneck by fully exploiting the potential of three-phase (gas–liquid–liquid) segmented flow to supply reagents after segmentation, hence facilitating fouling free multi-step flow syntheses. The reactor design and materials selection address the operation challenges inherent to gas–liquid–liquid flow and reagent addition into segments allowing for a wide range of flow rates, flow ratios, temperatures, and use of continuous phases (no perfluorinated solvents needed). This “Lego®-like” reactor platform comprises elements for three-phase segmentation and sequential reagent addition into fluid segments, as well as temperature-controlled residence time modules that offer the flexibility required to translate even complex nanomaterial synthesis protocols to flow. To demonstrate the platform's versatility, we chose a fouling prone multi-step synthesis, i.e., a water-based partial oxidation synthesis of iron oxide nanoparticles. This synthesis required I) the precipitation of ferrous hydroxides, II) the addition of an oxidation agent, III) a temperature treatment to initiate magnetite/maghemite formation, and IV) the addition of citric acid to increase the colloidal stability. The platform facilitated the synthesis of colloidally stable magnetic nanoparticles reproducibly at well-controlled synthetic conditions and prevented fouling using heptane as continuous phase. The biocompatible particles showed excellent heating abilities in alternating magnetic fields (ILP values >3 nH m2 kgFe−1), hence, their potential for magnetic hyperthermia cancer treatment. The platform allowed for long term operation, as well as screening of synthetic conditions to tune particle properties. This was demonstrated via the addition of tetraethylenepentamine, confirming its potential to control particle morphology. Such a versatile reactor platform makes it possible to translate even complex syntheses into flow, opening up new opportunities for material synthesis.
Introduction
Flow chemistry approaches to synthesise advanced materials became (and remained) popular over the last decade. The interest in flow reactors is mostly due to 1) the advantageous synthetic conditions made possible, e.g., by fast mixing, improved heat and mass transfer, precise temperature control, and precisely timed reagent addition; 2) their inherent continuous operation mode, providing opportunities for large-scale production; 3) the reduction of manual steps reducing the operator effect on reproducibility.1–7 Their ease of automation is also why flow reactors are in demand for high-throughput screening as well as self-optimising reactor platforms.8–13 Not only for fine chemical, but also polymer as well as micro and nanoparticle synthesis, such platforms combining flow reactors, intelligent algorithms and real-time material characterisation can pave the way to fully computer aided chemical science tuning molecular or particle properties.14–19The commonly used flow reactors which were developed for wet-chemical syntheses of small and large molecules, however, are often unsuitable for fouling prone syntheses, and nano or micro particle syntheses in particular. This is because (nano) particle formation, and any formation of solid matter from solution, is likely to foul the reactor walls. It is worth mentioning that fouling is not only a problem for wet-chemical processes forming solid matter deliberately. Less commonly reported is that many synthetic procedures for molecules (especially >500 kDa), can form intermediates or side products of different solubility that are likely to solidify and foul the reactor.
The common premise of flow-chemistry for large scale production and high-throughput-screening is that after altering synthetic conditions (e.g., by updating flow rates or temperatures), steady state operation is established, which (strictly speaking) does not exist for fouling reactors. Accumulations of reagents, products or any kind of intermediates at the reactor wall are likely to impede the synthesis, i.e., change the synthetic conditions over time. This could either be due to affected heat or mass transfer rates, inconsistent conversion, or altered reaction pathways due to reactions involving the deposited material. The inherently high (reactor wall) surface area-to-volume ratio of the commonly used microfluidic and millifluidic flow reactors makes them inherently vulnerable to such changes. Therefore, versatile non-fouling flow reactors are vital for the development and production of advanced (solid) materials, and in particular, for high-throughput screening or self-optimising synthetic procedures in the rapidly emerging field of digital chemistry.
Single phase flow reactors (as most commonly used for molecular synthesis) have been used for (nano) particle synthesis without fouling (e.g., using special reactor designs working for rapid particle formation,20,21 electrostatic repulsion by the wall material by tuning pH values,22,23 or acoustophoretic focusing – demonstrated for microparticles24). Nevertheless, the success of single-phase systems is the exception. Most non-fouling flow reactor concepts for nanoparticle synthesis (which is the focus in the following) use liquid–liquid segmentation, i.e., two immiscible liquids where the reactive (dispersed) phase is compartmentalised and shielded from the wall by the continuous phase.25 Which of the immiscible liquids becomes the continuous phase is determined by the preferential wetting of the wall. Hence, aqueous chemistry can be made non-fouling in plastic capillaries with a non/less-polar liquid preventing interactions with the reactor wall.
In addition, the secondary flows in droplets (dispersed phase is spherical) or slugs (dispersed phase extends to the reactor wall – without contact) can improve mixing, especially at low flow rates, where single phase modules suffer from high axial dispersion and diffusion limited mass transfer. Although gas–liquid segmentation also improves mixing and reduces axial dispersion, it does not prevent fouling. The gas is the dispersed phase and the reactive liquid phase remains in contact with the wall. This is why liquid–liquid segmented flow reactors (here including droplet microfluidics)26–28 facilitate robust long term operation making it key for large-scale production and high-throughput screening of nanoparticle synthesis.
A significant drawback of liquid–liquid segmentation is the lack of versatility. The difficulty of feeding new reagent solution to the reactive phase after segmentation, limits flow syntheses using liquid–liquid segmentation to simple synthetic protocols with a single reagent addition step. In some cases this limitation can be circumvented by providing all reagents before segmentation (possible if fouling is not expected/is minimal before segmentation)29–31 or reagent mixing where segmentation happens.32–36 Nevertheless, full flexibility to translate nanoparticle synthesis into flow requires novel non-fouling flow reactors facilitating multiple reagent addition steps when/wherever needed.
The potential of gas–liquid–liquid segmentation to achieve this was demonstrated first by Nightingale et al.37 The authors showed that feeding an inert gas (which constitutes a 3rd phase) helps to space droplets/slugs uniformly and suppress new droplet formation when feeding new reagent solution to previously formed liquid droplets/slugs. This work showed that gas–liquid–liquid segmentation can be an effective method to repeatedly add controlled quantities of reagent post segmentation and showcased its versatility for a multistep quantum dot synthesis at low flow rates (<200 μl min−1). Three-phase segmented flow was generated in a customised polytetrafluoroethylene (PTFE) element and subsequent reagent addition into the liquid droplets/slugs formed was facilitated through a fused silica tubing piercing a capillary for subsequent reagent addition into the liquid droplets formed.
The potential for flow rates of several ml min−1 was outlined by Nightingale et al.37 and confirmed later by Wong et al.38 showing a larger scale flow synthesis of palladium nanoparticles at ∼5 ml min−1 (∼10 litre per day) using a single addition step in a reactor made from off-the-shelf parts. The same group studied the scalability of gas–liquid–liquid segmented flow,39,40 and demonstrated stable operation (not considering reagent addition into droplets) at channel diameters up to ∼3 mm. At larger diameters the Bond number (Bo), which represents the ratio of hydrostatic and Laplace pressure, exceeds unity and gravitational forces become dominant, causing de-wetting and breakage of the organic film surrounding the gas bubbles. From studies on gas–liquid segmented flow, this film thickness is known to depend on the capillary number (Ca), i.e., the ratio of viscous drag and capillary forces across the gas–liquid interface. Hence, the flow rates and fluid properties affect the film thickness, which increases with higher Ca.41,42
More recently, Abdel-Latif et al. showed how three-phase flow reactors facilitate a robust two stage quantum dot nanoparticle synthesis that can be operated autonomously to tune particle/optical properties via robotic experimentation.43 Their set-up comprised a customised polyether ether ketone (PEEK) element for gas–liquid–liquid segmentation and a standard T-connector for subsequent reagent addition. Using a similar flow reactor, Volk et al. demonstrated that multiphase flow reactors can be used to disperse two reactive phases, showing four-phase segmented flow reactors for ligand exchange of quantum dots.44
Although gas–liquid–liquid flow reactors demonstrated sufficient flexibility to translate complex multi-step syntheses into flow without fouling, their applications remain rare. The reason is the challenges regarding the operation stability for medium flow rates (0.1–5 ml min−1), different residence times (0.1–10 min) between reagent addition steps, varying flow rate ratios (dispersed phase/new reagent solution), temperature ramps, reactions with rapid particle formation, and not least, the start-up procedure.
This work presents a modular three-phase “Lego®-like” reactor platform which was specifically designed for gas–liquid–liquid segmented flow to overcome these obstacles, hence offering a new powerful tool for material flow syntheses. The platform includes customised reactor elements made of hydrophobic plastics (significantly improving the robustness of three-phase segmented flow operations) and residence time modules which allowed to tune the droplet/slug sizes, the residence time between reagent addition steps and temperature, and to better monitor three-phase segmentation and subsequent reagent addition steps. The versatility of this platform was demonstrated for a complex multistep nanoparticle synthesis, i.e., partial oxidation synthesis of iron oxide nanoparticles (IONPs), requiring medium residence times (1–5 min) at elevated temperatures (70 °C), operation, which is also prone to fouling as particles and intermediates form rapidly. The platform made it possible to reproducibly and continuously synthesise colloidally stable magnetic nanoparticles using heptane as continuous phase (no need for perfluorinated solvents) and to conveniently explore or screen the synthetic conditions, which was shown by varying additive concentrations to control particle morphology.
Development of modular three-phase flow reactors
Three-phase reactor platform
The modular three-phase reactor platform comprised elements for droplet formation (droplet generator; note that we don't distinguish between droplets and slugs in the following) and for reagent addition into droplets (T-units), stands to arrange these elements neatly, as well as temperature-controlled residence time modules, that consist of perfluoroalkoxy alkanes (PFA) tubing, 1 mm inner diameter (ID) coiled around a metal stand immersed in a heated water bath to maintain the reaction temperature set.The droplet generator is where the three phases (gas, and the dispersed and continuous liquid phase, i.e., the aqueous solution and heptane) meet and was made of highly transparent polycarbonate for better visibility (see Fig. 1a) as well as hydrophobic fluorinated ethylene propylene (FEP). The inlet channels for the continuous and dispersed phase (1 mm ID) were placed around a central metal nozzle feeding the gas phase (1 mm outer diameter (OD) and 0.8 mm ID). This nozzle was adjustable in order to control the cross section at the droplet generator's outlet channel (see Fig. 1b), following recent concepts for bubble size control in Taylor flow.45 The cross section limits the extension of gas bubbles, and with it, the bubble size,45,46 hence, it controls the spacing between aqueous droplets which is crucial for robust reagent addition downstream. Three-phase segmentation was completed (gas and aqueous phase were fully dispersed in the organic/continuous phase) just after a gas bubble formed when using the hydrophobic FEP droplet generator. In the less hydrophobic polycarbonate element, segmentation was completed in the PFA tubing (see Fig. 1c and the video Droplet_generation_video provided in the ESI†).
 | ||
| Fig. 1 (a) Schematic of droplet generator for three-phase segmentation with (b) adjustable gas nozzle to control the gas bubble size. (c) Three-phase droplet generator made of transparent polycarbonate in operation (1st aqueous stream with blue dye) and 3 steps of the droplet formation (see Fig. S1† for more details). (d) Schematic of T-unit for reagent addition into aqueous droplets. (e) T-Unit made of FEP in operation showing 4 addition steps (2nd aqueous stream with reddish dye, see Fig. S2 and S3† for more details). (f) Stand with 2 columns allowing to slot and mount the reactor elements. | ||
The T-units for reagent addition into droplets (see Fig. 1d) were also made from FEP with a main channel (for the three-phase flow to pass) of 1 mm ID to match the ID of the tubing used. The channel for reagent addition (2nd aqueous stream) had to be adapted to the feed rates to avoid, i) the formation of new droplets at high feed rates (a small diameter yields higher velocities which can cause the gas bubbles to break) or ii) flow instabilities at low flow rates (larger diameters can destabilise the three-phase segmentation in the main channel). The T-units used in this study had a 0.6–0.7 mm reagent addition channel. For better visibility of the reagent addition step, the T-units were thinned out alongside the main channel (see Fig. 1e and the videos Droplet_addition_1 and Droplet_addition_2 provided in the ESI†).
It is important to highlight that robust operation was achieved using heptane as continuous phase and not the commonly used fluorinated oils. This is a significant improvement in terms of cost, waste management, and safety for which the use of hydrophobic T-units was crucial. Adding reagent solution into droplets of three-phase segmented flow was possible using less hydrophobic polycarbonate and polyacrylic T-units (especially at higher continuous phase flow rates), but operations were more vulnerable. Once the aqueous phase came in contact with the less hydrophobic wall material, it could not be removed fully and the residues interrupted the three-phase segmentation (see Fig. S4† showing unsuccessful addition using a polycarbonate T-unit).
The T-units' customised channel dimensions and the hydrophobic FEP were crucial to robustly add reagent solution into the aqueous droplets at 1st![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2nd aqueous stream flow rate ratios of up to 1:1 (see the videos Droplet_addition_varying_feedrates, Droplet_addition_feedrates_lower, and Droplet_addition_feedrates_higher provided in the ESI†). This almost doubling of the dispersed phase volume in a single reagent addition step is remarkable, considering the batch equivalent of rapidly doubling the vessel liquid volume. Furthermore, the platform allowed a large range of the total flow rate, showing robust reagent addition into droplets for aqueous flow rate ratios < 1:1 at all flow rates tested (1st aqueous stream 0.2–0.8 ml min−1, see ESI† section 2.2).
:2nd aqueous stream flow rate ratios of up to 1:1 (see the videos Droplet_addition_varying_feedrates, Droplet_addition_feedrates_lower, and Droplet_addition_feedrates_higher provided in the ESI†). This almost doubling of the dispersed phase volume in a single reagent addition step is remarkable, considering the batch equivalent of rapidly doubling the vessel liquid volume. Furthermore, the platform allowed a large range of the total flow rate, showing robust reagent addition into droplets for aqueous flow rate ratios < 1:1 at all flow rates tested (1st aqueous stream 0.2–0.8 ml min−1, see ESI† section 2.2).
All inlets and outlets ports of the platform elements had a 1/4 (inch)-28 (threads per inch) flat bottom thread to match standard fluidic connectors. When connected to 1 mm ID (1.56 mm OD) PFA tubing via flangeless ferrules, the three-phase segmentation showed no disruptions. To arrange the set-ups neatly, all elements could be slotted into customized stands (see Fig. 1f).
All reactive aqueous solutions and heptane, i.e., the inert continuous phase used, were fed using syringe pumps equipped with 50 ml gas-tight glass syringes allowing for long operation times. Nitrogen gas was fed via a mass flow controller (not a pressure controller) to maintain the gas flow rate set when the system pressures fluctuated, for example during the start-up period before reaching steady state operation.
The details of the platform components, the materials and equipment used are provided in the ESI† (see ESI section 2.1) together with details on the manufacturing of the droplet generators and T-units, images of the platform elements and the set-up in operation (see ESI† section 2.2 and the videos Platform_operation_1 and Platform_operation_2 provided in the ESI†), studies of the flow rate range (see Fig. S6–S8†), as well as detailed recommendations for operation procedures (see ESI† section 2.3).
Nanoparticle chemistry
IONPs were synthesised via a partial oxidation synthesis (also known as oxidative precipitation or hydrolysis of Fe2+ or oxidation of ferrous hydroxide).47–49 It is an alternative water-based method to the common co-precipitation methods, where ferric (Fe3+) and ferrous (Fe2+) salts are precipitated simultaneously (most likely forming the final iron oxide phase via complex phase transitions);50,51 hence, suffering from limited shape and particle size control, especially for particles >10 nm (required for many biomedical applications).52–55 Partial oxidation syntheses are known to yield larger IONPs (usually >30 nm).49,56 They are initiated by precipitating a ferrous salt via base addition to form a ferrous hydroxide (Fe2+), which is then oxidised by an oxidation agent (Fe2+ partially oxidises to Fe3+), most commonly a nitrate,49 at elevated temperatures (60–100 °C). This multistep synthesis offers additional flexibility to control the magnetite (Fe3O4)/maghemite (γ-Fe2O3) formation. Therefore, partial oxidation syntheses were used to modify the IONP size and morphology via proteins found in magneto-tactic bacteria.57,58 Recent studies demonstrated that instead of proteins also inexpensive additives, such as tetraethylenepentamine (TEPA), can be used to control IONP size and morphology.59 This could provide another control handle to fine-tune the properties of >10 nm IONPs.Despite being a biocompatible water-based synthesis, the poor colloidal stability of IONPs obtained from classical partial oxidation protocols limits their use for biomedical applications. However, some studies demonstrated that the colloidal stability can be improved by adding ethanol and sulphuric or polymers, such as polyethylenimine to the reagent solutions.60–62 Still, reports on partial oxidation control via additives are not always consistent and are hard to compare due to varying procedures. Hence, systematic studies covering a broad synthesis parameter space are required to identify the critical parameters.
Characterisation
The particle diameter and the hydrodynamic diameter of the synthesised IONPs were obtained using transmission electron microscopy (TEM) with a JEOL 1200 EX, and dynamic light scattering (DLS) using a Beckman Coulter DelsaMax-Pro. The IONP crystal structure was identified by X-ray diffraction (XRD) using a Malvern Instruments PANalytical X'Pert3 equipped with a CoKα radiation source. The heating potential of the particles in an alternating magnetic field was determined at a frequency of 488 kHz and a field strength of 308 Oe (= 25 kA m−1). Details of these procedures have been described previously.63,64 The particle iron concentration, i.e., the concentration of Fe in the form of particles (mgFe–IONP mlsol−1) was obtained via microwave plasma atomic emission spectroscopy using an Agilent 4210 MP-AES. For the analysis, IONP solutions of known volume were washed with de-ionised (DI) water (resistivity ≥ 15 MΩ cm−1) and decanted magnetically. After subsequent digestion in aqua regia, the samples were diluted with DI water (250 fold) to match the instrument sensitivity range. Assuming all Fe from the precursor (mgFe–FeSO4 mlsol−1, concentration known) converted to magnetic IONPs (mgFe–IONP mlsol−1, concentration measured), the conversion was defined as mgFe–IONP mlFe–FeSO4−1. Images and videos (recorded at 240 fps) showing the droplet formation and reagent addition into droplets using blue and red aqueous solutions were taken on a LED backlight with an antiglare diffuser.Fouling free partial oxidation flow synthesis
Initial batch studies
Common partial oxidation batch protocols report hours-long oxidation at elevated temperatures. As this corresponds to residence times that limit the flow reactor throughout, the particle formation kinetics were studied in batch prior designing the reactor. The colour change of the synthesis solution and magnetic separability of particles formed at different temperatures (tested using a hand-held magnet) showed that magnetite/maghemite formed within 3–8 min at 70 °C (see ESI† section 3.1 and Fig. S9†). Hence, residence times of several minutes can be sufficient for partial oxidation flow syntheses, not least due the rapid heating.Previous studies on co-precipitation syntheses showed how adding a citric acid solution after magnetite/maghemite formation yields highly colloidally stable IONPs.22,63 Following the same concept of incrementally increasing the citric acid concentration did improve the colloidal stability. However, IONPs dissolved before achieving colloidal stability (here defined by no sign of IONP sedimentation within a day). Moreover, the timing of citric acid addition was studied and shown to be crucial. These observations together with TEM and XRD analysis (confirming the synthesis of magnetite/maghemite) indicate that the colloidal stability improves when adding citric acid just after magnetite/maghemite formation (see ESI† section 3.2 and Fig. S10 and S11†).
Flow synthesis
Learning from the initial batch studies (see ESI† section 3), the partial oxidation synthesis was translated to flow as follows. The base and oxidation agent were combined in one solution and sulphuric acid was added to the precursor solution for a better stability against oxidation and to increase the colloidal stability. Also, the concentrations were adjusted to bring flow rates to a similar order of magnitude. All solutions were prepared fresh for each synthesis using water kept under N2 purging before use. Table S2† lists the details of all chemicals used including product numbers.
Fig. 2 outlines the partial oxidation flow synthesis in the three-phase reactor platform. The 1st aqueous solution contained the base and oxidation agent (0.125 M KOH, 0.625 M KNO3), the 2nd aqueous solution the precursor (0.1 M FeSO4, 0.005 M H2SO4) and the 3rd aqueous solution the stabiliser (0.3 M citric acid solution). Three-phase segmentation in the FEP droplet generator formed droplets of the 1st aqueous solution (0.4 ml min−1) separated by nitrogen gas bubbles (0.4 mln min−1), both shielded from the wall by heptane (0.15 ml min−1). The 1st droplet reagent addition in a T-unit added the 2nd aqueous solution (0.2 ml min−1) to the droplets, initiating the ferrous hydroxide precipitation. Oxidation happened in the subsequent temperature controlled (70 °C) residence time module with 8 m of tubing (between the 1st and 2nd droplet reagent addition) resulting in a ∼5 min residence time accounting for the gas expansion at the flow rates and temperatures used. Just after this oxidation step which formed magnetite/maghemite, the 2nd droplet reagent addition in a T-unit added the 3rd aqueous solution (0.2 ml min−1) into the droplets containing now the black IONP solution. The samples were subsequently collected into vials, where the nitrogen bubbles escaped. The heptane formed a thin film at the surface (aqueous phases:heptane ≥ 0.6:0.15) which could be removed easily via a pipette. Samples were collected only during steady state operation. Therefore, sample collection was paused for at least two residence times after any change in flow rates.
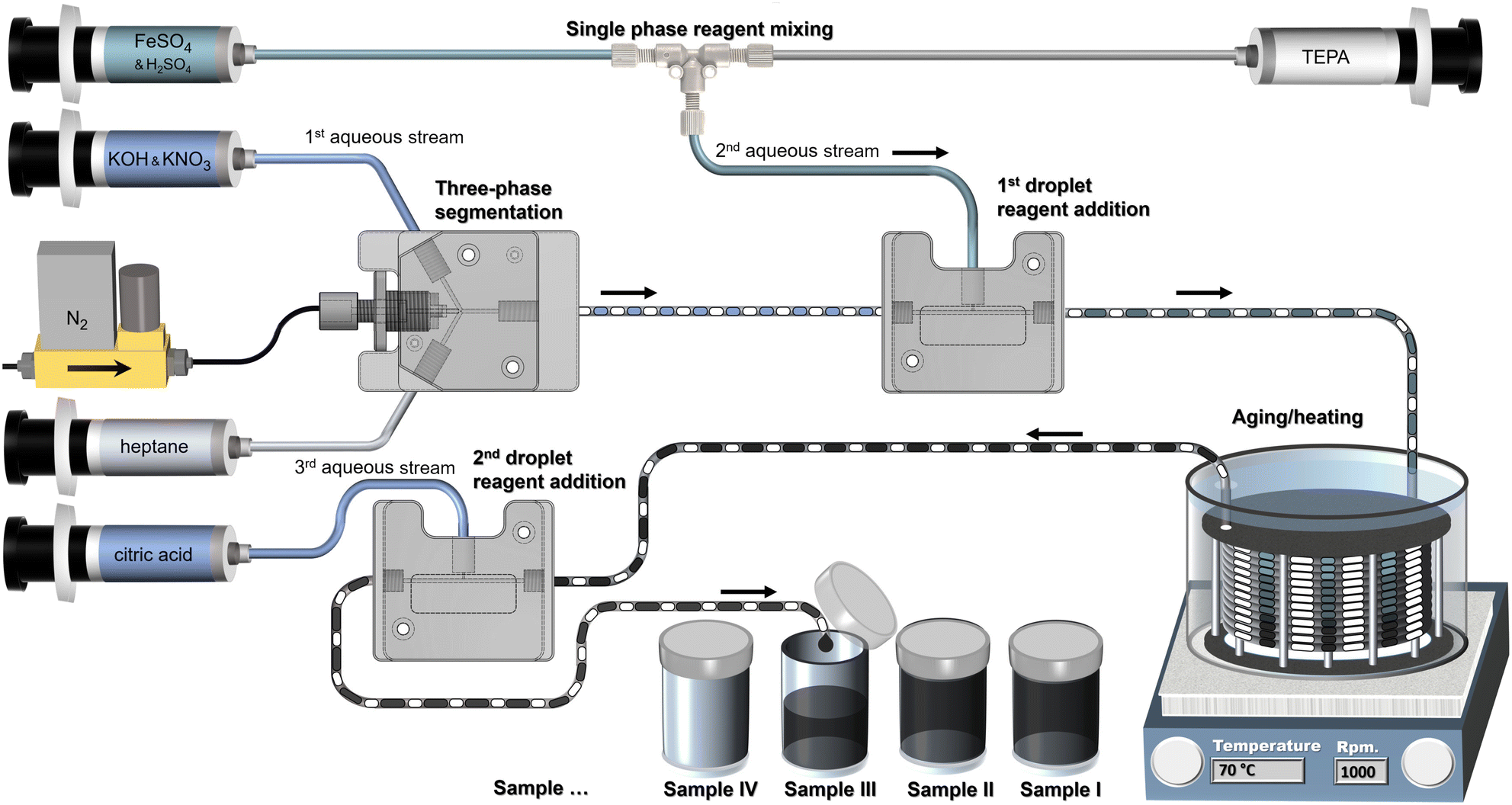 | ||
| Fig. 2 Schematic of continuous multistep partial oxidation synthesis of iron oxide nanoparticles using the three-phase reactor platform varying the tetraethylenepentamine to iron sulphate ratio. | ||
The effect of TEPA on IONP size and morphology was examined by feeding 0–0.04 ml min−1 of 0.016 M TEPA solution to the precursor solution (single phase reagent mixing).
For the flow reactor described no back pressurisation was required. Without the T-unit for the 2nd droplet reagent addition, however, this would have required to stabilise the three-phase segmentation due to the expansion of the gas phase in the heated residence time module. Also, the flow synthesis was typically operated for ∼2 h (maximum time using 50 ml syringes) after reaching steady state (∼10 min). Starting with low gas flow rates and low temperatures for the residence time module made it easier to establish steady state operation. The ESI† provides detailed recommendations to establish steady state operation (see ESI† section 2.3). There was no sign of reactor fouling or three-phase segmentation instabilities throughout the process. This is remarkable considering how fouling prone the synthesis was using a single phase system (see ESI† section 4 and Fig. S12†).
Flow synthesis results
IONP solutions produced without TEPA, as well as for three TEPA concentrations, i.e., feeding 0, 0.02, and 0.04 ml min−1 of the TEPA solution into the 2nd aqueous phase (single phase reagent mixing with precursor solution fed at 0.2 ml min−1) were sampled. The TEPA solution feed rates were low (compensated by a high concentration) in order not to affect the residence time and mixing with the previously formed droplets (i.e., during 1st droplet reagent addition). The synthesis at 0.02 ml min−1 TEPA solution resulted in a molar ratio of TEPA:Fe = 1:62.5 (corresponding to 1:12.5 active amino group to iron), i.e., the same ratio that was shown recently to modify IONP morphologies for a co-precipitation synthesis,59 and at 0.04 ml min−1 to TEPA:Fe = 1:31.3. The TEPA to iron ratio was increased further by halving the precursor solution flow rate from 0.2 to 0.1 ml min−1, resulting in a 2nd aqueous phase flow rate of 0.104 ml min−1, yielding TEPA:Fe = 1:15.6.
A clear effect of TEPA, however, became apparent from TEM analysis only for TEPA:Fe = 1:15.6 (see Fig. 3b). The latter showed the expected change in morphology,59,65,66i.e., facetted octahedral crystals instead of the spherical morphologies observed for lower TEPA:Fe ratios. The particle iron concentration was comparable for all IONP solutions synthesised at 0.2 ml min−1 precursor solution with 1.11, 1.07 and 1.03 mgFe–IONP mlsol−1 for 0, 0.02 and 0.04 ml min−1 of the TEPA solution, and 0.55 mgFe–IONP mlsol−1 (at 0.04 ml min−1 TEPA solution) after halving the precursor solution feed rate. This corresponds to precursor conversions (assuming no other product than IONPs were formed) of 80%, 79%, 78% and 73% and showing a decrease with increasing TEPA concentration. The slightly lower conversions for the highest TEPA concentration were attributed to net reduction of IONP growth rate due to facet specific interactions with TEPA. The ≤80% conversions were anticipated, as the citric acid in the 3rd aqueous stream solution can partially dissolve formed IONPs.22 In general, the IONPs synthesised in flow were smaller compared to the partial oxidation syntheses in batch (∼DTEM = 30–50 nm). This is a familiar trend when translating temperature sensitive nanoparticle synthesis into flow67–69 and could be ascribed to the faster heating rates (room temperature to 70 °C within seconds) in flow yielding higher nucleation rates. In addition, the flow synthesis described evades hours-long oxidation steps (at elevated temperatures), which reduces the likelihood of aggregation. The IONP solutions showed excellent colloidal stability after ultra-sonication with hydrodynamic diameters of Dh = 102, 101, 96, and 101 nm for syntheses without TEPA and at TEPA:Fe ratios of 1:62.5, 1:31.3, and 1:15.6 respectively. This was attributed to the smaller IONP sizes of DTEM ≈ 30 nm compared to batch partial oxidation procedures (with more particle diameters exceeding the superparamagnetic limit),70 as well as the combined stabilisation effects of sulphuric and citric acid. This shows that a water-based synthesis can yield IONPs >10 nm which are colloidally stable.
 | ||
| Fig. 3 (a) IONP solutions synthesised in flow using the three-phase reactor platform. (b) TEM analysis of IONPs synthesised at different TEPA:Fe ratios. (c) Corresponding heating profiles (of washed solutions) with ILP values in the legend. | ||
Tests of the IONPs' heating ability in alternating magnetic fields (see Fig. 3c) showed promising results. The IONPs synthesised in flow yielded an intrinsic loss power (ILP),71i.e., a parameter used to compare heating abilities of magnetic nanoparticles at different field strengths and frequencies, of 3.1, 3.4, 3.3 and 2.8 nH m2 kgFe−1 without TEPA and at TEPA:Fe ratios of 1:62.5, 1:31.3, and 1:15.6 respectively. This corresponds to specific absorption rates (SARs) of 913, 1002, 972 and 825 W gFe−1 for the magnetic field strength and frequency used. The lowest SAR/ILP values for the highest TEPA concentration (despite comparable size and the highly crystalline appearance) could be attributed to the reduced volume of the octahedral IONPs, i.e., a spherical particle with the same DTEM would have a higher volume. The flow synthesis yielded colloidally stable IONPs with ILP values >3 nH m2 kgFe−1 (without any optimisation for their heating abilities), which is considered high for water-based syntheses.72,73 Furthermore, the flow synthesis was reproducible as particle iron concentrations, conversions, hydrodynamic diameters and heating rates were in good agreement with repeated synthesis at all TEPA:Fe ratios (see reproducibility study in the ESI† section 5 and Fig. S13†). This indicates the potential of this water-based, green, and low-cost flow synthesis to produce heating agents for magnetic hyperthermia treatment.
Summary and perspectives
Despite the potential of non-fouling gas–liquid–liquid segmented flow reactors to translate complex (nano) material syntheses to flow, there has been less than a handful of showcases. This is because adding reagents into droplets/slugs robustly is limited by the operation range of continuous and disperse phase flow rates, number of reagent addition steps, reagent addition volumes, solid phase formation, residence times, operating pressures, and temperatures. For this reason, we designed an easy-to-use “Lego®-like” modular flow reactor platform for three-phase segmented operation, comprising elements for droplet/slug generation and reagent addition into droplets. The platform's robustness and flexibility were due to the movable gas injection nozzle (to control the droplet spacing), hydrophobic plastics (for robust three-phase flow without using fluorinated oils as continuous phase), thinned walls over the channels (for visual inspection in less transparent plastic elements), standard fluidic connectors (guaranteeing simplicity and modularity) and temperature-controlled residence time modules. We also provide operating recommendations to establish steady state operation quickly and guarantee robust long term operation.The potential of this platform to translate complex nanoparticle (and material) syntheses into flow was demonstrated for a partial oxidation synthesis of IONPs and their stabilisation, comprising multiple reagent addition steps for the initial ferric hydroxides precipitation, oxidation agent addition, citric acid addition, as well as a temperature treatment. This multistep synthesis was operated for hours in this platform without any sign fouling, in contrast to single-phase flow reactors showing immediate fouling and clogging after minutes. In addition, the synthetic parameter space was explored, changing the TEPA to iron ratio during synthesis, which showed the ability of this bioinspired additive to produce more faceted (and slightly smaller) particles. The observed IONPs size reduction was assumed to be the reason why no positive effect of TEPA on IONPs heating abilities in alternating magnetic fields was found. Due to its flexibility and inherent non-fouling nature, the reactor platform opens unique opportunities for high-throughput synthesis screening. Although not fully exploited in this work, screening the synthetic conditions allowed to develop a partial oxidation flow synthesis yielding colloidally stable IONPs (colloidal stability is an issue for partial oxidation protocols) through a fine-tuned citric acid addition step. The IONPs produced showed remarkable heating abilities for a (biocompatible) water-based synthesis with ILP values >3 nH m2 kgFe−1.
Another opportunity for nanoparticle flow synthesis the three-phase reactor platform offers is to provide oxidation (e.g., oxygen or nitrous oxide) or reducing agents (e.g., hydrogen or carbon monoxide) through the gas phase. The latter can also accommodate gaseous byproducts (which are common for thermal decomposition syntheses) preventing the formation of new gas bubbles and the disruption of segmented flow. Furthermore, the volume occupied by the gas in the three-phase segmented flow reduces the amount of solvent for the continuous phase in liquid–liquid segmented flow reactors, allowing for more sustainable operation.
The commonly used single phase or liquid–liquid segmented flow-chemistry platforms do not exhibit the flexibility required for complex nanoparticle flow synthesis. Therefore, the platform presented allows nanoparticle development to finally benefit from the high-throughput screening and self-optimisation technologies established for small molecule synthesis.
Associated content
Further details on the flow reactor platform, the set-ups used, as well as reactor and particle characterisation are provided in the ESI.† The ESI† provides videos showing i) three phase droplet generation, ii) reagent addition into droplets and iii) the platform in operation. All material is available online and free of charge.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
MOB, AG, NTKT and SS thank the Centre for Nature Inspired Engineering (CNIE) for financial support (EPSRC “Frontier Engineering” Award EP/K038656/1) through an Inspiration Grant. The authors also thank the EPSRC UK for financial support (EP/M015157/1) through the Manufacturing Advanced Functional Materials (MAFuMa) scheme.References
- V. Sebastian, S. A. Khan and A. A. Kulkarni, J. Flow Chem., 2017, 7, 96–105 CrossRef CAS.
- P. Sagmeister, R. Lebl, I. Castillo, J. Rehrl, J. Kruisz, M. Sipek, M. Horn, S. Sacher, D. Cantillo, J. Williams and C. O. Kappe, Angew. Chem., 2021, 133, 8220–8229 CrossRef.
- A. Sivo, R. De Souza Galaverna, G. R. Gomes, J. C. Pastre and G. Vile, React. Chem. Eng., 2021, 6, 756–786 RSC.
- R. L. Hartman and K. F. Jensen, Lab Chip, 2009, 9, 2495–2507 RSC.
- M. Guidi, P. H. Seeberger and K. Gilmore, Chem. Soc. Rev., 2020, 49, 8910–8932 RSC.
- K. Kusada and H. Kitagawa, Mater. Horiz., 2022, 9, 547–558 RSC.
- J. Sui, J. Yan, D. Liu, K. Wang and G. Luo, Small, 2020, 16, 1902828 CrossRef CAS PubMed.
- R. W. Epps, K. C. Felton, C. W. Coley and M. Abolhasani, Lab Chip, 2017, 17, 4040–4047 RSC.
- L. Bezinge, R. M. Maceiczyk, I. Lignos, M. V. Kovalenko and A. J. DeMello, ACS Appl. Mater. Interfaces, 2018, 10, 18869–18878 CrossRef CAS PubMed.
- A. A. Volk, R. W. Epps and M. Abolhasani, Adv. Mater., 2021, 33, 2004495 CrossRef CAS PubMed.
- B. Pinho and L. Torrente-Murciano, React. Chem. Eng., 2020, 5, 342–355 RSC.
- S. Li, Y. Meng, Y. Guo, T. Liu, S. Stavrakis, P. D. Howes and A. J. DeMello, J. Mater. Chem. C, 2021, 9, 925–933 RSC.
- K. Abdel-Latif, R. W. Epps, C. B. Kerr, C. M. Papa, F. N. Castellano and M. Abolhasani, Adv. Funct. Mater., 2019, 29, 1970157 CrossRef.
- S. T. Knox, S. J. Parkinson, C. Y. P. Wilding, R. A. Bourne and N. J. Warren, Polym. Chem., 2022, 13, 1576–1585 RSC.
- N. Cherkasov, Y. Bai, A. J. Expósito and E. V. Rebrov, React. Chem. Eng., 2018, 3, 769–780 RSC.
- M. O. Besenhard, P. Neugebauer, C.-D. Ho and J. G. Khinast, Cryst. Growth Des., 2015, 15, 1683–1691 CrossRef CAS.
- J. A. Bennett and M. Abolhasani, Curr. Opin. Chem. Eng., 2022, 36, 100831 CrossRef.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed.
- M. Y. S. Ibrahim and M. Abolhasani, Nat. Commun., 2022, 13, 2441 CrossRef CAS PubMed.
- L. Norfolk, A. Rawlings, J. Bramble, K. Ward, N. Francis, R. Waller, A. Bailey and S. Staniland, Nanomaterials, 2019, 9, 1729 CAS.
- Z. Whiteley, H. M. K. Ho, Y. X. Gan, L. Panariello, G. Gkogkos, A. Gavriilidis and D. Q. M. Craig, Nanoscale Adv., 2021, 3, 2039–2055 Search PubMed.
- M. O. Besenhard, A. P. LaGrow, A. Hodzic, M. Kriechbaum, L. Panariello, G. Bais, K. Loizou, S. Damilos, M. M. Cruz, N. T. K. Thanh and A. Gavriilidis, Chem. Eng. J., 2020, 125740 CAS.
- J. L. Perry and S. G. Kandlikar, Microfluid. Nanofluid., 2008, 5, 357–371 Search PubMed.
- Z. Dong, D. F. Rivas and S. Kuhn, Lab Chip, 2019, 19, 316–327 CAS.
- Y. He, K.-J. Kim and C. Chang, Nanomaterials, 2020, 10, 1421 CAS.
- A. M. Nightingale, J. H. Bannock, S. H. Krishnadasan, F. T. F. O'Mahony, S. A. Haque, J. Sloan, C. Drury, R. McIntyre and J. C. DeMello, J. Mater. Chem. A, 2013, 1, 4067–4076 CAS.
- S. Abalde-Cela, P. Taladriz-Blanco, M. G. De Oliveira and C. Abell, Sci. Rep., 2018, 8, 2440 Search PubMed.
- Y. Ding, P. D. Howes and A. J. DeMello, Anal. Chem., 2020, 92, 132–149 CAS.
- S. Damilos, I. Alissandratos, L. Panariello, A. N. P. Radhakrishnan, E. Cao, G. Wu, M. O. Besenhard, A. A. Kulkarni, C. Makatsoris and A. Gavriilidis, J. Flow Chem., 2021, 11, 553–567 CAS.
- C. H. Kwak, S. M. Kang, E. Jung, Y. Haldorai, Y. K. Han, W. S. Kim, T. Yu and Y. S. Huh, J. Ind. Eng. Chem., 2018, 63, 405–410 CAS.
- I. Lignos, L. Protesescu, D. B. Emiroglu, R. MacEiczyk, S. Schneider, M. V. Kovalenko and A. J. DeMello, Nano Lett., 2018, 18, 1246–1252 CAS.
- A. M. Nightingale, S. H. Krishnadasan, D. Berhanu, X. Niu, C. Drury, R. McIntyre, E. Valsami-Jones and J. C. DeMello, Lab Chip, 2011, 11, 1221–1227 CAS.
- S. Duraiswamy and S. A. Khan, Small, 2009, 5, 2828–2834 CAS.
- X. Luo, P. Su, W. Zhang and C. L. Raston, Adv. Mater. Technol., 2019, 4, 1900488 CAS.
- K. Kumar, A. M. Nightingale, S. H. Krishnadasan, N. Kamaly, M. Wylenzinska-Arridge, K. Zeissler, W. R. Branford, E. Ware, A. J. DeMello and J. C. DeMello, J. Mater. Chem., 2012, 22, 4704 CAS.
- C. D. Ahrberg, J. W. Choi and B. G. Chung, Beilstein J. Nanotechnol., 2018, 9, 2413–2420 CAS.
- A. M. Nightingale, T. W. Phillips, J. H. Bannock and J. C. DeMello, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- W. K. Wong, S. K. Yap, Y. C. Lim, S. A. Khan, F. Pelletier and E. C. Corbos, React. Chem. Eng., 2017, 2, 636–641 CAS.
- D. Karan and S. A. Khan, React. Chem. Eng., 2019, 4, 1331–1340 CAS.
- S. K. Yap, W. K. Wong, N. X. Y. Ng and S. A. Khan, Chem. Eng. Sci., 2017, 169, 117–127 CAS.
- M. Wörner, Theor. Found. Chem. Eng., 2020, 54, 3–16 Search PubMed.
- M. Mac Giolla Eain, V. Egan and J. Punch, Int. J. Heat Fluid Flow, 2013, 44, 515–523 Search PubMed.
- K. Abdel-Latif, R. W. Epps, F. Bateni, S. Han, K. G. Reyes and M. Abolhasani, Adv. Intell. Syst., 2021, 3, 2000245 Search PubMed.
- A. A. Volk, R. W. Epps, D. Yonemoto, F. N. Castellano and M. Abolhasani, React. Chem. Eng., 2021, 6, 1367–1375 CAS.
- S. D. Svetlov and R. S. Abiev, Chem. Eng. J., 2018, 354, 269–284 CAS.
- G. N. Ahn, S. Taniguchi, T. Aoyama, S. Hasebe, D. P. Kim and O. Tonomura, Chem. Eng. J., 2020, 385, 123492 CAS.
- L. Panariello, G. Wu, M. O. Besenhard, K. Loizou, L. Storozhuk, N. T. K. Thanh and A. Gavriilidis, Materials, 2020, 13, 1019 CAS.
- T. Asimakidou, A. Makridis, S. Veintemillas-Verdaguer, M. P. Morales, I. Kellartzis, M. Mitrakas, G. Vourlias, M. Angelakeris and K. Simeonidis, Chem. Eng. J., 2020, 393, 124593 CAS.
- F. Vereda, J. de Vicente and R. Hidalgo-Alvarez, J. Colloid Interface Sci., 2013, 392, 50–56 CAS.
- T. Ahn, J. H. Kim, H.-M. Yang, J. W. Lee and J.-D. Kim, J. Phys. Chem. C, 2012, 116, 6069–6076 CAS.
- A. P. LaGrow, M. O. Besenhard, A. Hodzic, A. Sergides, L. K. Bogart, A. Gavriilidis and N. T. K. Thanh, Nanoscale, 2019, 11, 6620–6628 CAS.
- S. S. Staniland, A. Rawlings, J. Bramble, J. Tolosa, O. Wilson, J. C. García-Martínez and C. Binns, in Frontiers of Nanoscience, Elsevier, 2014, vol. 6, pp. 85–128 Search PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CAS.
- P. Sangaiya and R. Jayaprakash, J. Supercond. Novel Magn., 2018, 31, 3397–3413 CAS.
- M. Mahmoudi and S. Laurent, Iron Oxide Nanoparticles for Biomedical Applications: Synthesis, Functionalization and Application, Elsevier, Amsterdam, 1st edn, 2017 Search PubMed.
- T. Granath, P. Löbmann and K. Mandel, Part. Part. Syst. Charact., 2021, 38, 2000307 CAS.
- Y. Amemiya, A. Arakaki, S. S. Staniland, T. Tanaka and T. Matsunaga, Biomaterials, 2007, 28, 5381–5389 CAS.
- A. E. Rawlings, J. P. Bramble, A. M. Hounslow, M. P. Williamson, A. E. Monnington, D. J. Cooke and S. S. Staniland, Chem. – Eur. J., 2016, 22, 7885–7894 CAS.
- L. Norfolk, K. Kapusta, D. Cooke and S. Staniland, Green Chem., 2021, 23, 5724–5735 CAS.
- M. P. Calatayud, C. Riggio, V. Raffa, B. Sanz, T. E. Torres, M. R. Ibarra, C. Hoskins, A. Cuschieri, L. Wang, J. Pinkernelle, G. Keilhoff and G. F. Goya, J. Mater. Chem. B, 2013, 1, 3607–3616 CAS.
- C. J. Perecin, B. M. Tirich, L. C. C. M. Nagamine, G. Porto, F. V. Rocha, N. N. P. Cerize and L. C. Varanda, Colloids Surf., A, 2021, 627, 127169 CAS.
- M. A. Vergés, R. Costo, A. G. Roca, J. F. Marco, G. F. Goya, C. J. Serna and M. P. Morales, J. Phys. D: Appl. Phys., 2008, 41, 10 Search PubMed.
- M. O. Besenhard, L. Panariello, C. Kiefer, A. P. LaGrow, L. Storozhuk, F. Perton, S. Begin, D. Mertz, N. T. K. Thanh and A. Gavriilidis, Nanoscale, 2021, 13, 8795–8805 CAS.
- L. Storozhuk, M. O. Besenhard, S. Mourdikoudis, A. P. LaGrow, M. R. Lees, L. D. Tung, A. Gavriilidis and N. T. K. Thanh, ACS Appl. Mater. Interfaces, 2021, 13, 45870–45880 CAS.
- A. E. Rawlings, L. A. Somner, M. Fitzpatrick-Milton, T. P. Roebuck, C. Gwyn, P. Liravi, V. Seville, T. J. Neal, O. O. Mykhaylyk, S. A. Baldwin and S. S. Staniland, Nat. Commun., 2019, 10, 1–9 CAS.
- J. M. Galloway, A. Arakaki, F. Masuda, T. Tanaka, T. Matsunaga and S. S. Staniland, J. Mater. Chem., 2011, 21, 15244 CAS.
- L. Uson, M. Arruebo, V. Sebastian and J. Santamaria, Chem. Eng. J., 2018, 340, 66–72 CAS.
- M. Taddei, D. A. Steitz, J. A. van Bokhoven and M. Ranocchiari, Chem. – Eur. J., 2016, 22, 3245–3249 CAS.
- J. Pellico, J. Ruiz-Cabello, I. Fernández-Barahona, L. Gutiérrez, A. V. Lechuga-Vieco, J. A. Enríquez, M. P. Morales and F. Herranz, Langmuir, 2017, 33, 10239–10247 CAS.
- Q. Li, C. W. Kartikowati, S. Horie, T. Ogi, T. Iwaki and K. Okuyama, Sci. Rep., 2017, 7, 1–7 Search PubMed.
- R. R. Wildeboer, P. Southern and Q. A. Pankhurst, J. Phys. D: Appl. Phys., 2014, 47, 495003 Search PubMed.
- I. Rubia-Rodríguez, A. Santana-Otero, S. Spassov, E. Tombácz, C. Johansson, P. De La Presa, F. J. Teran, M. D. P. Morales, S. Veintemillas-Verdaguer, N. T. K. Thanh, M. O. Besenhard, C. Wilhelm, F. Gazeau, Q. Harmer, E. Mayes, B. B. Manshian, S. J. Soenen, Y. Gu, Á. Millán, E. K. Efthimiadou, J. Gaudet, P. Goodwill, J. Mansfield, U. Steinhoff, J. Wells, F. Wiekhorst and D. Ortega, Materials, 2021, 14, 1–37 Search PubMed.
- K. El-Boubbou, Nanomedicine, 2018, 13, 929–952 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2lc00892k |
| This journal is © The Royal Society of Chemistry 2023 |