
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bifunctional intermetallic PdZn nanoparticle-loaded deficient TiO2 nanosheet electrocatalyst for electrochemical water splitting†
Keerti M.
Naik
 *,
Kanaru
Hashisake
,
Eiji
Higuchi
and
Hiroshi
Inoue
*
*,
Kanaru
Hashisake
,
Eiji
Higuchi
and
Hiroshi
Inoue
*
Department of Applied Chemistry, Graduate School of Engineering, Osaka Metropolitan University (formally Osaka Prefecture University), Sakai, Osaka 599-8531, Japan. E-mail: keertiraman@gmail.com; inoue-chem@omu.ac.jp
First published on 22nd November 2022
Abstract
Catalysts for electrochemical water splitting are receiving remarkable consideration, and raw materials and methods for preparing catalysts are important. Through an impregnation method using low-temperature heating, a concise technique for the preparation of layered TiO2−x NSs incorporating PdZn nanoparticles (NPs) has been established in this work. With an overpotential of 64 mV at 10 mA cm−2 and a low Tafel slope of 68 mV dec−1 in 1 M KOH at ambient temperature, the synthesized PdZn NP-decorated TiO2−x NSs (PdZn/TiO2−x NSs) showed better catalytic activity for the hydrogen evolution reaction (HER). The same was observed for the oxygen evolution reaction (OER); the PdZn/TiO2−x NSs showed satisfactory electrocatalytic performance, delivering a current density of 10 mA cm−2 at an overpotential of 0.46 V and a Tafel slope of 115 mV dec−1. Furthermore, the PdZn/TiO2−x NS electrocatalyst also exhibited high stability in a constant voltage electrochemical water splitting operation for 35 h. The synergistic impact of TiO2−x NSs and PdZn, higher conductivity, and large electrochemical active surface area are all factors contributing to the electrocatalyst's improved HER and OER performance. The highly active electrocatalyst demonstrated in this work, as well as its simple preparation method, will encourage the development of other transition metal catalysts supported on carbon-free nanostructured supports for future large-scale applications in electrochemical water splitting.
Introduction
The worldwide energy issues and ecological contamination have inspired extreme research on creating inexhaustible and clean energy.1 Electrochemical water splitting is a promising innovation to deliver hydrogen, which is an energy source that is sustainable, secure and harmless for the ecosystem.2,3 Electrochemical water splitting is a feasible way to achieve the hydrogen evolution reaction (HER) on the cathode and the oxygen evolution reaction (OER) on the anode in a mole proportion of 2.4 The overall efficiency, however, is strictly constrained by the sluggish kinetics of the four-electron transfer process for the OER, leading to a larger driving cell voltage of 1.8–2.0 V than the theoretical thermodynamic requirement of 1.23 V in water splitting.5 Highly active OER catalysts are consequently significant in bringing down the energy hindrance and speeding up the electrode reaction.6,7 At present, novel metal oxides (like IrO2 and RuO2) are prominent as the most productive catalysts for the OER, as is Pt for the HER, but they are not suitable options for large scale applications because of their immense cost and scarcity. Thus, substantial research attempts have been devoted to developing and manufacturing bifunctional electrocatalysts, particularly those using bimetallic materials for water splitting.8,9Carbon-based materials have been widely utilized as supports because of their high conductivity; however, the corrosion of carbon-based materials, and its metal composites (Ni, Co, etc. in acidic medium), can bring down the general performance of the catalysts.10,11 Recently, much effort has been devoted to developing non-carbon electrocatalysts particularly by utilizing 3d transition metal oxides.12,13 Distinguishable changes in the OER activity of these materials have been observed because of the crystallinity, surfaces, heterostructures and morphology of nanoparticles.12,13 Consequently, developing a dynamic and stable bimetallic electrocatalyst-stacked metal oxide for general water splitting is vital and is one of the predominant challenges in this field of research.
Among various inorganic materials (e.g., Ti, In, Al, Si, and W oxides, nitrides, and carbides) broadly considered as planned supports,14 titanium dioxide shows amazing reliability in an oxidizing environment. In addition, it is well-known that strong metal–support interaction (SMSI) is observed between the catalyst metals and the TiO2 support, which confirmed that 4f binding energy of Pt in Pt/TiO2 is lower than that of Pt in Pt/C.15 This interaction improves the catalytic performance and stability of the materials. In particular, the insulating nature of the TiO2 support prevents the electron transport required for the electrochemical reactions. The catalytic performance can be enhanced by doping highly conductive components with TiO2 or utilizing TiO2 nanomaterials with high surface area.15–20 Doping with Ti3+ and creating oxygen vacancies result in the arrangement of non-stoichiometric TiO2 and creation of TiO2−x phases, which are broadly used as photocatalysts.18 This band gap reduction increases the conductivity and is advantageous for the manufacture of high-performance catalysts. In our previous study, we focused on optimizing the interaction of Pt with metal oxide supports via improving the conductivity and the SMSI effect to enhance catalytic activity.20
Nowadays, Pd-based catalysts have attracted extensive research interest because of their excellent methanol-tolerance and high catalytic activity in alkaline media, although the catalytic activity of pure Pd is not enough to replace Pt in fuel cells.21,22 According to the Sabatier principle,23 the catalytic activity of Pd-based catalysts for the ORR can be controlled by changing the inter-atomic distance of Pd–Pd (strain impact) and in this manner altering the binding attraction between oxygen molecules and the active sites of the catalysts. Adding foreign metals (such as Zn, Cu, Fe, Co or Ni) with different lattice constants into Pd has been demonstrated as a viable approach to change the Pd–Pd inter-atomic distance,24 which plays a vital role in the electrocatalytic performance. In any case, the formed bimetallic compounds often have strong arrangements, in which the foreign metal atoms are arbitrarily distributed.25 The activity of Pd-based bimetallic electrocatalysts is primarily affected by the Pd/M ratio, total metal loading, surface structural characteristics, particle size, crystal orientation and surface structural defect. In addition to the strain (lattice concentration) effect, the chemisorptive properties of Pd can moreover be modified through electron exchange between the Pd and the instant metal (ligand impact), expanding the d-band vacancies or bringing down the d-band center. A PdZn alloy is known to be an inter(bi)metallic compound. The electronic structure of the bimetallic PdZn is completely different from that of the constituents and similar to that of pure Cu.26 Interestingly, the bimetallic PdZn catalyst showed high catalytic activity in methanol dehydrogenation, CO2 hydrogenation to methanol, reverse water-gas shift reaction and glycerol hydrogenolysis.26–28
The current work focuses on the appropriateness of bimetallic PdZn supported on conductive TiO2−x nanosheets (PdZn/TiO2−x NSs) as an effective bifunctional electrocatalyst for both the OER and HER as well as for the overall water splitting in alkaline solution. Moreover, in this work, a simple methodology was proposed for the large-scale generation of PdZn/TiO2−x NSs by impregnating TiO2 NSs with equimolar Pd acetic acid and Zn acetic acid, taken after heating at a low temperature. The PdZn/TiO2−x NS catalyst shows excellent HER performance, delivering a current density of 10 mA cm−2 with an overpotential of 64 mV in 1 M KOH and a low Tafel slope of 68 mV dec−1. As for OER catalysis, the PdZn/TiO2−x NS catalyst exhibited impressive electrocatalytic activity with an overpotential of 0.46 V to obtain a current density of 10 mA cm−2 and a Tafel slope of 115 mV dec−1. Furthermore, the PdZn/TiO2−x NS catalyst exhibited better stability during the long-term constant voltage operation of both the HER and OER. This enhanced performance of the PdZn/TiO2−x NS catalyst is related to the highly interactive, larger surface areas of TiO2−x NS and bimetallic PdZn.
Experimental section
Synthesis of a-TiO2 NSs
Based on the previous work, anatase TiO2 (a-TiO2) NSs were prepared using a simple hydrothermal process.15 In a dry Teflon-lined autoclave, Ti(OC4H9)4 (Sigma-Aldrich) and 50 wt% HF solution (Wako Pure Chemicals) were mixed at ambient temperature, and after that maintained at 180 °C for 24 h. Caution! Because of its severe corrosiveness and contact toxicity, hydrofluoric acid should be handled with extra care. Fluorine has an atomic ratio of 1.0 with titanium. The final product was washed with alcohol and water just after the completion of the hydrothermal reaction and thereafter oven dried at 80 °C.Synthesis of PdZn/TiO2−x NSs by the impregnation process
Palladium acetate (8.01 mg) (Wako Pure Chemicals), zinc acetate (9.8 mg) (Wako Pure Chemicals) and TiO2 NS powder (67.5 mg) were mixed in THF solution (75 mL) (Wako Pure Chemicals) in an eggplant flask, and ultrasonically dispersed for 20 min. Afterwards, the solution was heat treated to around 70 °C to evaporate THF and then the solid residue was annealed in a tubular furnace at 400 °C for 3 h in an (Ar + H2) mixed gas (97 + 3%) at a flow rate of 30 mL min−1, as illustrated in Scheme 1. The finally obtained black powder was named PdZn/TiO2−x NSs. For comparison, PdZn/C and Pt/C were prepared by the same impregnation process by employing carbon black powder as the support. Also, K2PtCl4 was used as a Pt precursor in the same procedure for the preparation of Pt/C.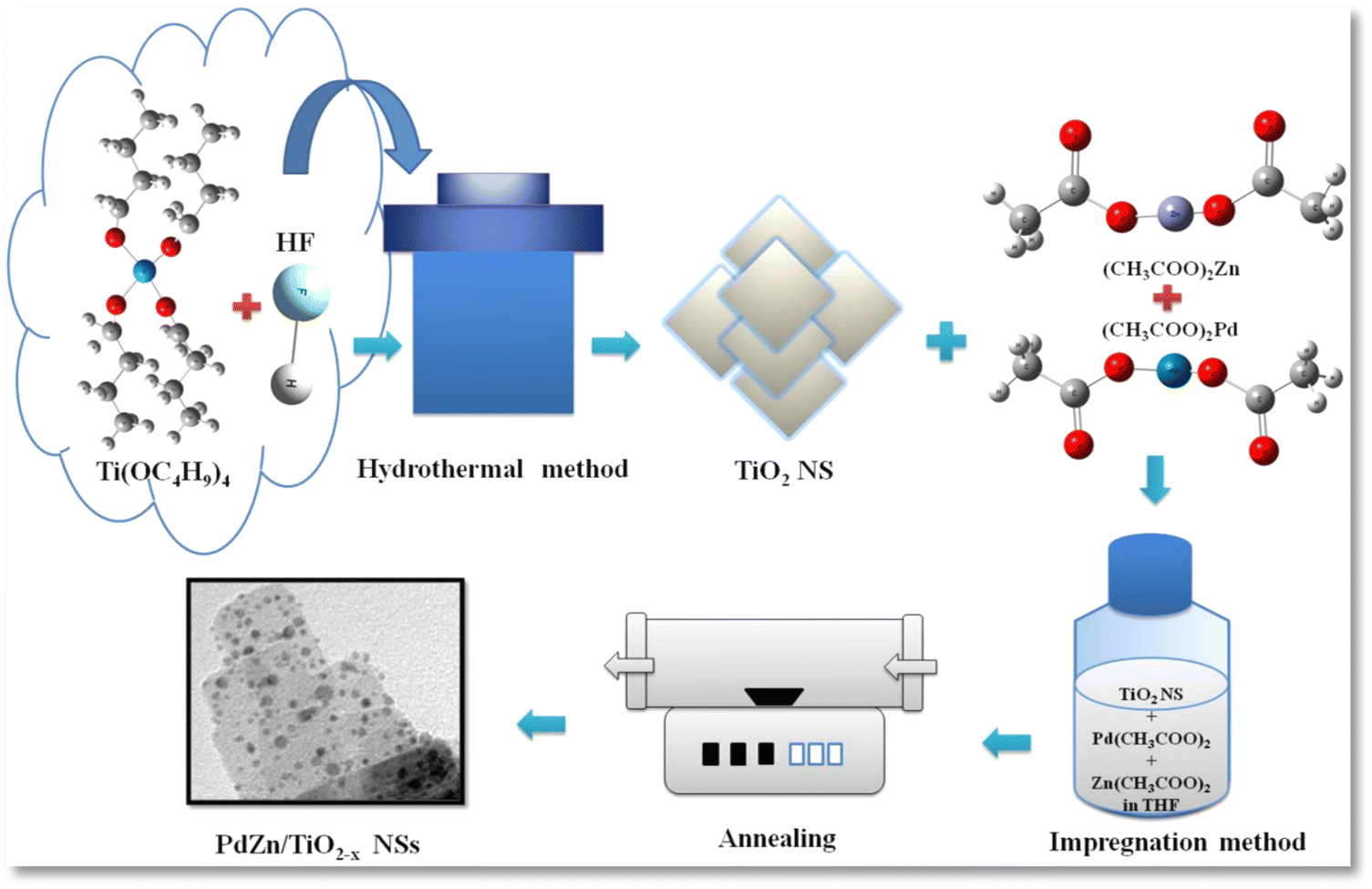 | ||
| Scheme 1 Schematic representation of the synthesis of PdZn/TiO2−x NSs. | ||
Physicochemical characterization
A Hitachi SU8230 scanning electron microscope and transmission electron microscopy (TEM) and high-resolution TEM (HR-TEM) were used to examine the morphologies of the synthesized electrocatalysts at 15 kV and 200 kV acceleration voltage, respectively. Structural information was obtained through X-ray diffraction (XRD) patterns by using a X-ray diffractometer (Shimadzu XRD-6100) with monochromatic Cu Kα radiation (λ = 0.1541 nm, 50 kV, 30 mA) in the 2θ range of 10–80°. A Jasco NRS-3100 Laser Raman spectrometer with a 532 nm green laser was used to record the Raman spectra of the samples at ambient temperature. A UV-Vis spectrophotometer (UV-2100PC, Shimadzu) was used to calculate the band gap of the materials in the spectral range of 200–800 nm. X-ray photoelectron spectroscopy (XPS, ECSA-3400, Shimadzu) operating at 10 kV and 20 mA was used to investigate the surface chemistry of the catalysts. The catalyst composition was determined using an atomic absorption spectrophotometer (AAS-6650, Shimadzu)Electrochemical measurements
Electrochemical estimations were carried out by utilizing a three-electrode setup of CHI 730 C electrochemical workstation. A Pt plate was utilized as the counter cathode. The reversible hydrogen electrode (RHE) and Hg/HgO electrode were used as the reference electrode, respectively. All the potentials reported in this work were changed over to the RHE scale using the relationship E(RHE) = E(Hg/HgO) + 0.059 pH + E°(Hg/HgO). Cyclic voltammograms (CVs) were recorded at 50 mV s−1 between 1.20 and 0.05 V in an Ar-saturated 0.5 M H2SO4 or 1 M KOH aqueous solution.The catalyst ink was prepared by ultrasonicating PdZn/TiO2−x NS powders in 1 mL of 2-propanol/water mixed solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 volume ratio), taken after by including 10 μL of 5 wt% Nafion solution and after that dropped (10 μL) on to a cleaned glassy carbon (GC, 5 mm in diameter) with a PdZn-loading of 81.0 μg cm−2. For HER measurements, 0.5 M H2SO4 and 1 M KOH aqueous solutions were used as the electrolyte, while for OER measurements 1 M KOH solution was used. For evaluation, PdZn/C and Pt/C were also prepared according to the above method with the exception of using Ketjen black (Lion Specialty Chemicals, EC300J) instead of TiO2−x NSs. For comparison, PdZn/C, Pt/C and commercial IrO2 were also explored under similar circumstances.
:4 volume ratio), taken after by including 10 μL of 5 wt% Nafion solution and after that dropped (10 μL) on to a cleaned glassy carbon (GC, 5 mm in diameter) with a PdZn-loading of 81.0 μg cm−2. For HER measurements, 0.5 M H2SO4 and 1 M KOH aqueous solutions were used as the electrolyte, while for OER measurements 1 M KOH solution was used. For evaluation, PdZn/C and Pt/C were also prepared according to the above method with the exception of using Ketjen black (Lion Specialty Chemicals, EC300J) instead of TiO2−x NSs. For comparison, PdZn/C, Pt/C and commercial IrO2 were also explored under similar circumstances.
Electrochemically active surface area (ECSA) of palladium cannot be accurately obtained through the redox of adsorbed atomic hydrogen due to the hydrogen absorption in palladium.29 Therefore, the ECSA of Pd-based catalysts is determined by using the charge (QR) required for the reduction of the surface oxide, PdO, and using the next relation,29
| ECSA = QR/Qref | (1) |
To evaluate HER and OER activity of catalyst-coated electrodes, hydrodynamic voltammograms (HVs) with a rotating disk electrode (RDE) were recorded at 5 mV s−1 from 0.1 to −0.5 V vs. RHE for the HER and 1.2 to 1.9 V vs. RHE for the OER in an Ar-saturated 0.5 M H2SO4 or 1 M KOH aqueous solution. Each RDE was rotated at 1600 rpm in order to eliminate the effects of gas bubbles during the HV measurement. Tafel plots were drawn with the help of Liner-sweep voltammograms (LSVs) for assessing the HER and OER kinetics of the catalyst. Electrochemical impedance spectroscopic data (Nyquist plots) for three electrodes were collected over a frequency range from 10 kHz to 0.1 Hz with an amplitude of 5 mV at different potentials with respect to the RHE. The chronoamperometric experiments were carried out to validate the catalyst stability.
The overall water splitting was performed by utilising the two-electrode arrangement in 1 M KOH solution. The geometric surface area of the GC substrates was 1.0 cm2, and the loading of the PdZn/TiO2−x NS catalyst for both electrodes was 0.1 mg cm−2.
Results and discussion
Morphological and structural characterization
The XRD pattern of PdZn/TiO2−x NSs is shown in Fig. 1a with the standard diffraction patterns of a-TiO2 (JCPDS-021-1272) and bimetallic PdZn (JCPD-006-0620). The small diffraction peaks centered at ca. 26.8°, 41.3°, 44° and 54.6° were assigned to the (001), (111), (200) and (002) planes in the standard diffraction pattern of bimetallic PdZn. The lattice parameters (a = 0.4 nm, b = 0.4 nm, c = 0.33 nm) were calculated with these diffraction peaks, and similar to the theoretical values (a = 0.41 nm, b = 0.41 nm, c = 0.34 nm) for the tetragonal structure of bimetallic PdZn.30 The high and sharp peaks assigned to TiO2−x NSs in Fig. 1a were also observed in the XRD pattern of TiO2 NSs (Fig. S1, ESI†), suggesting that TiO2−x NSs have the same crystal structure of TiO2 NSs. The calculated lattice parameters of a = 0.37 nm and c = 0.94 nm from the diffraction peaks assigned to TiO2−x NSs were similar to the theoretical values of a-TiO2 (a = 0.3785 nm and c = 0.9514 nm). These results suggest that the anatase structure of TiO2 NSs was also retained in TiO2−x NSs. The XRD pattern of PdZn/C (Fig. S2, ESI†) also suggests the formation of intermetallic PdZn on the carbon support.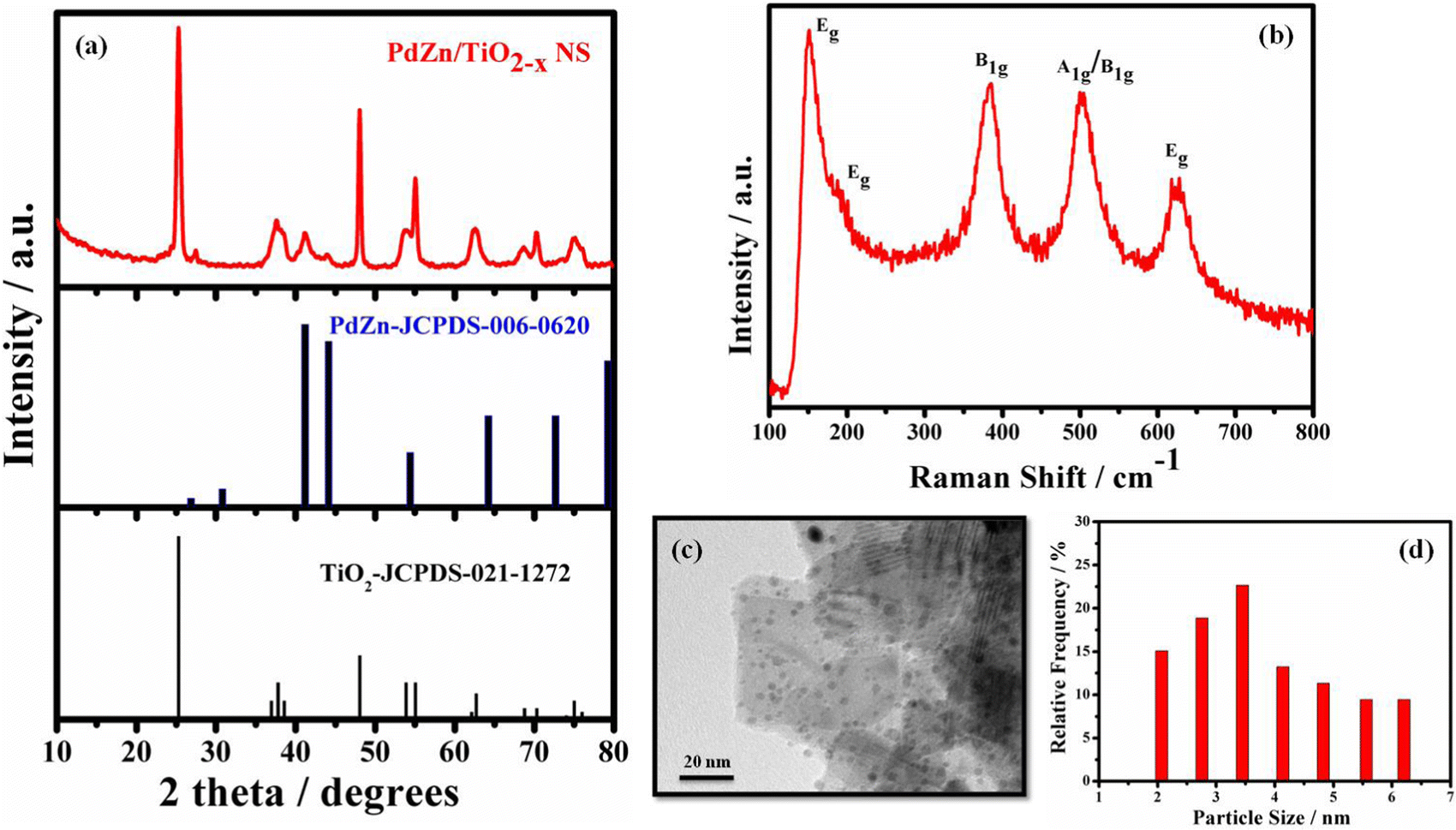 | ||
| Fig. 1 (a) XRD pattern of PdZn/TiO2−x NSs and standard XRD patterns of PdZn (JCPDS-006-0620) and TiO2 (JCPDS-021-1272), (b) Raman spectrum, (c) TEM image and (d) particle size distribution histogram of PdZn/TiO2−x NSs. | ||
Raman spectroscopy was used to provide further information about the structural change in the TiO2−x NSs during PdZn NP loading. The active modes of peaks at 150.4, 190.2, 384.5, 502.8 and 625.7 cm−1 in the Raman spectra of PdZn/TiO2−x NSs (Fig. 1b) are ascribed to the Eg, Eg, B1g, A1g or B1g, and Eg vibration modes of a-TiO2, respectively. Fig. S3 (ESI†) shows the Raman spectrum of TiO2 NSs for comparison. The external vibration of the Ti–O bond blue-shifted the Eg mode at 148.6 cm−1 for TiO2 NSs by around 1.8 cm−1 compared to PdZn/TiO2−x NSs, which may be directly related to the presence of oxygen deficiencies or O-vacancies on the TiO2−x NS surface for PdZn/TiO2−x NSs.31,32
The TEM image shows that the PdZn/TiO2−x NSs exhibit a nanoparticle decorated nanosheets structure (Fig. 1c). Similar features also seen for the PdZn/C and Pt/C samples (Fig. S4, ESI†). It is also confirmed from the images that the PdZn NPs have a size of ∼3.5 nm and are evenly scattered on the TiO2−x NSs plane (Fig. 1d).
XPS analysis allowed one to reveal the composition and the oxidation states of all elements present in the PdZn/TiO2−x NS catalyst. As observed in Fig. 2a, in the high-resolution Ti 2p core level spectrum of the PdZn/TiO2−x NSs doublet peaks appeared at 460.0 and 466.0 eV, which were deconvoluted to doublet peaks at 459.7 and 465.4 eV for Ti3+ and those at 460.4 and 466.1 eV for Ti4+. The analysis shows the presence of Ti3+ in PdZn/TiO2−x NSs, which is obtained from the reduced Ti4+ during heat treatment.
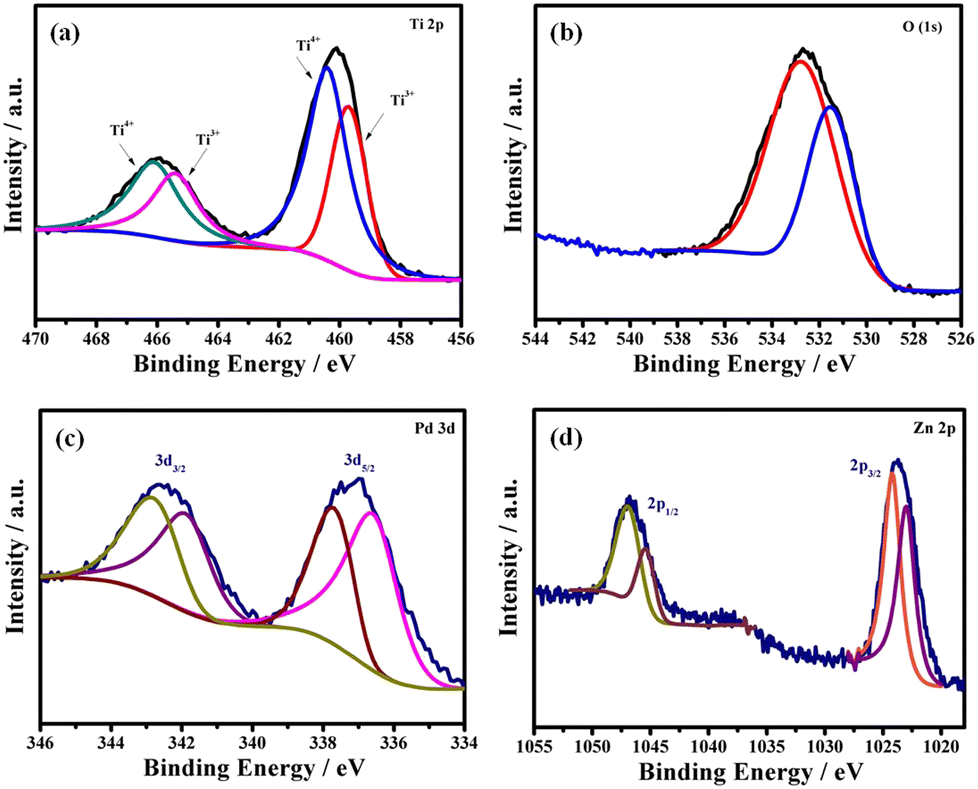 | ||
| Fig. 2 (a) Ti 2p, (b) O 1s, (c) Pd 3d and (d) Zn 2p core level spectra of PdZn/TiO2−x NSs. | ||
The O 1s spectrum of PdZn/TiO2−x NSs shows a peak at around 532.6 eV, as shown in Fig. 2b. The deconvoluted peak at 531.5 eV is attributed to the Ti–O–Ti band from the TiO2 crystal lattice, while that at 532.8 eV is assigned to non-lattice or oxygen vacancy in TiO2 NS.33
Fig. 2c shows doublet peaks at 336.9 and 342.5 eV ascribed to metallic Pd 3d5/2 and 3d3/2, respectively in the Pd 3d core level spectrum of PdZn/TiO2−x NSs. Deconvoluting these two peaks into two pairs of doublets was possible. These spectra clearly show that the metallic Pd doublets for PdZn/TiO2−x NSs changed to a higher binding energy than the Pd/C. Partial electron transfer from Pd to Ti4+ in TiO2−x or/and Zn might indicate the positive binding energy shift for Pd. In general, partial electron loss causes an increase in the binding energy in the core level spectrum of metallic elements. The d-band center shift relative to the Fermi level is directly connected to the binding energy shift.34 Due to the partial oxidation of the Pd surfaces, the fitting data for PdZn/TiO2−x NSs (Fig. 2c) also revealed peaks for Pd2+ at 336.6 and 341.9 eV including the metallic Pd peaks.35,36
The high-resolution Zn 2p spectra are given in Fig. 2d. The Zn 2p1/2 and Zn 2p3/2 doublet peaks might be deconvoluted into a pair of two sub peaks. Zn(II) species were assigned two peaks at 1045.4 eV and 1023.1 eV, whereas metallic Zn was assigned two peaks at 1046.9 eV and 1024.2 eV.37 The higher shift of the metallic Pd 3d peaks as well as the higher shift of the Zn 2p peaks clearly indicates the formation of electronic interconnection among Pd and Zn atoms in the PdZn/TiO2−x NSs catalyst, which could be important in the electrocatalytic reaction because the reactants and product adsorption energies can be adjusted.38,39
Hydrogen evolution activity
The electrocatalytic activity of PdZn/TiO2−x NSs for the HER was evaluated through LSV. Fig. 3a shows HVs at a rotation speed of 1600 rpm for PdZn/TiO2−x NS-, PdZn/C-, TiO2 NS-, TiO2−x NS- and Pt/C-loaded GC electrodes in an Ar-saturated 0.5 M H2SO4 aqueous solution. The PdZn/C- and PdZn/TiO2−x NS-loaded GC electrodes showed an onset potential of about 0 V vs. RHE, which is similar to the Pt/C electrode. The PdZn/C catalyst showed a low overpotential of 17 mV at a geometric cathodic current density of 10 mA cm−2, which is smaller than Pt/C (25 mV). This suggests that the bimetallic PdZn NPs have the same HER activity as Pt NPs. In addition, the overpotential at 10 mA cm−2 for the PdZn/TiO2−x NS was 40 mV, which is close to that of the Pt/C electrode.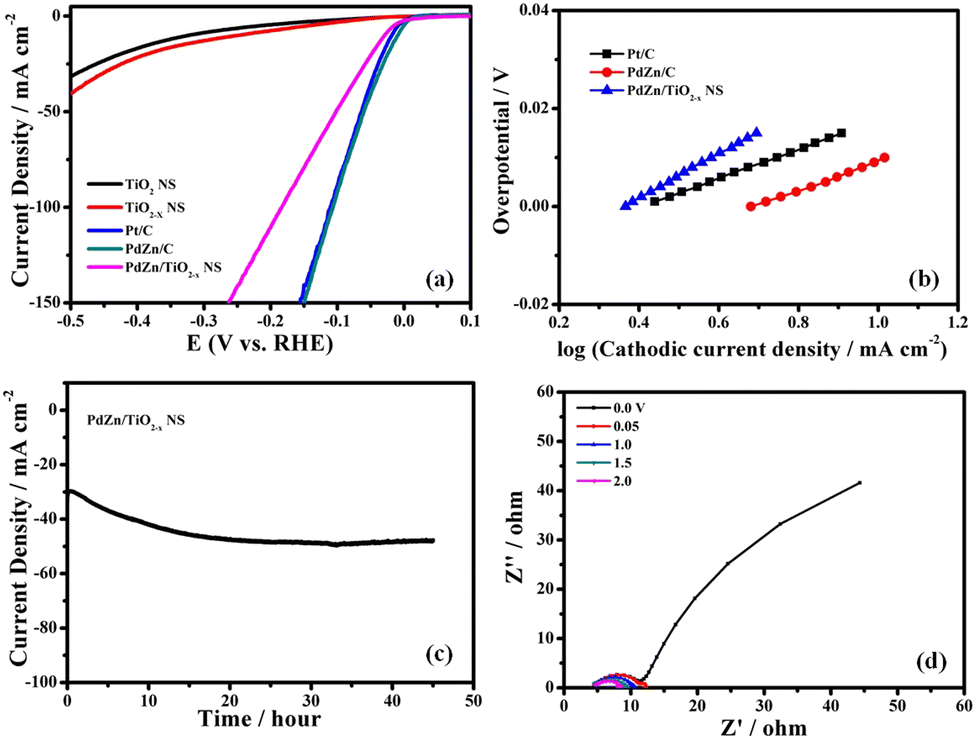 | ||
| Fig. 3 (a) Hydrodynamic voltammograms at 1600 rpm of PdZn/TiO2−x NSs, PdZn/C, TiO2−x NSs, TiO2 NSs and Pt/C, (b) Tafel plots of PdZn/TiO2−x NSs, PdZn/C and Pt/C, (c) change in current density with time in the potentiostatic HER measurement at −0.05 V vs. RHE, and (d) Nyquist plots of PdZn/TiO2−x NSs at different cathodic overpotentials in an Ar-saturated 0.5 M H2SO4 solution. Temperature: 25 °C. | ||
For PdZn/TiO2−x NS-, PdZn/C- and Pt/C-loaded GC electrodes, Tafel plots were prepared with their HVs in Fig. 3a, and given in Fig. 3b. Each fitted to the Tafel equation: η = blogj + a, where η is the overpotential, j is current density, b is the Tafel slope, and a is a constant.40 From Fig. 3b, the Tafel slopes of the PdZn/TiO2−x NS-, PdZn/C-, and Pt/C-loaded GC electrodes are 40, 30 and 29 mV dec−1, respectively. The possible HER elemental reactions in acidic solution are represented as Volmer reaction:41
| H+ + e− → Hads | (2) |
| Hads + H+ + e− → H2 | (3) |
| Hads + Hads → H2 | (4) |
The Tafel slope decided by the rate-limiting step of the HER is an intrinsic property of electrocatalysts. For the most part, when the rate-limiting step of the HER is the Volmer process (reaction), the Tafel slope ought to be 120 mV dec−1, though the Heyrovsky or Tafel process is the rate-limiting step with a Tafel slope of 40 or 30 mV dec−1.42–45 The Tafel slope of the PdZn/C-loaded GC electrode was almost the same as that of the Pt/C-loaded GC electrode, suggesting that the rate-determining step of the HER on the bimetallic PdZn surface was the Tafel reaction, which was the same as that of the Pt/C-loaded GC electrode. Moreover, the Tafel slope of PdZn/TiO2−x NSs is close to the Tafel reaction; so the HER for PdZn/TiO2−x NSs proceeds through the Volmer–Tafel mechanism, in which the Tafel reaction is rate-determining.
The electrochemical stability of the catalyst is a significant factor for viable catalysts. Potentiostatic HER measurements were carried out to confirm the durability of the PdZn/TiO2−x NS catalyst during the HER in 0.5 M H2SO4 at −0.05 V vs. RHE for 45 h. As revealed in Fig. 3c, the PdZn/TiO2−x NS catalyst showed outstanding stability. The HV of the PdZn/TiO2−x NS catalyst was recorded before and after the potentiostatic HER measurement at −0.05 V vs. RHE for 45 h, and both voltammograms are quite similar to each other, as shown in Fig. S5 (ESI†), which also designates the superior durability of the catalyst.
Electrochemical impedance spectroscopy (EIS) was used to know more about the HER kinetics on the PdZn/TiO2−x NS catalyst. Fig. 3d show the Nyquist plots of PdZn/TiO2−x NSs at different cathodic overpotentials in an Ar saturated 0.5 M H2SO4 solution. Each Nyquist plot fitted into a simple Randle equivalent circuit. In this model, the x-intercept shows Rs, and this is assigned to the electric resistance of the solution and wires and the contact resistance between the GC electrode and the catalysts. Based on the semicircle at medium frequencies, the charge-transfer resistance Rct was determined. The smaller the Rct, the faster the HER kinetics. Zw represents the Warburg impedance. In Fig. 3d, the Rct of the PdZn/TiO2−x NSs reduced when the cathodic overpotential increased from 50 mV to 250 mV, which was confirmed by the decrease of the diameter of the semicircles. Fig. S6 (ESI†) shows the Nyquist plots of the PdZn/TiO2−x NS catalyst at −0.05 V vs. RHE before and after the potentiostatic HER measurement at −0.05 V vs. RHE for 45 h. In this figure, only a few changes were observed in the Nyquist plots of the PdZn/TiO2−x NSs before and after the long-term potentiostatic HER measurement. These results indicate that the bimetallic PdZn NPs had outstanding durability and the excellent catalytic activity in acidic media, providing new types of catalyst materials for the HER.
The CVs measured at 50 mV s−1 in Ar-saturated 0.5 M H2SO4 solution are shown in Fig. S7 (ESI†). The electrochemically active surface area (ECSA) of PdZn/TiO2−x NSs was calculated with the charge related to reduction peak of PdO in the potential range between 0.5 and 0.9 V shown in Fig. S7 (ESI†), and found to be 1.68 and 1.71 cm2 for PdZn/TiO2−x NSs and PdZn/C, respectively.
The HER activity of the PdZn/TiO2−x NS catalyst was also investigated in 1 M KOH solution (pH = 14) to know the catalytic performance of the material in a wide pH range (Fig. 4a). The overpotential at 10 mA cm−2 for the PdZn/TiO2−x NS catalyst was determined to be 64 mV, which was larger than that in 0.5 M H2SO4 solution (40 mV) and similar to that of Pt/C (74 mV), suggesting the amazing HER activity of the PdZn/TiO2−x NS catalyst even in alkaline medium. Fig. 4b shows the Tafel slopes of the PdZn/TiO2−x NSs and Pt/C catalysts, which were 68 and 66 mV dec−1, respectively, signifying that the PdZn/TiO2−x NS catalyst has a similar HER pathway and rate-determining step to Pt/C.
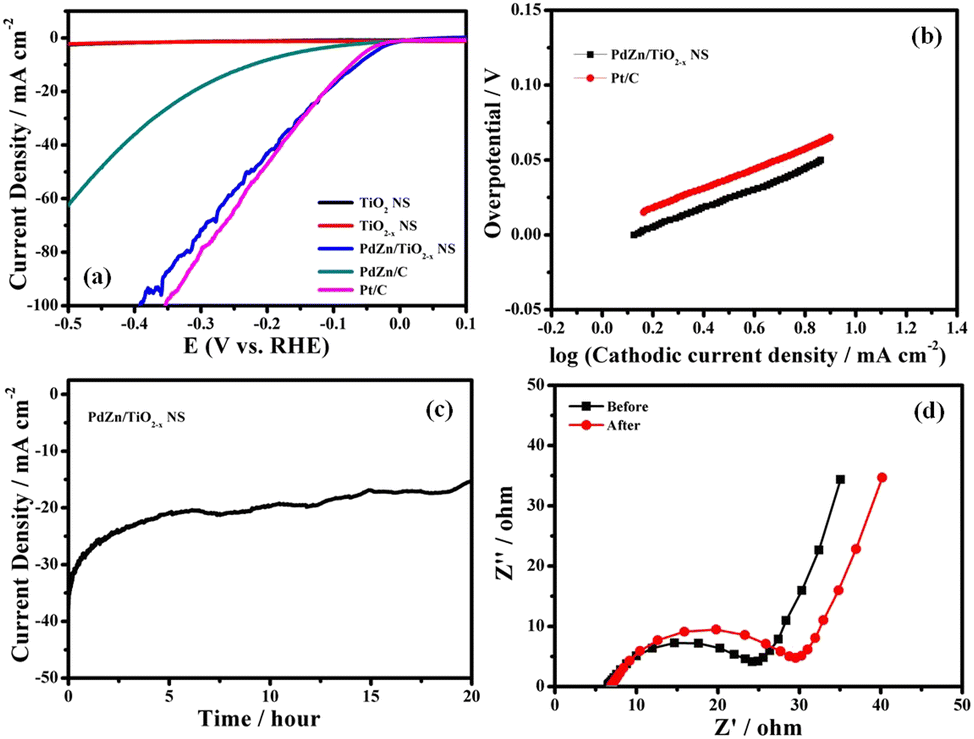 | ||
| Fig. 4 (a) Hydrodynamic voltammograms at 1600 rpm of PdZn/TiO2−x NSs, PdZn/C, TiO2−x NSs, TiO2 NSs and Pt/C, (b) Tafel plots of PdZn/TiO2−x NSs and Pt/C, (c) change in current density with time in the potentiostatic HER measurement at −0.1 V vs. RHE, and (d) Nyquist plots of PdZn/TiO2−x NSs at −0.1 V vs. RHE before and after the potentiostatic HER measurement at −0.1 V vs. RHE for 20 h. Electrolyte: Ar-saturated 1 M KOH solution. Temperature: 25 °C. | ||
The durable stability of the PdZn/TiO2−x NS catalyst with regard to the HER was evaluated by time course of current density in the potentiostatic HER measurement at −0.1 V vs. RHE for 20 h in 1 M KOH solution at 25 °C. As demonstrated in Fig. 4c, the PdZn/TiO2−x NS catalyst showed outstanding stability even in alkaline solution. The HVs at 1600 rpm of the PdZn/TiO2−x NS catalyst before and after the potentiostatic HER measurement at −0.1 V vs. RHE for 20 h are given in Fig. S5 (ESI†). In this figure, both voltammograms had limited changes, which too demonstrates the amazing durability of the PdZn/TiO2−x NS catalyst. Moreover, the Nyquist plots of PdZn/TiO2−x NSs before and after the durability measurement (Fig. 4d) exhibited that there was a small change in the Rct values, which also indicates high stability of the PdZn/TiO2−x NS catalyst in alkaline solution. The structure reduces the contact resistance between the nanoparticles while providing fast mass transfer channels to transport the reactants to each reaction site and discharge the products from the catalyst particles. Recent literature also supports the comparable reaction mechanisms.46,47 These results demonstrate higher HER activity and durability of the proposed catalyst in wide pH ranges.
Oxygen evolution activity
The OER electrocatalytic activity of the PdZn/TiO2−x NS catalyst was examined in an Ar-saturated 1 M KOH aqueous solution at 25 °C. Fig. 5a shows HVs at 1600 rpm for PdZn/TiO2−x NSs, PdZn/C, IrO2, TiO2−x NSs and TiO2 NSs. To obtain an anodic current density of 10 mA cm−2, the PdZn/TiO2−x NS catalyst required an overpotential of 460 mV. Furthermore, the PdZn/C and IrO2 showed overpotentials of 430 and 390 mV to reach a current density of 10 mA cm−2, respectively. The electrocatalytic kinetics for the OER with different catalysts was obtained using the corresponding Tafel plots (Fig. 5b), and the Tafel slopes for the PdZn/TiO2−x NS, PdZn/C and IrO2 catalysts were 115, 113 and 94 mV dec−1, respectively, suggesting that each catalyst can competently oxidize water. The OER performance of the PdZn/TiO2−x NS electrode is not the best in alkaline media, but it is acceptable to some of the bimetallic catalysts as well as many metal catalysts.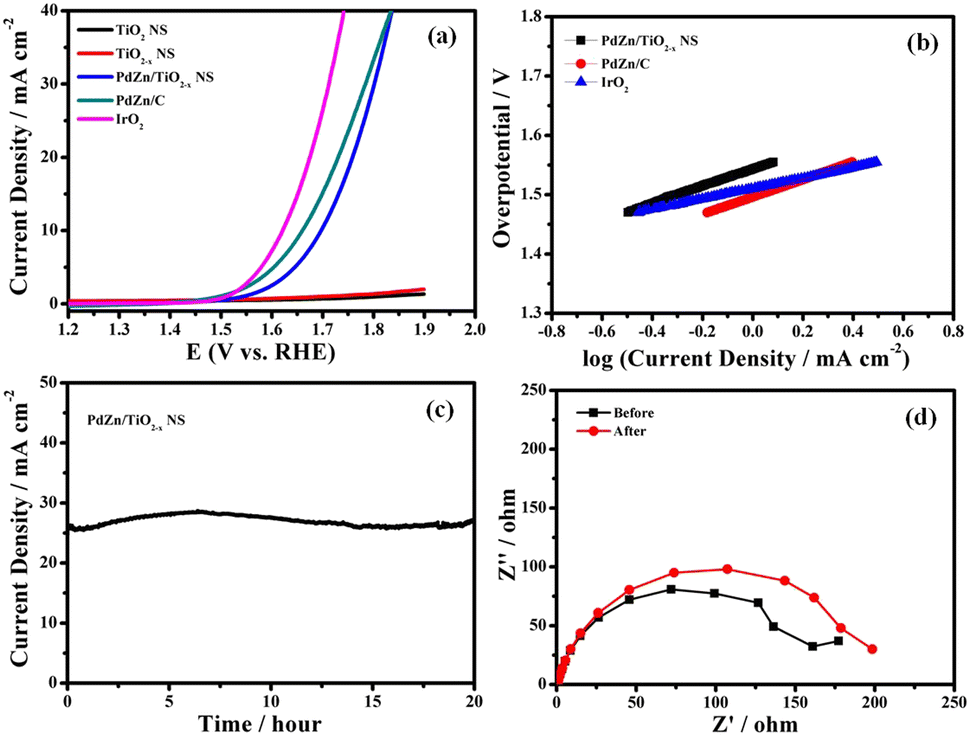 | ||
| Fig. 5 (a) Hydrodynamic voltammograms at 1600 rpm of PdZn/TiO2−x NSs, PdZn/C, TiO2−x NSs, TiO2 NSs and IrO2, (b) Tafel plots of PdZn/TiO2−x NSs, PdZn/C and IrO2, (c) change in current density with time in the potentiostatic OER measurement at 1.8 V vs. RHE, and (d) Nyquist plots of PdZn/TiO2−x NSs at 1.8 V vs. RHE before and after the potentiostatic OER measurement at 1.8 V vs. RHE for 20 h. Electrolyte: Ar-saturated 1 M KOH solution. Temperature: 25 °C. | ||
A plausible mechanism for electrocatalytic water splitting reaction on PdZn/TiO2−x NS in alkaline solutions can be represented by the following equations.48,49
| OH− → Pd–OHads + e− | (5) |
| Pd–OHads + OH− → Pd–O*ads + H2O + e− | (6) |
| Pd–O*ads + OH− → Pd–OOHads + e− | (7) |
| Pd–OOHads + OH− → O2 + H2O + e− | (8) |
In the first step (eqn (5)), hydroxide in the alkaline solution is oxidized at the Pd sites of PdZn NPs at an appropriate potential to adsorbed OH (OHads). Then, OHads is oxidized to an oxygen atom (O*ads) and a proton, which couples with OH− in the alkaline solution to form water (eqn (6)), and one-electron oxidation of O*ads and the following coupling with OH− results in the formation of a peroxide group (OOHads) at the Pd sites (eqn (7)). Finally, the oxygen molecule is formed by the oxidation of OOHads followed by the subsequent release of a proton (eqn (8)). The rate-determining step is the conversion from O*ads to OOHads, which can occur by the recombination of the adsorbed OH group over PdZn and O atoms on to the TiO2−x NS surface.
The long-term durability of the PdZn/TiO2−x NS catalyst was investigated using the potentiostatic technique. As shown in Fig. 5c, the anodic current density with time in a potentiostatic OER measurement at 1.8 V vs. RHE is so stable, and the change in the anodic current density is negligible over 20 h, demonstrating excellent OER durability. It is essential to note that the PdZn/TiO2−x NS catalyst is more stable in the OER than highly active OER catalysts reported previously,50,51 which may mainly originate from the robust contact between PdZn NPs and TiO2−x NSs, and good corrosion resistance of TiO2−x NSs. Nyquist plots of the PdZn/TiO2−x NS-loaded GC electrode before and after the potentiostatic OER measurement at 1.8 V vs. RHE for 20 h are the confirmation for a small change in the Rct value (Fig. 5d). The experimental results demonstrates the better OER performance and durability of the PdZn/TiO2−x NS catalyst. Thus, the excellent durability, along with the better OER activity of PdZn/TiO2−x NS in alkaline solution, will open up a new type of material for the electrochemical OER (Table S1, ESI†).
Overall water splitting
Based on the catalytic performance of the PdZn/TiO2−x NSs for the OER and HER in 1 M KOH, it can be expected that the catalyst is applied as an excellent electrocatalyst for overall water splitting as a bifunctional electrode catalyst. The LSV curves for two-electrode cells with different catalysts were recorded with a scan rate of 5 mV s−1 from 1.0 to 2.0 V in an Ar-saturated 1 M KOH electrolyte at 25 °C, and summarized in Fig. 6a. The cell with a commercial Pt/C cathode and an IrO2 anode (Pt/C//IrO2) and the cell with the PdZn/C cathode and PdZn/C anode (PdZn/C//PdZn/C) were also tested, and compared with the cell with the PdZn/TiO2−x NS cathode and PdZn/TiO2−x NS anode (PdZn/TiO2−x NS//PdZn/TiO2−x NS). The PdZn/TiO2−x NS//PdZn/TiO2−x NS cell is capable of giving an anodic current density of 10 mA cm−2 with an overpotential of 1.74 V in 1 M KOH solution at 25 °C during the overall water splitting process, which is comparable to that of the several reported bifunctional catalysts.52,53 The difference in the cell voltage between the PdZn/TiO2−x NS//PdZn/TiO2−x NS cell and Pt/C//IrO2 cell at 10 mA cm−2 are 1.74 V to 1.67 V. Fig. 6b clearly exhibits that the PdZn/TiO2−x NS//PdZn/TiO2−x NS maintained a current density of around 30 mA cm−2 over 35 h with a constant voltage of 2.0 V, clearly indicating that the PdZn/TiO2−x NS catalyst had high durability for long-term electrochemical water splitting.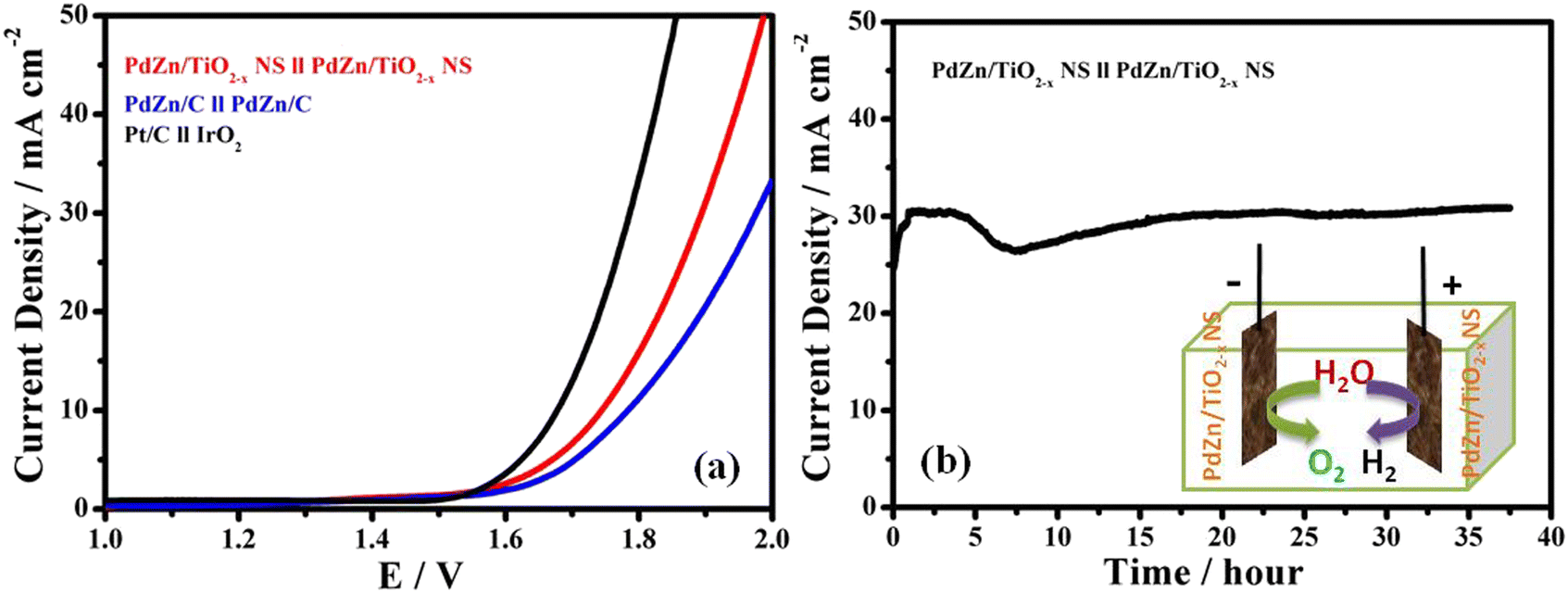 | ||
| Fig. 6 (a) Polarization curves of the PdZn/TiO2−x NS//PdZn/TiO2−x NS, PdZn/C//PdZn/C, and Pt/C//IrO2 cells, and (b) time course of anodic current density for the PdZn/TiO2−x NS//PdZn/TiO2−x NS cell in the long-term electrochemical water splitting at 2.0 V in Ar-saturated 1 M KOH solution at 25 °C. | ||
According to the abovementioned experimental results, the following factors may be responsible for the good catalytic activity and stability of the bifunctional PdZn/TiO2−x NSs catalyst: (i) The electrochemically active surface area of the nanosheets are more readily accessible, which is advantageous for mass transfer, exposing active sites, and promoting gas release. (ii) based on the high electrical conductivity and strong metal support interaction in the catalyst, electrons can pass through the substrate quickly to dissociate H2O and OH− adsorbed on the nanoparticles to nanosheets and simultaneously produce H2 and O2 and (iii) The SMSI effect generates a channel that is advantageous for electron conversion throughout the entire structure between the PdZn NP and TiO2−x NSs support.
Conclusions
In summary, TiO2−x NS was used as an alternative support for an electrocatalyst in the water splitting reaction. An easy methodology has been established for the preparation of the layered TiO2−x NS supported on PdZn by an impregnation process including low temperature annealing. The two-dimensional (2D) TiO2−x NS supported PdZn exhibited greater HER catalytic performance delivering a current density of 10 mA cm−2 at an overpotential of 64 mV in 1 M KOH and a Tafel slope of 68 mV dec−1. During the OER process, the same PdZn/TiO2−x NS catalyst showed acceptable electrocatalytic activity with an overpotential of 1.69 V to obtain a current density of 10 mA cm−2 with a Tafel slope of 115 mV dec−1. The chronoamperometric experiments revealed the high durability of the catalyst in alkaline solution for more than 20 h without any changes in its activity in both OER and HER processes. The PdZn/TiO2−x NSs catalyst seems to handle a supplied potential of 1.74 V at a current density of 10 mA cm−2 and offer durable steadiness for overall water splitting in alkaline solution as the electrolyte. In comparison to the IrO2 or Pt catalyst, PdZn/TiO2−x NSs are less expensive, durable and corrosion resistant in alkaline solution. This new work favours an opening for encouraging investigation to reasonably develop more carbon free supported bimetallic nanostructures utilizing individual transition metals for the efficient electrochemical water splitting process.Author contributions
K. N. and H. I. designed the research; K. N and K. H. performed the preparation of catalysts; K. N., K. H., and E. H. preformed and analyzed spectroscopic and electrochemical experiments; H. I. supervised the work; K. N. and H. I. wrote the paper; all authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We acknowledge the financial support from the (Grants in Aid for scientific research (KAKENHI)) Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT) under the ‘Multifunctional durable nanostructured redox electrocatalysts on conducting titanium oxide support’ project (Project Number 18F18372). Keerti M. Naik is grateful to the Japan Society for the Promotion of Sciences (JSPS) Standard Postdoctoral Fellowship.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 Search PubMed.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- L. Yan, L. Cao, P. Dai, X. Gu, D. Liu, L. Li, Y. Wang and X. Zhao, Adv. Funct. Mater., 2017, 27, 1703455 CrossRef.
- X. Li, X. Hao, A. Abudula and G. Guan, J. Mater. Chem. A, 2016, 4, 11973–12000 RSC.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 Search PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- B. Klahr, S. Gimenez, F. F. Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294–4302 CrossRef CAS PubMed.
- Y. Tan, H. Wang, P. Liu, Y. Shen, C. Cheng, A. Hirata, T. Fujita, Z. Tang and M. Chen, Energy Environ. Sci., 2016, 9, 2257–2261 RSC.
- X. Zhao, P. Pachfule, S. Li, J. R. J. Simke, J. Schmidt and A. Thomas, Angew. Chem., Int. Ed., 2018, 57, 8921–8926 Search PubMed.
- L. An, L. Huang, P. Zhou, J. Yin, H. Liu and P. Xi, Adv. Funct. Mater., 2015, 25, 6814–6822 CrossRef CAS.
- X. Ge, Y. Liu, F. W. T. Goh, T. S. A. Hor, Y. Zong, P. Xiao, Z. Zhang, S. H. Lim, B. Li, X. Wang and Z. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 12684–12691 CrossRef CAS PubMed.
- E. Oakton, D. Lebedev, M. Povia, D. F. Abbott, E. Fabbri, A. Fedorov, M. Nachtegaal, C. Coperet and T. J. Schmidt, ACS Catal., 2017, 7, 2346–2352 CrossRef CAS.
- C. Xiao, Y. Li, X. Lu and C. Zhao, Adv. Funct. Mater., 2016, 26, 3515–3523 CrossRef CAS.
- S. Sharma and B. G. Pollet, J. Power Sources, 2012, 208, 96–119 CrossRef CAS.
- K. M. Naik, E. Higuchi and H. Inoue, Nanoscale, 2020, 12, 11055–11062 Search PubMed.
- W. Yuan, J. Li, L. Wang, P. Chen, A. Xie and Y. Shen, ACS Appl. Mater. Interfaces, 2014, 6, 21978–21985 CrossRef CAS PubMed.
- Z. Lian, W. Wang, G. Li, F. Tian, K. S. Schanze and H. Li, ACS Appl. Mater. Interfaces, 2017, 9, 16959–16966 Search PubMed.
- A. Naldoni, M. Altomare, G. Zoppellaro, N. Liu, S. Kment, R. Zbořil and P. Schmuki, ACS Catal., 2018, 9, 345–364 CrossRef PubMed.
- K. M. Naik, T. Hamada, E. Higuchi and H. Inoue, ACS Appl. Energy Mater., 2021, 4, 12391–12402 CrossRef CAS.
- K. M. Naik, E. Higuchi and H. Inoue, J. Power Sources, 2020, 455, 227972 Search PubMed.
- H. Liu, C. Koenigsmann, R. R. Adzic and S. S. Wong, ACS Catal., 2014, 4, 2544–2555 CrossRef CAS.
- Y. Z. Lu, Y. Y. Jiang, X. H. Gao, X. D. Wang and W. Chen, J. Am. Chem. Soc., 2014, 136, 11687–11697 CrossRef CAS PubMed.
- A. J. Medford, A. Vojvodic, J. S. Hummelshoj, J. Voss, F. A. Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Norskov, J. Catal., 2015, 328, 36–42 Search PubMed.
- G. Bampos, S. Bebelis, D. I. Kondarides and X. Verykios, Top. Catal., 2017, 60, 1260–1273 Search PubMed.
- Y. Yan, J. S. Du, K. D. Gilroy, D. Yang, Y. Xia and H. Zhang, Adv. Mater., 2017, 29, 1605997 Search PubMed.
- A. P. Tsai, S. Kameoka, K. Nozawa, M. Shimoda and Y. Ishii, Acc. Chem. Res., 2017, 50, 2879–2885 Search PubMed.
- X. Li, G. Liu, D. Xu, X. Hong and S. C. E. Tsang, J. Mater. Chem. A, 2019, 7, 23878–23885 RSC.
- Q. Sun, S. Wang and H. Liu, ACS Catal., 2017, 7, 4265–4275 CrossRef CAS.
- K. M. Naik, K. Hashisake, T. Hamada, E. Higuchi and H. Inoue, J. Mater. Chem. A, 2022, 10, 13987–13997 RSC.
- Z. X. Chen, K. M. Neymann, A. B. Gordienko and N. Rösch, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 075417 Search PubMed.
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 Search PubMed.
- Z. Wang, C. Yang, T. Lin, H. Yin, P. Chen, D. Wan, F. Xu, F. Huang, J. Lin, X. Xie and M. Jiang, Energy Environ. Sci., 2013, 6, 3007–3014 RSC.
- M. Imran, A. B. Yousaf, X. Zhou, Y. F. Jiang, C. Z. Yuan, A. Zeb, N. Jiang and A. W. Xu, J. Phys. Chem. C, 2017, 12, 1162–1170 Search PubMed.
- M. Wakisaka, S. Mitsui, Y. Hirose, K. Kawashima, H. Uchida and M. Watanabe, J. Phys. Chem. B, 2006, 110, 23489–23496 CrossRef CAS PubMed.
- H. He, J. Chen, D. Zhang, F. Li, X. Chen, Y. Chen, L. Bian, Q. Wang, P. Duan, Z. Wen and X. Lv, ACS Catal., 2018, 8, 6617–6626 Search PubMed.
- X. Wang, Y. F. Jiang, Y. N. Liu and A. W. Xu, New J. Chem., 2018, 42, 19901–19907 RSC.
- Y. Cheng, S. Lu, W. Xu, H. Wen and J. Wang, J. Mater. Chem. A, 2015, 3, 16774–16784 RSC.
- T. T. V. Doan, J. Wang, K. C. Poon, D. C. L. Tan, B. Khezri, R. D. Webster, H. Su and H. Sato, Angew. Chem., Int. Ed., 2016, 55, 6842–6847 CrossRef CAS PubMed.
- G. Wang, J. Guan, L. Xiao, B. Huang, N. Wu, J. Lu and L. Zhuang, Nano Energy, 2016, 29, 268–274 CrossRef CAS.
- K. M. Naik and S. Sampath, Electrochim. Acta, 2018, 292, 268–275 CrossRef CAS.
- Z. Chen, D. Cummins, B. N. Reinecke, E. Clark, M. K. Sunkara and T. F. Jaramillo, Nano Lett., 2011, 11, 4168–4175 Search PubMed.
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881–17888 CrossRef CAS PubMed.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- T. R. Hellstern, J. Kibsgaard, C. Tsai, D. W. Palm, L. A. King, F. A. Pedersen and T. F. Jaramillo, ACS Catal., 2017, 7, 7126–7130 CrossRef CAS.
- J. Yang, G. Ning, L. Yu, Y. Wang, C. Luan, A. Fan, X. Zhang, Y. Liu, Y. Dong and X. Dai, J. Mater. Chem. A, 2019, 7, 17790–17796 Search PubMed.
- X. Guo, X. Wan, Q. Liu, Y. Li, W. Li and J. Shui, eScience, 2022, 2, 304–310 CrossRef.
- Z. Li, W. Wang, Q. Qian, Y. Zhu, Y. Feng, Y. Zhang, H. Zhang, M. Cheng and G. Zhang, eScience, 2022, 2, 416–427 Search PubMed.
- B. M. Szyja and R. A. V. Santen, Phys. Chem. Chem. Phys., 2015, 17, 12486–12491 RSC.
- I. C. Man, H. Y. Su, F. C. Vallejo, A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- S. Duan, S. Chen, T. Wang, S. Li, J. Liu, J. Liang, H. Xie, J. Han, S. Jiao, R. Cao, H. L. Wang and Q. Li, Nanoscale, 2019, 11, 17376–17383 RSC.
- X. Cao, L. Wang, G. Ding, R. Yu and B. Geng, Nanoscale, 2020, 12, 19404–19412 RSC.
- X. Yan, K. Li, L. Lyu, F. Song, J. He, D. Niu, L. Liu, X. Hu and X. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 3208–3214 CrossRef CAS PubMed.
- Z. Peng, D. Jia, A. M. Al-Enizi, A. A. Elzatahry and G. Zheng, Adv. Energy Mater., 2015, 5, 1402031 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00904h |
| This journal is © The Royal Society of Chemistry 2023 |