
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of phosphonates on lanthanide separation for surface-grafted porous zirconia†
Miho
Otaki
 *a,
Topi
Suominen
a,
Valtteri
Suorsa
a,
Sami
Hietala
b and
Risto T.
Koivula
a
*a,
Topi
Suominen
a,
Valtteri
Suorsa
a,
Sami
Hietala
b and
Risto T.
Koivula
a
aDepartment of Chemistry – Radiochemistry Unit, FI-00014 University of Helsinki, A. I. Virtasen Aukio 1, P.O. Box 55, Helsinki, Finland. E-mail: miho.otaki@helsinki.fi
bDepartment of Chemistry, FI-00014 University of Helsinki, A. I. Virtasen Aukio 1, P.O. Box 55, Helsinki, Finland
First published on 9th December 2022
Abstract
The effect of phosphonate groups on selective lanthanide sorption for a series of inorganic–organic hybrid materials was investigated. Four functional ligands with an increasing number of aminomethylenephosphonate groups were attached to the porous zirconia matrix via the post-synthetic grafting method. Successful surface grafting was confirmed by FTIR and 31P MAS-NMR spectroscopies. All the synthesized hybrids showed a lanthanide uptake of about 100 μmol g−1 at pH 3. The sorption studies revealed that the hybrids of ligands with a larger number of phosphonate groups possess higher selectivity, as one with the largest number showed a separation factor of over 80 for La3+ and Lu3+. Fixed-bed column experiments also demonstrated the materials’ favored uptake of Lu3+ over La3+ at pH 3, in addition to the reusability and applicability to the actual separation settings. These results propose strategies for the selection of organic ligands for hybrid sorbent synthesis.
Introduction
Lanthanides (Ln) exhibit a variety of photophysical, magnetic, and nuclear properties that offer the possibility of a wide range of applications.1 They have been utilized as critical raw materials for batteries, permanent magnets, and fluid cracking catalysts. In addition, recently, the interest in the medical use of radiolanthanides for both diagnostic and therapeutic purposes has also grown.2 It is expected that the demand for lanthanides is likely to increase exponentially in the following decades. Since lanthanides are often found in trace amounts, elaborate separation and preconcentration are required before being utilized. However, owing to the similar physicochemical properties, the separation of individual lanthanides is very challenging.3One of the promising techniques for lanthanide separation known now is liquid–liquid extraction (solvent extraction), which has been the state-of-art technology for industrial recovery of lanthanides.4 However, due to the limited separation efficiency, the separation process requires repetitive extraction steps to achieve the desired purity, and thus a large amount of hazardous and radioactive liquid waste is generated during the purification process.5 In recent years, extraction chromatography has emerged as a prominent tool in the separation and purification of radiolanthanides in particular, which inherits the advantages of the selectivity of solvent extraction and the ease of chromatographic operation.6 In extraction chromatography, an organic extractant is adsorbed onto inert support to function as the stationary phase, and thus it is possible to reduce the amount of organic liquid waste produced during the purification process.7 Currently, composite polymeric resin beads encapsulating extractants have been reported to show good stability and high uptake values and thus are considered a good option for the method.8 However, extraction chromatography still possesses several drawbacks such as the slow uptake kinetics and low throughput8 which prevent its industrial application.
Solid-phase extraction technique with highly selective materials requires fewer liquid phases than conventional liquid–liquid extraction, and handling fewer liquid phases can realize simpler and faster separation. According to previous research, inorganic–organic hybrid materials have been successfully applied to the separation of group 3 and f-block elements, by combining an inorganic solid matrix and an organic functional ligand.9–13 For this purpose, silica is commonly studied as an inorganic matrix. However, silica-based materials are not stable in acidic media.14 On the other hand, metal(IV) oxides such as zirconium oxides have been reported as a promising class of inorganic support materials with high resistance to acids, oxidation, and radiation.15–17 Also, their porous structure is synthetically alterable, leading to the size selection of target ions.
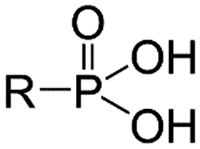
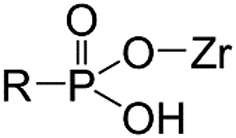
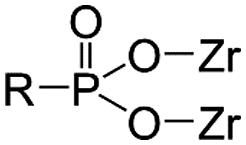
As functional ligands, organophosphorus compounds are highly promising coupling molecules that allow strong anchoring of organic groups to inorganic solids.18,19 Utilizing organophosphorus coupling agents would realize homogenous ligand layer grafting and the resulting M–O–P–C and M–O–P–O–C linkages (where M–metal, O–oxygen, P–phosphorus, and C–carbon) are highly stable.20 Because of these benefits, zirconium phosphonate has been reported as a promising class of materials that are particularly suitable for nuclear applications due to their stability in acid solutions and resistance to ionizing radiation.21
One of the promising examples is amino tris(methylphosphonic acid)-incorporated zirconium oxide (Zr-ATMP). It has been confirmed that Zr-ATMP shows intra-lanthanide selectivity (e.g., a separation factor of 2.7 for La and Eu),10 and by tuning the conditions of synthesis, it is possible to control its porosity, chemical composition, sorption properties and thermochemical stability.22,23 Zr-ATMP also possesses high radiolytic stability due to its multiple Zr–O–P linkages, attributed to polyphosphonate ATMP.24 Although it is clear that Zr-ATMP has several advantages, especially in terms of morphological flexibility and stability, the selectivity is still low.
Thus, to find a clue for better selectivity, the effect of phosphonate groups on lanthanide sorption was studied. We have synthesized certain inorganic–organic hybrid sorbents using a porous zirconia matrix and a series of phosphonate ligands with an increasing number of aminomethylenephosphonate groups (Chart 1). This selection is rationalized by the fact that all the four ligands exhibit high complexation with trivalent metal cations,25,26 and some of them have been applied for hybrid synthesis,27 though systematic studies on their intra-lanthanide separation capability have not been carried out yet. For the functionalization of zirconia, the post-synthetic grafting method was employed,28 since it is believed that surface grafting via this method does not alter the porous structure of zirconia so that the difference in the sorption behaviour of each hybrid is assumed to reflect only the nature of the ligand.
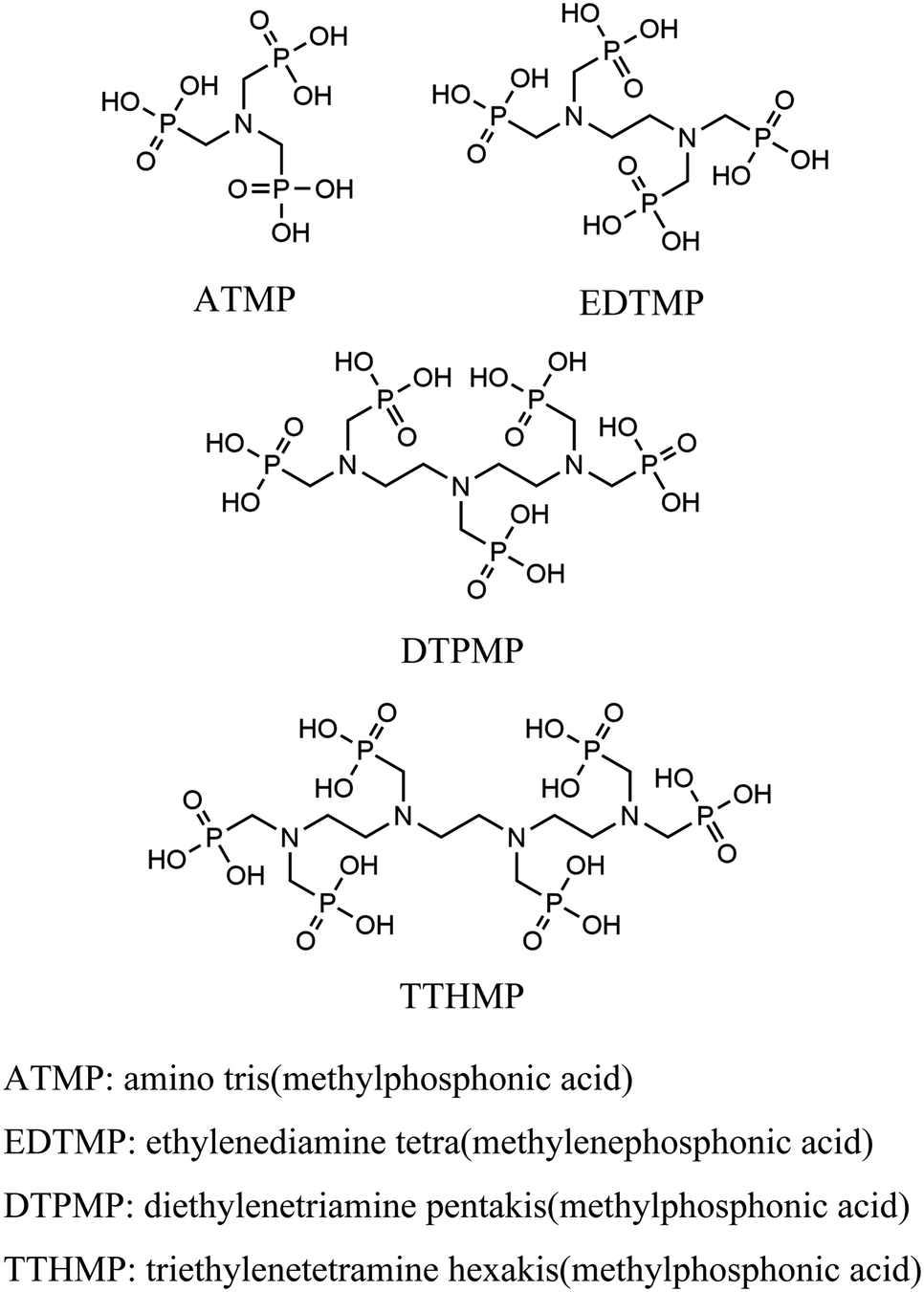 | ||
| Chart 1 Structures of ligands. | ||
Experimental
Synthetic procedures
Instrumentation for structural and compositional characterization
The powder XRD patterns were obtained using Malvern Panalytical X’Pert3 Powder diffractometer with a monochromatic CuKα X-ray source (λ = 1.5405980 Å) operating at 40 kV and 40 mA. The FTIR spectra over the range of 400–4000 cm−1 were recorded using a Bruker Alpha FTIR spectrometer with a single reflection attenuated total reflection (ATR) mode. The crystal surface morphology was investigated using a Hitachi S-4800 FE-SEM (field-emission SEM). To prevent the surface charge effects, the samples were coated with a 10.9 nm layer of carbon. The nitrogen adsorption–desorption isotherms were obtained at 77 K using a Quantachrome Instruments Autosorb iQ after degassing at 200 °C. The surface areas were estimated according to the Brunauer–Emmett–Teller (BET) method, and pore volumes and pore size distributions were calculated using density functional theory (DFT). The compositional rates of carbon and nitrogen of the hybrid materials were analyzed by combustion methods using a Thermo Scientific Flash 2000 elemental analyzer. The simultaneous thermogravimetry analysis (TGA) and differential scanning calorimetry (DSC) were performed using a Netzsch STA 449F3 Jupiter with a heating rate of 10 °C min−1 under an air gas flow of 20 mL min−1. The liquid-state 1H and 31P NMR spectra of the synthesized ligand were obtained using a 400 MHz Bruker Avance Neo 400 spectrometer. The solid-state 31P magic angle spinning (MAS) NMR spectra of the hybrid materials were collected using a Bruker Avance III 500 MHz spectrometer equipped with a 4 mm H/X/Y MAS probe. The samples were filled into a 4 mm zirconia rotor and spun at a rate of 10 kHz to separate the spinning sidebands with the main resonance. 31P spectra were recorded at room temperature by 1H decoupling using a 5 s recycle delay of up to 8500 scans. The 31P chemical shifts were referenced to external 85% H3PO4 at 0 ppm. The peak deconvolutions were made via peak fitting iteration algorithms assuming a Lorentz distribution of peaks using the IGOR pro program (WaveMetrics, Inc.).Batch sorption study
The uptake and selectivity of Ln3+ were studied in a batch mode. 20 ± 0.2 mg of the material was placed in a 20 mL polyethylene vial with 10 mL of the Ln3+-containing solution. The samples were equilibrated for 72 h by constant rotary mixing (50 rpm). The samples were then centrifuged using a Thermo Fisher Scientific Heraeus Megafuge 1.0R with 2773 × g for 10 minutes and filtered through Pall Laboratory Acrodisc 0.2 μm PVDF syringe filters. The filtered solution was pipetted for concentration/activity determination. The equilibrium pH was measured from the remaining filtrate using a Thermo Scientific Orion 3 Star pH Benchtop Meter. All the batch operations followed this procedure.The distribution coefficient (Kd, mL g−1), a ratio of the distribution of ions between the solution and the solid material, was calculated as per eqn (1).
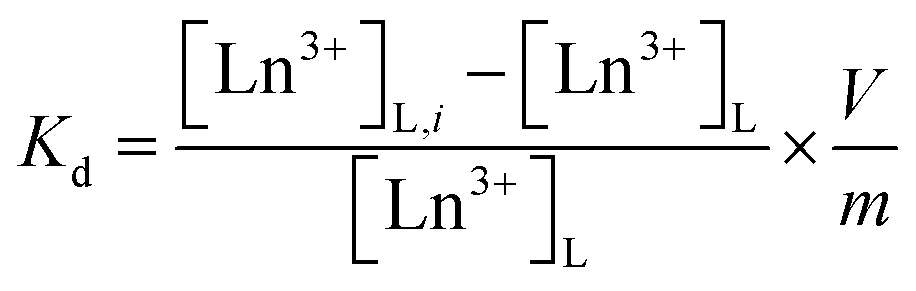 | (1) |
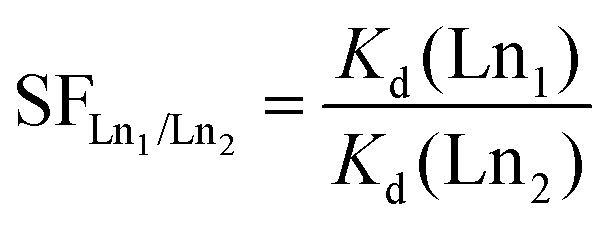 | (2) |
Column operation
 | (3) |
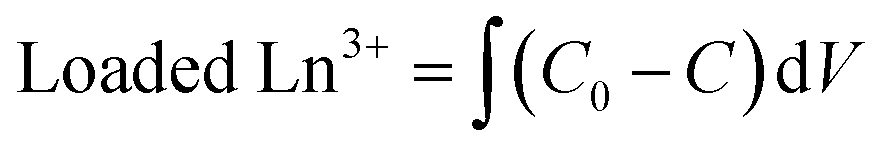 | (4) |
Results and discussion
Characterization of hybrid materials
For hybrid synthesis, 1 M hydrochloric acid was used since the resulted materials showed higher sorption than ones synthesized at lower concentrations (Table S1, ESI†). It is assumed that a high concentration of acid promoted esterification between phosphonic acid and hydroxyl groups on the zirconia surface, as reported in the case of phosphates and zirconia.30 Surface grafting was confirmed by comparing the IR spectra of the bare porous zirconia with those of hybrid materials. The spectra of the hybrids showed a broad band at 900–1200 cm−1 attributed to P–O stretching vibrations, with the P–OH vibration at the lower end of the region (Fig. 1).10,23 The band in 1130–1150 cm−1 is indicative of a P–O group that is coordinated to a metal centre (i.e. Zr–O–P).31,32 These phosphorous-originated bands were not observed in the spectrum of the bare zirconia SG85, suggesting that in all the hybrid materials, phosphonate binding was successfully formed on the zirconia matrix. On the other hand, all the XRD patterns of the hybrid materials were similar to that of the original SG85 zirconia with a monoclinic structure (Fig. S1, ESI†). SEM images also did not show recognizable differences, except SG85-EDTMP, which had a smaller particle size than the others most likely due to grinding by a magnetic stirrer during hybrid synthesis (Fig. S2, ESI†). In addition, all the bare zirconia and hybrids showed similar nitrogen sorption isotherms (Fig. S3, ESI†) with characteristics of type IV of the IUPAC classification, which indicates the presence of the mesoporous structure.33 The differences in BET surface areas, pore volumes, and size distributions among the samples were limited (Fig. S4 and Table S2, ESI†). These results imply that hybridization occurred only on a very thin layer on the surface of zirconia and the macroscopic structures were not changed.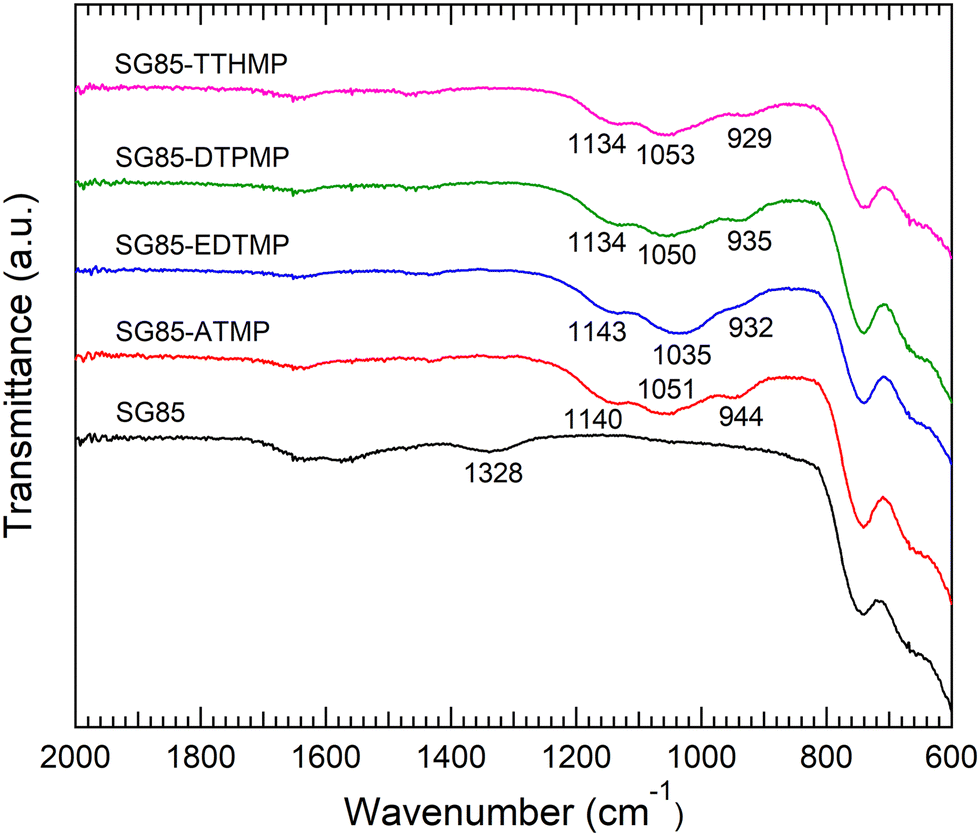 | ||
| Fig. 1 FTIR spectra of the bare porous zirconia and synthesized hybrid materials. | ||
The combustion elemental analysis also supports this observation, since the compositional rates of carbon and nitrogen (Table 1) were low in all materials (C: <2%, N: <1%). The n(C)/n(N) ratios (n(A) stands for the atomic content of A) for each material were all close to a theoretical value of 3. The deviation from the theoretical value most likely implies the measurement uncertainty and the partial degradation of the ligand. The estimation of the level of surface functionalization was made using the nitrogen content assuming that the structure of the aminomethylenephosphonate groups in the ligand was preserved. The level of the ligand loading was normalized relative to the surface area of the zirconia matrix. The loading level (0.8–1.5 molecules per nm2) and its decrease along with the complexity of the phosphonic acid ligand molecules were consistent with the previous report.28 TGA gave similar curves for all the hybrids; the weight decreased by approximately 4% continuously up to 600 °C (Fig. 2(a), derivative TG: Fig. S5, ESI†) Considering the result of the bare zirconia (SG85), the weight loss below 200 °C is attributed mainly to the loss of moisture and hydrated water, and at higher temperatures the decomposition of organic ligands seems to be the dominant reaction. The faster weight loss of SG85 at below 130 °C, which is attributed to the loss of loosely bound water molecules, suggests that the surface of the bare zirconia is more hydrophilic than those of the hybrid materials due to the lack of ligands. The DSC curves were also similar for most of the hybrids, while SG85-EDTMP, only which was synthesized in a different way, showed a slightly steeper increase of exothermic heat flow after 600 °C (Fig. 2(b)).
| Material | C wt% | N wt% | n(C)/n(N) | Ligand loading (molec. per nm2) |
|---|---|---|---|---|
| SG85-ATMP | 0.72 ± 0.18 | 0.28 ± 0.06 | 3.04 | 1.19 |
| SG85-EDTMP | 1.80 ± 0.06 | 0.68 ± 0.02 | 3.08 | 1.48 |
| SG85-DTPMP | 1.61 ± 0.06 | 0.60 ± 0.04 | 3.15 | 0.87 |
| SG85-TTHMP | 1.88 ± 0.02 | 0.77 ± 0.02 | 2.84 | 0.86 |
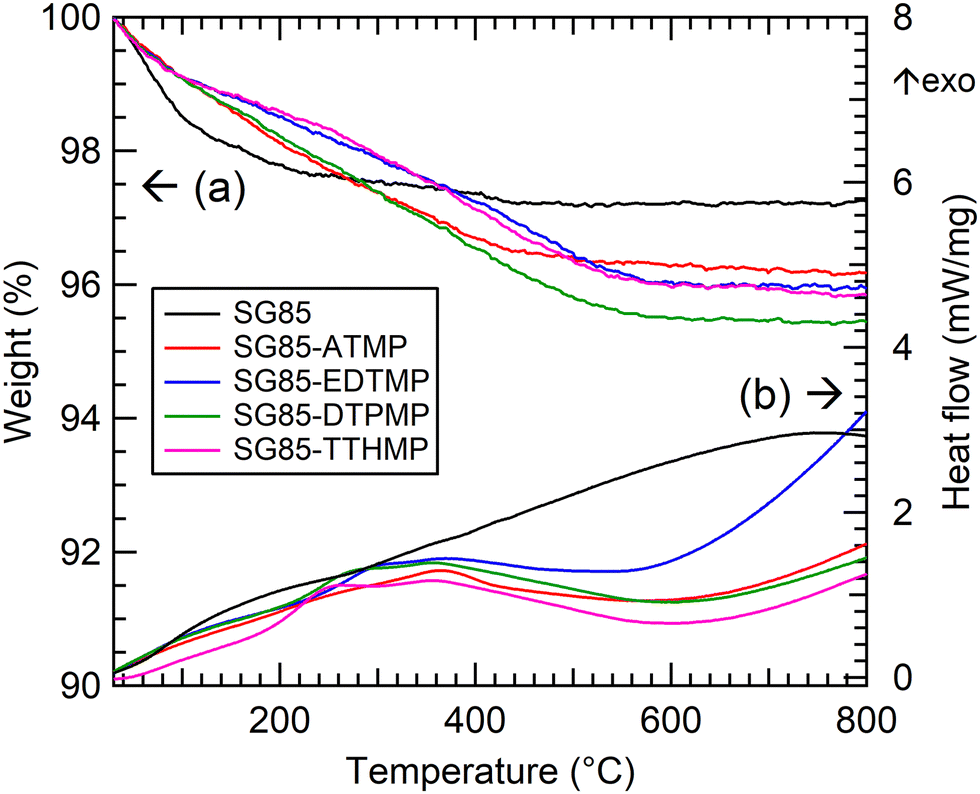 | ||
| Fig. 2 Thermal analysis of the bare porous zirconia and synthesized hybrid materials. (a) TGA and (b) DSC. | ||
Solid-state 31P MAS-NMR is one of the most definitive methods for phosphorus speciation analysis and it is possible to assign the 31P chemical shifts to phosphonate groups with different coordination states (Fig. 3). The quantification results in Table 2 illustrate the relative speciation of the phosphonate groups. The deconvoluted Lorentzian peaks in the range from 6 to 8 ppm are attributed to “free” phosphonate groups or phosphonate that is not coordinated by Zr,22 while the corresponding peaks of ligands before grafting appear at 8–12 ppm (Fig. S6, ESI†). The upfield shift of 3–4 ppm upon grafting is assumed to be caused by weak interactions between the phosphonate groups and nearby Zr atoms, as a similar shift has been observed in previous studies.34,35 It is noteworthy that a relatively higher number of free phosphonate groups were observed in most of the hybrids since the retention of free phosphonate groups is important for the sorption, which originates from the coordination of phosphonate groups with Ln3+ ions. The peaks in the range of 0 to 4 ppm and from −6 to −2 ppm were assigned to mono- and bidentate species of phosphonic acid on zirconia.22 These species are considered to result primarily from esterification with surface hydroxyls of zirconium oxide and are expected to have peaks in the upfield region at 5–15 ppm from those of the free acid.35,36 Finally, the peaks at 17–20 ppm were assigned to phosphonate groups in which phosphonate oxygen atoms are coordinated to more than one metal cation,22 as it has been reported that increasing the degree of coordination of phosphonate oxygen atoms results in a downfield shift of the signals of the phosphonate groups.37 Protonation states of the ligands were not determined, since in the MAS spectra, anisotropic changes of the 31P NMR chemical shift tensor of phosphonates, which are caused by deprotonation as well as noncovalent interactions, are averaged out, resulting in a less obvious shift of isotropic signals.38 Although the hybrid materials except SG85-EDTMP were synthesized by the same functionalization method, each material showed a different ratio of binding modes with Zr (Table 2). Compared to the hybrid of ATMP, which is the smallest ligand in this study, the number of free phosphonate groups in those of larger ligands was higher, assumably because only one or two phosphonate groups in one ligand more likely coordinated with zirconia due to the steric hindrance. In addition, when the ligand is long enough in the stretched configuration, it has a higher possibility to bind through each of the phosphonate end groups within the curved surface of a pore,28 as it was observed that SG85-TTHMP has fewer free phosphonate groups but more monodentate ones than SG85-DTPMP.
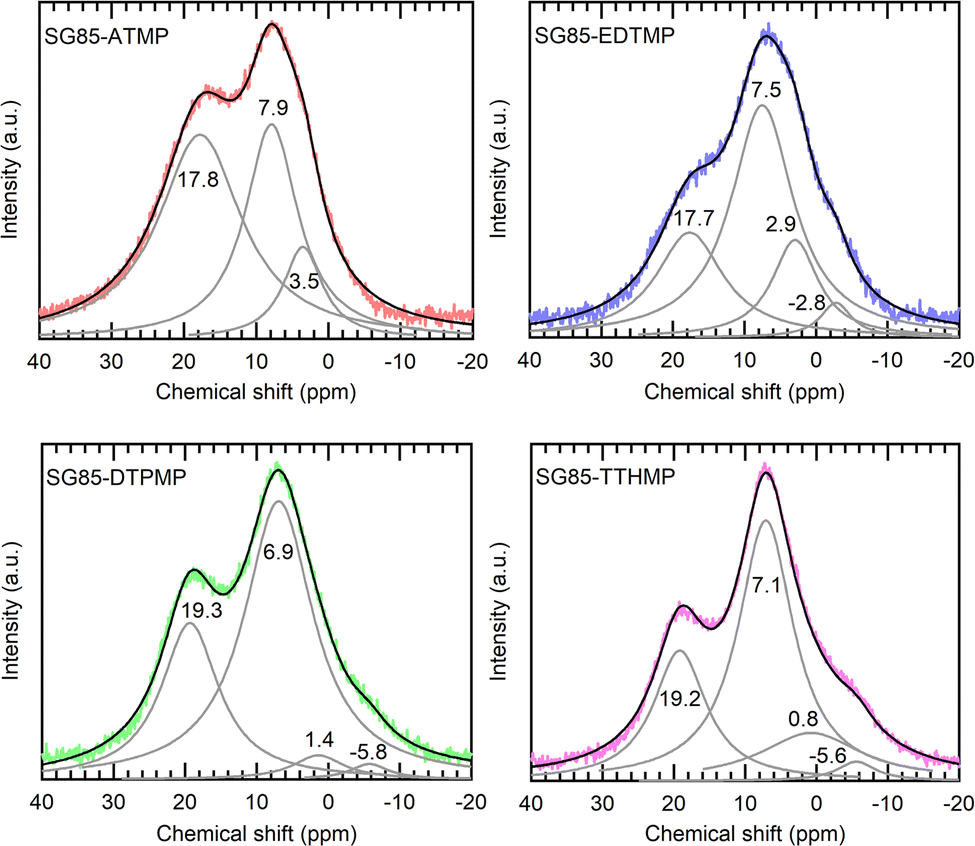 | ||
| Fig. 3 Solid-state 31P MAS NMR spectra of the synthesized hybrid materials with peak deconvolution. | ||
| Chemical shift | 17–20 ppm | 6–8 ppm | 0–4 ppm | −6–−2 ppm |
|---|---|---|---|---|
|
|
|
|
|
|
| SG85-ATMP | 53.2% | 35.5% | 11.3% | 0.0% |
| SG85-EDTMP | 26.0% | 53.6% | 16.5% | 4.0% |
| SG85-DTPMP | 29.9% | 64.0% | 4.1% | 1.9% |
| SG85-TTHMP | 25.5% | 53.7% | 18.0% | 2.7% |
Sorption behavior
The sorption of Lu3+ on the raw porous zirconia matrix (SG85) was negligible when the pH is below 3.5 (Fig. 4), and thus the hybrids were hereinafter studied at a lower pH of 3.5, although the high complexation yield of the grafted ligands with Ln3+ is expected when the pH is higher than 5.26 From the %sorption (Fig. 4), the uptake at pH 3 was estimated for SG85-ATMP, -EDTMP, -DTPMP, and -TTHMP as 110, 89, 117, and 100 μmol g−1, respectively. As reported in the literature,28 post-synthetic grafting of ATMP on mesoporous zirconium titanate resulted in the uptake of about 5 μmol g−1 for Gd3+ in 0.01 M HNO3. Compared to this value, the tested material with the same ligand (SG85-ATMP) showed over ten times higher uptake of 62 μmol g−1 for Lu3+ at pH 2. This significantly higher uptake cannot be explained only by the slightly higher affinity of the heavier lanthanide Lu than that of Gd, and thus we suggest that one reason could be the larger mean pore size of the matrix, which was 9–10 nm in this study (Table S2, ESI†) while 3.57 nm in the literature. It is hypothesized that more ligands are attached to the pore structure when the pore size is larger, and Ln3+ ions are efficiently “trapped” to the pore by these ligands, and thus the materials in this study showed higher uptake.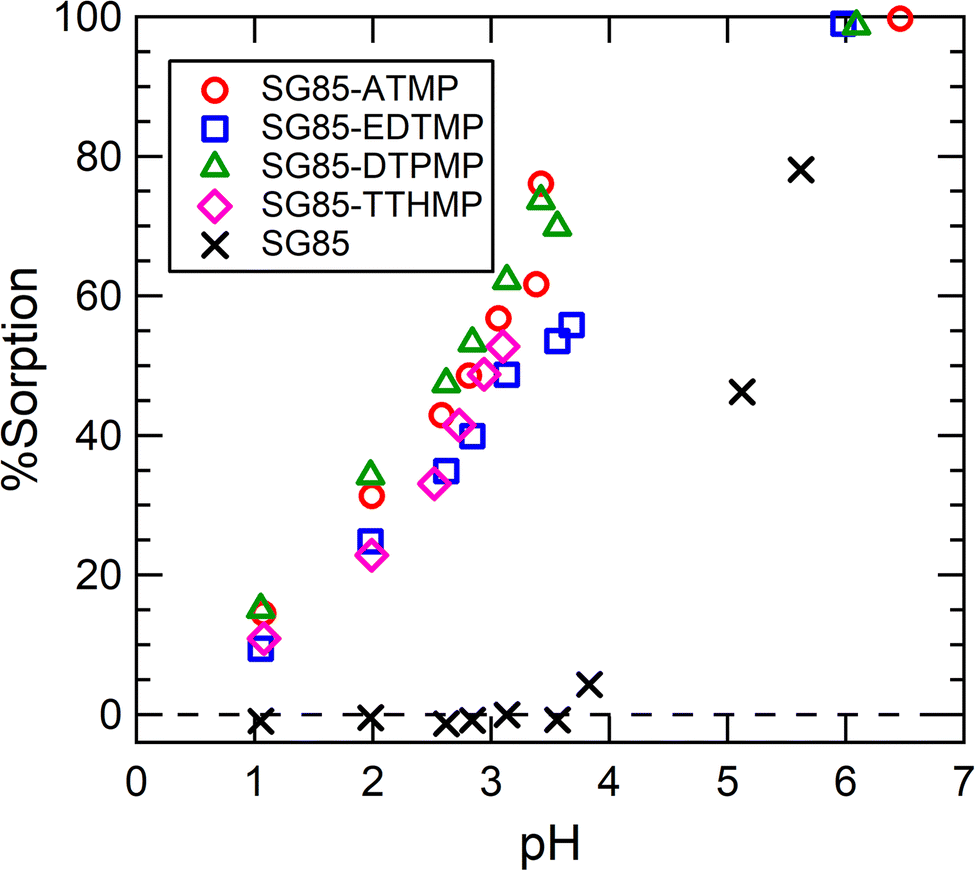 | ||
| Fig. 4 Lu3+ uptake as a function of equilibrium pH. Initial solution: 0.4 mM stable Lu(NO3)3 with a 177Lu radiotracer. The uncertainties of %sorption were in the range of 0.1–0.3% (2σ). | ||
Intra-lanthanide separation tests
K d values as a function of the ionic radius of each Ln3+ ion are shown in Fig. 5. It can be roughly said that Kd values increased with the decreasing ionic radius with all the hybrid materials. This trend is commonly observed for phosphonate ligands39 and is consistent since the decreasing ionic radius from La to Lu (lanthanide contraction) results in an increase in the charge density and stronger ion–dipole interactions between Ln3+ ions and the ligand donor groups.40 The increasing trend of Kd was more obvious in the heavier lanthanide region. The uptake can be considered as a result of the competition of the hydration of Ln3+ ions and their complex formation with grafted ligands. For the complexation reactions of lanthanides, particularly when multidentate ligands are involved, the main driving force for complex formation is the large positive entropy change, because the enthalpy change is either exothermic or endothermic but generally small.1 Thus, the coordination number of aqua ions and ligands towards Ln3+ ions is also one of the important factors deriving the variation of uptake. According to the literature, aqueous trivalent ions of light lanthanides (La–Nd) form nonahydrates, and those of heavy lanthanides (Tb–Lu) octahydrates, while the coordination numbers of lanthanides between Nd and Tb are transitional between 8 and 9.41,42 This coordination change in hydrated Ln3+ ions is assumed to be related to the increase of Kd in the heavier lanthanides region, although a detailed argument cannot be made since the coordination number after the complexation with grafted ligands is not known. | ||
| Fig. 5 K d values of each lanthanide at equilibrium pH 3.0–3.1 as a function of the ionic radius of each Ln3+ ion. Initial solution: 0.04 mM equimolar mixture of Ln3+ (La–Lu excluding Pm) in 10 mM NaNO3. | ||
The increase of Kd values did not show a linear dependence on Ln contraction, but instead, the tetrad effect was observed.43,44 This effect originates from the stability variation due to the 4f electron configuration in ground states. The trend of Kd had discontinuities at Gd3+ and between Ho3+ and Er3+, which correspond to half and three-quarter filling of the 4f electron shell of Ln3+. The discontinuity was not clear at the position of the quarter filling (between Nd3+ and Pm3+) because the data of Pm3+ was missing due to its atomic instability (only radioactive isotopes). The convex curves of the tetrad effect seen in this study imply the covalency of the Ln–O bond on the surface of the material is stronger than that in the aquo complex of Ln3+.45,46 Additionally, the greater incremental increase of Kd for the heavy lanthanides was found in the longer ligand-grafted materials. This sharper increasing trend of Kd is possibly due to the size of the cavity that formed upon surface grafting. Each ligand forms a cavity with the porous zirconia surface, where ligands and the zirconia surface synergistically trap Ln3+ ions having the right volume to fit the coordination environment.47,48 Thus, when longer ligands are grafted, the hybrids are assumed to have a stronger preference for smaller ions.
It is also noticeable that SG85-TTHMP showed the lower sorption of the light and middle Ln relative to the other materials. By comparing the uptake at pH 3 of each material (Fig. 4), it is reasonable that the total Kd values of SG85-TTHMP are lower than, at least those of SG85-ATMP and -DTPMP. In addition, when a material with high selectivity for heavier Ln, which seems to be the case of SG85-TTHMP, is tested in a mixed solution of all the lanthanides of equal concentrations, the Kd values for lighter Ln are lower than those of less-selective materials.
The high separation factors of 87.4 ± 9.8 for Lu/La and 59.3 ± 6.7 for Yb/La were observed in SG85-TTHMP (Fig. 6(a)). The separation factor for Lu/La of SG85-DTPMP was also relatively high as 39.1 ± 1.8. Further analysis of other Ln pairs suggests that the hybrid materials of shorter ligands (ATMP and EDTMP) show better selectivity for light lanthanides, while those of longer ligands (DTPMP and TTHMP) for heavy lanthanides (Fig. 6(b)). The separation between middle-sized lanthanides seems less efficient with any of the materials. In terms of adjacent lanthanide pairs, selectivity was more pronounced in Ce/La (e.g., 2.3 ± 0.4 for SG85-TTHMP) and Yb/Tm (e.g., 2.7 ± 0.1 for SG85-TTHMP) than other combinations (Fig. 6(c)). The variation of the selectivity resulted from multiple causes such as the strength of ion–dipole interactions between Ln3+ ions and ligands and the coordination number in the Ln3+-aquo and -ligand complex,1 as discussed in the previous section.
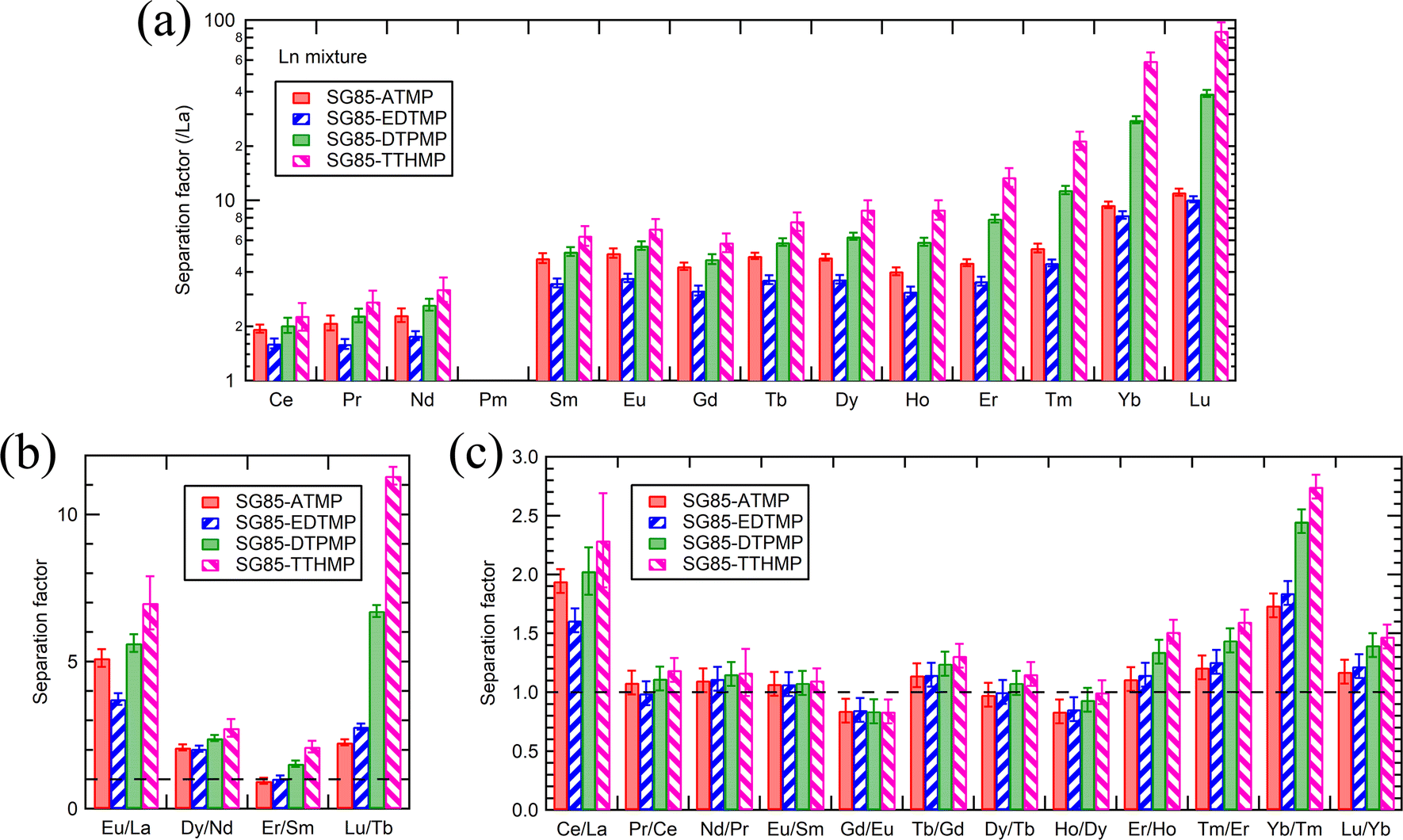 | ||
| Fig. 6 (a) Separation factors between La and other Ln excluding Pm, (b) pairs of light, medium, and heavy Ln, and (c) adjacent Ln pairs, at equilibrium pH 3.0–3.1. Initial solution: 0.04 mM equimolar mixture of Ln3+ (La–Lu excluding Pm) in 10 mM NaNO3. | ||
Separation factors determined in binary solutions (Fig. 7) showed a similar trend to that with the Ln3+ mixture. However, there were clear differences between the values. For example, the separation factor for Lu/La of SG85-TTHMP was 29.8 ± 2.7 with the binary solution while 87.4 ± 9.8 with the Ln3+ mixture solution. This difference may come from the competitive sorption among other Ln ions, which is more obvious in the Ln3+ mixture than in binary solutions. Thus, it should be noted that values of the separation factor may largely vary depending on the composition of the liquid phase.
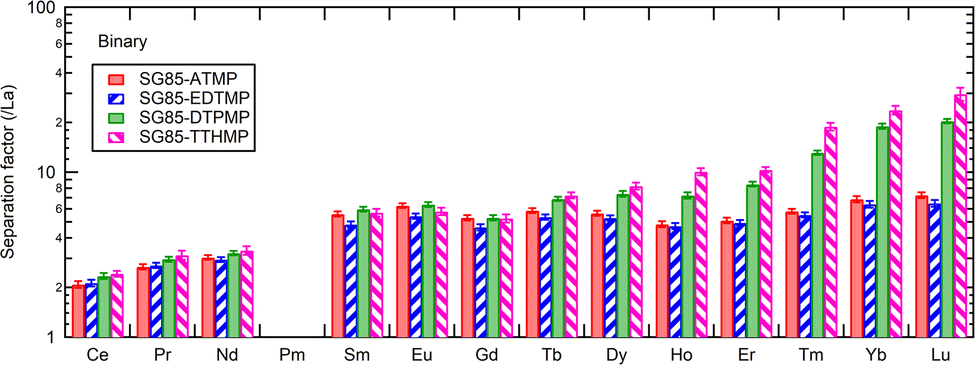 | ||
| Fig. 7 Separation factors between La and other Ln excluding Pm at equilibrium pH 3.0–3.1. Initial solution: 0.2 mM equimolar mixture of La3+ and one of the other stable lanthanides (Ce–Lu excluding Pm) in 10 mM NaNO3. | ||
Compared to the values from recent studies (Table 3), the materials of this study, in particular SG85-DTPMP and SG85-TTHMP, exhibited comparative or better selectivity than other reagents for solid-phase extraction, although still less selective than that for liquid–liquid extraction methods.
| Target Ln pair | Ref. | |||||
|---|---|---|---|---|---|---|
| La–Eu | La–Lu | Nd–Dy | Tb–Lu | Yb–Lu | ||
| Liquid–liquid extraction | ||||||
| D2EHPA | 305 | 49 | ||||
| TODGA/TBP/IL | 1622 | 4.8 | 50 | |||
| TOPS 99 | 483.3 | 51 | ||||
| HEHAPP | 1.93 | 52 | ||||
| Extraction chromatography | ||||||
| HEH[EHP] resin | 1.8 | 53 | ||||
| Solid-phase extraction | ||||||
| Functionalized silica KIT-6-1,2-PA | 65.8 | 47 | ||||
| Functionalized silica Si–Ti alkylphosphate | ∼3 | 11 | ||||
| Zr phosphate | 1.8 | 54 | ||||
| Zr-ATMP | 2.7 | 10 | ||||
| SG85-ATMP | 5.1 | 11.1 | 2.1 | 2.3 | 1.2 | |
| SG85-EDTMP | 3.7 | 10.1 | 2.0 | 2.8 | 1.2 | |
| SG85-DTPMP | 5.6 | 39.1 | 2.4 | 6.7 | 1.4 | |
| SG85-TTHMP | 7.0 | 87.4 | 2.7 | 11.3 | 1.5 | |
Separation studies on the fixed-bed column system.
The applicability of SG85-DTPMP and SG85-TTHMP for selective Ln3+ sorption in a fixed bed column system was investigated, since batch studies indicated that these two were the best materials, in particular for the separation of La3+ and Lu3+. The breakthrough of La3+ and Lu3+ started at 33 BV and 80 BV, respectively, for SG85-DTPMP (Fig. 8(a)), compared to 27 BV and 70 BV for SG85-TTHMP (Fig. 8(d)). The full breakthroughs of La3+ were achieved at 47 BV for SG85-DTPMP and 38 BV for SG85-TTHMP, and after this, the La3+ concentration exceeded the initial feed concentration by about 40–50% due to elution caused by the uptake of Lu3+. These breakthrough curves demonstrate that both materials favoured the Lu3+ uptake over La3+. The change in pH (Fig. 8(b) and (e)) can be divided into three phases which correspond to the phases (1) before the breakthrough, (2) after La3+ but before the Lu3+ breakthrough, and (3) after Lu3+ breakthrough. The pKa values of alkyl amino-phosphonic acid in aqueous solutions are known to be around 2.5 and 7.55 Thus, the majority of phosphonate groups of the grafted ligands are supposed to exist as monoanionic at pH 3. The coordination by deprotonated phosphonate groups is considered to play a major role in the Ln3+ uptake. This uptake apparently leads to further deprotonation of the ligands (in order to stabilize the coordination) which corresponds to the pH changes of the three stages of the column run. Elution studies (Fig. 8(c) and (f)) show that neither 0.1 M nor 0.5 M HNO3 could elute the La and Lu as separate phases.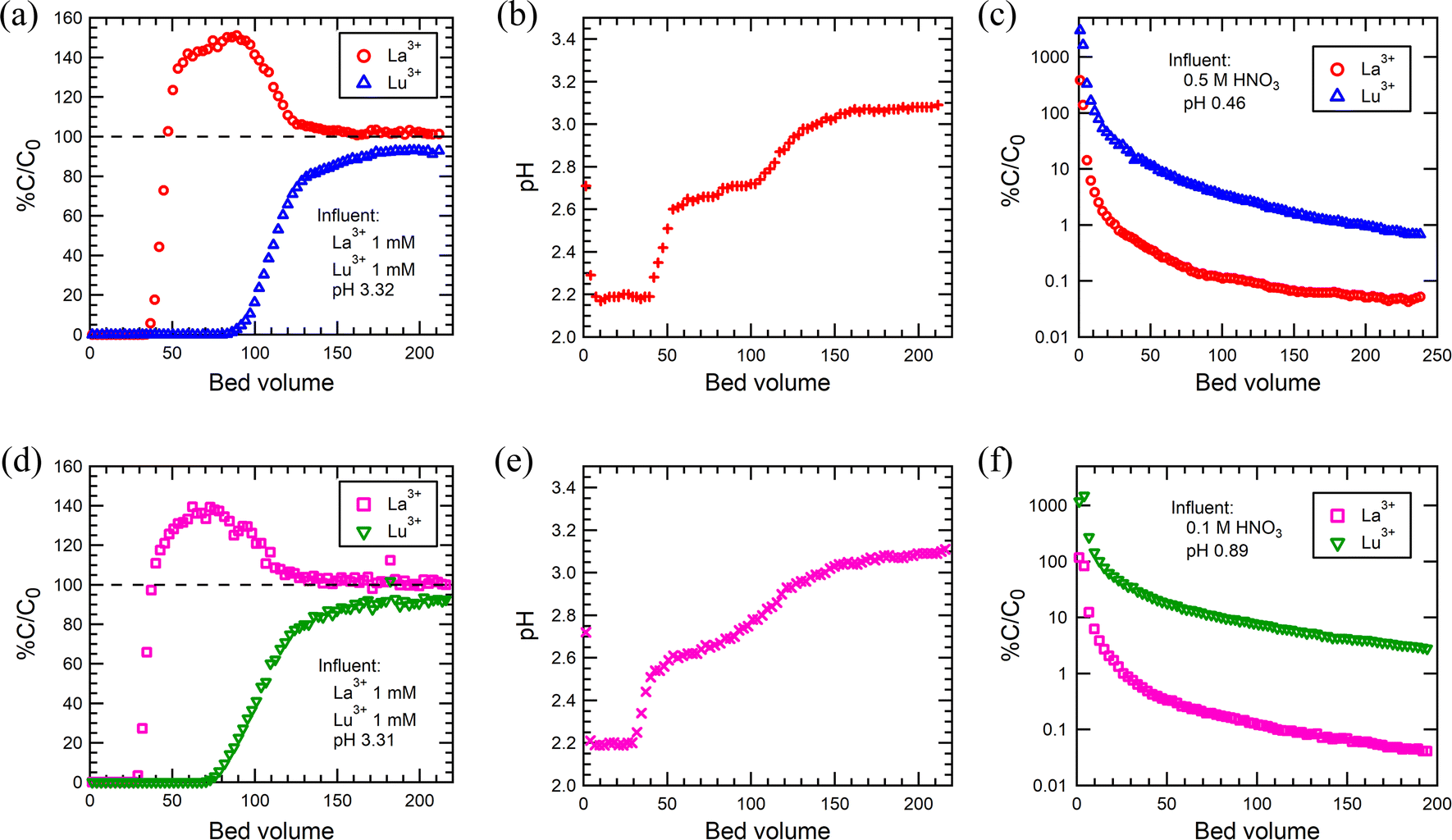 | ||
| Fig. 8 (a) Breakthrough curves of La3+ and Lu3+, (b) pH changes of the effluent, and (c) eluting curves for SG85-DTPMP. The corresponding results for SG85-TTHMP are shown in (d–f). Loading solution: 1 mM equimolar La3+–Lu3+ (pH 3.3). | ||
Column regeneration studies
The regeneration of the sorbent material is important from an economic and environmental point of view, and thus regeneration studies were carried out to evaluate the reusability of the materials. After five loading–eluting cycles, uptake decreased by 26% for SG85-DTPMP and 36% for SG85-TTHMP (Table 4). These results indicate that although the materials seem to lose some of their uptake performance, it is possible to regenerate them without losing their performance drastically and thus they could be reused multiple times. The loss of capacity after regeneration is assumed to be due to the loss of ligands. To investigate this, TGA/DSC was performed on the as-synthesized materials and ones after 5-cycle column runs (Fig. S7, ESI†). The faster weight loss at below 130 °C was observed for the used materials, implying that the used materials have more hydrophilic surfaces, which is similar to that of the bare zirconia (Fig. 2). This could be an indication of the loss of the organic ligands, although the difference was minor and further studies are needed for confirmation.| Cycle | SG85-DTPMP | SG85-TTHMP | ||
|---|---|---|---|---|
| Uptake (μmol BV−1) | %Retention | Uptake (μmol BV−1) | %Retention | |
| 1st | 137.3 ± 0.8 | 122.9 ± 1.2 | ||
| 2nd | 114.7 ± 3.1 | 83.6 ± 2.3 | 105.6 ± 2.1 | 85.9 ± 1.9 |
| 3rd | 110.9 ± 2.1 | 80.8 ± 1.6 | 91.8 ± 3.3 | 74.7 ± 2.8 |
| 4th | 104.2 ± 1.3 | 75.9 ± 1.1 | 82.6 ± 1.2 | 67.2 ± 1.2 |
| 5th | 101.7 ± 2.0 | 74.1 ± 1.6 | 79.1 ± 3.5 | 64.4 ± 2.9 |
Conclusions
In this work, aminophosphonate-based hybrid materials were successfully synthesized by a post-synthetic grafting method using functional ligands and a porous zirconia matrix. All the obtained materials showed a high uptake of lanthanides of roughly 100 μmol g−1 at pH 3, which is significantly higher than those of similar materials previously reported. The uptake would be most likely even higher at a higher pH, considering the better complexation yield of the grafted ligands. The separation studies revealed that all the hybrid materials have the potential for intra-lanthanide separation, and the selectivity is improved as the number of phosphonate groups becomes larger. In particular, the one with a ligand containing most phosphonate groups among the tested, SG85-TTHMP, showed high selectivity with a separation factor for Lu/La of 87.4 ± 9.8. The selectivity of SG85-DTPMP and SG85-TTHMP was comparative or better than those of other reagents for solid-phase extraction in recent studies. The selective sorption was more remarkable for lanthanides of smaller ionic radii and ligands with larger availability of phosphonate groups due to the increase of ion–dipole interactions. The coordination number in the Ln3+ complex and the tetrad effect are also assumed to be important factors for the observed variation in selectivity. The selectivity of SG85-DTPMP and SG85-TTHMP was also confirmed by a fixed-bed column operation to favour the uptake of Lu3+ over La3+. The regeneration studies showed that these materials have the potential to be reused multiple times, even though some of the uptake performance was lost during five regeneration cycles. The observed pH changes of the effluent while loading imply further deprotonation of phosphonate groups of the grafted ligands to stabilize the ligand–Ln3+ coordination. This lab-scale column study implies the possible application of these materials for a selective cartridge process for lanthanide separation in actual industrial settings. In summary, the detailed comparison of the synthesized materials in this study provides further insights into the sorption characteristics of phosphonate-based hybrids. In addition, the hybrid materials composed of the porous zirconia matrix and organic ligands with a larger number of phosphonate groups realized high selectivity and stability, which possibly proposes strategies for designing selective sorbent synthesis. Further studies on eluting agents will be made since the elution process also play an important role in selective separation and recovery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to the results is funded by the Academy of Finland (USEMA project, decision no. 316091). SEM imaging was performed in the ALD centre Finland research infrastructure. M.O. thanks Ms Gudrun Silvennoinen for combustion elemental analysis.Notes and references
- S. Cotton, Lanthanide and Actinide Chemistry, John Wiley & Sons Ltd., West Sussex, 2006 Search PubMed.
- C. S. Cutler, C. J. Smith, G. J. Ehrhardt, T. T. Tyler, S. S. Jurisson and E. Deutsch, Cancer Biother. Radiopharm., 2000, 15, 531–545 CrossRef CAS PubMed.
- T. Cheisson and E. J. Schelter, Science, 2019, 363, 489–493 CrossRef CAS PubMed.
- E. O. Opare, E. Struhs and A. Mirkouei, Renewable Sustainable Energy Rev., 2021, 143, 110917 CrossRef CAS.
- H. P. Neves, G. M. D. Ferreira, G. M. D. Ferreira, L. R. De Lemos, G. D. Rodrigues, V. A. Leão and A. B. Mageste, Sep. Purif. Technol., 2022, 282, 120064 CrossRef.
- E. P. Horwitz, D. R. McAlister and M. L. Dietz, Sep. Sci. Technol., 2006, 41(10), 2163–2182 CrossRef CAS.
- B. Chen, M. He, H. Zhang, Z. Jiang and B. Hu, Phys. Sci. Rev., 2017, 2(4), 20160057 Search PubMed.
- S. A. Ansari and P. K. A. Mohapatra, J. Chromatogr. A, 2017, 1499, 1–20 CrossRef CAS PubMed.
- J. Florek, F. Chalifour, F. Bilodeau, D. Larivière and F. Kleitz, Adv. Funct. Mater., 2014, 24, 2668–2676 CrossRef CAS.
- J. Veliscek-Carolan, T. L. Hanley and V. Luca, Sep. Purif. Technol., 2014, 129, 150–158 CrossRef CAS.
- W. Zhang, D. Avdibegović, R. Koivula, T. Hatanpää, S. Hietala, M. Regadío, K. Binnemans and R. Harjula, J. Mater. Chem. A, 2017, 5, 23805–23814 RSC.
- Y. Hu, J. Florek, D. Larivière, F.-G. Fontaine and F. Kleitz, Chem. Rec., 2018, 18, 1261–1276 CrossRef PubMed.
- J. Florek, S. Giret, E. Juère, D. Larivière and F. Kleitz, Dalton Trans., 2016, 45, 14832–14854 RSC.
- S. E. Mourabit, M. Guillot, G. Toquer, J. Cambedouzou, F. Goettmannb and A. Grandjean, RSC Adv., 2012, 2, 10916–10924 RSC.
- A. Clearfield, Solvent Extr. Ion Exch., 2000, 18(4), 655–678 CrossRef CAS.
- J. Nawrocki, C. Dunlap, A. McCormick and P. W. Carr, J. Chromatogr. A, 2004, 1028(1), 1–30 CrossRef CAS PubMed.
- J. Nawrocki, M. Rigney, A. McCormick and P. W. Carr, J. Chromatogr. A, 1993, 657(2), 229–282 CrossRef CAS PubMed.
- G. Guerrero, P. H. Mutin and A. Vioux, Chem. Mater., 2001, 13(11), 4367–4373 CrossRef CAS.
- P. H. Mutin, G. Guerrero and A. Vioux, C. R. Chim, 2003, 6(8–10), 1153–1164 CrossRef CAS.
- P. H. Mutin, G. Guerrero and A. Vioux, J. Mater. Chem., 2005, 15, 3761–3768 RSC.
- K. J. Gagnon, H. P. Perry and A. Clearfield, Chem. Rev., 2012, 112(2), 1034–1054 CrossRef CAS PubMed.
- J. Veliscek-Carolan, A. Rawal, V. Luca and T. L. Hanley, Microporous Mesoporous Mater., 2017, 252, 90–104 CrossRef CAS.
- J. Veliscek-Carolan, A. Rawal, D. T. Oldfield, G. J. Thorogood and N. M. Bedford, ACS Appl. Nano Mater., 2020, 3, 3717–3729 CrossRef CAS.
- V. Luca and J. Veliscek-Carolan, Phys. Chem. Chem. Phys., 2020, 22, 17027–17032 RSC.
- S. Lacour, V. Deluchat, J.-C. Bollinger and B. Serpaud, Talanta, 1998, 46(5), 999–1009 CrossRef CAS PubMed.
- S. Chakraborty, T. Das, P. R. Unni, H. D. Sarma, G. Samuel, S. Banerjee, M. Venkatesh, N. Ramamoorthy and M. R. Pillai, Nucl. Med. Commun., 2002, 23(1), 67–74 CrossRef CAS PubMed.
- Y. Zhu, T. Ren and Z. Yuan, Catal. Sci. Technol., 2015, 5, 4258–4279 RSC.
- C. S. Griffith, M. De Los Reyes, N. Scales, J. V. Hanna and V. Luca, ACS Appl. Mater. Interfaces, 2010, 2(12), 3436–3446 CrossRef CAS PubMed.
- K. Moedritzer and R. R. Irani, J. Org. Chem., 1966, 31(5), 1603–1607 CrossRef CAS.
- W. A. Schafer, P. W. Carr, E. F. Funkenbusch and K. A. Parson, J. Chromatogr. A, 1991, 587(2), 137–147 CrossRef CAS.
- G. H. Dahl and B. P. Block, Inorg. Chem., 1967, 6(8), 1439–1443 CrossRef CAS.
- M. C. Zenobi, C. V. Luengo, M. J. Avena and E. H. Rueda, Spectrochim. Acta, Part A, 2010, 75(4), 1283–1288 CrossRef PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87(9–10), 1051–1069 CrossRef CAS.
- M. M. Gómez-Alcántara, A. Cabeza, P. Olivera-Pastor, F. Fernandez-Moreno, I. Sobrados, J. Sanz, R. E. Morris, A. Clearfield and M. A. G. Aranda, Dalton Trans., 2007, 2394–2404 RSC.
- X.-Z. Lin and Z.-Y. Yuan, Eur. J. Inorg. Chem., 2012, 2661–2664 CrossRef CAS.
- W. Gao, L. Dickinson, C. Grozinger, F. G. Morin and L. Reven, Langmuir, 1996, 12(26), 6429–6435 CrossRef CAS.
- D. Massiot, S. Drumel, P. Janvier, M. Bujoli-Doeuff and B. Bujoli, Chem. Mater., 1997, 9(1), 6–7 CrossRef CAS.
- I. G. Shenderovich and H.-H. Limbach, Solids, 2021, 2, 139–154 CrossRef CAS.
- R. Chiarizia, D. R. McAlister and A. W. Herlinger, Sep. Sci. Technol., 2005, 40(1–3), 69–90 CrossRef CAS.
- K. L. Nash and M. P. Jensen, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneider Jr. and L. Eyring, Elsevier, Amsterdam, 2000, ch 180, vol. 28, pp. 311–371 Search PubMed.
- F. H. Spedding, J. A. Rard and A. Habenschuss, J. Phys. Chem., 1977, 81(11), 1069–1074 CrossRef CAS.
- E. N. Rizkalla and G. R. Choppin, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneider Jr. and L. Eyring, Elsevier, Amsterdam, 1991, ch. 103, vol. 15, pp. 393–442 Search PubMed.
- I. Kawabe, Geochem. J., 1992, 26, 309–335 CrossRef CAS.
- I. Kawabe and A. Masuda, Geochem. J., 2001, 35(4), 215–224 CrossRef CAS.
- I. Kawabe, Geochem. J., 1999, 33(4), 267–275 CrossRef CAS.
- M. Majdan, A. Gładysz-Płaska, S. Pikus, D. Sternik, O. Maryuk, E. Zięba and P. Sadowski, J. Mol. Struct., 2004, 702, 95–102 CrossRef CAS.
- Y. Hu, E. Drouin, D. Larivière, F. Kleitz and F.-G. Fontaine, ACS Appl. Mater. Interfaces, 2017, 9(44), 38584–38593 CrossRef CAS PubMed.
- Y. Hu, L. C. M. Castro, E. Drouin, J. Florek, H. Kählig, D. Larivière, F. Kleitz and F.-G. Fontaine, ACS Appl. Mater. Interfaces, 2019, 11(26), 23681–23691 CrossRef CAS PubMed.
- M. Mohammadi, K. Forsberg, L. Kloo, J. Martinez De La Cruz and Å. Rasmuson, Hydrometallurgy, 2015, 156, 215–224 CrossRef CAS.
- A. N. Turanov, V. K. Karandashev and M. Boltoeva, Hydrometallurgy, 2020, 195, 105367 CrossRef CAS.
- S. Radhika, B. N. Kumar, M. L. Kantam and B. R. Reddy, Sep. Purif. Technol., 2010, 75(3), 295–302 CrossRef CAS.
- S. Kuang, Z. Zhang, Y. Li, H. Wei and W. Liao, J. Rare Earths, 2018, 36(3), 304–310 CrossRef CAS.
- E. P. Horwitz, D. R. McAlister, A. H. Bond, R. E. Barrans and J. M. Williamson, Appl. Radiat. Isot., 2005, 63(1), 23–36 CrossRef CAS PubMed.
- J. Xu, R. Koivula, W. Zhang, E. Wiikinkoski, S. Hietala and R. Harjula, Hydrometallurgy, 2018, 175, 170–178 CrossRef CAS.
- L. D. Freedman and G. O. Doak, Chem. Rev., 1957, 57(3), 479–523 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of measurement methods and further characterization results. See DOI: https://doi.org/10.1039/d2ma00928e |
| This journal is © The Royal Society of Chemistry 2023 |